

INTRODUCTION
Waldenström’s macroglobulinemia (WM) is a low-grade malignant monoclonal proliferation of immunoglobulin M (IgM)-producing lymphocytes capable of exhibiting plasmacytoid or plasma cell differentiation [1]. According to the World Health Organization’s classification of neoplastic diseases of hematopoietic and lymphoid tissues, WM is classified as lymphoplasmacytic lymphoma.
Clinical features of WM may include cytopenia, bleeding, lymphadenopathy, splenomegaly, and, most commonly, a hyperviscosity syndrome related to high serum levels of monoclonal IgM. Central nervous system (CNS) complications arise in approximately 25% of WM cases, usually as a result of the hyperviscosity syndrome or an immune-related neuropathy [1–2]. Rare cases of WM involving direct CNS infiltration by malignant cells have been referred to as Bing-Neel Syndrome (BNS). To date, limited data exist regarding BNS. The authors describe a rare case of BNS presenting as a cerebellar mass with associated brainstem infiltration and subacute neurologic deterioration.
CASE REPORT
Presentation
A 53-year-old right-handed man with a history of WM presented with dizziness, nausea, and vomiting. Magnetic resonance imaging (MRI) of the brain showed a cerebellar mass with vasogenic edema (Figure 1). Surgical excision was recommended based on the patient’s symptoms from increased intracranial pressure and local mass effect. The patient underwent posterior fossa craniectomy and gross total tumor resection. Intraoperatively, a plane was developed around the mass at the brain–tumor interface, facilitating tumor removal. The pathology report indicated a lymphoproliferative process with extensive necrosis (Figure 2).
Figure 1.
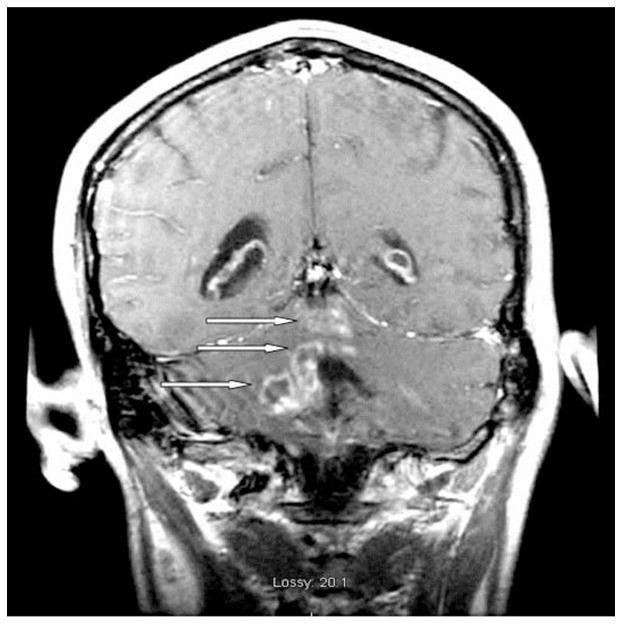
Preoperative brain MRI shows a cerebellar mass with vasogenic edema.
Figure 2.

Pathology slides indicate a lymphoproliferative process with extensive necrosis. (A) extensive necrotic tissue with histiocytic-lymphocytic and plasmacytic inflammatory infiltrate; typical large lymphoid B cells stain positively for CD20 (B) and PAX-5 (C) and rarely for CD138 (D).
Past History
The patient’s clinical course initially began with progressive weight loss and fatigue beginning 2 years before his presentation with the cerebellar mass. Anemia was diagnosed, and bone marrow biopsy revealed 30% infiltration with a plasmacytic non-Hodgkin’s lymphoma consistent with a diagnosis of multiple myeloma. Serology revealed a hemoglobin of 8.3 (g/dL), erythrocyte sedimentation rate of 137 (mm/hr), gamma globulin of 2.5 (g/dL), and a lambda light chain of 354 (mg/L). Computed tomographic scans of the abdomen and pelvis and a chest x-ray were within normal limits.
The patient completed two cycles of rituximab and fludarabine. During the third cycle, infusion was terminated due to the onset of dyspnea and flushing. Rituximab was discontinued, and fludarabine was continued for four additional cycles. At that time, the gamma globulin level was 2.7 (g/dL), with a lambda light chain level of 248 (mg/dL).
One year into treatment, the patient presented with an involuntary tremor of the right hand, headaches, visual difficulties, and bilateral plantar paresthesias. A bone marrow biopsy revealed extensive infiltration of small lymphocytes with partial plasmacytoid lymphocytic differentiation in 60% of the smear, consistent with the diagnosis of WM. Immunohistochemistry revealed that the lymphoid infiltrate was positive for PAX-5, CD20, and CD79a. Flow cytometry revealed two abnormal populations. The first population of cells expressed the following cell surface markers: CD45, HLADR, CD19, lambda light chain, CD5 (heterogeneous), CD20, CD22, CD38 (dim), and CD52. This was consistent with lymphoplasmacytic lymphoma, or WM. The second abnormal population expressed CD38, CD138, CD45, and lambda light chain and was negative for CD56, consistent with multiple myeloma.
The patient began therapy with lenalidomide and dexamethasone followed by six cycles of a CHOP chemotherapy regimen (cyclophosphamide, vincristine, adriamycin, prednisone), with partial disease remission but continued constitutional symptoms. Twenty-one months into treatment, the patient had an M-spike of 2.37 (g/dL), IgM of 3790 (mg/dL), and free light chain serum viscosity of 2.9 (cP). Repeat bone marrow biopsy revealed extensive persistent small lymphocyte type lymphoma with occasional plasmacytoid lymphocytes. Monoclonal B cell genetic rearrangement peaks measured by polymerase chain reaction (PCR) assay on bone marrow aspirate revealed: 295–296 base pairs (bp) with IHG-FR1, 111–112 bp with IGH-FR3, 199 and 289–290 bp with IGK-A, and 281–282 bp with IGK-B. Treatment continued with a combination of rituximab and bendamustine.
Postoperative Course
Following the patient’s craniectomy for cerebellar tumor removal 2 years into treatment, further chemotherapy was suspended to allow recovery from surgery. The mass consisted of extensive necrotic tissue with histiocytic-lymphocytic and plasmacytic inflammatory infiltrate (Figure 2A), a fibroblastic reaction, and focal areas consisting of a mature plasma cell lymphoproliferative neoplasm. The vast majority of the cell population consisted of CD68+ histiocytes with a smaller population of CD3+ T cells. Monoclonal B cell genetic rearrangement peaks were detected by PCR assay as follows: 147 and 201 bp with IGK-A, and 234–236 and 282 bp with IGK-B. No definite monotypic plasma cell population could be isolated by in-situ hybridization for kappa and lambda light chain immunoglobulin; however, the presence of atypical large lymphoid B cells stained positively for CD20 (Figure 2B) and PAX-5 (Figure 2C), and rarely for CD138 (Figure 2D), and partially positive for BCL-6.
Three months postoperatively, the patient suffered further neurologic deterioration characterized by dysphagia, dysarthria, and increasing ataxia. Examination revealed a new right peripheral facial nerve palsy. MRI of the brain demonstrated edema of the brainstem, with evidence of new contrast enhancement extending from the deep cerebellum into the brachium pontis and pons (Figure 3). A cerebrospinal fluid (CSF) sample obtained was positive for mature B-cell lymphoproliferative neoplasm. Flow cytometry revealed a monotypic B-cell population that stained positively for CD45 and CD19 markers and carried a surface lambda light chain restriction similar to the restriction previously reported in the patient’s bone marrow, with similar monoclonal peaks as observed in tissue blocks prepared from the resected cerebellar mass. A course of palliative radiation therapy was initiated, however, the patient died before its completion.
Figure 3.

Brain MRI obtained 3 months postoperatively shows brainstem edema, with evidence of new contrast enhancement extending from the deep cerebellum into the brachium pontis and pons.
DISCUSSION
BNS has been defined as an infiltration of the CNS by malignant plasmacytoid lymphocytes in the setting of WM [1–3]. BNS can present in either diffuse or tumoral forms. In the diffuse form, lymphoplasmacytoid cells are found in the leptomeningeal space, pons, medulla, and periventricular white matter. MRI often indicates leptomeningeal enhancement. Patients may present with memory loss, headaches, and confusion. In the tumoral form, lymphoplasmacytoid cells form a defined mass within the CNS. MRI usually shows either a discrete intracranial mass or nodular lesions. Clinical presentation is characterized by focal neurologic deficit or seizures [1–2].
The diagnosis of BNS can be made on the basis of imaging studies, CSF analysis, and biopsy histology. Some cases of BNS have been diagnosed on the basis of imaging and CSF analysis alone [1]. However, cases of WM with CNS complications unrelated to infiltration of the primary neoplasm have often been misdiagnosed as BNS [4]. In some cases, primary CNS lymphoma or transformation of a large B-cell lymphoma can mimic BNS. Thus, definitive diagnosis usually requires biopsy of the CNS lesion to exclude the possibility of other pathologic conditions coexistent with WM [3].
The patient described here developed two monoclonal neoplastic B-cell lines consistent with both multiple myeloma and WM. Treatment with rituximab and fludarabine produced a shift in the predominant pattern of the malignant cell line from a plasmacytic infiltrate characteristic of non-Hodgkin’s lymphoma to extensive infiltrate of small lymphocytes with partial plasmacytoid lymphocytic differentiation. This response correlates with the clinical picture of multiple myeloma amenable to treatment coexistent with underlying treatment-refractory WM. Additionally, intrathecal rituximab therapy has been described to treat Bing-Neel Syndrome and, although not administered to our patient, should remain a consideration in similar clinical scenarios [5].
Our patient’s initial neurologic deterioration involved involuntary right hand tremor, headaches, visual difficulties, and bilateral plantar paresthesias. Although such symptoms can occur in the setting of elevated serum viscosity and anemia of chronic illness, the direct effects of cerebellar and brainstem involvement account largely for the neurologic picture in this patient.
Immunohistochemical data suggests the patient’s lymphopoietic malignancies are of common origin. The monoclonal B cell genetic rearrangements of 199 bp in IGK–A, and 281–282 bp in IGK–B taken from bone marrow aspirate matched rearrangements of 201 bp in IGK- A, and 282 BP in IGK-B from the resected cerebellar tumor. CSF analysis performed 1 month after tumor resection indicated the presence of mature lymphoproliferative B cells. The presence of monoclonal peaks found in common between the population of malignant B cells within the resected cerebellar lesion and those within the CSF further suggest a common origin. Furthermore, immunophenotypic analysis revealed similar light chain restrictions between the neoplastic monotypic B cells isolated from the CSF and malignant B cells isolated from the bone marrow. Thus, laboratory data evidence strongly support the possibility of a single population of malignant B cells as the etiology of neurological involvement in this disease.
Evidence presented here suggests that our patient suffered from the tumoral form of BNS. Nevertheless, we cannot exclude the possibility that his cerebellar tumor could have originated from underlying multiple myeloma, which we and others have shown can appear within the CNS [6]. In such a case, his neurologic disease could have represented a large-cell transformation. However, genetic linkage between malignant B cells isolated from the resected tumor and from the bone marrow makes such a transformation unlikely.
CONCLUSION
BNS can be difficult to treat, and optimal therapy remains controversial [3]. We decided on palliative radiation therapy as the primary treatment following tumor resection. Although BNS is a rare condition, it should be considered in the differential diagnosis of patients with lymphoproliferative disorders presenting with intracranial mass lesions.
Direct CNS invasion can occur in Waldenstrom’s macroglobulinemia.
Biopsy is usually required to diagnose Bing-Neel Syndrome.
Bing-Neel Syndrome lesions typically originate from monoclonal B cell lines.
Acknowledgments
Funding
This work was supported by Roswell Park Cancer Institute and National Cancer Institute (NCI) grant #P30 CA016056.
We gratefully thank Alice Mohr, N.P., for her invaluable technical assistance with reporting this case.
Abbreviations
- BNS
Bing-Neel Syndrome
- bp
base pairs
- CNS
central nervous system
- CSF
cerebrospinal fluid
- IgM
immunoglobulin M
- MRI
magnetic resonance imaging
- PCR
polymerase chain reaction
- WM
Waldenström’s macroglobulinemia
Footnotes
Ethics
The patient in this report was cared for in accordance with the precepts established by the Helsinki Declaration, and informed consent was obtained from him.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grewal JS, Brar PK, Sahijdak WM, Tworek JA, Chottiner EG. Bing-Neel syndrome: a case report and systematic review of clinical manifestations, diagnosis, and treatment options. Clin Lymphoma Myeloma. 2009;9:462–466. doi: 10.3816/CLM.2009.n.091. [DOI] [PubMed] [Google Scholar]
- 2.Zhang S, Gottardi-Littell NR, Nayar R, De Frias D. Cytology features of Bing-Neel syndrome. Acta Cytol. 2009;53:240–243. doi: 10.1159/000325135. [DOI] [PubMed] [Google Scholar]
- 3.Malkani RG, Tallman M, Gottardi-Littell N, Karpus W, Marszalek L, Variakojis D, Kaden B, Walker M, Levy RM, Raizer JJ. Bing-Neel syndrome: an illustrative case and a comprehensive review of the published literature. J Neurooncol. 2010;96:301–312. doi: 10.1007/s11060-009-9968-3. [DOI] [PubMed] [Google Scholar]
- 4.Fintelmann F, Forghani R, Schaefer PW, Hochberg EP, Hochberg FH. Bing-Neel Syndrome revisited. Clin Lymphoma Myeloma. 2009;9:104–106. doi: 10.3816/CLM.2009.n.028. [DOI] [PubMed] [Google Scholar]
- 5.Kolbaske S, Grossmann A, Benecke R, Wittstock M. Progressive gait ataxia and intention tremor in a case of Bing-Neel syndrome. J Neurol. 2009;256:1366–1368. doi: 10.1007/s00415-009-5107-5. [DOI] [PubMed] [Google Scholar]
- 6.Reddy N, Karampelas I, Chanan-Khan A, Fenstermaker R, Padmanabhan S. Aggressive relapse of multiple myeloma with intracerebral extension and associated hemorrhage. Leuk Lymphoma. 2007;48:1228–1230. doi: 10.1080/10428190701272371. [DOI] [PubMed] [Google Scholar]