

Abstract
N-acetylcysteine (NAC) is being investigated as an antioxidant for several conditions including traumatic brain injury, but the mechanism by which it crosses membrane barriers is unknown. We sought to understand how the transporter inhibitor, probenecid, affects NAC pharmacokinetics and to evaluate the interaction of NAC with transporters.
Juvenile Sprague-Dawley rats were administered NAC alone or in combination with probenecid intraperitoneally. Plasma and brain samples were collected serially and NAC concentrations were measured. Transporter studies were conducted with human embryonic kidney-293 cells that overexpress organic anion transporter (OAT)1 or OAT3 and with human multi-drug resistance-associated protein (MRP)1 or MRP4 membrane vesicles.
NAC area under the curve was increased in plasma (1.65-fold) and brain (2.41-fold) by probenecid. The apparent plasma clearance was decreased by 65%. Time- and concentration-dependent NAC uptake that was inhibitable by probenecid was observed with OAT1 and OAT3. No uptake of NAC was observed with MRP1 or MRP4.
Our results indicate for the first time that NAC is substrate for OAT1 and OAT3 and that probenecid increases NAC plasma and brain exposure in vivo. These data provide insight regarding how NAC crosses biological barriers and suggest a promising therapeutic strategy to increase NAC exposure.
Keywords: transporter, pharmacokinetics, antioxidant, inhibitor, organic anion tranporter, n-acetylcysteine, probenecid
Introduction
N-acetylcysteine (NAC) is a thiol-containing, acetylated derivative of cysteine that has been in clinical use for over 50 years (Rushworth and Megson, 2014, Samuni et al., 2013, Cotgreave, 1997). It acts as an antioxidant by directly scavenging reactive oxygen and nitrogen radicals via its thiol group, as well as by acting as a cysteine donor in the biosynthesis of glutathione, the most abundant intracellular antioxidant (Samuni et al., 2013, Atkuri et al., 2007, Dodd et al., 2008). NAC is FDA-approved for prevention of liver toxicity due to acetaminophen/paracetamol overdose and as a mucolytic for patients such as those with cystic fibrosis or chronic bronchitis (Grandjean et al., 2000, Prescott et al., 1979, Rushworth and Megson, 2014, Hurst et al., 1967). It has also been investigated as a cytoprotective agent in several conditions including traumatic brain injury (Hoffer et al., 2013), contrast-induced nephropathy, cancer chemotherapy, cardiovascular disease, diabetes, human immunodeficiency virus infection, neuropsychiatric disorders, and heavy metal toxicity (reviewed here, (Cotgreave, 1997, Dodd et al., 2008, Rushworth and Megson, 2014). NAC is a hydrophilic molecule (log D = −5.4; (Giustarini et al., 2012)) that is approximately 30% renally eliminated (Borgstrom et al., 1986). Despite its extensive clinical and research use, an understanding of how NAC crosses certain biological barriers and its interactions with membrane transporters remains incomplete. Additionally, controversy exists regarding the extent to which NAC can enter the central nervous system (CNS) through the blood-brain barrier (BBB) (Samuni et al., 2013).
Our group is investigating the use of NAC in combination with probenecid, the prototypical organic anion transporter inhibitor, in the treatment of pediatric traumatic brain injury (Margulies et al., 2016). We proposed that a potentially synergistic interaction between NAC and probenecid exists to enhance antioxidant capacity of the brain after injury. While NAC acts as donor of cysteine, the rate limiting substrate in biosynthesis of glutathione, probenecid has been shown to prevent active efflux of glutathione and its conjugates by inhibiting MRP transporters (Versantvoort et al., 1995). Probenecid is known to increase the plasma and CNS exposure of a number of drugs such as acyclovir, zidovudine and bumetanide by inhibiting transport systems in renal tubules, blood brain barrier (BBB), and/or blood-cerebrospinal fluid (CSF) barrier (Laskin et al., 1982, Wong et al., 1993, Sawchuk and Hedaya, 1990, Donovan et al., 2015). The objectives of the current study were to evaluate the effect of probenecid on the brain and plasma exposure of NAC, and to assess the mechanism through which their interaction may be mediated.
Materials and Methods
Animals
All studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Healthy, male Sprague-Dawley rats (n=48), were purchased from Harlan Laboratories (Indianapolis, Indiana, USA) and acclimated for one week prior to study initiation when rats were 17–18 days old. The animals were allowed free access to food and water and were housed on a 12 h light/dark cycle.
Drug Administration and Pharmacokinetic Sampling
Rats were administered a single dose of either 163 mg/kg of NAC (Sigma-Aldrich, St. Louis, MO, USA) alone or in combination with 150 mg/kg of probenecid (Sigma-Aldrich, St. Louis, MO, USA) via intraperitoneal injection. Both drugs were prepared by dissolution in NaOH and HCl was added to adjust the pH to ~7.4. The administration volume was 168μL to 258μL for NAC, and 84μL to 122.5μL for probenecid or vehicle control depending on the animal weight. Blood was collected at 0.5, 1, 2, 4, 6 and 8 h after injection by cardiac puncture into heparinized tubes, centrifuged immediately to isolate plasma, and frozen at −80°C for drug analysis. Similarly, brain tissue was harvested following decapitation at each time point and brain hemispheres were isolated carefully to avoid contamination with blood. Hemispheres were then homogenized by sonication in brain homogenization buffer (containing 137mM of NaCl, 2.7mM of KCl, 10mM of Na2HPO4, 1.8mM KH2PO4 and 1mM of EDTA dissolved in H2O and adjusted to pH of 7.4) and an aliquot of the homogenate was used for LC-MS/MS analysis. Four rats were studied per time point in each group. Concentration-time data is reported as mean ± SEM.
Drug Analysis by LC-MS/MS
Total NAC concentrations were quantified in plasma and brain tissue using ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Deuterated internal standard (d3-NAC, Cambridge Isotopes (Andover, MA) was added to plasma (50 μL) or brain homogenate (50 μL) and samples were reduced by adding dithiothreitol, derivatized with n-ethylmaleimide (NEM), and proteins were precipitated with acetonitrile. The supernatant was dried out under nitrogen and reconstituted with water before injecting onto the UPLC-MS/MS system. Chromatographic separation was achieved using a porous graphitic carbon hypercarb column 3.0 μm (1.0 X 100 mm) and an acetonitrile-formic acid gradient (A: 0.5% formic acid in water and B: 0.1% formic acid in acetonitrile; 95% A and 5% B for 1 min followed by a linear gradient to 30% B from 1–3 min, a step to 85% B from 3–5 min, then column re-equilibration to initial conditions for 3 min for a total 8 min run time) on an Accela series UPLC system (Thermo, San Jose, CA). MS/MS detection of NAC-NEM (m/z 433.1→304.2) was performed on a Thermo TSQ Quantum Ultra mass spectrometer with a heated electrospray source (Thermo, San Jose, CA). Calibration curves were linear from 100–10,000 ng/mL (r2 > 0.995). Inter-day accuracy (% bias) and precision (RSD) determined at the concentrations of 300, 2000, and 7000 ng/mL ranged from −1.11% to 10.5% and 1.88% to 7.63%, respectively. All samples were diluted as necessary to be within the linear range of the assay.
Pharmacokinetic Analysis
Pharmacokinetic parameters were estimated using standard noncompartmental methods (WinNonlin Phoenix, Certara, St. Louis, MO). Area under the curve (AUC) profiles for plasma and brain concentrations were calculated using the trapezoidal rule. The AUC from the last measured time point to infinity (AUC0-inf) was estimated by dividing the last measured concentration by the elimination rate constant. Apparent plasma clearance was calculated by dividing the dose given by AUC0-inf.
Cell Uptake Studies
Human embryonic kidney cell lines stably transfected with human OAT3 (HEK-OAT3), human OAT1 (HEK-OAT1), and the corresponding HEK-empty vector (HEK-EV, control cells) were kindly provided by Professor Kathleen Giacomini (University of California, San Francisco, CA, USA). Cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) high glucose (4.5g/L) supplemented with L-glutamine (Life Technologies, Long Island, NY, USA), 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), Hygromycin-B (100 μg/mL, Corning, Corning, NY) and penicillin-streptomycin (100 μg/mL, Mediatech Inc., Manassas, VA) at 37°C in a humidified atmosphere with 5% CO2. Cells were plated in 6-well, poly-D-lysine coated tissue culture plates (Corning, Corning, NY, USA) 48–72 hours before the uptake study. On the day of the experiment, medium was removed and cells were washed twice with warm Dulbecco’s Phosphate Buffered Saline containing calcium and magnesium (DPBS, Mediatech Inc., Manassas, VA, USA). Cells were then incubated in a DPBS solution containing 5mM of the transporter activator glutaric acid (Acros Organics, Geel, Belgium) for 30 minutes at 37°C. This solution was then replaced with incubation buffer (containing 128mM NaCl, 4.73mM KCl, 1.25mM CaCl2, 1.25mM MgSO4 and 5mM HEPES in H2O and adjusted to pH 7.4) for 10 minutes as previously described (Erdman et al., 2006). Uptake was initiated by adding 14C–Acetyl-L-Cysteine (14C-NAC; specific activity 0.0581 Ci/mmol or 0.054 Ci/mmol; Moravek Biochemicals, Brea, California) and unlabeled NAC (Sigma-Aldrich, St. Louis, MO, USA) followed by incubation at 37°C for the indicated period of time. Uptake studies were terminated by removing the incubation solution and washing cells with ice-cold DPBS three times. Cells were then lysed with a solution of 0.1N NaOH containing 0.1% sodium dodecyl sulfate. Lysate radioactivity was then measured using a LS 6500 Scintillation System (Beckman Coulter, Brea, CA). Protein concentration was also assayed using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) per the manufacturer’s protocol. The resultant uptake rates were normalized for protein content to account for differences in cell number between plates. Inhibition experiments were performed as described above except that cells were pre-incubated with 200 μM of probenecid in incubation media for 5 minutes. For the concentration-dependent uptake study, varying amounts of unlabeled NAC were added to the loading buffer containing a fixed amount of radiolabeled NAC to give total NAC concentrations ranging from 1 μM to 10 mM. All experiments were conducted in triplicate and means ± SD were calculated. Uptake activity due to OAT1 and OAT3 was calculated by subtracting NAC uptake in HEK-EV cells from that of HEK-OAT1 and HEK-OAT3 at each of the corresponding concentrations. Michaelis-Menten kinetic analysis of the uptake data was conducted using Prism 5.04 (Graphpad Software, La Jolla, CA). Data were first plotted and then the best-fit curve was fit using the following equation: V = Vmax · [S]/(Km + [S]), where V is the rate of uptake activity, Vmax is the maximum rate of uptake activity, [S] is the substrate concentration and Km is the concentration of the substrate where rate of uptake activity is half of the maximum.
Vesicular Uptake Assay
Human MRP1 and MRP4 membrane vesicles, negative control vesicles, and ABC Transport Assay Reagent Kit were purchased from Life Technologies (Carlsbad, CA, USA). A positive control substrate, 3H-estradiol-17β-D- glucuronide (E217βG; 41.4 Ci/mmol) was obtained from Perkin Elmer, Inc. (Boston, MA, USA). MultiScreenHTS 96-well glass fiber filter plates were purchased from EMD-Millipore Corporation (Billerica, MA, USA). ATP-dependent and independent uptake was measured using either 50 μg of control, MRP1, or MRP4 baculovirus-transfected Sf9 insect cells. Assays were performed at 37°C in a final volume of 50 μl in a reaction mixture containing either 4 mM ATP or AMP, 2 mM glutathione, 10 μM cold E217βG, 200 nM of 3H-E217βG (for MRP1) or 100 nM of 3H-E217βG (for MRP4). The reaction was initiated by adding vesicles to reaction mix, incubating for 0.5, 1, 2.5, or 5 minutes, and stopped by adding 200 μl ice-cold buffer. Samples were added to glass fiber filter plates and rapid filtration was performed using a Bio-Rad vacuum filtration unit. Wells were washed 5 times, filters collected, and counted using the liquid scintillation counter. Additional experiments were performed using varying concentrations of the known MRP1/MRP4 substrate E217βG (0.25–100 μM) for 0.5 minutes as described above. Radioactivity counts in wells without membrane vesicles were subtracted to account for any 3H-E217βG bound to the filter. ATP-dependent uptake was determined by subtracting uptake in presence of AMP from uptake in the presence of ATP at each concentration. Michaelis-Menten kinetic analysis was conducted using as performed in the cellular uptake experiments. NAC assays were similarly performed using MRP1 or MRP4 vesicles with ATP or AMP, using 15 μM cold NAC and 10 μM of radiolabeled NAC for a final concentration of 25 μM. ATP- or AMP-dependent NAC uptake rate was compared an F-test to determine if there was an ATP-dependent, transporter specific uptake of NAC.
Statistical Analysis
Between groups comparisons were made using unpaired Student’s t tests. p < 0.05 was considered significant. All analyses were conducted using Graphpad Prism 5.04 (Graphpad Software, La Jolla, CA).
Results
Probenecid increased NAC plasma and brain exposure and decreased apparent plasma clearance
The impact of probenecid co-administration on NAC plasma and brain levels was first investigated. NAC plasma concentrations were increased significantly in the presence of probenecid starting at 2 h after administration compared to when NAC was given alone (Figure 1A). Moreover, the NAC AUC was increased by 1.65 fold by probenecid co-administration (Figure 1B). Probenecid reduced apparent plasma clearance of NAC by 65% (CL/F: 1113±29 vs 674±8.2 mL/h/kg; p < 0.001).
Figure 1.

NAC plasma concentration-time profile and total plasma exposure (AUC) with or without co-administration of probenecid. Sprague-Dawley rats were administered a dose of either NAC (163 mg/kg) alone or in combination with probenecid (150 mg/kg) intraperitoneally and serial NAC plasma concentrations were measured. (A) Probenecid increased NAC plasma concentration-time profile after 2h (mean±SEM, n=4 per time point; *p<0.05). (B) Probenecid increased NAC plasma AUC0–inf by 1.65-fold (mean±SEM; p<0.01).
Similarly, NAC brain concentration-time profile was significantly increased by probenecid (Figure 2A). Differences were observed starting from 1 h up to the 4 h time point. While NAC levels remained measurable until the last sampling time point (8 h) in the group that received both NAC and probenecid, NAC levels were below the limit of quantificaiton at the 6 h and 8 h time points in the group that received NAC only. Figure 2B shows that NAC total brain exposure was signifcantly increased by 2.46 fold by probenecid co-administration. Interestingly, the proportional increase in NAC total brain exposure with co-administration of probenecid was higher than that observed in plasma (2.46 vs 1.65 fold increase, respectively).
Figure 2.

NAC brain concentration–time profile and total brain exposure (AUC) of NAC, with or without co-administration of probenecid. Sprague-Dawley rats were administered a dose of either NAC (163 mg/kg) alone or in combination with probenecid (150 mg/kg) intraperitoneally and brain hemispheres were collected at several time points for NAC analysis. (A) Probenecid increased NAC brain concentrations starting from 1 h up to the 4 h time point. NAC levels were measurable until the last sampling time (8 h) in the group that received both NAC and probenecid, but were below limit of quantification at 6 h and 8 h in the group that received NAC only (mean±SEM, n=4 per time point, *p<0.05). (B) Probenecid increased brain AUC0–inf of NAC 2.46-fold (mean±SEM; p<0.01).
NAC uptake by OAT3 and OAT1
To understand the mechanism of interaction between NAC and probenecid, we examined the time- and concentration-dependent uptake of NAC by transporters known to be inhibited by probenecid. OAT3- and OAT1-mediated transport activities were evaluated using cell lines that stably express those transporters and their negative controls (cell lines stably transfected with the empty vector). Figure 3 shows that the uptake of NAC by OAT3- and OAT1-overexpressing cells is linear up to at least 15 and 42.5 (latest time point measured) minutes, respectively. At 25 μM NAC, update rates were 37.7±1.9 and 4.2±0.1 pmol/mg/min respectively. Subsequent concentration dependent uptake studies were then examined within the established linear range, 25 minutes for OAT1 and 10 minutes for OAT3. NAC uptake by either of the transporter overexpressing cell lines was concentration-dependent, but did not reach saturation even at very high substrate concentration of 10 mM (Figure 4). Figure 5 shows that probenecid significantly decreased NAC uptake by both OAT1 (59% decrease, n = 3; p < 0.01) and OAT3 (93.9% decrease, n = 3; p < 0.001). There was no difference in NAC uptake by control cells in the presence or absence of probenecid (n = 3; N.S.).
Figure 3.

Time-dependent Uptake of NAC by OAT1- and OAT3-overexpressing cells. HEK-OAT1 (A), HEK-OAT3 (B) and HEK-EV (C) cells were incubated in transport buffer containing 25 mM of NAC traced with 14C-acetyl-L-cysteine at 37 °C for designated incubation times. Each value represents the mean ±SD of three determinations.
Figure 4.

Concentration-dependent NAC uptake by OAT1- and OAT3-overexpressing cells. HEK-OAT3 (A) and HEK-OAT1 (B) cells were incubated in transfer buffer for 10 min (HEK-OAT3), 25 min (HEK-OAT1) with increasing concentrations of NAC traced with 14C-acetyl-L-cysteine. Nonspecific uptake was accounted for by subtracting uptake activity in HEK-EV cells from that of HEK-OAT1 or HEK-OAT3. The rate of uptake of NAC did not reach saturation even at high substrate concentrations. Each value represents the mean ±SD of three determinations.
Figure 5.
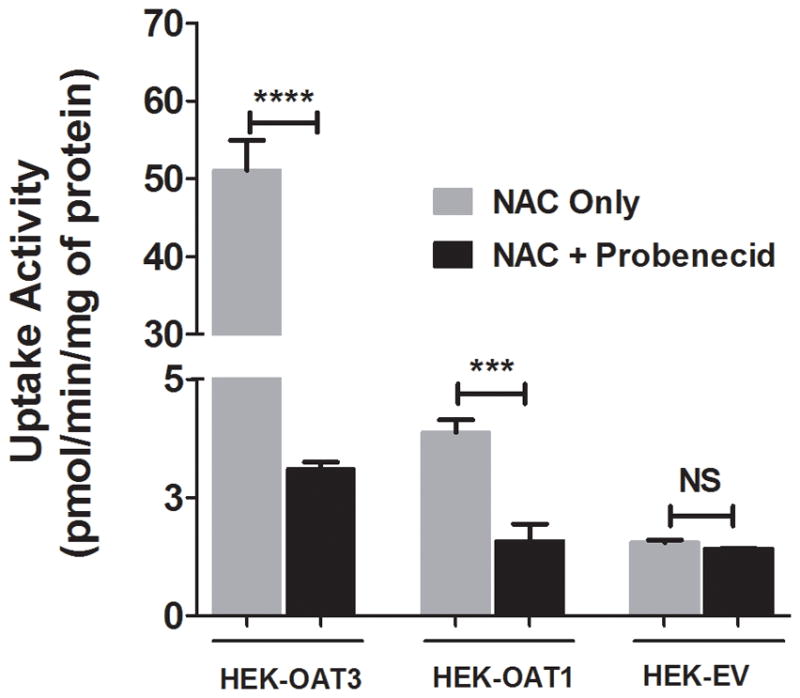
Inhibition of OAT1- and OAT3-mediated NAC uptake by probenecid. Uptake of 25 mM of NAC traced with 14C-acetyl-L-cysteine was evaluated in human OAT1-, OAT3- and EV-HEK291 cells with or without 200 mM of probenecid for 10 min (HEK-OAT3), 25 minutes (HEK-OAT1) and 25 minutes (HEK-EV). NAC uptake activity was reduced in HEK-OAT1 and HEK-OAT3, but not HEK-EV cells following pre-incubation with probenecid. Each value represents the mean ±SD of three determinations. **p<0.01. ****p<0.001.
To evaluate whether NAC is a substrate for probenecid-sensitive efflux transporters that are expressed in the renal proximal tubules and in the brain, NAC uptake in MRP1- and MRP4-overexpressing membrane vesicles was evaluated. However, ATP-dependent, transporter-mediated uptake rate of NAC by either of the transporters was not statistically different from that of AMP-dependent (i.e., negative control) (Figure 6 C and D). This is despite observing saturable uptake of E217βG, a well established substrate for both transporters, as a positive control (Figure 6 A and B).
Figure 6.

Effect of MRP1 or MRP4 on NAC uptake in membrane vesicles. ATP-dependent uptake of E217βG by membrane vesicles expressing MRP1 (A) or MRP4 (B) was determined by substracting uptake (30 s at 37 °C) in the presence of AMP from that in the presence of ATP. Time-course of 25 mM NAC (15 mM cold+10 mM 14C-NAC) uptake was not significantly different in presence of AMP or ATP by either MRP1 (C) and MRP4 (D) membrane vesicles. Each value represents the mean ±SD of at least two determinations.
Discussion
We report the novel finding that probenecid increases NAC plasma and brain exposure in vivo (Figures 1 and 2), which suggests that NAC is a likely putative transporter susbstrate. Further, we demostrated that NAC is an OAT1 and OAT3 (Figures 3, 4 and 5), but not an MRP1 and MRP4 (Figure 6), substrate. NAC has been touted as a potential therapeutic agent for a number of ailments, including CNS injuries, but the process by which this hydrophilic molecule (log D = −5.4; (Giustarini et al., 2012)) crosses certain biological barriers is poorly understood.
NAC plasma exposure was significantly increased and its apparent clearance was signficantly decreased by probenecid co-administration in the rat(Figure 1). Probenecid is a well-established organic anion transport inhibitor and has been used both clinically and preclinically (in rats) to determine the role of OAT1 and OAT3 in the clearance of renally-excreted drugs. Human and rat forms of both OAT3 and OAT1 show overlapping substrate specificity and comparable level of activity (Minematsu et al., 2008, Kikuchi et al., 2003). This is suggestive that these transporters mediate the uptake of NAC into the renal tubule for eventual secretion into the urine. In order to determine if NAC is substrate for OAT1 and/or OAT3, we conducted an in vitro uptake study of NAC with HEK-293 cell lines that overexpress human OAT1 or OAT3 transporters.
In vitro uptake and inhibition experiments confirm that NAC is a substrate for OAT1 and OAT3(Figures 3, 4, and 5). However, OAT1 and OAT3 concentration-dependent uptake did not reach saturation even after incubation with 10 mM of NAC (Figure 5). One possible explanation for the apparent lack of saturability is that NAC may exist in various chemical forms following admininistration. Once NAC enters the systemic circulation, it exists in a reduced form and in various oxidized forms that are products of redox reactions between NAC, cysteine, glutathione and proteins (Olsson et al., 1988, Harada et al., 2001). Consistent with the possibility that some conjugate forms of NAC could be the substrates for OAT1 and OAT3, previous reports have demonstrated that endogenous and exogenous mercapturic acid conjugates of NAC as well as methylmercury conjugates of NAC are substrates of OAT1 in Xenopus Laevis oocytes that overexpress rat Oat1 (Koh et al., 2002, Pombrio et al., 2001). Further investigation is warranted in order to elucidate which among the different froms of NAC are substrates of OAT1 and OAT3. Furthermore, since probenecid is also known to inhibit organic anion transporting polypeptide(OATP) transporters, albeit less potently (Maeda et al., 2014, Tahara et al., 2006, Shimizu et al., 2005, Minematsu et al., 2008), future studies should investigate whether NAC or any of its derivatives are substrates of the OATPs.
Adequate brain penetration is of paramount importance for treating CNS pathologies such as traumatic brain injury. Prior to our study, contradictory reports have been published regarding the capacity of NAC to cross the BBB. Following administration of a single dose of NAC IP or IV into the tail vein of mice, NAC levels in the brain and spinal cord are below the lower limit of quantification (McLellan et al., 1995, Zhou et al., 2015). However, Offen et al administered NAC and N-acetylcysteine amide both IP and orally and were able to detect N-acetylcysteine amide but not NAC in the brain of mice (Offen et al., 2004). Similarly, low levels of NAC were measurable in the brain of mice after IP administration and were significantly increased with coadministration of lipopolysaccharide (Erickson et al., 2012). Route of administration also appears to impact NAC brain distribution. Farr et al reported that NAC crosses BBB readily and accumulated in the brain after administration via jugular vein in mice (Farr et al., 2003) while Neuwelt et al showed that NAC can cross the BBB when given into the carotid artery in rats (Neuwelt et al., 2001).
In the current study, we show that NAC is detectable in the brain of juvenile rats after single IP administration using ultra sensitive methods; but that brain clearance of NAC is rapid with undetectable levels by 4 h (Figure 2). Importantly, we also report that NAC brain penetration can be enhanced with probenecid coadministration. NAC brain exposure increased by nearly 2.5 fold and an additional 46% over that expected by the increase in plasma exposure alone, suggesting a possible transporter effect at the BBB or blood-CSF barrier. In addition to renal tubules, OAT3 is expressed in the basolateral side of the endothelial cells on the BBB and epithelial cells of the choroid plexus. Uptake transporters such as OAT3 that are expressed in the basolateral side of the endothelial cells on the BBB and the apical side of the choroid plexus epithelial cells that constitute the blood-CSF barrier have been implicated as initiators of a vectorial transport process that transfers molecules out of the brain into the circulating blood (Kusuhara and Sugiyama, 2005). Such a mechanism has been proposed in the clearance of PGE2, zidovudine and bumetanide from the brain tissue through the choroid plexus and/or BBB (Sawchuk and Hedaya, 1990, Wong et al., 1993, Tachikawa et al., 2012, Donovan et al., 2015). Inhibitors of OAT3 decreased the clearance of PGE2, zidovudine, and bumetanide from the CSF and increased levels of these molecules in the brain. The enhanced NAC brain exposure following administration of OAT3 inhibitor probenecid could be explained by the same mechanism. Although we explored the possibility of involvement of MRP1 and MRP4 given their known expression in the brain and probenecid-sensitivity (Löscher and Potschka, 2005), our in vitro data suggest that NAC is not a substrate of these transporters (Figure 6).
Recently, clinical modulation of efflux transporters at the BBB as a therapeutic strategy to increase CNS drug levels has been ruled improbable (Kalvass et al., 2013). The findings in this study are provocative in that they support the possibility of increasing the brain exposure of certain drugs by inhibiting uptake transporters at the BBB and/or choroid plexus using an FDA-approved drug with a favorable safety profile. A Phase I/II study is underway (ClinicalTrials.gov Identifier #NCT01322009) to test the safety of NAC and probenecid co-administration in children with TBI as a first step to evaluate this potential therapeutic strategy.
Limitations to the study should be noted. The NAC LC-MS/MS assay used employs sample reduction and derivitization to achieve high sensitivity to detect NAC in biological matrices. While this approach is ideal to measure overall exposure following NAC administration, it cannot distinguish potential downstream forms of NAC such as conjugates. Similarly, an inherant limitation to radiolabeled-based assays is they cannot differentiate between parent molecules such as NAC and conjugates or degradation products. The work was also conducted in control animals and it is also possible or even likely that NAC pharmacokinetics and/or brain penetration may differ is disease conditions or following injury. Future work will focus on interactions between probenecid and OAT1/3 on NAC renal clearance, systemic exposure and brain penetration following traumatic brain injury.
Conclusion
In summary, we report that co-administration of probenecid reduces the clearance of NAC and increases both plasma and brain levels of NAC in the rat. In vitro data reveal that the mechanism of interaction between probenecid and NAC include transport by OAT1 and OAT3. To the best of our knowledge, this is the first study to report the interaction of NAC with OAT1 and OAT3 transporters, and the effect of probenecid on the NAC pharmacokinetics mediated through these transporters. With expanding interest in the use of NAC as an antioxidant therapy for several pathologies, including those within the CNS, these findings have particular relevance for understanding how NAC crosses certain biological barriers. Repurposing the FDA-approved transporter inhibitor, probenecid, is a promising therapeutic strategy to increase NAC exposure.
Acknowledgments
The authors would like to thank Professor Kathleen Giacomini (University of California, San Francisco, CA, USA), who provided us with the human embryonic kidney cell lines stably transfected with human OAT3 (HEK-OAT3), human OAT1 (HEK-OAT1), and the corresponding HEK-empty vector (HEK-EV, control cells).
Footnotes
Declaration of Interest
This work was supported by the National Institutes of Health NS069247 and TR000146. The authors have no conflicts of interest related to this study.
References
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7:355–9. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgstrom L, Kagedal B, Paulsen O. Pharmacokinetics of n-acetylcysteine in man. Eur J Clin Pharmacol. 1986;31:217–22. doi: 10.1007/BF00606662. [DOI] [PubMed] [Google Scholar]
- Cotgreave IA. N-acetylcysteine: Pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–27. [PubMed] [Google Scholar]
- Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: Pharmacology and clinical utility. Expert Opin Biol Ther. 2008;8:1955–62. doi: 10.1517/14728220802517901. [DOI] [PubMed] [Google Scholar]
- Donovan MD, O’Brien FE, Boylan GB, Cryan JF, Griffin BT. The effect of organic anion transporter 3 inhibitor probenecid on bumetanide levels in the brain: An integrated in vivo microdialysis study in the rat. J Pharm Pharmacol. 2015;67:501–10. doi: 10.1111/jphp.12341. [DOI] [PubMed] [Google Scholar]
- Erdman AR, Mangravite LM, Urban TJ, Lagpacan LL, Castro RA, de la Cruz M, Chan W, Huang CC, Johns SJ, Kawamoto M, Stryke D, Taylor TR, Carlson EJ, Ferrin TE, Brett CM, Burchard EG, Giacomini KM. The human organic anion transporter 3 (oat3; slc22a8): Genetic variation and functional genomics. Am J Physiol Renal Physiol. 2006;290:F905–12. doi: 10.1152/ajprenal.00272.2005. [DOI] [PubMed] [Google Scholar]
- Erickson MA, Hansen K, Banks WA. Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood-brain barrier: Protection by the antioxidant n-acetylcysteine. Brain Behav Immun. 2012;26:1085–94. doi: 10.1016/j.bbi.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants alpha-lipoic acid and n-acetylcysteine reverse memory impairment and brain oxidative stress in aged samp8 mice. J Neurochem. 2003;84:1173–83. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- Giustarini D, Milzani A, Dalle-Donne I, Tsikas D, Rossi R. N-acetylcysteine ethyl ester (nacet): A novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem Pharmacol. 2012;84:1522–33. doi: 10.1016/j.bcp.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Grandjean EM, Berthet P, Ruffmann R, Leuenberger P. Efficacy of oral long-term n-acetylcysteine in chronic bronchopulmonary disease: A meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22:209–21. doi: 10.1016/S0149-2918(00)88479-9. [DOI] [PubMed] [Google Scholar]
- Harada D, Naito S, Kawauchi Y, Ishikawa K, Koshitani O, Hiraoka I, Otagiri M. Determination of reduced, protein-unbound, and total concentrations of n-acetyl-l-cysteine and l-cysteine in rat plasma by postcolumn ligand substitution high-performance liquid chromatography. Anal Biochem. 2001;290:251–9. doi: 10.1006/abio.2000.4980. [DOI] [PubMed] [Google Scholar]
- Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by n-acetyl cysteine: A double-blind, placebo controlled study. PLoS One. 2013;8:e54163. doi: 10.1371/journal.pone.0054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst GA, Shaw PB, LeMaistre CA. Laboratory and clinical evaluation of the mucolytic properties of acetylcysteine. Am Rev Respir Dis. 1967;96:962–70. doi: 10.1164/arrd.1967.96.5.962. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Polli JW, Bourdet DL, Feng B, Huang SM, Liu X, Smith QR, Zhang LK, Zamek-Gliszczynski MJ. Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The itc evidence-based position. Clin Pharmacol Ther. 2013;94:80–94. doi: 10.1038/clpt.2013.34. [DOI] [PubMed] [Google Scholar]
- Kikuchi R, Kusuhara H, Sugiyama D, Sugiyama Y. Contribution of organic anion transporter 3 (slc22a8) to the elimination of p-aminohippuric acid and benzylpenicillin across the blood-brain barrier. Journal of Pharmacology and Experimental Therapeutics. 2003;306:51–58. doi: 10.1124/jpet.103.049197. [DOI] [PubMed] [Google Scholar]
- Koh AS, Simmons-Willis TA, Pritchard JB, Grassl SM, Ballatori N. Identification of a mechanism by which the methylmercury antidotes n-acetylcysteine and dimercaptopropanesulfonate enhance urinary metal excretion: Transport by the renal organic anion transporter-1. Mol Pharmacol. 2002;62:921–6. doi: 10.1124/mol.62.4.921. [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Sugiyama Y. Active efflux across the blood-brain barrier: Role of the solute carrier family. NeuroRx. 2005;2:73–85. doi: 10.1602/neurorx.2.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin OL, de Miranda P, King DH, Page DA, Longstreth JA, Rocco L, Lietman PS. Effects of probenecid on the pharmacokinetics and elimination of acyclovir in humans. Antimicrob Agents Chemother. 1982;21:804–7. doi: 10.1128/aac.21.5.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Progress in Neurobiology. 2005;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Maeda K, Tian Y, Fujita T, Ikeda Y, Kumagai Y, Kondo T, Tanabe K, Nakayama H, Horita S, Kusuhara H, Sugiyama Y. Inhibitory effects of p-aminohippurate and probenecid on the renal clearance of adefovir and benzylpenicillin as probe drugs for organic anion transporter (oat) 1 and oat3 in humans. Eur J Pharm Sci. 2014;59:94–103. doi: 10.1016/j.ejps.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Margulies S, Anderson G, Atif F, Badaut J, Clark R, Empey P, Guseva M, Hoane M, Huh J, Pauly J, Raghupathi R, Scheff S, Stein D, Tang H, Hicks M. Combination therapies for traumatic brain injury: Retrospective considerations. J Neurotrauma. 2016;33:101–12. doi: 10.1089/neu.2014.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan LI, Lewis AD, Hall DJ, Ansell JD, Wolf CR. Uptake and distribution of n-acetylcysteine in mice: Tissue-specific effects on glutathione concentrations. Carcinogenesis. 1995;16:2099–106. doi: 10.1093/carcin/16.9.2099. [DOI] [PubMed] [Google Scholar]
- Minematsu T, Hashimoto T, Aoki T, Usui T, Kamimura H. Role of organic anion transporters in the pharmacokinetics of zonampanel, an alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor antagonist, in rats. Drug Metab Dispos. 2008;36:1496–504. doi: 10.1124/dmd.107.019828. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA, Pagel MA, Hasler BP, Deloughery TG, Muldoon LL. Therapeutic efficacy of aortic administration of n-acetylcysteine as a chemoprotectant against bone marrow toxicity after intracarotid administration of alkylators, with or without glutathione depletion in a rat model. Cancer Res. 2001;61:7868–74. [PubMed] [Google Scholar]
- Offen D, Gilgun-Sherki Y, Barhum Y, Benhar M, Grinberg L, Reich R, Melamed E, Atlas D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J Neurochem. 2004;89:1241–51. doi: 10.1111/j.1471-4159.2004.02428.x. [DOI] [PubMed] [Google Scholar]
- Olsson B, Johansson M, Gabrielsson J, Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized n-acetylcysteine. Eur J Clin Pharmacol. 1988;34:77–82. doi: 10.1007/BF01061422. [DOI] [PubMed] [Google Scholar]
- Pombrio JM, Giangreco A, Li L, Wempe MF, Anders MW, Sweet DH, Pritchard JB, Ballatori N. Mercapturic acids (n-acetylcysteine s-conjugates) as endogenous substrates for the renal organic anion transporter-1. Mol Pharmacol. 2001;60:1091–9. doi: 10.1124/mol.60.5.1091. [DOI] [PubMed] [Google Scholar]
- Prescott LF, Illingworth RN, Critchley JA, Stewart MJ, Adam RD, Proudfoot AT. Intravenous n-acetylcystine: The treatment of choice for paracetamol poisoning. Br Med J. 1979;2:1097–100. doi: 10.1136/bmj.2.6198.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushworth GF, Megson IL. Existing and potential therapeutic uses for n-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014;141:150–9. doi: 10.1016/j.pharmthera.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of n-acetylcysteine. Biochim Biophys Acta. 2013;1830:4117–29. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Sawchuk RJ, Hedaya MA. Modeling the enhanced uptake of zidovudine (azt) into cerebrospinal fluid. 1. Effect of probenecid. Pharm Res. 1990;7:332–8. doi: 10.1023/a:1015854902915. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, Sugiyama Y. Contribution of oatp (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metabolism and Disposition. 2005;33:1477–1481. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- Tachikawa M, Ozeki G, Higuchi T, Akanuma S, Tsuji K, Hosoya K. Role of the blood-cerebrospinal fluid barrier transporter as a cerebral clearance system for prostaglandin e(2) produced in the brain. J Neurochem. 2012;123:750–60. doi: 10.1111/jnc.12018. [DOI] [PubMed] [Google Scholar]
- Tahara H, Kusuhara H, Maeda K, Koepsell H, Fuse E, Sugiyama Y. Inhibition of oat3-mediated renal uptake as a mechanism for drug-drug interaction between fexofenadine and probenecid. Drug Metabolism and Disposition. 2006;34:743–747. doi: 10.1124/dmd.105.008375. [DOI] [PubMed] [Google Scholar]
- Versantvoort CHM, Bagrij T, Wright KA, Twentyman PR. On the relationship between the probenecid-sensitive transport of daunorubicin or calcein and the glutathione status of cells overexpressing the multidrug resistance-associated protein (mrp) International Journal of Cancer. 1995;63:855–862. doi: 10.1002/ijc.2910630617. [DOI] [PubMed] [Google Scholar]
- Wong SL, Van Belle K, Sawchuk RJ. Distributional transport kinetics of zidovudine between plasma and brain extracellular fluid/cerebrospinal fluid in the rabbit: Investigation of the inhibitory effect of probenecid utilizing microdialysis. J Pharmacol Exp Ther. 1993;264:899–909. [PubMed] [Google Scholar]
- Zhou J, Coles LD, Kartha RV, Nash N, Mishra U, Lund TC, Cloyd JC. Intravenous administration of stable-labeled n-acetylcysteine demonstrates an indirect mechanism for boosting glutathione and improving redox status. J Pharm Sci. 2015;104:2619–26. doi: 10.1002/jps.24482. [DOI] [PubMed] [Google Scholar]