

Abstract
Smooth muscle cells (SMCs) and the extracellular matrix (ECM) are intimately associated in the aortic wall. Fbln4SMKO mice with a smooth muscle cell-specific deletion of the Fbln4 gene, which encodes the vascular ECM component fibulin-4, develop ascending aortic aneurysms that have increased abundance of angiotensin converting enzyme (ACE); inhibiting angiotensin II signaling within the first month of life prevents aneurysm development. We used comparative proteomics analysis of Fbln4SMKO aortas from postnatal day (P) 1 to P30 mice to identify key molecules involved in aneurysm initiation and expansion. At P14, the actin depolymerizing factor cofilin was dephosphorylated and thus activated, and at P7, the abundance of slingshot-1 phosphatase (SSH1), an activator of cofilin, was increased, leading to actin cytoskeletal remodeling. Also by P7, biomechanical changes and underdeveloped elastic lamina-SMC connections were evident and the abundance of early growth response-1 (Egr1), a mechanosensitive transcription factor that stimulates ACE expression, was increased, which was before the increases in ACE abundance and cofilin activation. Postnatal deletion of Fbln4 in SMCs at P7 prevented cofilin activation and aneurysm formation, suggesting that these processes required disruption of elastic lamina-SMC connections. Phosphoinsitide 3-kinase (PI3K) is involved in the angiotensin II-mediated activation of SSH1 and administration of PI3K inhibitors from P7 to P30 decreased SSH1 abundance and prevented aneurysms. These results suggest that aneurysm formation arises from abnormal mechanosensing of SMCs resulting from the loss of elastic lamina-SMC connections and from increased SSH1 and cofilin activity, which may be potential therapeutic targets for treating ascending aortic aneurysms.
Introduction
Thoracic aortic aneurysms (TAAs) can be either non-syndromic or syndromic, and are associated with a high risk of mortality from dissection and/or rupture. Substantial efforts have gone into identifying the genes and signaling pathways involved in TAAs in humans (1). Genetic analysis of TAA has revealed that mutations in extracellular matrix (ECM) proteins, including fibrillin-1(2), type III alpha 1-collagen (3) and fibulin-4 (4,5), lead to the formation of aneurysms, along with various symptoms including skeletal, cutaneous and connective tissue defects. Furthermore, activation of transforming growth factor β (TGFβ) in the vascular wall, by loss of proper tethering of TGFβ and/or abnormal compensatory increase in TGFβ activity, lead to aortic aneurysms in Marfan syndrome and related disorders (6–8). Whether altered signal transduction is involved in other types of aneurysms characterized by compromised ECM is not completely understood. The ECM also affects the phenotype of smooth muscle cells (SMCs) (9,10). Mutations in genes encoding SMC contractile proteins, including SMC-specific myosin heavy chain (Myh11) (11) and α-actin (ACTA2) (12), and type I cGMP-dependent protein kinase (PRKG1) (13) are responsible for non-syndromic familial TAAs and dissection in humans. A heterozygous mutant allele of MYH11 or ACTA2 might serve as a dominant negative to impair the force-generating machinery of SMCs (11) and a gain-of-function mutation of PRKG1 may reduce SMC contractility (13). Both mutations affect the contraction of SMCs in the ascending aorta, a region where cells are constantly exposed to high outflow pressure.
We have previously shown that SMC-specific fibulin-4 knockout mice (Fbln4SMKO) develop ascending aortic aneurysms characterized by disruption of the elastic laminae and increased proliferation of SMCs (9). Proliferation of SMCs precedes aneurysm formation, and is associated with increased phosphorylation of extracellular signal-regulated kinase (ERK) and decreased expression of SMC differentiation markers in the aortas of the mutant mice. In addition, abnormal increases in angiotensin converting enzyme (ACE) abundance and subsequent activation of angiotensinII (AngII) signaling in the aortic wall are responsible for driving the aneurysm phenotype (14). In this model, aneurysms are completely prevented by administration of an ACE inhibitor or angiotensin II type 1 receptor (AT1R) blocker (ARB) within the first month of life. ARB treatment initiated after establishment of the aneurysms does not reverse the aneurysm phenotype, indicating that the signal(s) required for maintenance of aneurysms may be independent of AngII-AT1R. The precise molecular events that occur within this therapeutic time window, however, have not been determined. The aim of the present study, therefore, was to determine the molecular pathways involved in initiation and expansion of aneurysms and to identify key molecules that potentially connect ECM defects, AngII signaling and aneurysm formation in Fbln4SMKO mice.
Results
Proteins with differential abundance were identified in Fbln4SMKO aortas during initiation and expansion of aneurysms
To determine the time points involved in the increase in AngII signaling and phenotypic changes in SMCs, we first examined the abundance of ACE and the phosphorylation of ERK in control and Fbln4SMKO aortas from postnatal day (P) 1 to P90. ACE amounts began to increase at P7 in Fbln4SMKO aortas and reached a two-fold increase at P30 compared to control mice (Fig. 1A, fig. S1). Consistent with previous findings, the phosphorylation of ERK was increased at P30. In addition, expression of SMC differentiation marker genes was either maintained (Acta2; which encodes smooth muscle α-actin) or increased (Cnn1, which encodes calponin 1; Myh11, which encodes the heavy chain of smooth muscle myosin; SM22, which encodes smooth muscle protein 22-α; Myocd, which encodes myocardin; and Srf, which encodes serum response factor) 2.0 to 3.5 fold in the Fbln4SMKO aorta at P1, followed by a sharp decrease after P7 (Fig. 1B), suggesting that Fbln4-deficient SMCs rapidly dedifferentiated after birth. Based on these data, we concluded that the first 30 days of the postnatal period is a critical time for the initiation and expansion of aneurysms.
Fig. 1. Proteomic analysis of ascending aortas during initiation and expansion of aneurysms.
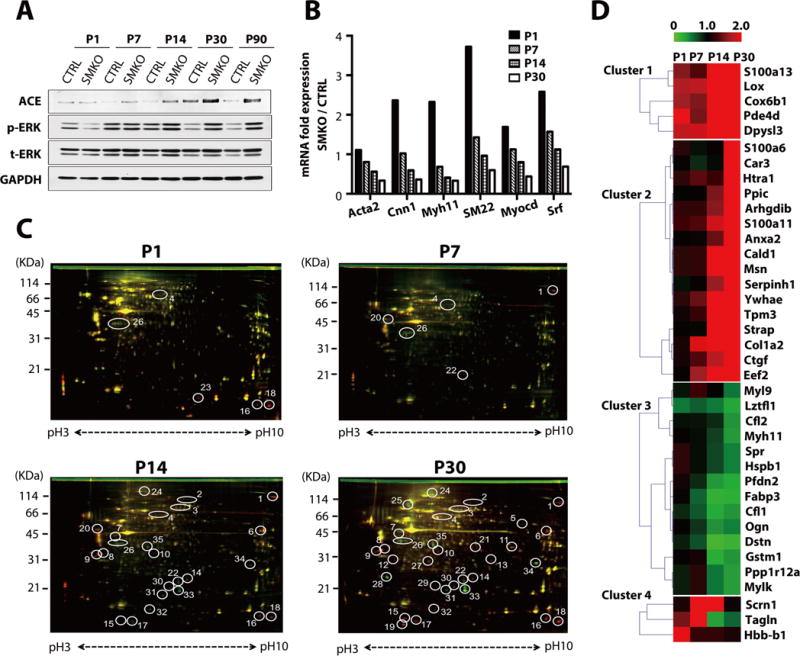
(A) Representative Western blots of ascending aortas from control and Fbln4SMKO (SMKO) mice. The experiment was performed three times with different pools of animals. The numbers of aortas used per genotype in each time point are: P1, n=5–12; P7, n=5–15; P14, n=5–12; P30, n=3–7; P90, n=2–6. See fig. S1 for quantification. (B) qPCR analysis of SMC-specific genes from ascending aortas of CTRL (pooled P1; n=12, P7; n=18, P14;n=9, P30; n=12) and SMKO (pooled P1; n=12, P7; n=18, P14; n=8, P30; n=11) mice performed in technical triplicate. (C) Representative 2D-DIGE using entire aortas (for P1: n=4 per genotype) or ascending aortas (P7: n=7, P14: n=5, P30; n=3 per genotype). Proteins with increased abundance in the SMKO aortas appear in red, proteins with decreased abundance appear in green, and those with similar abundance appear in yellow. Circled spots with numbers indicate more than ± 2.0 fold changes between CTRL and SMKO in 3 independent experiments. (D) Heat map showing identified proteins divided into four clusters according to the expression patterns during postnatal development. Red: increased abundance in SMKO aortas, green: decreased abundance in SMKO aortas.
To identify key molecules involved in the aneurysm development, we compared the protein profiles of control and Fbln4SMKO aortas harvested at P1 (before aneurysms develop) to those harvested at P30 (when aneurysms have been established) by two-dimensional differential gel electrophoresis (2D-DIGE) (15). Many proteins showed changes in abundance after P14 (Fig. 1C) and we subjected each protein spot with >2-fold difference in three independent experiments to mass spectrometric analysis. The identified proteins were classified into four clusters according to the temporal changes in their abundance (Fig. 1D, Table 1, and fig. S2A). The abundance of 21 proteins was increased (2.0 to 5.7 fold) and the abundance of 14 proteins was decreased (−2.0 to −6.9 fold) in Fbln4SMKO aortas compared to control. Consistent with previous findings (9), we found that the abundance of Myh11 and Tagln (also known as SM22) was decreased at P30 (Fig. 1D). Other SMC proteins such as Ppp1r12a (MYPT1; myosin phosphatase-targeting subunit 1) and Mylk (myosin light chain kinase) also showed decreased abundance in Fbln4SMKO aortas compared to control. In contrast, the abundance of ECM-related molecules such as LOX (lysyl oxidase), Col1a2 (collagen type 1 alpha 2 chain), CTGF (connective tissue growth factor) and Htra1 (serine peptidase 1) were increased as the aneurysms expanded (fig. S2A). Gene ontology analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) revealed that a substantial number of genes were involved in actin cytoskeleton organization, including Cald1 (caldesmon 1) and cofilins (fig. S2B). We further validated the changes in abundance at the mRNA level by qPCR analysis (fig. S3).
Table 1.
Identification of proteins #1–35 by Orbitrap Velos or Q Exactive mass-spectrometer.
| Spot | Protein | Accession No | Symbol | MWa) | p1b) | Peptidesc) | % coveraged) |
|---|---|---|---|---|---|---|---|
| 1 | Collagen 1 alpha-2 chain | NP_031769 | Col1A2 | 129.6 | 9.3 | 7 | 5.9 |
| 2 | Elongation factor 2 | NP_031933 | Eef2 | 95.3 | 6.4 | 14 | 14.0 |
| 3 | Moesin | NP_034963 | Msn | 66.5 | 5.9 | 51 | 70.9 |
| Caldesmon-1 | NP_663550 | Cald1 | 60.5 | 7.0 | 46 | 69.2 | |
| 4 | Dihydropyrimidinase-like 3 | NP_001129558 | Dpysl3 | 61.9 | 6.0 | 12 | 26.4 |
| 5 | Serine protease HTRA1 | NP_062510 | Htra1 | 51.2 | 7.8 | 22 | 45.0 |
| 6 | Serpin H1 | NP_001104514 | Serpinh1 | 46.5 | 8.9 | 13 | 30.0 |
| 7 | Serine-threonine kinase receptor-associated protein | NP_035629 | Strap | 38.4 | 5.0 | 4 | 16.9 |
| 8 | Tropomyosin 3 | NP_071709 | Tpm3 | 32.8 | 4.2 | 26 | 60.2 |
| 9 | 14-3-3 protein epsilon | NP_033562 | Ywhae | 29.2 | 4.6 | 28 | 74.9 |
| 10 | Protein-lysine6-oxidase | NP_034858 | Lox | 46.7 | 8.7 | 6 | 15.8 |
| 11 | Annexin A2 | NP_031611 | Anxa2 | 38.6 | 7.6 | 5 | 32.4 |
| 12 | Rho dissociation inhibitor 2 | NP_031512 | Arhgdib | 22.9 | 5.0 | 13 | 57.0 |
| 13 | Carbonic anhydrase 3 | NP_031632 | Car3 | 29.4 | 6.9 | 6 | 24.6 |
| 14 | Peptidyl-prolyl cis-trans isomerase C | NP_032934 | Ppic | 22.8 | 7.0 | 2 | 11.8 |
| 15 | Protein S100-A11 | NP_058020 | S100a11 | 11.0 | 5.3 | 6 | 51.0 |
| 16 | Phosphodiesterase 4D, cAMP-specific (Fragment) | NP_035186 | Pde4d | 84.5 | 4.8 | 2 | 2.3 |
| 17 | Protein S100-A13 | NP_033139 | S100a13 | 17.7 | 6.2 | 5 | 43.9 |
| 18 | Cytochrome c oxidase subunit 6B1 | NP_079904 | Cox6b1 | 10.1 | 9.0 | 2 | 11.6 |
| 19 | Protein S100-A6 | NP_035443 | S100a6 | 10.1 | 5.3 | 2 | 16.9 |
| 20 | Secernin-1 | NP_081544 | Scrn1 | 46.3 | 4.7 | 7 | 15.2 |
| 21 | Connective tissue growth factor | NP_034347 | Ctgf | 38.6 | 7.6 | 3 | 12.1 |
| 22 | Transgelin (SM-22) | NP_035656 | Tagln | 22.5 | 8.9 | 17 | 63.2 |
| 23 | Hemoglobin subunit beta-1 | NP_032246 | Hbb-b1 | 15.7 | 7.1 | 7 | 49.7 |
| 24 | Myosin, light polypeptide kinase | NP_647461 | Mylk | 213.6 | 5.9 | 14 | 15.2 |
| Protein phosphatase 1 regulatory subunit 12A | NP_082168 | Ppp1r12a | 111.8 | 5.5 | 6 | 6.3 | |
| 25 | Myh11 protein | NP_038635 | Myh11 | 227.1 | 5.4 | 19 | 10.4 |
| 26 | Leucine zipper transcription factor-like 1 | NP_201579 | Lztfl1 | 34.7 | 5.1 | 6 | 21.5 |
| 27 | Sepiapterin reductase | NP_035597 | Spr | 27.9 | 5.9 | 6 | 37.4 |
| Heat shock protein beta-1 | NP_038588 | Hspb1 | 23.0 | 6.1 | 5 | 31.4 | |
| 28 | Myosin regulatory light polypeptide 9 | NP_742116 | Myl9 | 19.9 | 4.8 | 6 | 33.7 |
| 29 | Cofilin-2 | NP_031714 | Cfl2 | 18.7 | 7.7 | 9 | 51.8 |
| 30 | Cofilin-1 | NP_031713 | Cfl1 | 18.6 | 8.2 | 8 | 51.8 |
| 31 | Prefoldin subunit 2 | NP_035200 | Pfdn2 | 16.5 | 6.2 | 7 | 45.5 |
| 32 | Fatty acid-binding protein, heart | NP_034304 | Fabp3 | 14.8 | 6.1 | 8 | 54.9 |
| 33 | Destrin | NP_062745 | Dstn | 18.5 | 8.1 | 8 | 49.1 |
| 34 | Glutathione S-transferase Mu | NP_034488 | Gstm1 | 26.0 | 7.7 | 11 | 47.2 |
| 35 | Mimecan (Osteoglycin) | NP_032786 | Ogn | 34.0 | 5.5 | 5 | 13.8 |
CPFP version 2.0.3 was used for database searching against the Uniprot mouse database.
Theoretical molecular weight (KDa).
Theoretical isoelectric point (pI).
Number of matched peptides.
Percentage of sequence coverage.
Cofilin was activated in the ascending aortas of Fbln4SMKO mice
Actin filament assembly and disassembly (dynamics) and reorganization are fundamental for various cellular activities (16,17). In SMCs, the contractile apparatus is organized such that the actin and myosin filaments run obliquely across the cell. At the cell surface, the actin filaments anchor into membrane-associated dense plaques, which are connected on the extracellular face of the membrane to elastin and microfibril extensions radiating from the surface of the elastic laminae (18). Proper actin dynamics and orientation are therefore critical so that SMCs can generate and distribute appropriate forces to the elastic laminae and across the vessel wall. Based on these reasons, we focused on cofilins for further analysis. Cofilin-1 and -2 belong to the ADF/cofilin family of actin depolymerizing factors which rapidly sever polymerized actin, thereby triggering in the disassembly of actin fibers (19). In addition to direct regulation of actin remodeling, cofilin is also involved in various cellular functions of SMCs, including PDGF-induced migration and neointima formation (20,21). QPCR analysis showed that the abundance of the mRNA encoding cofilin-2 (muscle cofilin) was decreased at P14 and P30, whereas that of the mRNA encoding cofilin-1 (non-muscle cofilin) was unchanged until P14 and then markedly increased at P30 (fig. S3). Cofilin is inactivated when phosphorylated by LIM kinases (LIMKs), thereby leading to decreased actin binding, severing and depolymerization (22). Western blotting revealed that the phosphorylation of cofilin started to decrease at P7, and was significantly decreased at P14 in Fbln4SMKO aortas, which coincided with thickening of the medial layers. Total cofilin abundance was similar in both genotypes (Fig. 2A). Consistently, immunostaining revealed that the phosphorylation of cofilin was markedly decreased in P30 Fbln4SMKO ascending aortas and to a lesser extent in the descending aorta (Fig. 2B, fig. S4). These data indicated that cofilin activity was significantly increased either by compromised phosphorylation or accelerated dephosphorylation.
Fig. 2. Activation of cofilin in SMKO ascending aorta.
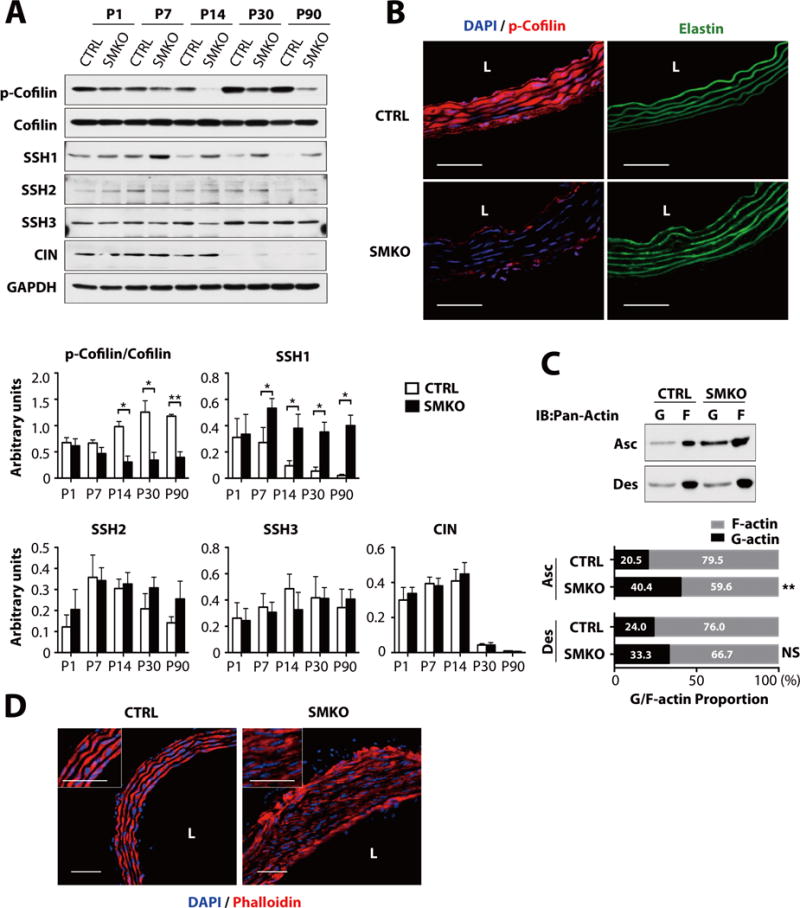
(A) Representative Western blot of ascending aortas of CTRL and SMKO. The experiment was performed three times with different pools of animals with similar results. N values as in Fig. 1A. *P < 0.05, **P < 0.01, unpaired t-test. SSH, slingshot; CIN, chronophin. (B) Cross sections of the ascending aorta at P30 (n=5 mice per genotypes) immunostained with phosphorylated (p)-cofilin (red) and DAPI (blue). Elastic laminae are green (autofluorescence). Bars are 50μm. (C) G and F actin for ascending and descending aortas at P30. Representative blot (upper) and quantification of G- and F-actin (bottom) are shown (n=5 aortas per genotype). Means of G-actin (black bar) and F-actin (gray bar) are shown in each bar. **P < 0.01, NS, not significant, unpaired t-test. (D) Cross sections of the ascending aorta at P30 from CTRL and SMKO (n=5 mice per genotype), immunostained with phalloidin (red) and DAPI (blue). Bars are 50 μm.
Because LIMKs are regulated by RhoA signaling, we examined if RhoA abundance was decreased in Fbln4SMKO aorta. Instead, Western blot analysis showed that total RhoA abundance began to increase in Fbln4SMKO ascending aortas starting at P7 (fig. S5A) and that active RhoA was also increased in Fbln4SMKO aortas (fig. S5B). Accordingly, phosphorylation of downstream molecules, including myosin light chain (MLC), myosin-binding subunit of myosin phosphatase (MYPT), myosin light chain kinase (MLCK), and LIMK, were also substantially increased in Fbln4SMKO aortas (figs. S5C, S5D), indicating that RhoA-LIMK signaling was activated in the mutant aorta, consistent with the increase in AngII signaling in the mutant aortic wall (14).
Several phosphatases, including the Slingshot (SSH) family (composed of SSH1, SSH2 and SSH3 in mammals) and chronophin (CIN), can dephosphorylate cofilin and restore its activity (23,24). We therefore examined if cofilin phosphatases were dysregulated in Fbln4SMKO aortas. The abundance of SSH1, but not that of SSH2, SSH3 or CIN, was significantly increased at P7 in Fbln4SMKO aortas, preceding the decrease in the phosphorylation of cofilin (Fig. 2A). These data suggested that cofilin was dephosphorylated by SSH1 in the vessel wall and that cofilin activity was increased in Fbln4SMKO aortas. Indeed, when overexpressed in rat vascular SMCs, wild-type SSH1, but not phosphatase-inactive SSH1 (SSH1-CS), dephosphorylated cofilin, confirming its phosphatase activity as previously described (Figs. S6A and S6B) (25). To further confirm these findings, monomeric actin (G-actin) and filamentous actin (F-actin) were examined (Fig. 2C). The ratio of G to F actin was significantly increased in ascending aortic extracts from Fbln4SMKO mice compared to control mice, whereas descending aortic extracts did not show statistically significant differences between genotypes (Fig. 2C). Accordingly, actin distribution was disrupted in the P30 Fbln4SMKO aortas compared to control aortas (Fig. 2D). These data suggested that increased cofilin activity led to an increase of G-actin and affected actin filament integrity, which could potentially contribute to the development of aneurysms in Fbln4SMKO mice.
Elastic lamina-SMC connections were disrupted and mechanical properties were altered in Fbln4SMKO aortas
The activation of cofilin and expansion of aneurysms prompted us to investigate the trigger for these changes. Cofilin can serve as a cellular mechanosensor in an in vitro reconstitution system (26). Therefore, we hypothesized that cofilin activation was induced as a result of abnormal mechanosensing and/or mechanotransduction in Fbln4SMKO SMCs. To test this hypothesis, we first examined the ultrastructure of the established aneurysmal wall at P90. Extensive connections were present between the elastic laminae and SMCs in control aortas (indicated by white arrows in Fig. 3A - CTRL, P90), but not in Fbln4SMKO aortas, which had large, irregular aggregates of elastin with a lacy appearance between SMCs instead of continuous elastic laminae (Fig. 3A - SMKO, P90). We next examined if similar changes were apparent in P7 aortas. Wild-type P7 aortas had near complete elastic laminae and early connections between the elastic laminae and SMCs, although they were not as fully established as those in adult aortas (Fig. 3A - CTRL, P7, white arrows). In the Fbln4SMKO aortas, the elastic laminae appeared to be fragmented into juxtaposed, individual elastin globules (Fig. 3A - SMKO, P7, arrowhead). In some areas, connections appeared to be disrupted due to the abnormal organization of the elastic laminae (Fig. 3A – SMKO, P7, white arrows).
Fig. 3. Disruption of elastic lamina-SMC connections and alteration of the mechanical properties of Fbln4SMKO aortas.
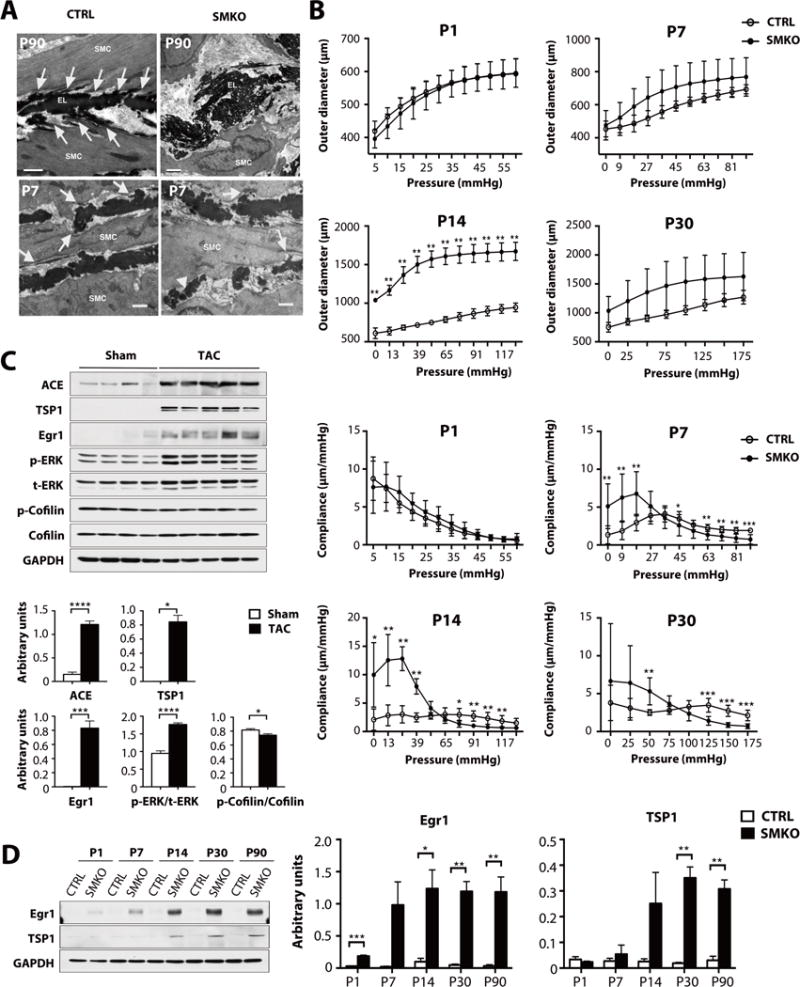
(A) Electron microscopy images from CTRL and SMKO ascending aortas at P90 and P7. Elastic lamina-SMC connections are well-formed in CTRL aortas (white arrows), whereas elastic laminae are disrupted and not connected to SMCs in the SMKO aorta at P90. Elastic laminae were also abnormal at P7 in SMKO vessels, with numerous globules of elastin rather than solid bands of elastin (white arrowhead) and less organized cell-elastin associations (white arrows). Bars are 1 μm. Image are representative of at least n=2 (CTRL) and n=3 (SMKO) mice per age. (B) Upper panel: Aortic pressure-outer diameter curves for P1, P7, P14 and P30 ascending aorta. SMKO aortas at P14 have significantly large outer diameter than CTRL. Lower panel: Aortic pressure-compliance curves for CTRL and SMKO ascending aortas. SMKO aortas show significant differences beginning at P7. N=5–8 mice per group. Bars are means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, Generalized estimating equation. (C) Western blots showing the abundance of ACE, TSP1, Egr1 and the phosphorylation of ERK are increased by transverse aortic constriction (TAC) in wild-type mice. N = 4 mice for Sham and 5 mice for TAC. Bars are means ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001. Exact Wilcoxon rank-sum test for comparison with TSP1 in sham group. All the other comparisons were done by unpaired t-test. (D) Western blots showing Egr1 and TSP1 abundance is increased in postnatal SMKO aortas. The experiment was performed three times with different pools of animals with similar results. N values as in Fig. 1A. *P < 0.05, **P < 0.01, ***P < 0.001, unpaired t-test.
We examined temporal changes in the biomechanical properties of the aortas by pressure-diameter analyses (Fig. 3B). The outer diameter was similar between the genotypes at P1 or P7; however, aneurysmal wall changes were detected at P14 (Fig. 3B, upper panel). Compliance, which is the inverse of stiffness, was significantly different between the genotypes from P7 and it increased at low pressures and decreased at or above physiological pressures in the mutants from P7 (Fig. 3B, lower panel).
The abundance of early growth response-1 (Egr1) was increased in SMKO aortas
We wondered how mechanical alteration and loss of elastic laminae-SMC connections were converted to intracellular signaling. Integrins and focal adhesion kinase (FAK) are mechanosensors that are found in the membrane-associated dense plaques of SMCs (27). The phosphorylation of FAK was transiently decreased in SMKO aortas at P1 but total abundance did not differ between the genotypes after P7 (fig. S7A). The phosphorylation of integrin-linked kinase (ILK) was similar between control and Fbln4SMKO aortas at all time points (figs. S7A and S7B). Egr1 is a zinc-finger transcription factor that is induced by AngII in cardiac fibroblasts and by mechanical stress in vascular SMCs (28,29), and which increases the abundance of thrombospondin-1 (TSP1) and ACE (30,31). We therefore examined if changes in the abundance of these molecules correlated with changes in mechanical stress in the aortic wall. Transverse aortic constriction (TAC) increases mechanical force and lead to aortic dilatation and moderate thickening of the medial layers, which is associated with an increase in the phosphorylation of ERK (32). Based on this finding, we tested if Egr1 could respond to mechanical stress applied to the ascending aortic wall by TAC in wild-type mice (33). Western blot analysis showed a significant increase in the abundance of Egr1, ACE and TSP1 in TAC-operated ascending aortas (Fig. 3C). These data suggested that Erg1, ACE and TSP1 are mechanical stress responsive genes in vivo. The phosphorylation of cofilin was also decreased slightly in TAC-operated animals compared to sham-operated animals (Fig. 3C). We next examined if mechanosensing of SMCs was altered in Fbln4SMKO aorta. The abundance of Egr1 was transiently increased at P1 and remained high in Fbln4SMKO aortas from P14, whereas that of TSP1 was significantly increased at P30, which was maintained until P90 (Fig. 3D). These results indicated that Fbln4SMKO aortas showed biochemical changes similar to pressure overload even under the normal blood pressure conditions that are present in 6–8 week old mutant mice (14).
Postnatal Fbln4 deletion did not lead to cytoskeletal changes and aneurysm formation in vivo
To confirm if structural defects involving the association of the SMCs with the elastic laminae contributed to the full development of the aneurysmal phenotype in vivo, we generated tamoxifen-inducible Fbln4 knockout mice by using SMA-Cre-ERT2, which induces Cre-recombinase in vascular SMCs (iSMKO: Fbln4KO/fl;SMA-Cre-ERT2+)(34). Mice were injected with tamoxifen for five consecutive days beginning at P7, a time point after which the elastic laminae have already been essentially assembled in the aortic wall (35). Tamoxifen-injected Fbln4+/+; Cre-or Cre+, Fbln4fl+; Cre− and Fbln4KO/+; Cre+ mice served as controls. In both control and iSMKO mice, aortas were normal and did not have dilatations or aneurysms (Fig. 4A). In addition, the descending aorta did not exhibit a tortuous appearance, which is characteristic of most mouse models with defective elastic laminae assembly (such as Fbln5−/− and Eln−/− mice)(36,37). Deletion of Fbln4 was confirmed by qPCR (Fig. 4B). To examine if molecular changes had occurred in the iSMKO aorta, we examined the abundance of mechanosensitive molecules as well as the phosphorylation of ERK1/2 and cofilin by Western blot analysis (Fig. 4C). Consistent with a normal phenotype, biochemical markers were similar between control and iSMKO aortas. Immunostaining of cross sections of the iSMKO and control aortas also showed intact elastic laminae and robust staining for phosphorylated cofilin (Fig. 4D). These data suggest that deletion of Fbln4 after the establishment of elastic lamina-SMC connections does not cause abnormal mechanosensing in SMCs under baseline conditions.
Fig. 4. Postnatal deletion of Fbln4 in vascular SMCs.
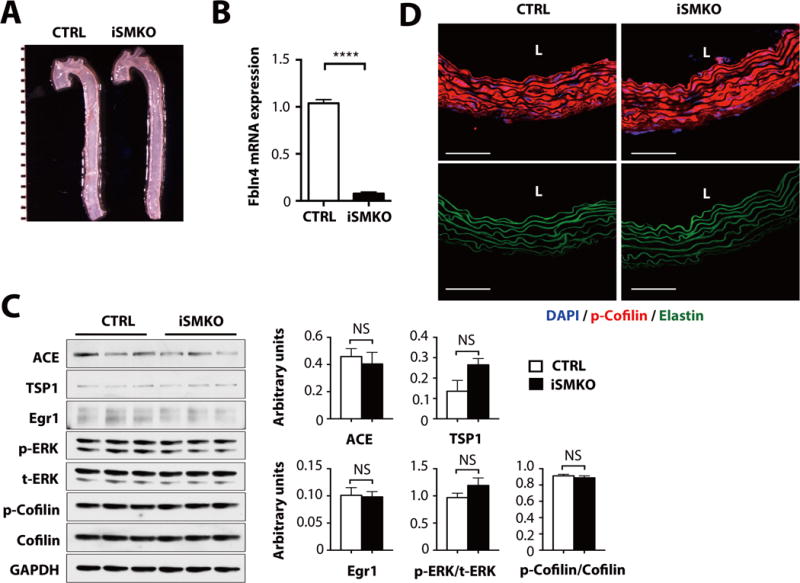
(A) Gross photos of CTRL and SMA-Cre-ERT2 (iSMKO) aortas at P60. Mice were injected with tamoxifen for five consecutive days beginning at P7. Images are representative of 17 mice per genotype. (B) qPCR analysis on aortas harvested from P60 CTRL and iSMKO mice (n=6 mice per genotype). ****P < 0.0001, unpaired t-test. (C) Representative Western blots of ascending aortas from CTRL and iSMKO mice (n=6 mice per genotype). NS, not significant. Exact Wilcoxon rank-sum test for comparison with p-ERK/t-ERK in CTRL group. All the other comparisons were done by unpaired t-test. (D) Cross sections of the ascending aorta from CTRL and iSMKO mice at P60 (n=5 mice per genotype) immunostained with p-cofilin (red) and DAPI (blue). Elastic laminae are green (autofluprescence). Bars are 50 μm.
Losartan effectively decreased SSH1 and cofilin, preventing aneurysm formation
We have previously shown that losartan reverses both the increase in the phosphorylation of ERK and the reduction of the expression of SMC differentiation markers in Fbln4SMKO aortas; we have also established a critical therapeutic time window (P7–P30) for preventing aneurysm development (14). To examine if Egr1, SSH1 and cofilin were associated with the therapeutic effects of losartan, we compared their abundance at P30 in aortas from Fbln4SMKO mice treated or not with losartan between P7 and P30. Losartan treatment markedly decreased the phosphorylation of ERK, partially prevented the increase in the abundance of Egr1 and SSH1, and partially restored the phosphorylation of cofilin (Fig. 5A). Consistently, losartan increased the phosphorylation of cofilin and improved the morphology of elastic fibers in Fbln4SMKO aortas to that detected in control vessels (Fig. 5B) and phalloidin staining showed well-organized actin assembly (Fig. 5C, compare to the phalloidin staining in Fig. 2D). These data suggested that losartan targeted mechanosensitive molecules downstream of AT1R between P7 and P30.
Fig. 5. The effects of losartan on cofilin activity and aneurysm formation.
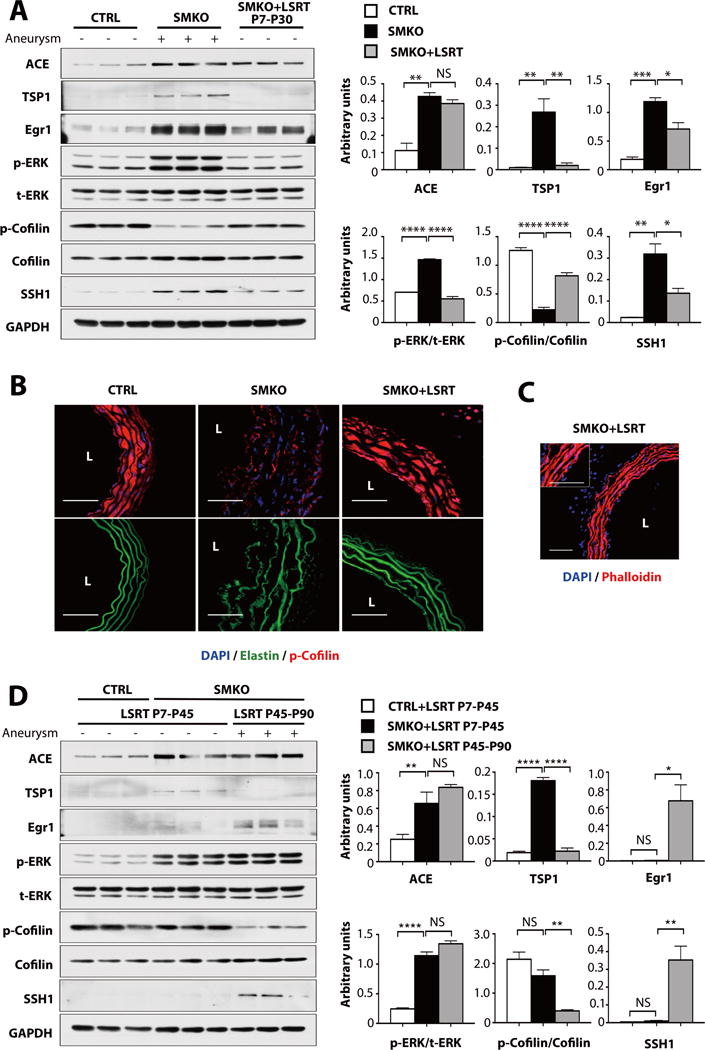
(A) The effects of losartan treatment from P7 to P30 on ACE, TSP1, Egr1, p-ERK, t-ERK, p-cofilin, cofilin and SSH1 abundance in SMKO aortas (pooled 3–6 aortas per sample; 9–17 mice per genotype and treatment). All animals were evaluated at P30. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA. (B–C) Cross sections of the ascending aorta from SMKO with losartan treatment from P7 to P30. L; lumen. n=5 mice per genotype and treatment. Bars are 50 μm. B. Immunostained with p-cofilin (red) and DAPI (blue). Elastic laminae are green (autofluorescence). C. Immunostained with phalloidin (red) and DAPI (blue). N=5 mice per treatment. (D) The effects of postnatal losartan treatment on aneurysm formation. All animals were evaluated at P90. n=3 mice per genotype and treatment. *P < 0.05, **P < 0.01, ****P < 0.0001, one-way ANOVA.
To further examine the relationship between losartan and downstream signaling pathways, we used two losartan treatment protocols in which we treated animals from P7 to P45 or P45 to P90 and harvested aortas from both groups at P90 (Fig. 5D), as previously described (14). Consistent with a previous report (14), aneurysms were completely prevented in Fbln4SMKO mice receiving losartan during the early postnatal period, even when losartan was withdrawn from P45 to P90 (LSRT P7–P45). Treatment starting at P45 did not rescue aneurysm formation under continuous administration of losartan up to P90 (LSRT P45–P90) (Fig. 5D). Surprisingly, the phosphorylation of ERK was increased in Fbln4SMKO aortas after losartan withdrawal. The abundance of Egr1 and SSH1 was decreased only in aortas from Fbln4SMKO mice which had been treated in the early postnatal period (P7–P45) and the phosphorylation of cofilin was increased to an amount comparable to that detected in control mice. Taken together, abnormal mechanosensing and activation of AngII-AT1R pathway led to the activation of cofilin through SSH1 between P7 and P30, which is consistent with the therapeutic window of losartan in Fbln4SMKO aortas.
Inhibition of the SSH1-cofilin pathway prevents aortic aneurysms in vivo
We finally asked if we could prevent aneurysm formation by pharmacologically inhibiting the SSH1-cofilin pathway. PI3K is involved in insulin-induced or AngII-induced activation of SSH1 in vitro (38–40). Therefore, we treated Fbln4SMKO mice with the PI3K inhibitors Wortmannin or LY294002 from P7 to P30 and examined the formation of aneurysms at P30. Neither drug affected body weight at P30 (fig. S8A). Although Wortmannin or LY294002 treatment successfully ameliorated aneurysm formation, tortuosity of the descending aorta was still evident (Fig. 6A, figs. S8B, S9A). In addition, aortas from these drug-treated Fbln4SMKO mice had smaller lumens compared to those from untreated mutants but thicker aortic walls compared to those of control animals, suggesting a balancing of wall tension by an increase in the thickness of aortic wall (Fig. 6B, fig. S9B). Biochemically, both inhibitors decreased the phosphorylation of AKT as well as that of ERK1/2 (Fig. 6C, fig. S9C), and the abundance of TSP1 and Egr1 was decreased in PI3K inhibitor-treated Fbln4SMKO aortas. As expected, SSH1 abundance was significantly decreased and the phosphorylation of cofilin was increased to the amount seen in control mice.
Fig. 6. The involvement of PI3K in aortic aneurysm formation.
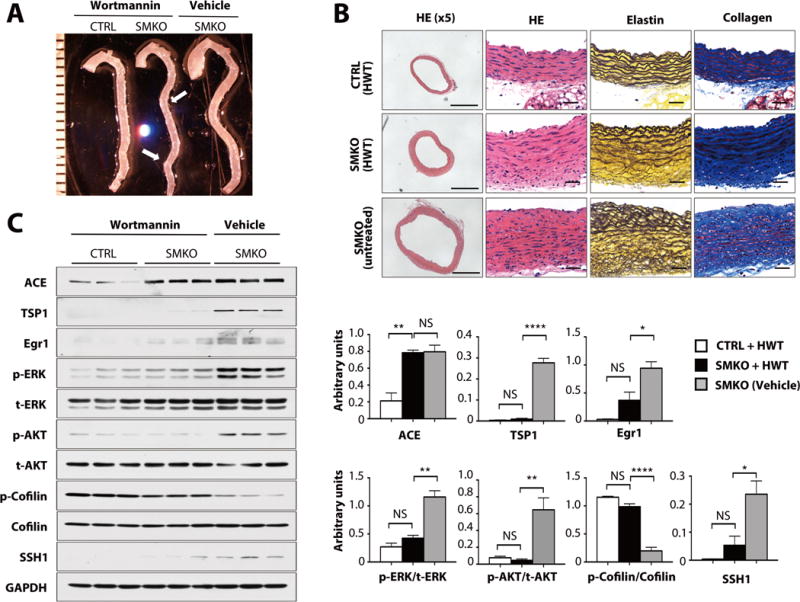
(A) Gross photos of CTRL and SMKO aortas with or without Wortmannin (HWT) treatment. Arrow shows a tortuous descending aorta. Images are representative of 11–16 mice per genotype. (B) Histological images of cross sections of the ascending aorta from HWT-treated CTRL, SMKO and untreated-SMKO mice stained with hematoxylin and eosin (HE), Hart’s (elastin) and Masson-Trichrome (collagen). Scale bars are 500 μm (x5) and 20 μm. Images are representative of 4 mice per genotype. (C) Western blots showing the effect of HWT treatment (pooled 3 aortas per sample) compared to vehicle-treated SMKO (pooled 2 aortas per sample). 6–9 mice per genotype and treatment. *P < 0.05, **P < 0.01, ****P < 0.0001, one-way ANOVA.
Several upstream signaling pathways regulate the phosphatase activity of SSH1 (41). Protein kinase D (PKD) inhibits SSH1 activity by phosphorylating SSH1 (42–44) and inducing its binding to the scaffold protein 14-3-3. Conversely, calcium-induced signaling induces SSH1 activation through the calcineurin-mediated dephosphorylation of SSH1 (45). In Fbln4SMKO aortas, the phosphorylation of PKD was unchanged compared to control aortas (fig. S10A) and activation of calcineurin as deduced by the abundance of its inhibitor RCAN1.4 remained increased even after inhibition of Wortmannin or LY294002 (fig. S10B–D). Taken together, these results suggested that cofilin is downstream of activation of the AT1R by AngII and regulated by PI3K-dependent increases in SSH1 abundance, and appears to be involved in aneurysm progression in Fbln4SMKO mice.
Discussion
Using a combination of 2D-DIGE and established mouse models of ascending aortic aneurysms, we investigated the molecular mechanism(s) driving aneurysm initiation and expansion. We showed that structural changes involving elastic laminae-SMC connections and alteration in mechanical properties of the aortic wall were associated with abnormal mechanosensing of SMCs as evidenced by increasing the abundance of mechanosensitive molecules Egr1, ACE and TSP1. Subsequent activation of the AngII-AT1R pathway lead to aneurysm formation in a manner that is dependent on PI3K and the SSH1-cofilin pathway (Fig. 7).
Fig. 7. A model illustrating a potential mechanism of aneurysm formation in Fbln4SMKO aortas.
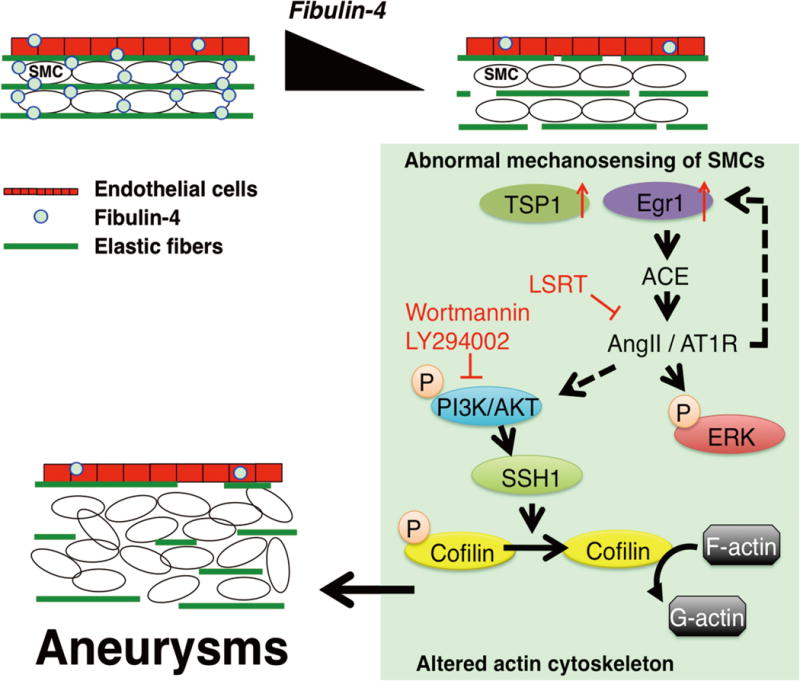
Absence of fibulin-4 in SMCs led to loss of elastic lamina-SMC connections and changes in the mechanical properties of the aorta. Abnormal mechanosensing of SMCs are indicated by increased Egr1, TSP1 and ACE abundance. Increased abundance of ACE leads AngII-mediated signaling and induces downstream events, including (i) increased abundance of Egr1 and establishment of a feed forward loop of AngII signaling, (ii) increased phosphorylation of ERK and proliferation of SMCs, and (iii) PI3K-dependent activation of cofilin through SSH1, leading to the aneurysm formation.
Aneurysm initiation was triggered by altered mechanosensing of Fbln4SMKO SMCs
Temporal expression analysis of Fbln4SMKO aortas before the initiation of aneurysmal changes revealed that differentiated Fbln4SMKO SMCs underwent progressive phenotypic changes from P7 to P30. This period coincides with the completion of elastic laminae formation and the development of mature elastic lamina-cell associations (18,35), as well as an increase in postnatal blood pressure under physiological conditions (46). In normal development of the aortic wall, fibrillin-1 microfibrils provide a scaffold for elastic lamina formation. From late embryogenesis, and during the few weeks of postnatal life, elastin is continually deposited onto this scaffold until mature elastic laminae are formed (18,35). The fibrillin-1 microfibrils also form connections between the elastic laminae and the SMC surface, which become increasingly infiltrated with elastin, anchoring on the cell membrane in regions occupied intracellularly by electron-dense plaques. Because the dense plaques are the sites of actin filament attachment, a continuum of tension is established from within the cell to the elastic laminae (35). Fbln4SMKO aortas exhibited alteration of biomechanical properties and distinct biochemical responses compared to wild-type aortas with an increase in the abundance of mechanosensitive molecules Egr1, ACE and TSP1. These observations suggest that tethering of SMCs to the elastic laminae is required for the proper distribution of increasing wall stress and homeostasis of cellular tension as well as maintenance of the differentiated status of SMCs. Loss of appropriate elastic lamina-SMC connections is instrumental for development of aneurysms, because neither deletion of Fbln4 in SMCs after the connections are formed nor induction of mechanosensitive genes alone without structural defects in wild-type aorta by TAC was sufficient to induce ascending aortic aneurysms in vivo.
Mice with reduced expression of fibrillin-1 (Fbn1mgR/mgR) develop dilated cardiomyopathy due to impaired mechanotransduction of cardiac myocytes, which involves integrin ß1 and FAK (47). Fbn1mgR/mgR cardiac myocytes exhibited nearly absent phosphorylation of FAK and increased phosphorylation of ERK1/2 and Smad2/3, effects that were rescued by losartan, but not by an ACE inhibitor or deletion of angiotensinogen. Because fibrillin-1 has an RGD domain that binds the cell surface integrin ß1(48), it was suggested that a primary defect was a loss of mechanotransduction mediated by fibrillin-1-cell interactions. In our study, loss of fibulin-4, in contrast, resulted in exaggerated mechanosensing in SMCs and activation of RhoA independently of integrins or FAK. Because fibulin-4 has no known cell surface receptors and because it potentiates binding between lysyl oxidase and tropoelastin in vitro (49), it is likely that fibulin-4 is involved in deposition of elastin and formation of elastic lamina-SMC connections, thereby indirectly regulating mechanotransduction mediated by cell-elastic lamina connections. Together, our findings support the involvement of altered mechanotransduction in development of aortic aneurysms in humans (50).
A feed forward loop involving ACE and angiotensin II leads to abnormal actin remodeling and aneurysm expansion
The earliest and consistent biochemical change in Fbln4SMKO aortas was the increased abundance of Egr1, followed by those of ACE and TSP1. Because Egr1 is a transcriptional activator of ACE and downstream target of AngII (29), it is likely that abnormal mechanosensing by SMCs is biochemically translated as the increased abundance of Egr1 followed by local increase of ACE, resulting in production of AngII and activation of the AngII-AT1R pathway. Our analysis also suggests that cofilin is downstream of the AngII-AT1R pathway and regulated by PI3K. The possibility that SSH1 is directly induced by Egr1 and activating cofilin-1 needs to be tested. Our observation that substantial mechanical and biochemical changes occur at P7 in Fbln4SMKO mice emphasizes the importance of a therapeutic time window that targets the establishment of proper connections between elastic lamina and SMCs for effective prevention of aortic aneurysms.
Actin filament remodeling by cofilin is regulated by the balance between phosphorylation and dephosphorylation through LIMK and cofilin phosphatases, respectively (51). Although the abundance of RhoA and LIMK was increased in the Fbln4SMKO aorta, which would be expected to enhance F-actin formation, the increase in actin polymerization appeared to be negated by increased cofilin activity. The reduced F-actin potentially impacts aneurysm expansion by inhibiting MRTF-A-dependent, CArG box-containing, SMC contractile gene expression such as Acta2, Myh11 and Cnn1, and decreasing the contractile forces of SMCs, as has been shown in mice carrying a mutation in MYH11R247C (52,53). Cofilin also increases mitochondria size and abundance through actin remodeling in a SRF-dependent manner (54). Active cofilin translocates to mitochondria together with BAX and induces apoptosis in cardiac myocytes independently of actin filaments (55). These observations indicate that activated cofilin exerts multiple cellular functions both in an actin-dependent and independent manner. The exact role of cofilin during aneurysm development, therefore, warrants further investigation.
Limitations and future prospective
Our study showed that the reduction in the abundance of SSH1 by PI3K inhibitors suppressed cofilin activity and prevented aortic aneurysms in Fbln4SMKO aortas. These results may offer a new potential strategy to treat ascending aortic aneurysms and lumen enlargement in humans. Clinical trials comparing the effects of losartan to β-blocker atenolol on aneurysm growth in Marfan patients show that losartan has no advantages over atenolol (56). Although the underlying disease mechanism is different between Marfan syndrome and fibulin-4 deficiency, it may be beneficial to target a specific intracellular signaling pathway downstream of angiotensin II. In addition, the timing of administration of losartan should be carefully considered to exert most efficient therapeutic effects. Our current studies fail to directly measure the SSH1 activity during aneurysm formation. More thorough studies to determine the specificity of the SSH-cofilin pathway in ascending aortic aneurysms will be necessary to establish drug regimens for precisely controlling and preventing aneurysm formation.
Materials and Methods
Mice
Fbln4SMKO mice were generated as described previously (9,14). Fbln4+/+, Fbln41/+ or Fbln4+/− mice containing SM22α-Cre (57) transgene were used as controls in this study. All mice were kept on a 12 h/12 h light/dark cycle under specific pathogen free condition and all animal protocols were approved by the IACUC of the University of Texas Southwestern Medical Center.
Western blot analysis
Aortas were harvested and perivascular adipose tissue was thoroughly removed. Aortas were divided into ascending thoracic (from the aortic root to immediately past the left subclavian artery) and descending thoracic aortas, then minced in liquid nitrogen by pestle and dissolved in RIPA lysis buffer (Sigma) containing 1% protease inhibitor (Sigma) and 1% phosphatase inhibitor (Sigma). The lysates were mixed with 3x SDS sample buffer with 2-mercaptoethanol and boiled at 95 °C for 5 min, and then were subjected to SDS-PAGE. Proteins were transferred to a PVDF membrane (Millipore) and immunoblotted with antibodies indicated in Supplemental Table S1. Membranes were then incubated with respective anti-mouse or anti-rabbit HRP-conjugated secondary antibody (1:1000, Bio-Rad) and visualized with chemiluminescence kit (Santa Cruz Biotechnology) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific). Quantification of proteins, normalized to GAPDH is shown in the graphs adjacent to Western blots and bars are means ± SEM.
Quantitative PCR
RNA was isolated from aortas at the indicated age using RNeasy Plus Micro Kit (QIAGEN) and 1 μg of total RNA was subjected to reverse transcription reactions using iScript™ Reverse Transcription Supermix (Bio-Rad). SYBR Green was used for Amplicon detection and gene expression was normalized to the expression of housekeeping genes β2-microglobulin (β2m) and GAPDH. PCR reactions were carried out in triplicate in a CFX96 real-time PCR detection system (Bio-Rad) with one cycle of 3 min at 95 °C, then 39 cycles of 10 sec at 95 °C and 30 sec at 55 °C. mRNA abundance was determined using the ddCt method and expressed relative to the mean dCt of controls. Primer sequences are provided in Supplemental Table S2.
2D-DIGE
Entire aortas from P1 (n=4 mice) and ascending aortas from P7 (n=7 mice), P14 (n=5 mice) and P30 (n=3 mice) per genotype (controls and Fbln4SMKO) were collected and combined per each time point and prepared for three independent experiments. Aortas were homogenized in lysis buffer containing 7M Urea, 2M Thiourea, 3% w/v 3-[(3-cholami-dopropyl)-dimethylamonio]-1-Propanesulfonate (CHAPS), and 1% TritonX-100. Fifty μg of individual sample group was labeled with Cy3 or Cy5, and equal amount of sample mix was labeled with Cy2 as the internal standard (15). Labeled samples were mixed and applied to the IPG strips (pH3-10, GE Healthcare life sciences) and first-dimension isoelectric focusing was performed using an Ettan IPGphor 3 (GE Healthcare life sciences). The strips were mounted on top of the 12.5% SDS-polyacrylamide gel for second dimensional protein separation by using an Ettan DALT six system (GE Healthcare life sciences). After SDS-PAGE, gels were scanned using a Typhoon Variable Mode Imager (GE Healthcare life sciences) and quantified using the DeCyder version 6.5 software (GE Healthcare life sciences).
Protein Identification
For spot picking, 500 μg of protein samples without fluorescent labeling were subjected to 2D-DIGE and the gel was stained with DeepPurple (GE Healthcare life sciences). Protein spots of interest were picked with an Ettan Spotpicker (GE Healthcare life sciences). These spots were then destained, reduced and alkylated, following which overnight digestion with trypsin was performed. The resulting tryptic peptides were extracted, dried and cleaned by a SpeedVac. The peptides were then dried again and reconstituted in 10 μL of water, 0.1% TFA. Samples were analyzed using either an Orbitrap Velos or Q Exactive mass-spectrometer (Thermo Fisher) coupled with identical Ultimate 3000 RSLCnano HPLC systems (Dionex, Sunnyvale CA). Samples were loaded onto 75 μm i.d. × 15 cm column and separated with a linear gradient from 0–40% buffer B, where buffer A is water with 0.1% formic acid (FA) and buffer B is CAN with 0.1% FA. CPFP version 2.0.3 was used for database searching against the Uniprot mouse database (58,59). All raw mass spectrometry data files have been deposited to the Mass spectrometry Interactive Virtual Environment (MassIVE; Center for Computational Mass Spectrometry at the University of California, San Diego) and can be accessed using the MassIVE ID MSV000079171. This raw data also links to the ProteomXchange consortium (http://proteomecentral.proteomexchange.org) with the identifier PXD002423. Supplemental Table S3 contains the list of accession file names of raw mass spectrometry data.
Hierarchical Cluster Analysis
Hierarchical cluster analyses were carried out using the Multiple Experimental Viewer (http://www.tm4.org/mev/). The proteins in a given cluster exhibited similar changes in abundance in a synergistic manner.
Immunostaining
Aortas were harvested and embedded in OCT (SAKURA Finetek USA Inc.) and snap frozen in liquid nitrogen. Cross sections of the mouse aorta were immediately fixed with 4% paraformaldehyde for 30 minutes, blocked in 5% normal serum in which secondary antibody was raised, containing 0.1% Triton-X for 1 hour at 37°C, and then incubated with primary antibodies at the following concentrations (Supplemental Table S1). Incubations were performed overnight at 4°C. After washing, high cross-absorbed Alexa Fluor 568-conjugated secondary antibodies (Invitrogen) were added at a dilution of 1:200 for 2 hours at 37°C. Slides were covered with Vectarshield containing DAPI (Vector Laboratories) and viewed under a LSM 510 (ZEISS) or Axio Observer (ZEISS).
G-actin/F-actin assay
The G/F-actin ratio was analyzed in control and Fbln4SMKO aortas using a commercially available kit (Cytoskeleton Inc) according to the manufacturer’s instructions. Briefly, harvested aortas were separated by ascending and descending regions and lysed with lysis buffer (kit provided) containing 1 mmol/L ATP to stabilize F-actin. The lysates were centrifuged at 100,000g for 1 hour at 37 °C, using Beckman ultracentrifuge. The G-actin containing supernatants were then separated from the F-actin containing pellets. The pellets were resuspended to the same volume as the supernatants, using ice-cold water containing 10 μmol/L Cytochalasin D (kit provided). Equivalent volumes of corresponding F- and G-actin fractions were loaded onto an SDS-PAGE gel and analyzed by Western blot with anti-pan-actin antibody (kit provided). G- and F-actin were quantified by using image-J software (NIH Image).
RhoA activity assay
RhoA activity was assessed by a pull-down assay according to the manufacturer’s instructions for a RhoA Activation Assay Biochem Kit (Cytoskeleton). Briefly, aortas from control and Fbln4SMKO mice were lysed in the lysis buffer provided in the kit. GTP-bound RhoA was then immunoprecipitated from cleared lysate with glutathione S-transferase-tagged Rhotekin-Rho-binding domain protein bound to glutathione agarose. The beads were washed and the immunoprecipitates were analyzed by Western blot using RhoA-specific monoclonal antibody provided in the kit. Blots were quantified by using image-J software (NIH Image).
Cell Culture, Transfection and Immunostaining
Rat vascular SMCs (Lonza) were maintained in DMEM/F12 supplemented with 20% fetal bovine serum and 0.1% GA-1000, containing 30 μg/ml Gentamicin and 15 ng/ml Amphotericin (Lonza). For immunostaining, cells were grown on 4-well chamber slide (Thermofisher) and the plasmid DNA encoding human Slingshot-1L (WT or CS, a phosphatase mutant) fused with YFP (generous gift from Dr. Kensaku Mizuno), were transfected into cells using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Forty eight to 72 hrs after transfection, cells were fixed in 100% Methanol at −20°C for 20 minutes, blocked with 2% BSA fraction V (Wako Chemical, Japan) containing 0.1% Tween-20 for 1 hour, and incubated with phospho-cofilin antibodies (Santa Cruz, 1:500) overnight at 4°C. Alexa Fluor 568-conjugated secondary antibodies (Invitrogen) were used at a dilution of 1:200 for 1 hour at 37°C. Slides were covered with Vectarshield containing DAPI (Vector Laboratories). Fluorescence images were obtained using a LSM700 (ZEISS).
Electron Microscopy
Aortas were dissected from P7 and P90 mice following cardiac perfusion with 3% glutaraldehyde in 0.1M sodium cacodylate and samples were prepared as previously described (9). Sections were viewed using a Tecnai 12 transmission electron microscope at 120kV and images were digitally captured. At least 2 wild-type and 3 mutant animals at each time point per genotype were used.
Transverse aortic constriction
Eleven-week old male C57BL/6 mice underwent TAC using a standard surgical protocol (33) with modifications. Briefly, anesthetized mice were placed in a spine position and aortic constriction was achieved by tying a 7-0 polypropylene suture against a 27 gauge blunt needle. For the sham group, the same operation was performed without ligating the aorta. Studies were carried out 3 weeks after TAC, prior to the maladaptive cardiac failure associated with TAC.
Generation of inducible SMC-specific fibulin-4 knockout mice
Transgenic mice have been previously described (34). Fbln4fl/KO mice carrying the SMA-Cre-ERT2 transgene were designated as inducible SMKO (iSMKO). Mice were injected with tamoxifen (0.1 mg/gBW) for five consecutive days beginning at P7, then aortas were harvested at P60 for detection of aortic aneurysms. Deletion of Fbln4 was confirmed by qPCR using aortas harvested at P60. The primers used are in Supplemental Table S2.
Losartan experiment
Losartan (0.6g/liter in drinking water, ad libitum, provided by Merck & Co. Inc. under material transfer agreement 38648) was administered during P7 to P30 or P45 and P45 to P90 as previously described (14).
Wortmannin or LY294002 treatment in vivo
Fbln4SMKO and control pups were divided into two groups: vehicle control and inhibitor treatment. Pups were injected intraperitoneally 17β-hydroxy analog Wortmannin (HWT; 0.25 mg/kg BW, Calbiochem), LY294002 (3.0 mg/kg BW, Calbiochem) or PBS (containing 0.05% DMSO) as previously reported(60–62). The treatments were continued everyday from P7 to P14, then every other day from P14 to P30. At P30, pups were sacrificed and aortas were harvested and evaluated for aneurysm phenotypes.
Histology and morphometric analysis
Aortas were harvested and perfusion-fixed with 4% paraformaldehyde and embedded in paraffin. Five-micrometer sections were stained with H&E for routine histology and Masson Trichrome staining for detection of collagen fibers. Cross-sections of the aorta were stained with Hart’s stain to visualize elastic fibers and images were digitally captured with Leica DM2000 microscope. Morphometric analysis was performed with NIH image J software as described previously (14).
Mechanical testing
Mechanical testing was performed as previously described (63). Briefly, each aorta was horizontally mounted in the mechanical test device (Myograph 110P, DMT, Denmark) in physiologic saline and stretched in the longitudinal direction to the approximate in vivo length and then was pressurized from 0 mmHg (5 mmHg for P1, because the wall was often collapsed at 0 mmHg) to 2–3 times the physiological blood pressure of the mouse (P1: 60 mmHg, P7: 90 mmHg, P14: 130 mmHg, and P30: 175 mmHg). The vessel was preconditioned before obtaining the experimental data, and changes in the outer diameter, internal pressure and axial force were recorded for analysis. The mechanical testing was completed within 3 days of dissection.
Statistical analysis
All experiments are presented as mean ± SEM except Fig. 3B where SD was used. Kolomogorov-Smirnov tests were conducted to examine if the data followed normal distribution. If the data followed a normal distribution, statistical significance was determined by unpaired t-test for two-group comparisons, and one-way analysis of variance (ANOVA) for comparison among three or more groups followed by Bonferroni’s correction for multiple comparison tests. If the normality assumption was violated, nonparametric tests were conducted. Exact Wilcoxon-rank sum tests were used for two-group comparison (Figs. 3C and 4C), and Kruskal-Wallis tests for comparisons among three or more groups followed by Bonferroni’s correction for multiple comparison tests (fig. S8B). Generalized estimating equation (GEE) approach was used for the analysis of repeated measurement data (Fig. 3B). P<0.05 denotes statistical significance.
Supplementary Material
Fig. S1. ACE abundance and ERK phosphorylation in Fbln4SMKO aortas after P7.
Fig. S2. Ontology analysis and proteins that showed changes in abundance during aneurysm development.
Fig. S3. qPCR analysis of genes encoding the proteins identified by 2D-DIGE.
Fig. S4. Phosphorylation of cofilin in Fbln4SMKO descending aortas.
Fig. S5. Activated RhoA signaling in Fbln4SMKO ascending aortas.
Fig. S6. Dephosphorylation of cofilin by SSH1 in rat vascular SMCs.
Fig. S7. Phosphorylation of FAK and ILK in ascending aortas.
Fig. S8. The effects of PI3K inhibitors on general growth and aneurysm development.
Fig. S9. The effect of LY294002 on aneurysm development in Fbln4SMKO mice.
Fig. S10. Evaluation of PKD phosphorylation and calcineurin activity in ascending aortas.
Table S1. Antibodies used in this study.
Table S2. qPCR primer sequences.
Table S3. List of links to raw data for mass spectrometry.
Acknowledgments
We thank G. Urquhart for technical assistance, M. Yanagisawa and A. Suzuki for sharing 2D-Ettan DALT system, P. Chambon and D. Metzger for providing SMA-Cre-ERT2 mice, and K. Mizuno and C. Wu for reagents. We also thank A. Lemoff and the Protein Chemistry Core Laboratory and J. Richardson and Molecular Pathology Core Laboratory at UT Southwestern, and the Facility for Electron Microscopy Research (FEMR) at McGill University for technical assistance, C. Ahn for advice on statistical analysis, B. Rothermel for discussion, and E. Olson for critical reading of the manuscript.
Funding: This work was supported by grants from NIH R01HL106305 (HY and ECD), NIH R01HL115560 and 105314 (JEW), and NIH institutional Training in Cardiovascular Research grant 5T32HL007360-34 and F32 HL122076-01 (CLP), American Heart Association (12EIA8190000 to HY), the Natural Sciences and Engineering Council of Canada (NSERC RGPIN 355710 to ECD), The Robert A. Welch Foundation (grant I-1850 to H.M), and the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant-in-Aid for Young Scientists (B) to YY).
Footnotes
Author contributions: YY designed and acquired experimental data, performed analysis and interpretation of data, and drafted the manuscript. CPL, JK, L-JR, Q-JZ, Z-PL, and HM designed, performed experiments, analyzed and interpretted the data. JEW and ECD contributed to design, analysis, and interpretation of the data, and editing the manuscript. HY represents a senior author of this manuscript contributing to its conception and design, acquisition of experimental data and interpretation of data, and writing the manuscript.
Competing interests: None.
Data and materials availability: Losartan was obtained from Merck & CO., Inc. under MTA 38648. All mass spectrometry data can be accessed at the Proteome Xchange with the identifier PXD002423.
References and Notes
- 1.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 2.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 1995;4 Spec No:1799–1809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- 3.Pope FM, Martin GR, Lichtenstein JR, Penttinen R, Gerson B, Rowe DW, McKusick VA. Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc Natl Acad Sci U S A. 1975;72:1314–1316. doi: 10.1073/pnas.72.4.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dasouki M, Markova D, Garola R, Sasaki T, Charbonneau NL, Sakai LY, Chu ML. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A:2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 5.Renard M, Holm T, Veith R, Callewaert BL, Ades LC, Baspinar O, Pickart A, Dasouki M, Hoyer J, Rauch A, Trapane P, Earing MG, Coucke PJ, Sakai LY, Dietz HC, De Paepe AM, Loeys BL. Altered TGFbeta signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur J Hum Genet. 2010;18:895–901. doi: 10.1038/ejhg.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 8.Doyle AJ, Doyle JJ, Bessling SL, Maragh S, Lindsay ME, Schepers D, Gillis E, Mortier G, Homfray T, Sauls K, Norris RA, Huso ND, Leahy D, Mohr DW, Caulfield MJ, Scott AF, Destree A, Hennekam RC, Arn PH, Curry CJ, Van Laer L, McCallion AS, Loeys BL, Dietz HC. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44:1249–1254. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, Davis EC, Chapman SL, Budatha M, Marmorstein LY, Word RA, Yanagisawa H. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res. 2010;106:583–592. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, Carey J, Wiktorowicz J, Tan FK, Feng XH, Pannu H, Milewicz DM. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88:520–529. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 12.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 13.Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, Boileau C, Coselli JS, Li Z, Leal SM, Shendure J, Rieder MJ, Bamshad MJ, Nickerson DA, T. A. C. R. C. Gen, L. National Heart, P. Blood Institute Grand Opportunity Exome Sequencing. Kim C, Milewicz DM. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93:398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Yamashiro Y, Papke CL, Ikeda Y, Lin Y, Patel M, Inagami T, Le VP, Wagenseil JE, Yanagisawa H. Angiotensin-converting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4-deficient mice. Sci Transl Med. 2013;5(183ra158):181–111. doi: 10.1126/scitranslmed.3005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashiro Y, Takei K, Umikawa M, Asato T, Oshiro M, Uechi Y, Ishikawa T, Taira K, Uezato H, Kariya K. Ectopic coexpression of keratin 8 and 18 promotes invasion of transformed keratinocytes and is induced in patients with cutaneous squamous cell carcinoma. Biochem Biophys Res Commun. 2010;399:365–372. doi: 10.1016/j.bbrc.2010.07.077. [DOI] [PubMed] [Google Scholar]
- 16.Rottner K, Stradal TE. Actin dynamics and turnover in cell motility. Curr Opin Cell Biol. 2011;23:569–578. doi: 10.1016/j.ceb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Zigmond SH. Signal transduction and actin filament organization. Curr Opin Cell Biol. 1996;8:66–73. doi: 10.1016/s0955-0674(96)80050-0. [DOI] [PubMed] [Google Scholar]
- 18.Davis EC. Immunolocalization of microfibril and microfibril-associated proteins in the subendothelial matrix of the developing mouse aorta. J Cell Sci. 1994;107 (Pt 3):727–736. doi: 10.1242/jcs.107.3.727. [DOI] [PubMed] [Google Scholar]
- 19.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 20.San Martin A, Lee MY, Williams HC, Mizuno K, Lassegue B, Griendling KK. Dual regulation of cofilin activity by LIM kinase and Slingshot-1L phosphatase controls platelet-derived growth factor-induced migration of human aortic smooth muscle cells. Circ Res. 2008;102:432–438. doi: 10.1161/CIRCRESAHA.107.158923. [DOI] [PubMed] [Google Scholar]
- 21.Torres RA, Drake DA, Solodushko V, Jadhav R, Smith E, Rocic P, Weber DS. Slingshot isoform-specific regulation of cofilin-mediated vascular smooth muscle cell migration and neointima formation. Arterioscler Thromb Vasc Biol. 2011;31:2424–2431. doi: 10.1161/ATVBAHA.111.232769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- 23.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 24.Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- 25.Kurita S, Watanabe Y, Gunji E, Ohashi K, Mizuno K. Molecular dissection of the mechanisms of substrate recognition and F-actin-mediated activation of cofilin-phosphatase Slingshot-1. J Biol Chem. 2008;283:32542–32552. doi: 10.1074/jbc.M804627200. [DOI] [PubMed] [Google Scholar]
- 26.Hayakawa K, Tatsumi H, Sokabe M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J Cell Biol. 2011;195:721–727. doi: 10.1083/jcb.201102039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morawietz H, Ma YH, Vives F, Wilson E, Sukhatme VP, Holtz J, Ives HE. Rapid induction and translocation of Egr-1 in response to mechanical strain in vascular smooth muscle cells. Circ Res. 1999;84:678–687. doi: 10.1161/01.res.84.6.678. [DOI] [PubMed] [Google Scholar]
- 29.Iwami K, Ashizawa N, Do YS, Graf K, Hsueh WA. Comparison of ANG II with other growth factors on Egr-1 and matrix gene expression in cardiac fibroblasts. Am J Physiol. 1996;270:H2100–2107. doi: 10.1152/ajpheart.1996.270.6.H2100. [DOI] [PubMed] [Google Scholar]
- 30.Grote K, Bavendiek U, Grothusen C, Flach I, Hilfiker-Kleiner D, Drexler H, Schieffer B. Stretch-inducible expression of the angiogenic factor CCN1 in vascular smooth muscle cells is mediated by Egr-1. J Biol Chem. 2004;279:55675–55681. doi: 10.1074/jbc.M406532200. [DOI] [PubMed] [Google Scholar]
- 31.Mungunsukh O, Marquez AP, Lee YH, Thiel G, Day RM. Characterization of the bovine angiotensin converting enzyme promoter: essential roles of Egr-1, ATF-2 and Ets-1 in the regulation by phorbol ester. Gene. 2008;421:81–88. doi: 10.1016/j.gene.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Kuang SQ, Geng L, Prakash SK, Cao JM, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol. 2013;33:2172–2179. doi: 10.1161/ATVBAHA.113.301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010 doi: 10.3791/1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wendling O, Bornert JM, Chambon P, Metzger D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18. doi: 10.1002/dvg.20448. [DOI] [PubMed] [Google Scholar]
- 35.Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 36.Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, Olson EN. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 37.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. The importance of elastin to aortic development in mice. Am J Physiol Heart Circ Physiol. 2010;299:H257–264. doi: 10.1152/ajpheart.00194.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishita M, Wang Y, Tomizawa C, Suzuki A, Niwa R, Uemura T, Mizuno K. Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase Slingshot and its role for insulin-induced membrane protrusion. J Biol Chem. 2004;279:7193–7198. doi: 10.1074/jbc.M312591200. [DOI] [PubMed] [Google Scholar]
- 39.Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell. 2009;20:2650–2660. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Homma Y, Kanno S, Sasaki K, Nishita M, Yasui A, Asano T, Ohashi K, Mizuno K. Insulin receptor substrate-4 binds to Slingshot-1 phosphatase and promotes cofilin dephosphorylation. J Biol Chem. 2014;289:26302–26313. doi: 10.1074/jbc.M114.565945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol. 2013;14:405–415. doi: 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peterburs P, Heering J, Link G, Pfizenmaier K, Olayioye MA, Hausser A. Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 2009;69:5634–5638. doi: 10.1158/0008-5472.CAN-09-0718. [DOI] [PubMed] [Google Scholar]
- 44.Barisic S, Nagel AC, Franz-Wachtel M, Macek B, Preiss A, Link G, Maier D, Hausser A. Phosphorylation of Ser 402 impedes phosphatase activity of slingshot 1. EMBO Rep. 2011;12:527–533. doi: 10.1038/embor.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem. 2005;280:12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- 46.Ishii T, Kuwaki T, Masuda Y, Fukuda Y. Postnatal development of blood pressure and baroreflex in mice. Auton Neurosci. 2001;94:34–41. doi: 10.1016/S1566-0702(01)00339-3. [DOI] [PubMed] [Google Scholar]
- 47.Cook JR, Carta L, Benard L, Chemaly ER, Chiu E, Rao SK, Hampton TG, Yurchenco P, Costa KD, Hajjar RJ, Ramirez F. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124:1329–1339. doi: 10.1172/JCI71059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, Dietz HC. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503:126–130. doi: 10.1038/nature12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horiguchi M, Inoue T, Ohbayashi T, Hirai M, Noda K, Marmorstein LY, Yabe D, Takagi K, Akama TO, Kita T, Kimura T, Nakamura T. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A. 2009;106:19029–19034. doi: 10.1073/pnas.0908268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116:1448–1461. doi: 10.1161/CIRCRESAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25:457–469. doi: 10.1016/j.cellsig.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Kuang SQ, Kwartler CS, Byanova KL, Pham J, Gong L, Prakash SK, Huang J, Kamm KE, Stull JT, Sweeney HL, Milewicz DM. Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res. 2012;110:1411–1422. doi: 10.1161/CIRCRESAHA.111.261743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007;316:1749–1752. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- 54.Beck H, Flynn K, Lindenberg KS, Schwarz H, Bradke F, Di Giovanni S, Knoll B. Serum Response Factor (SRF)-cofilin-actin signaling axis modulates mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:E2523–2532. doi: 10.1073/pnas.1208141109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiang SY, Ouyang K, Yung BS, Miyamoto S, Smrcka AV, Chen J, Heller Brown J. PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci Signal. 2013;6:ra108. doi: 10.1126/scisignal.2004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Selamet Tierney ES, Levine JC, Atz AM, Benson DW, Braverman AC, Chen S, De Backer J, Gelb BD, Grossfeld PD, Klein GL, Lai WW, Liou A, Loeys BL, Markham LW, Olson AK, Paridon SM, Pemberton VL, Pierpont ME, Pyeritz RE, Radojewski E, Roman MJ, Sharkey AM, Stylianou MP, Wechsler SB, Young LT, Mahony L, I. Pediatric Heart Network Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371:2061–2071. doi: 10.1056/NEJMoa1404731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trudgian DC, Thomas B, McGowan SJ, Kessler BM, Salek M, Acuto O. CPFP: a central proteomics facilities pipeline. Bioinformatics. 2010;26:1131–1132. doi: 10.1093/bioinformatics/btq081. [DOI] [PubMed] [Google Scholar]
- 59.Trudgian DC, Mirzaei H. Cloud CPFP: a shotgun proteomics data analysis pipeline using cloud and high performance computing. J Proteome Res. 2012;11:6282–6290. doi: 10.1021/pr300694b. [DOI] [PubMed] [Google Scholar]
- 60.Teranishi F, Takahashi N, Gao N, Akamo Y, Takeyama H, Manabe T, Okamoto T. Phosphoinositide 3-kinase inhibitor (wortmannin) inhibits pancreatic cancer cell motility and migration induced by hyaluronan in vitro and peritoneal metastasis in vivo. Cancer Sci. 2009;100:770–777. doi: 10.1111/j.1349-7006.2009.01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest. 2001;108:1387–1395. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duan W, Aguinaldo Datiles AM, Leung BP, Vlahos CJ, Wong WS. An anti-inflammatory role for a phosphoinositide 3-kinase inhibitor LY294002 in a mouse asthma model. Int Immunopharmacol. 2005;5:495–502. doi: 10.1016/j.intimp.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 63.Le VP, Knutsen RH, Mecham RP, Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ Physiol. 2011;301:H221–229. doi: 10.1152/ajpheart.00119.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ACE abundance and ERK phosphorylation in Fbln4SMKO aortas after P7.
Fig. S2. Ontology analysis and proteins that showed changes in abundance during aneurysm development.
Fig. S3. qPCR analysis of genes encoding the proteins identified by 2D-DIGE.
Fig. S4. Phosphorylation of cofilin in Fbln4SMKO descending aortas.
Fig. S5. Activated RhoA signaling in Fbln4SMKO ascending aortas.
Fig. S6. Dephosphorylation of cofilin by SSH1 in rat vascular SMCs.
Fig. S7. Phosphorylation of FAK and ILK in ascending aortas.
Fig. S8. The effects of PI3K inhibitors on general growth and aneurysm development.
Fig. S9. The effect of LY294002 on aneurysm development in Fbln4SMKO mice.
Fig. S10. Evaluation of PKD phosphorylation and calcineurin activity in ascending aortas.
Table S1. Antibodies used in this study.
Table S2. qPCR primer sequences.
Table S3. List of links to raw data for mass spectrometry.