

Abstract
A practical procedure for 11CN-labeling of native peptides has been developed. The process involves two sequential Pd-mediated cross-coupling reactions at the cysteine residue of a peptide and operates under mild conditions. The method was shown to be highly chemoselective for cysteine over other potentially nucleophilic residues and the radiolabelled products were synthesized and purified in less than 15 minutes. Appropriate for biomedical applications, the method could be used on extremely small scale (20 nmol) with high radiochemical yield. The success of the protocol stems from the use of a Pd-reagent based on a dihaloarene, which enables direct “nucleophile-nucleophile” coupling of the peptide and [11C]-cyanide by temporal separation of nucleophile addition.
Graphical Abstract
Positron emission tomography (PET) is a sensitive molecular imaging technique that can be used to study the biochemical behavior of molecules in vivo.1 Among various classes of radiotracers for PET imaging, radiolabeled peptides have gained considerable attention for their potential to measure the distribution and pharmacokinetics of peptide-based therapeutics and their potential to serve as imaging biomarkers for therapy. In fact, the favorable pharmacokinetic behavior and high specificity with their respective target receptors make them exceptionally well-suited as imaging agents for certain applications, for example in oncology.2 The demonstrated success and future promise of peptide-based imaging agents have stimulated a real need to increase the number and diversity of efficient methods for tagging peptides with radioactive functional groups.3
Chemical modification of peptides imposes unique challenges associated with the need for mildness of conditions and high chemoselectivity due to the existence of a variety of reactive functional groups present in amino acid side chains.4 Moreover, challenges with radiochemical modification of peptides are exacerbated by the use of short-lived. Short isotope half-life of course leads to time-sensitive radiosynthesis that requires a high reaction rate, and also dictates that the reaction profile is amenable to rapid purification of the desired radioactive product. Methods that function in both capacities have been developed by using radiometals, 18F, and more rarely 11C. Previous methods can be broadly divided into three categories: 1) introduction of a radionuclide to a prosthetic group in a peptide; 2) reaction of a peptide with a prosthetic group, that is pre-labeled with a radionuclide; and 3) direct labeling of a native peptide (Figure 1).
Figure 1.

Strategies for Peptide Radiolabeling.
In the first approach, a peptide of interest is pre-functionalized with a prosthetic group, which usually chelates a radiometal5,6 or forms a covalent bond with [18F]-fluoride (Figure 1, a).7,8 This approach, however, inevitably involves multiple synthetic operations on delicate peptide substrates, which diminishes the practicality of the method. Alternatively, a prosthetic group can be treated with the radionuclide to generate a radiochemical entity prior to introduction to the peptide (Figure 1, b).9,10 Despite the advantage of avoiding exposure of sensitive peptide substrates to harsh radiochemical reaction conditions (e.g., basic fluoride), the system mandates intensive maneuvering of the radioactive material, which includes time-sensitive synthetic and purification processes. Most importantly, these aforementioned methods involve major structural modifications of the peptide target, which may result in a change in biological activity of the radiotracer.
In contrast, a synthetic strategy, which allows for direct attachment of a radionuclide or a radiochemical entity that can be easily generated from an automated synthesis module onto an unmodified peptide, should serve as an ideal tool to access peptide radiotracers (Figure 1, c).11 The reported protocols of this category, however, usually require harsh reaction conditions (e.g., elevated temperature, use of a media with extreme pH, and/or the employment of a large quantity of the peptide substrate), or are limited by the available positions of radiolabelling. Therefore wide applications to complex systems are hampered.
Recently, we have demonstrated that Pd-based oxidative addition complexes supported by biarylphosphine ligands are able to mediate challenging cross-coupling reactions in highly specialized settings. Using this strategy, mild and efficient [11C]-cyanation of aryl (pseudo)halides12 (Figure 2, a) and arylation of a cysteine residue in native peptides13 (Figure 2, b) have been realized. We envisioned that a combination of these two approaches might find utility as a direct radiolabeling method for unmodified peptides. We hypothesized that the use of a doubly electrophilic organometallic entity (A) would be able to adjoin the precious radionuclide and equally important peptide substrate (Figure 2, c). Using this type of electrophile (A), we are able to fully exploit the nucleophilic nature of the desired reaction components (i.e., [11C]-cyanide and the thiol of cysteine) without rendering either one of them electrophilic by relying on oxidative conditions. This approach, however, could potentially suffer from inefficient generation of the desired product, as a slight bias in reaction kinetics or stoichiometry could result in the exclusive formation of symmetric coupling products (B and C).14
Figure 2.

Design Principle and Synthetic Strategy.
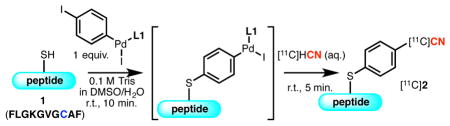
We reasoned that a sequential introduction of the nucleophilic components and the use of an appropriate organometallic reagent could resolve the problem. Instead of initiating the overall process with a binary electrophile A, a mono-organometallic compound originating from a dihaloarene (D) (Figure 2, d), could be employed. Exposure of D to a cysteine-containing peptide precursor results in C–S bond formation, and provide intermediate (E) and ligand bound Pd(0) species (F) (step a). Intermediate F is then able to undergo in situ oxidative addition with E to form another electrophilic mono-organometallic species (G). Provided that the oxidative addition proceeds with a significantly differentiated rate compared to that of C–S bond formation, the generation of the new electrophile G should occur without significantly interfering with the initial coupling.15 Electrophile G can, after addition of [11C]-cyanide, be converted to the final product H via “transmetallation” and subsequent reductive elimination (step b).
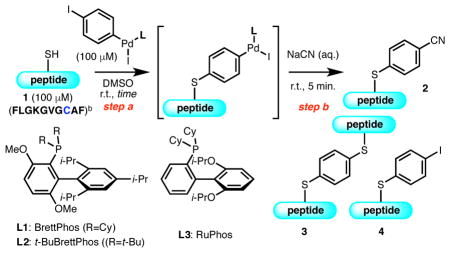
To evaluate our hypothesis, we studied the introduction of a non-radioactive cyanide source to model peptide 1 bearing an intrachain cysteine residue (Table 1). The peptide substrate was first treated with an oxidative addition complex derived from 1,4-diiodobenzene in DMSO, followed by an aqueous solution of NaCN. In the presence of the Pd complex supported by BrettPhos (L1), an efficient ligand for the [11C]-cyanation,12 the desired double cross-coupling product (2) was consistently observed in 34% average yield (Table 1, entry 1). Importantly, throughout the investigation double coupling product 3 and the intermediate from the first cross-coupling 4 were observed as the only peptidic side products, which were easily separable from the desired product. With the cyanide source as the limiting reagent, a condition that is usually employed in radiosynthesis, the product was obtained in consistently high yield, suggesting application to radiosynthesis was possible (Table 1, entry 2–3). Other variations in the reaction conditions, such as the use of an excess amount of Pd complex (Table 1, entry 4), elongated reaction time for step a (Table 1, entry 5), or buffered reaction media, a condition that should allow for wider application of the method, (Table 1, entry 6), did not significantly affect the outcome. The use of another effective ligand for [11C]-cyanation, t-BuBrettPhos (L2), also gave a high level of reactivity for the desired transformation (Table 1, entry 7).12 In contrast, the use of RuPhos, the ligand employed in cysteine arylation,13 was much less effective and provided 4 as the major product (Table 1, entry 8). As oxidative addition of LPd(0) (F) and ArX occurs rapidly (Figure 2, d), we attribute the formation of 4 to the regeneration of Ar–I from G under conditions where cyanation is inefficient.16 Therefore, ligand effects on both cysteine arylation (Figure 2, d, step a) and cyanide transfer to G (Figure 2, d, step b) contribute to the efficiency of the overall process to form radiolabelled product 2.
Table 1.
Non-radioactive Cyanation of a Peptide.a
| ||||
|---|---|---|---|---|
| entry | L | reaction time of step a (min.) | stoichimotetry of NaCN (equiv.) | yield (%)c |
| 1 | L1 | 10 | 1 | 34d |
| 2 | L1 | 10 | 0.2 | 46 |
| 3 | L1 | 10 | 0.1 | 61 |
| 4e | L1 | 10 | 1 | 31 |
| 5 | L1 | 30 | 1 | 33 |
| 6f | L1 | 10 | 1 | 31 |
| 7 | L2 | 10 | 1 | 64 |
| 8 | L3 | 10 | 1 | 13 |
Reaction conditions: 1 (20 nmol), Pd complex (20 nmol), DMSO (200 μL), r.t., 10–30 min.; NaCN (aq.) in 10 μL H2O, 5 min.
AcNH-FLGKGVGCAF-CO2H.
Determined by integration of HPLC peak area with calibration.
Average of three runs.
Results with 1.5 equivalent of Pd complex.
Result with 0.1 M Tris buffer in DMSO/H2O (9:1) in place of DMSO.
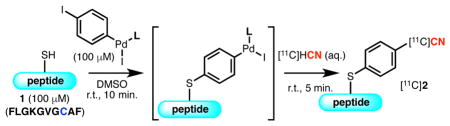
The optimized conditions were tested in a radiosynthesis using aqueous [11C]HCN as a [11C]-cyanide source (Table 2). While the results with the non-radioactive cyanide source were successfully extended to the radioactive system using L1 (Table 2, entry 1),17 the Pd complex supported by L2 or L3 showed significantly reduced reactivity (Table 2, entries 2 and 3). The contrast in reactivity compared to the non-radioactive system exemplifies the challenges associated with the extension of a synthetic method to a radiochemical setting. Nonetheless, the use of L1 gave good reactivity under both non-radioactive and radiosynthetic conditions, and thus was chosen as the ligand for subsequent studies.
Table 2.
[11C]-Cyanation of a Peptide.a
| ||
|---|---|---|
| entry | L | RCY (%) |
| 1 | L1 | 50 |
| 2 | L2 | 7 |
| 3 | L3 | < 5 |
Reaction conditions: 1 (20 nmol), Pd complex (20 nmol), DMSO (200 μL), r.t., 10 min.; [11C]HCN in H2O (20 μL, 1–10 mCi), r.t., 5 min.
Next, we assessed the robustness of the method by evaluating the radiosynthetic efficiency of the reaction in the presence of water or with reduced amount of the peptide substrate (Table 3). While decrease in reactivity was observed with increased water content in the reaction media, up to 10 vol% of water was tolerated providing the radiolabeled product in a practically useful yield (Table 3, entries 1–3). The effect of substrate concentration was also tested (Table 3, entries 4–7). The concentration of the peptide substrate could be reduced to 10 μM without significant loss of reactivity (Entries 4–7). It is expected that the low mass requirement of the peptide precursor will be advantageous in terms of simplifying the purification process of the radiolabelled product, and allowing for the radiolabelling of peptides with limited availability. In addition, we verified that the cysteine residue is the location of radiolabelling through a control experiment. When the reaction was set up utilizing an alternative peptide containing a serine residue in place of cysteine, no labeled peptide product was detected (Table 3, entry 8).
Table 3.
Robustness of the Method.a
| |||
|---|---|---|---|
| entry | [peptide] (μM) | DMSO/H2O | RCY (%) |
| 1 | 500 | 90/10 | 19 |
| 2 | 500 | 33/67 | 3 |
| 3 | 500 | 5/95 | 4 |
| 4 | 250 | 90/10 | 22 |
| 5 | 50 | 90/10 | 20 |
| 6 | 10 | 90/10 | 19 |
| 7 | 2 | 90/10 | < 1 |
| 8b | 500 | 90/10 | 0 |
Reaction conditions: 1 (1.0 equiv.), Pd complex (1.0 equiv.), 0.1 M Tris buffer in DMSO/H2O (200 μL), r.t., 10 min.; [11C]HCN in H2O (20 μL, 1–10 mCi), r.t., 5 min.
Result with AcNH-FLGKGVGSAF-CO2H.
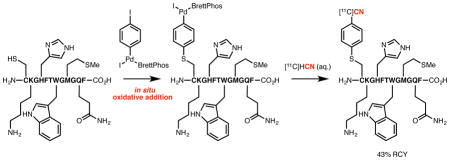
The method exhibits high chemoselectivity for the radiolabeling of the cysteine residue in the presence other potentially nucleophilic functional groups (Figure 3). In addition to the N-terminal amine and C-terminal carboxylic acid, nucleophilic side chain functional groups, such as an amino group (5), an imidazole (5), an indole (5), a thioether (5), an amide (5), a carboxylic acid (6), a guanidine (6), and a hydroxyl group (6), are all tolerated. It is also noteworthy that the position of the reactive cysteine residue is not limited to the intrachain location. It can be positioned either at the N-terminus (5) or C-terminus (6) without impeding the efficiency of the process.18 Moreover, a peptide containing the RGD sequence, a crucial motif in developing PET probes for tumor imaging,19 could be successfully modified.
Figure 3. Functional Group Compatibility of the Method.a.

aReaction conditions: 5 (H2N-CKGHFTWGMGQF-CO2H) or 6 (H2N-GRGDSPC-CO2H) (20 nmol), Pd complex (20 nmol), DMSO (200 μL), r.t., 10 min.; [11C]HCN in H2O (20 μL, 1–10 mCi), r.t., 5 min.
Finally, the protocol was applied to a high-activity synthesis to demonstrate the practicality of the strategy (Figure 4). In the presence of 20 mCi of [11C]-cyanide, the radiolabelled product was obtained in 10% non-decay corrected radiochemical yield after HPLC purification. The absolute radioactivity (2.0 mCi) and the specific activity (1.0 Ci/μmol) were sufficiently high for in vivo imaging and the overall process, including the purification, took only 15 minutes, which is highly suitable for clinical practice. Importantly, the purified product contained only 1.2 ppb of Pd, which is well below the accepted limit, based on ICP-MS analysis.20
Figure 4.

High Activity Synthesis.
In summary, we have developed a one-pot protocol for 11CN-labeling of cysteine-containing peptides based on the combination of Pd-mediated cyanation and cysteine arylation. This strategy exploits the nucleophilicity of both the peptide and labeling reagent, [11C]-cyanide, which enables “nucleophile-nucleophile coupling” in a direct manner. The method tolerates a wide range of reactive functional groups due to its mild reaction conditions and high chemoselectivity. A key feature of this method is that it can be successfully used on an extremely small amount of the peptide precursor (20 nmol), which not only simplifies purification, but is also economically advantageous. The ease of operation of this method should allow for the development of 11C-peptide PET probes and also the evaluation of the therapeutic efficacy of peptides through PET imaging.
Supplementary Material
Acknowledgments
One of the authors (H.G.L.) thanks Professor Bradley Pentelute (MIT) for his unconditional support and encouragement. Research reported in this publication was supported by the National Institutes of Health (NIH) under award number R01GM46059 (S.L.B.) and S10RR017208 (J.M.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We acknowledge the Department of Energy (DE-SC0008430) and the Phyllis and Jerome Lyle Rappaport MGH Research Scholar Award. We also acknowledge the National Center for Research Resources (P41RR14075) and the Athinoula A. Martinos Center for Biomedical Imaging for resource support. Dr. Ekaterina Vinogradova (MIT) is acknowledged for performing preliminary experiments. We also thank Dr. Nicolas White (MIT) and Dr. Mycah Uehling (MIT) for assistance with the preparation of the manuscript.
Footnotes
Notes
The authors declare the following competing financial interest: MIT has obtained or has filed patents on some of the ligands/precatalysts that are described in the paper from which S.L.B. and former/current coworkers receive royalty payments.
Experimental details and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Ametamey SM, Honer M, Schubiger PA. Chem Rev. 2008;108:1501. doi: 10.1021/cr0782426.Miller PW, Long NJ, Vilar R, Gee AD. Angew Chem Int Ed. 2008;47:8998. doi: 10.1002/anie.200800222.Placzek MS, Zhao W, Wey HY, Morin TM, Hooker JM. Semin Nucl Med. 2016;46:20. doi: 10.1053/j.semnuclmed.2015.09.001.Weissleder R. Science. 2006;312:1168. doi: 10.1126/science.1125949.Gambhir SS. Nat Rev Cancer. 2002;2:683. doi: 10.1038/nrc882.
- 2.For reviews, see: Richter S, Wuest F. Molecules. 2014;19:20536. doi: 10.3390/molecules191220536.Tolmachev V, Stone-Elander S. Biochim Biophys Acta. 2010;1800:487. doi: 10.1016/j.bbagen.2010.02.002.Aloj L, Morelli G. Curr Pharm Des. 2004;10:3009. doi: 10.2174/1381612043383511.
- 3.(a) Fani M, Maecke HR, Okarvi SM. Theranostics. 2012;2:481. doi: 10.7150/thno.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Okarvi SM. Med Res Rev. 2004;24:357. doi: 10.1002/med.20002. [DOI] [PubMed] [Google Scholar]
- 4.For reviews, see: Hu Q, Berti F, Adamo R. Chem Soc Rev. 2016;45:1691. doi: 10.1039/c4cs00388h.Boutureira O, Bernardes GJL. Chem Rev. 2015;115:2174. doi: 10.1021/cr500399p.Lau YH, de Andrade P, Wu Y, Spring DR. Chem Soc Rev. 2015;44:91. doi: 10.1039/c4cs00246f.Baslé E, Joubert N, Pucheault M. Chem Biol. 2010;17:213. doi: 10.1016/j.chembiol.2010.02.008.
- 5.For reviews, see: Brasse D, Nonat A. Dalton Trans. 2015;44:4845. doi: 10.1039/c4dt02911a.Cutler CS, Hennkens HM, Sisay N, Huclier-Markai S, Jurisson SS. Chem Rev. 2013;113:858. doi: 10.1021/cr3003104.
- 6.Chelation strategies generally have an advantage in terms of the small amount of precursor peptide required. In several cases where a peptide is employed for human imaging (e.g. [68Ga]DOTA-TOC) the amount of precursor used is so small that the labelled and unlabelled peptide need not be separated in the preparation of the administered dose. This is in contrast to most, if not all, of the methods used with labelled organic molecules.
- 7.(a) Mu L, Hohne A, Schubiger PA, Ametamey SM, Graham K, Cyr JE, Dinkelborg L, Stellfeld T, Srinivasan A, Voigtmann U, Klar U. Angew Chem Int Ed. 2008;47:4922. doi: 10.1002/anie.200705854. [DOI] [PubMed] [Google Scholar]; (b) Schirrmacher R, Bradtmoller G, Schirrmacher E, Thews O, Tillmanns J, Siessmeier T, Buchholz HG, Bartenstein P, Wangler B, Niemeyer CM, Jurkschat K. Angew Chem Int Ed. 2006;45:6047. doi: 10.1002/anie.200600795. [DOI] [PubMed] [Google Scholar]; (c) Laverman P, McBride WJ, Sharkey RM, Goldenberg DM, Boerman OC. J Labelled Comp Radiopharm. 2014;57:219. doi: 10.1002/jlcr.3161. [DOI] [PubMed] [Google Scholar]; (d) Li Y, Liu Z, Lozada J, Wong MQ, Lin KS, Yapp D, Perrin DM. Nucl Med, Biol. 2013;40:959. doi: 10.1016/j.nucmedbio.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 8.For an example of 11C-based radiolabeling of this category, see: Cornilleau T, Audrain H, Guillemet A, Hermange P, Fouquet E. Org Lett. 2015;17:354. doi: 10.1021/ol503471e.
- 9.For selected examples of this category, see: Chiotellis A, Sladojevich F, Mu L, Herde AM, Valverde IE, Tolmachev V, Schibli R, Ametamey SM, Mindt TL. Chem Commun. 2016;52:6083. doi: 10.1039/c6cc01982j.Jacobson O, Yan X, Ma Y, Niu G, Kiesewetter DO, Chen X. Bioconjugate Chem. 2015;26:2016. doi: 10.1021/acs.bioconjchem.5b00278.Way JD, Bergman C, Wuest F. Chem Commun. 2015;51:3838. doi: 10.1039/c5cc00182j.Marik J, Sutcliffe JL. Tetrahedron Lett. 2006;47:6681.
- 10.Notably, most of these approaches still require synthetic modifications of peptide precursors.
- 11.To the best of our knowledge there are only two reports that meet these requirements. Chin J, Vesnaver M, Bernard-Gauthier V, Saucke-Lacelle E, Wängler B, Wängler C. Amino Acids. 2013;45:1097. doi: 10.1007/s00726-013-1562-5.Hanyu M, Takada Y, Hashimoto H, Kawamura K, Zhang MR, Fukumura T. J Pep Sci. 2013;19:663. doi: 10.1002/psc.2546. During the preparation of the manuscript, another report that meets these criteria has been published. Anderson TL, Nordeman P, Christoffersen HF, Audrain H, Antoni G, Skrydstrup T. Angew Chem Int Ed. doi: 10.1002/anie.201700446.
- 12.Lee HG, Milner PJ, Placzek MS, Buchwald SL, Hooker JM. J Am Chem Soc. 2015;137:648. doi: 10.1021/ja512115s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vinogradova EV, Zhang C, Spokoyny AM, Pentelute BL, Buchwald SL. Nature. 2015;526:687. doi: 10.1038/nature15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Recently, an elegant approach that addresses related issues with kinetic discrimination of nucleophiles has been reported. Fyfe JWB, Fazakerley NJ, Watson AJB. Angew Chem Int Ed. 2017;56:1249. doi: 10.1002/anie.201610797.
- 15.Related multiple cross-coupling with a single nucleophile has been reported. Larrosa I, Somoza C, Banquy A, Goldup SM. Org Lett. 2011;13:146. doi: 10.1021/ol1027283.
- 16.The regeneration of an aryl iodide from the corresponding Pd oxidative addition complex supported by a related ligand has been documented. Milner PJ, Maimone TJ, Su M, Chen J, Müller P, Buchwald SL. J Am Chem Soc. 2012;134:19922. doi: 10.1021/ja310351e.
- 17.Other radioactive species in the reaction mixture are not originating from the peptide substrate, as was confirmed by a control experiment using identical conditions with the peptide substrate left out. See Supporting Information for details.
- 18.Some reported methods rely on the combined reactivity of the side chain and N-terminal amine. See Ref. 11b.
- 19.(a) Shi J, Wang F, Liu S. Biophys Rep. 2016;2:1. doi: 10.1007/s41048-016-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ruoslahti E. Annu Rev Cell Dev Biol. 1996;697(20) doi: 10.1146/annurev.cellbio.12.1.697. For guidelines of residual metal contents, see: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003587.pdf. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.