

Abstract
Background: Mutations of the human K-Ras 4B (K-Ras) G protein are associated with a significant proportion of all human cancers. Despite this fact, a comprehensive analysis of K-Ras interactions is lacking. Our investigations focus on characterization of the K-Ras interaction network. Materials and Methods: We employed a biotin ligase-tagging approach, in which tagged K-Ras proteins biotinylate neighbor proteins in a proximity-dependent fashion, and proteins are identified via mass spectrometry (MS) sequencing. Results: In transfected cells, a total of 748 biotinylated proteins were identified from cells expressing biotin ligase-tagged K-Ras variants. Significant differences were observed between membrane-associated variants and a farnesylation-defective mutant. In pancreatic cancer cells, 56 K-Ras interaction partners were identified. Most of these were cytoskeletal or plasma membrane proteins, and many have been identified previously as potential cancer biomarkers. Conclusion: Biotin ligase tagging offers a rapid and convenient approach to the characterization of K-Ras interaction networks.
Keywords: Proximity mapping, pancreatic cancer, subcellular localization
Mutations of the human Ras proteins, H-Ras, N-Ras, and K-Ras are associated with a third of all human cancers, and K-Ras itself accounts for over 80% of these mutations (1-7). These 21-kDa G proteins bind guanosine nucleotides, and when in the GTP-bound form activate effector proteins, promoting cell growth (1-9). Mutation of K-Ras glycine residue twelve to an aspartic acid (G12D) renders the protein less sensitive to GTP hydrolysis, and thus constitutively active (5,10-11). In contrast, serine-to-arginine mutation at K-Ras residue 17 (S17N) yields a protein that preferentially binds GDP, and is defective for effector protein activation (12-13). Post-translational modifications are also critical to Ras protein function (1,5-6,14-19). For the dominant K-Ras 4B splice variant (hereafter referred to simply as K-Ras), these involve farnesylation at cysteine residue 185 (C185), proteolytic cleavage of the three C-terminal residues, and carboxymethylation of the now C-terminal C185 (19-20). The K-Ras farnesyl moeity, and a string of lysine residues just upstream of C185 in the Ras hypervariable region (HVR) are important contributors to K-Ras membrane binding, and a C185S mutation impairs this process (1,5-6,17-19).
The intracellular trafficking and signal transduction schemes for wild-type (WT) K-Ras have been studied extensively (1-20). After post-translational processing steps, WT K-Ras traffics via incompletely understood pathways to cell plasma membranes (PMs), where it localizes to lipid raft domains near receptor tyrosine kinases (RTKs; (21)). RTK activation recruits adapter proteins and guanine exchange factors (GEFs). Recruitment of GEFs to the vicinity of K-Ras allows the GEF to catalyze the release of bound (usually GDP) nucleotide, which facilitates subsequent K-Ras activation through binding of GTP (22-4). Activated K-Ras can be deactivated through the assistance of GTPase-activating proteins (GAPs; (22-4)). If K-Ras is not inactivated, it can trigger signal transduction through its association with a variety of effector proteins. One such effector protein is Raf, which triggers the RAF-MEK-ERK signal transduction cascade (1-9,25-9). A second K-Ras effector pathway is the PI3K-AKT cascade (1-9). Other effectors have been shown to interact with Ras proteins, but the physiological roles of these interactions have yet to be completely elucidated (30-40).
Given the prominent role of K-Ras in many cancers, a comprehensive analysis of its interaction network is of interest. Surprisingly, a current compendium of K-Ras physical interactions, based on yeast two-hybrid, co-immunoprecipitation (co-IP), and pull-down experiments lists a total of only forty unique interactions (41). This small tally is due, in part, to the difficulties in the analysis of interactions with membrane proteins, using conventional methods. To extend previous studies, we have developed a system to analyze K-Ras protein interactions using the promiscuous Escherichia coli biotin ligase BirA* (42-4). With BirA*, biotin is activated to a biotinyl-5’-AMP moiety that is released prematurely by the enzyme, and reacts with lysine residues on nearby proteins in a proximity-dependent fashion, estimated to be in the range of 100-200Å (43). To characterize the K-Ras protein interaction network, we have analyzed four different BirA*-K-Ras (BirA*Ras) fusion proteins. These included WT, S17N, G12D variants, as well as a farnesylation-defective mutant that also carries the G12D mutation (C185S/G12D). When expressed in transfected cells, the BirA*Ras proteins biotinylated themselves, plus multiple other cellular proteins. Biotinylated proteins were isolated and identified by liquid chromatagraphy (LC) and dual mass spectrometry (MS/MS) (42-4). Relative to mocks, a total of 748 biotinylated proteins were identified from cells expressing BirA*Ras variants, with significant differences between membrane-associated variants and the farnesylation-defective mutant. We also expressed the G12D BirA*Ras protein in pancreatic cancer cells that express an endogenous G12D mutant K-Ras protein. Studies with these cells identified fifty-six putative K-Ras interaction partners, including EGFR, integrin β1, and mucin 13. Overall, the identification of putative K-Ras interaction partners should facilitate the characterization of K-Ras interaction networks, and may help guide efforts at therapeutic intervention.
Materials and Methods
Recombinant DNA constructs. BirA*Ras constructs use the phage-ubc-nls-ha-tdPCP-GFP vector backbone to express the BirA*Ras fusion proteins. The fusion protein coding regions derive from the humanized Myc epitope tagged promiscuous Escherichia coli BirA* biotin ligase (42-4) fused in frame to the N-terminal coding region of the human KRas4B cDNA. WT and mutant KRas4B coding regions derive from pENTR-PAmCherry1-K-Ras4B subclones (20), and the sequences of the mutated KRas4B residues are GAT for G12D, AAT for S17N, and TCT for C185S. For all BirA*Ras constructs, the sequence of the BirA*-KRas4B junction is as follows: AAG CTC GAC ATG, where the AAG represents the last (lysine) codon of BirA* and the ATG represents the initiation codon of KRas4B. The BirA*-KRas4B coding regions were inserted into the phage-ubc-nls-ha-tdPCP-GFP backbone as NotI-BamHI fragments, placing the BirA*Ras coding regions downstream of the CMV immediate early promoter (45).
Other constructs employed included the plasmid Bluescript -SK (-SK; (46)), which was used in mock-transfections; the previously described psPAX2 (47-8) and pVSV-G (44,49) constructs for lentivirus vector infections; and expression clones for PAmCherry-KRas4B and Flag epitope tagged Raf-1 proteins. The expression plasmid for the PAmCherry-KRas4B chimera, pcDNA3-PAmCherry1-KRas (20), uses the CMV immediate early promoter to drive expression. Expression plasmids for the WT and R89L (29) Raf-1 proteins used Raf-1 coding regions cloned as BamHI-XbaI fragments into the corresponding pcDNA3.1 sites (50). The Raf-1 open reading frames (ORFs) were preceded by dual FLAG tags.
Cell culture, transfections, focus forming assays, and derivation of AsPC-1 cell lines. HEK293T (51) and NIH 3T3 (52) cells were maintained and transfected as described previously (44,47,49). For transfections involving the detection of biotinylated proteins, cells were supplemented with 50 μM biotin (Sigma B4501) at 24 h post-transfection. For focus forming assays (53-5), NIH 3T3 cells on 10 cm plates were transfected or infected (44,47,49) with the different BirA*Ras constructs and grown for 10-14 d prior to staining (53-5). After staining, plates were washed five times for 1 min with phosphate-buffered saline (PBS), and photographed for foci counting (53-5). For derivation of stable pancreatic cancer cell lines expressing the G12D Bir*Ras protein, AsPC-1 (56) cells were infected with the G12D BirA*Ras lentivirus vector, seeded at limiting dilution in 96 well plates, expanded, and screened for BirA*Ras expression with fluorescent streptavidin as described below. Positive clones were screened by immunoblot detection of G12D BirA*Ras and the endogenous G12D K-Ras proteins as described below. AsPC-1 clones C3, D1, and D2 were selected for further analysis.
Electrophoresis and blotting. For cellular protein analysis, transfected cells were washed in cold PBS, and samples were electrophoresced and electroblotted as described previously (44,47,49). Protein filter blot detection followed our previously described protocols (44,47,49), using a mouse anti-Myc monoclonal antibody (9E10; BD Pharmingen #551101), a mouse anti-Flag monoclonal antibody (M2; Sigma F1804), a mouse anti-PAmCherry antibody (Clontech #632543), or a mouse anti-K-Ras antibody (Santa Cruz Biotech. #F234) as primary antibodies; or a streptavidin-alkaline phosphatase conjugate (Invitrogen #434322) for biotinylated protein detection. Protein levels were quantified densitometrically using NIH ImageJ software (57).
Membrane-cytosol fractionation. Cell membrane and cytosolic fractions were prepared as described previously (58). Aliquots (10%) of membrane and cytosolic fractions were subjected to electrophoresis, blotting, and quantification as described above. Fractionation results were plotted as percentages of proteins measured in the membrane fractions.
Fluorescence localization of proteins. Cells were processed for fluorescence localization of proteins as described previously (44), using the anti-Myc primary antibody at a 1:1,000 dilution for BirA*Ras protein detection, and a 1:1,000 dilution of Alexa Fluor 488-streptavidin for biotinylated protein detection. Cells were imaged and photographed as described previously (44), and Pearson’s correlation coefficients (59), as well as profile plots were calculated using NIH Image J software (57).
Isolation of biotinylated proteins, and mass spectrometry. Biotinylated proteins from cells were isolated at 4˚C through using a modification (44) of previous procedures (42-3). Protein fractions isolated from streptavidin bead purifications were processed for LC-MS/MS analysis by the Oregon Health & Science University Proteomics Shared Resource. LC-MS/MS analysis was performed as described before (44,60) on the AsPC-1 cell line and the C3, D1, and D2 clones, the HEK293T-transfected S17N and C185S/G12d samples, and twice on the HEK293T mock, WT and G12D samples. Sequest was used to search MS2 Spectra against the Sprot human FASTA protein database, with added sequences for E. coli BirA*, concatenated sequence-reversed entries to estimate error thresholds, and 179 common contaminant sequences and their reversed forms. The database processing was performed with python scripts have been described previously (44,61-2). Scores of histograms for reversed matches were used to estimate peptide false discovery rates (FDR) and set score thresholds for each peptide class that achieved the desired protein FDR (approximately 2-3%) for each sample. Background proteins present in mock samples were removed if the averaged spectral hit numbers for mock samples exceeded one tenth of the averaged spectral hit numbers for BirA*Ras-transfected samples. For AsPC-1 samples, background proteins that yielded fewer than five spectral hits for the C2 plus D1 clones also were removed. Following the exclusion of background proteins, signal levels in transfected cell samples were normalized to spectral hit counts for the transfected K-Ras spectral hit values so as account for transfection efficiencies. After normalization, values for duplicate LC-MS/MS runs were averaged, and normalized spectral count numbers were rounded to the nearest integer. A table of all biotinylated cellular biotinylated proteins identified in HEK293T-transfected samples is available on request, and these proteins were classified based on their protein class ontologies using the Panther classification system (63). Table I lists all non-background proteins identified in sequencing of the AsPC-1 parent and clones.
Table I. Biotinylated proteins in AsPC-1 cells.
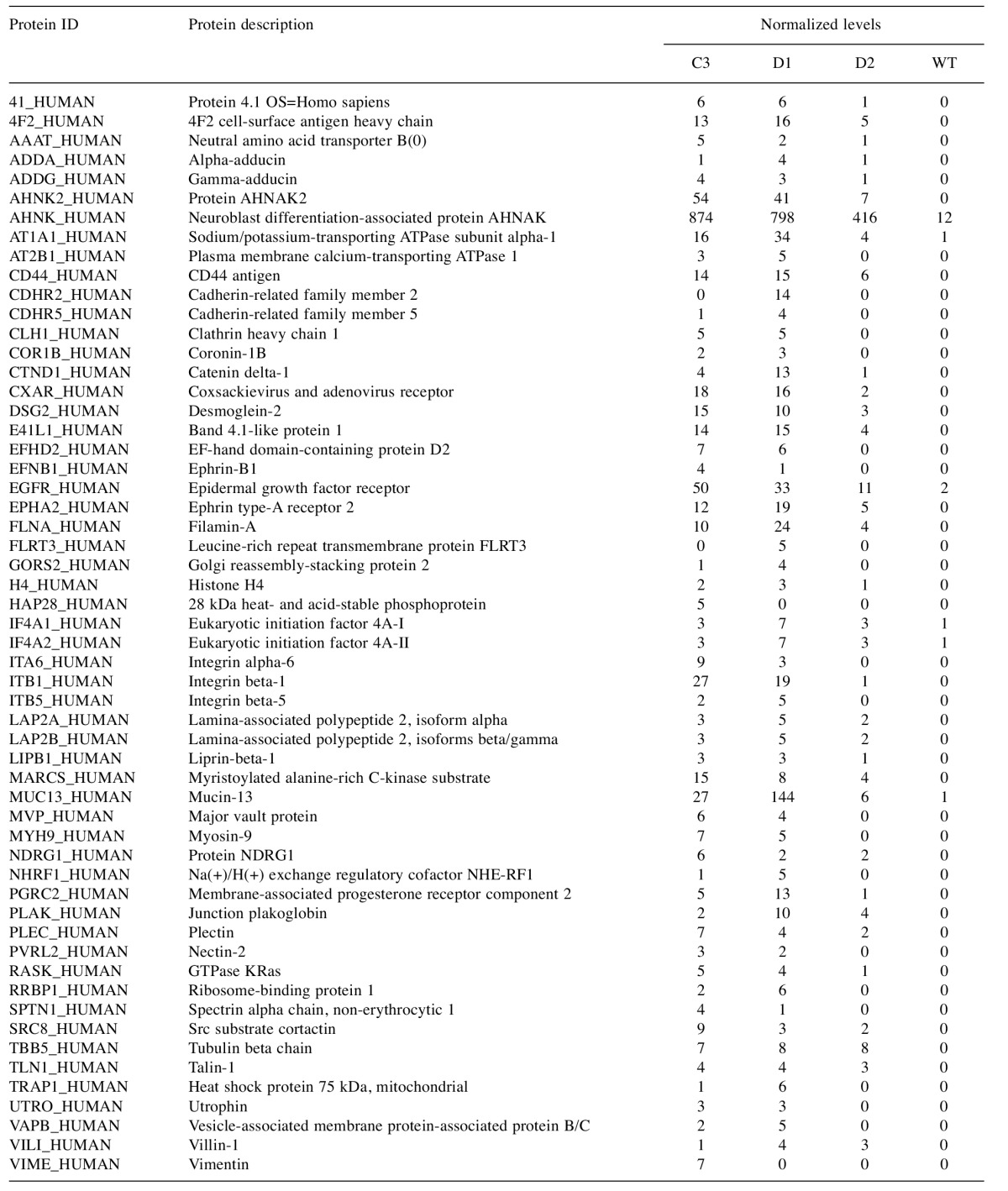
The list of proteins preferentially biotinylated in AsPC-1 clones expressiong the G12D BirA*Ras protein, after background subtraction is tabulated in the order of UniProt ID names (left column). The second column gives protein descriptions, and the remaining four columns give normalized MS signals for the BirA*Ras-expressing C3, D1, and D2 AsPC-1 clones, and for the AsPC-1 (WT) parent.
Results
Expression and localization of BirA*Ras proteins. To examine the network of proteins that interact with K-Ras, we adapted the promiscuous biotin ligase (BirA*) approach, in which proteins that associate with a BirA* fusion protein are subject to biotinylation (42-4). To do so, BirA*-K-Ras (BirA*Ras) fusion protein expression vectors were constructed. Each encoded BirA* fusion protein carried an N-terminal Myc epitope tag and the Escherichia coli biotin ligase BirA* upstream of the K-Ras coding region to avoid interfering with K-Ras post-translational C-terminal processing steps (1,5-6,14-20). Four BirA*Ras variants were chosen for analysis: WT, the constitutively active G12D mutant, the inactive S17N mutant, and C185S/G12D double mutant that is defective for farnesylation, but also carries the G12D mutation (10-11).
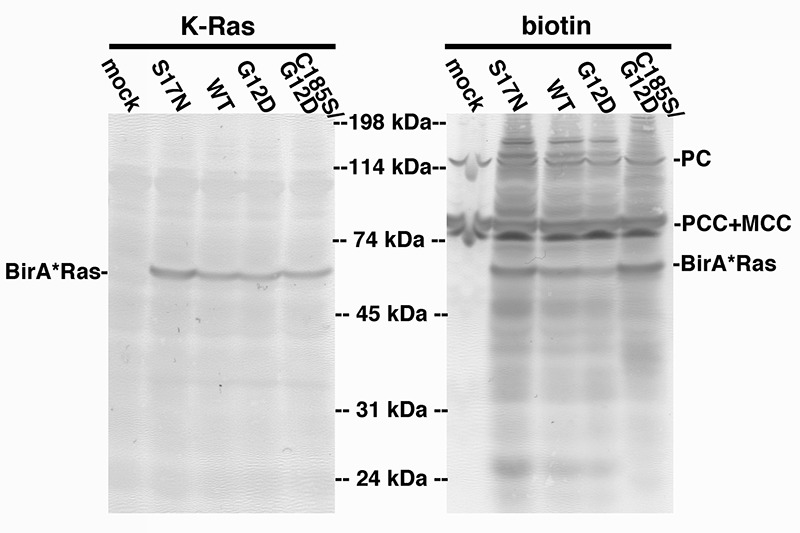
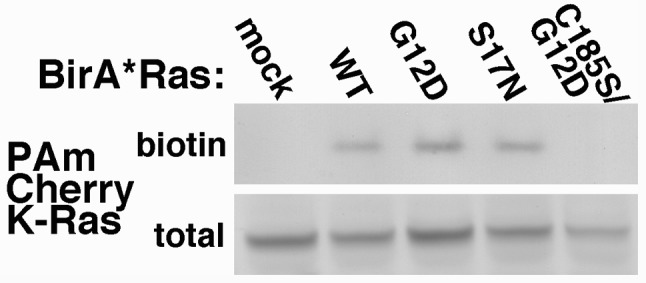
BirA*Ras expression levels and biotinylation properties initially were analyzed in transfected human HEK293T cells (51), because these cells have proven to be reliably and reproducibly transfectable (44,48-9). As a first test, proteins from transfected and mock-transfected (mock) cells were fractionated by electrophoresis and blotted to detect BirA*Ras (K-Ras) proteins by virtue of their Myc epitope tags, or biotinylated (biotin) proteins, using a streptavidin conjugate. As shown in Figure 1, with all transfected cell samples, but not in the mock cell sample, we observed Myc epitope tag-reactive bands at about 58 kDa, corresponding to the BirA*Ras proteins. Of the biotinylated proteins (Figure 1, right panel), in the mock-transfected lane a protein at about 130 kDa and a pair of proteins at about 80 kDa were detected: these correspond to the endogenously biotinylated mitochondrial proteins pyruvate carboxylase (PC), and the alpha chains of the proprionyl-CoA (PCC) and methylcrotonyl-CoA (MCC) carboxylases (44,64-5). Importantly, in transfected cells, the biotinylated BirA*Ras proteins were additionally detected, as were numerous cellular proteins, as indicated by the smears of bands in these lanes.
Figure 1. Expression and biotinylation of BirA*Ras proteins. Lysate samples from mock-transfected (mock) cells, or cells transfected with BirA*Ras constructs expressing S17N, wild type (WT), G12, or C185/G12D proteins were fractionated by electrophoresis, and electroblotted onto nitrocellulose filters. BirA*Ras proteins (left panel) were detected using an anti-Myc tag primary antibody and an alkaline phosphatase-conjugated secondary antibody, while biotinylated proteins were detected with alkaline phosphatase-conjugated streptavidin. Note that in mock-transfected cells, the endogenously biotinylated proteins pyruvate carboxylase (PC; 130 kDa) and the alpha chains of proprionyl-CoA carboxylase (PCC; 80 kDa) and methylcrotonyl-CoA carboxylase (MCC; 80 kDa) were detected. Note also that multiple additional biotinylated proteins, including the BirA*Ras proteins, were detected in transfected cells.
We next examined the subcellular localizations of BirA*Ras and its biotinylation products using fluorescence microscopy. As a control, we first imaged mock-transfected cells using both an anti-Myc tag antibody (to detect BirA*Ras) and fluorescently tagged streptavidin (to detect biotinylated targets). As expected no signal was detected for BirA*Ras (data not shown), and very low signals were detected with streptavidin at the default 10 ms exposure time. However, long exposure times (300 ms) permitted the visualization of the endogenously biotinylated proteins in mock-transfected cells (data not shown). Because the majority of these proteins are mitochondrial (44,64-5), localization patterns of endogenously biotinylated proteins were consistent with mitochondrial localization, although we did not co-stain with alternative mitochondrial markers to test this assumption.
With exposure times of 10 ms, proteins in mock-transfected cells were barely detected (data not shown), making it possible to visualize BirA*Ras and biotinylated proteins specifically in BirA*Ras protein-expressing cells. As shown in Figure 2, detection of BirA*Ras proteins showed heterogeneous staining patterns for the G12D, WT and S17N proteins, with an enrichment towards cell edges, consistent with PM staining. In some images, cytoplasmic rings of fluorescence were observed, suggestive of intracellular vesicle staining (data not shown). As compared to the other three BirA*Ras proteins, the C185S/G12D proteins appeared less at the cell surface, and more in the nucleus. Within the nucleus, staining often appeared reduced in what appear to be nucleolar spots, but this relative reduction in staining may be due to more limited reagent access to nucleolar compartments (66). The differences in cell surface versus internal staining for G12D, WT and S17N versus C185S/G12D proteins could be observed in profile plots of the immunostained proteins (Figure 2, right column). In these plots, relative fluorescence levels were graphed from one cell edge to the opposite edge; and the red line plots correspond to the cells indicated with the white bars in the left column panels, while the black line plots represent average profile plots for multiple cells. Comparison of profile plots clearly showed an intracellular enrichment of the C185S/G12D variant, and an apparent PM enrichment for the other BirA*Ras proteins.
Figure 2. Localization of BirA*Ras and biotinylated proteins in transfected cells. Cells expressing the indicated BirA*Ras proteins were processed to detect the BirA*Ras proteins with an anti-Myc antibody (K-Ras; red), and a fluorescently tagged streptavidin reagent to detect biotinylated proteins (biotin; green). Exposure times for biotinylated proteins were set at 10 ms, to minimize imaging of endogenously biotinylated proteins. The merge column shows the overlap BirA*Ras (K-Ras) and biotinylated protein (biotin) signals. Pearson’s correlation coefficients, determined from 20 cells for each sample, indicated high degree of co-localization and were as follows: G12D, 0.84±0.06; WT, 0.82±0.06; S17N, 0.86±0.04; C185S/G12D, 0.93±0.03. In the rightmost column (profile), fluorescence brightness profiles across cells, from one cell edge (0) to the opposite cell edge (100) were plotted. The red line plots derive from the cells indicated with white lines in the K-Ras column. The black line plots were averaged from a minimum of 13 cells for each sample, and indicate that G12D, WT, and S17N BirA*Ras proteins preferentially localized to cell edges, while the C185S/G12D protein appeared enriched intracellularly.
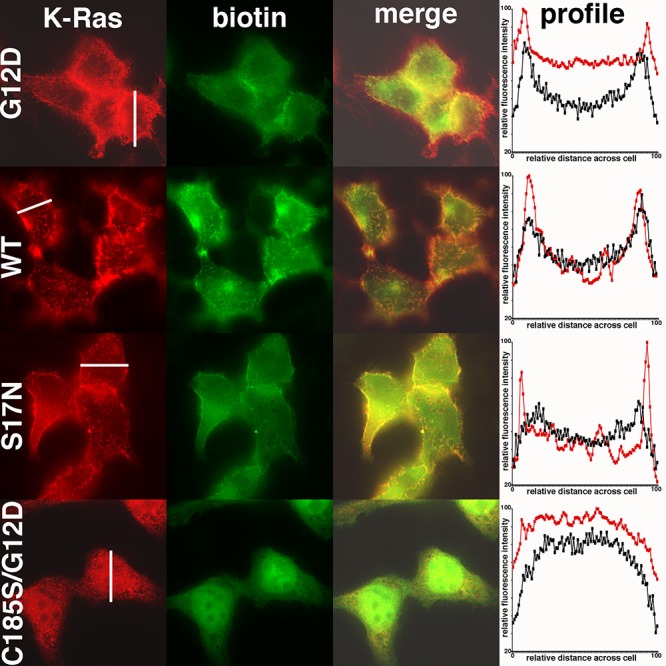
Staining patterns of biotinylated proteins were similar to their BirA*Ras counterparts, and merged images (merge) showed a large degree of co-localization (Figure 2). The degree of co-localization was calculated using Pearson’s correlation coefficients (59), which range from -1.0 (oppositely correlated) to +1.0 (completely correlated). Importantly, Pearson’s correlation coefficients indicated a high degree of BirA*Ras protein and biotinylated protein colocalization, with values of between 0.82 and 0.93 (Figure 2, legend). In summary, these results indicate that biotinylated proteins localized with the BirA*Ras proteins, at least at the level of conventional fluorescence microscopy; and that, as anticipated, the C185S/G12D protein showed reduced levels of cell surface association.
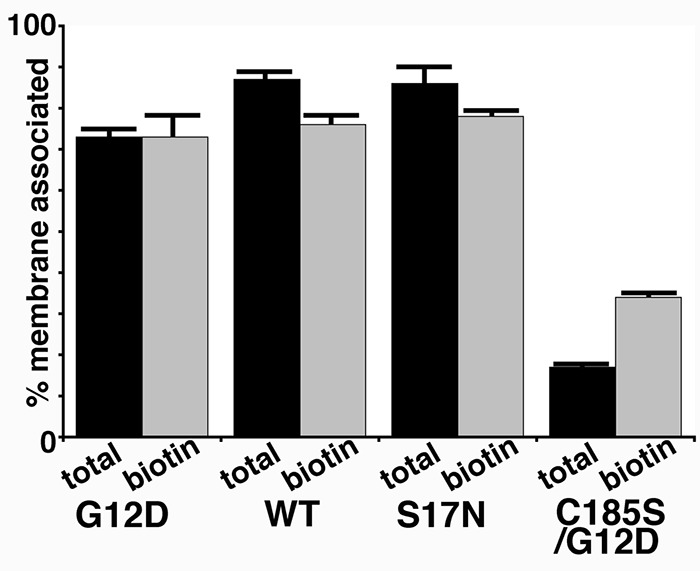
To assess cytosol and membrane localizations of BirA*Ras proteins in an independent fashion, we performed fractionation studies on cells expressing the four BirA*Ras variants. To do so, cytosol and membrane fractions were separated by centrifugation and subjected to blot detection to determine total BirA*Ras protein levels in each fraction. From these data, percentages of membrane associations were calculated (Figure 3). Importantly, the membrane fraction of biotinylated proteins showed the same trend as that of total BirA*Ras, indicating that membrane-bound BirA*Ras preferentially biotinylated membrane-bound targets, whereas cytosolic (C185S/G12D) BirA*Ras preferentially biotinylated cytosolic targets. Nevertheless, a small percentage of C185S/G12D BirA*Ras remained membrane bound, and this population appears to have been more efficient in biotinylating proximal proteins than the cytosolic C185S/G12D fraction, as indicated by the greater membrane association of biotinylated proteins BirA*Ras.
Figure 3. Membrane association of BirA*Ras proteins. Cells expressing the indicated BirA*Ras proteins were fractionated into cytosolic and membrane fractions, and BirA*Ras proteins from the fractions were quantified after electrophoresis and blotting to detect all the BirA*Ras proteins (total; using an anti-Myc primary antibody) and biotinylated BirA*Ras proteins (biotin; using a streptavidin conjugate). The percentages of membrane association for the proteins were determined by dividing membrane fraction signals by membrane plus cytosol signals. Results represent an average of two independent experiments.
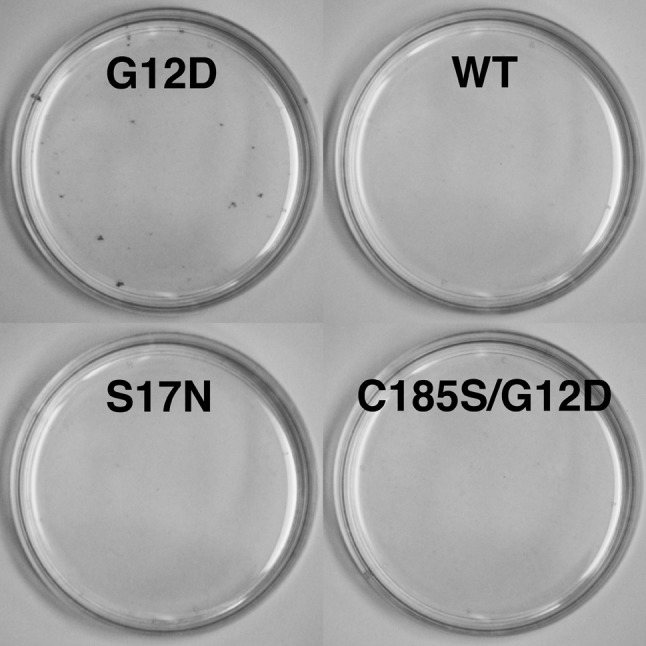
BirA*Ras protein activities, and biotinylation of K-Ras and Raf-1 proteins in trans. Previous studies have shown that the G12D K-Ras expression in NIH 3T3 (3T3) cells was sufficient to induce focus formation, but that expression of WT K-Ras and other variants was not (53-5). To test the focus-forming activities of the BirA*Ras proteins, 3T3 cells were transfected with the BirA*Ras expression constructs and scored for focus formation. As shown in Figure 4, and consistent with predictions, the G12D BirA*Ras protein indeed did induce focus formation, while the other variants did not. In two separate 3T3 transfection experiments, we observed 17 and 20 foci for G12D, and no foci for the other BirA*Ras variants. We also delivered the four BirA*Ras variants to target 3T3 cells by lentivirus infection, and again observed no foci in cells infected with the WT, S17N, or C185S/G12D variants, but 41 foci in G12D-infected 3T3 cells. These results support the notion that BirA* fusion to the N-termini of K-Ras proteins did not abrogate their expected cell proliferative activities.
Figure 4. Focus formation of BirA*Ras proteins. NIH 3T3 fibroblast cells were transfected with the indicated BirA*Ras constructs, grown for 10 days, and stained for focus formation. As shown, the G12D BirA*Ras protein yielded foci whereas the other variants did not. In three separate experiments, G12D BirA*Ras gave 17, 20, and 41 foci, while no foci were observed for the other variants.
Recent investigations have implicated that Ras proteins have the capacity to oligomerize, with dimers as perhaps a preferred oligomerization state (17,20). We examined this phenomenon by analysis of the capabilities of BirA*Ras proteins to biotinylate K-Ras proteins in trans. To do so, cells were mock-transfected or transfected with our four BirA*Ras variants along with an expression construct for a chimera of PAmCherry plus WT K-Ras (PAmCherry1-KRas). Total protein (total) and column-bound biotinylated (biotin) samples were fractionated by electrophoresis and immunoblotted with an anti-PAmCherry antibody to detect PAmCherry1-KRas proteins. As shown in Figure 5, PAmCherry1-KRas proteins were detected in all cell lysates, but biotinylated PAmCherry1-KRas proteins were not detected in cells that had either no BirA*Ras expression, or expression of the C185S/G12D protein. In contrast, WT, G12D, and S17N BirA*Ras proteins did biotinylate PAmCherry1-KRas proteins in trans (Figure 5, top panel). These results support the notion that a wild-type K-Ras HVR region is required for K-Ras oligomerization by targeting proteins to a membrane where oligomerization can occur. It also is possible that the lack of lipid modification alters the structure of the HVR, making it incapable of interaction with PAmCherry1-KRas (1-9,17,20). Our results also suggest that K-Ras proteins can occupy similar membrane locations, regardless of their GTP binding status.
Figure 5. Biotinylation of K-Ras proteins in trans. The indicated BirA*Ras proteins were co-expressed in cells with a WT PAm-Cherry K-Ras protein, after which biotinylated proteins in lysates were purified on streptavidin columns. Total lysate (total) and strepatvidin columnbound (biotin) samples were fractionated electrophoretically, and immunoblotted with an anti-PAmCherry antibody to detect PAm-Cherry K-Ras proteins.
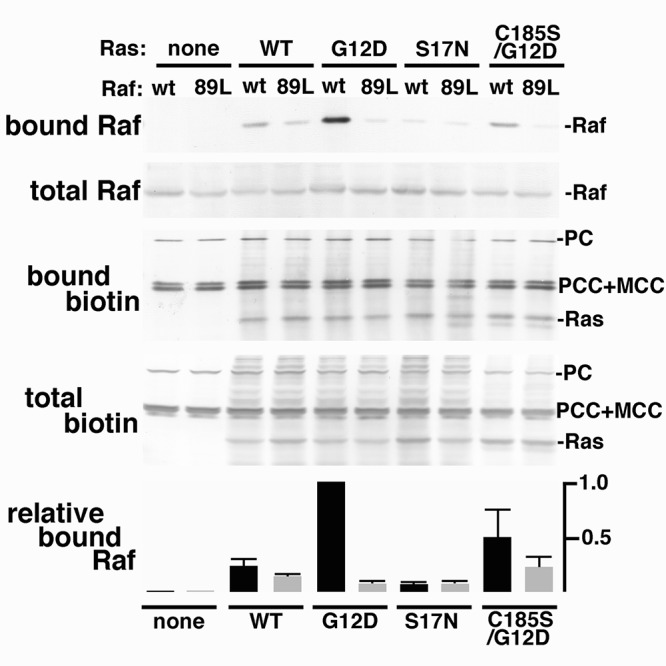
In addition to testing K-Ras biotinylation in trans, it also was of interest to analyze BirA*Ras biotinylation of one of its effector proteins, Raf-1. As noted in the background, Raf proteins bind to activated K-Ras (1-9,25-9). Previously, a single mutation at Raf-1 codon arginine 89 (R89L) was shown to impair Raf binding to activated Ras (Ras-GTP; (29)). Thus, to probe BirA*Ras-Raf interactions, cells were co-transfected with expression constructs for the BirA*Ras variants, plus vectors for FLAG epitope-tagged WT or R89L (89L) Raf-1 proteins. At three days post-transfection, total lysate samples (total) were collected, and biotinylated proteins were purified on streptavidin columns (bound). Total and bound samples were subjected to detection of biotinylated proteins (biotin) and of FLAG-tagged Raf-1 proteins (Raf). Consistent with Figure 1, cells transfected with only the Raf-1 vectors (Ras: none) showed only the endogenously biotinylated proteins (PC, PCC, MCC) on the biotin detection blots (Figure 6). When cells received one of the BirA*Ras expression plasmids, the biotinylated BirA*Ras (Ras) proteins also were detected, by virtue of their self-biotinylation activities. Because the FLAG-tagged Raf-1 proteins migrated so closely to the PCC and MCC proteins, it was not possible to distinguish them on the biotin blots. However, blots probed with the anti-Flag antibody readily detected approximately equal amounts of either WT or 89L Raf-1 proteins in all Flag-Raf-1-transfected cells. Significant differences in biotinylated bound Raf-1 proteins were observed for the different BirA*Ras co-transfections. Notably, no biotinylated Raf-1 was detected in the absence of BirA*Ras expression, and very little was detected with the activation-impaired S17N mutant. In contrast, constitutively active G12D BirA*Ras proteins efficiently biotinylated WT Raf-1, and to a much lower degree, R89L Raf-1. WT Bir*Ras proteins also biotinylated Raf-1, albeit with a lower preference for WT Raf-1 than G12D BirA*Ras. Interestingly, the farnesylation-defective C185S/G12D variant also biotinylated Raf-1, again with a preference for WT versus R89L Raf-1. While our results are consistent with the notion that Raf-1 can not bind to inactive K-Ras (Ras-GDP), and that R89L Raf-1 is impaired for Ras binding (29), they also suggest that the farnesylation-minus, but GTP-binding C185S/G12D variant is capable of Raf-1 binding. These results are consistent with previous observations that a G12V/C186S H-Ras mutant forms a stable complex with Raf in the cytosol (84).
Figure 6. Biotinylation of C-Raf proteins. Wild-type (wt) or mutant R89L (89L) C-Raf proteins were expressed alone in cells (none), or co-expressed with the indicated BirA*Ras proteins. Total protein lysates were collected and biotinylated proteins were purified on streptavidin columns. Total and bound samples were fractionated electrophoretically and blotted to detect Raf with an anti-Flag primary antibody, and a secondary alkaline phosphatase-conjugated antibody, or biotinylated proteins with alkaline phosphatase-conjugated streptavidin. In the top panels, Raf and Ras proteins are as indicated, as are the endogenously biotinylated pyruvate carboxylase (PC), proprionyl-CoA carboxylase (PCC), and methylcrotonyl-CoA carboxylase (MCC) proteins. The bottom panel indicates bound C-Raf protein levels, relative to bound wt C-Raf protein levels from cells coexpressing G12D BirA*Ras, averaged from two independent experiments.
Identification of cellular K-Ras-interacting proteins. Identification of cellular K-Ras-interacting proteins was achieved by purification of biotinylated proteins from mock transfected cells and from cells expressing the four BirA*Ras variants. Purified proteins were protease-digested, and subjected to liquid chromatography dual mass spectrometry (LC-MS/MS) sequencing. From all mock and BirA*Ras samples, a total of 1,557 proteins were identified. Proteins that appeared in mock samples at levels greater than one tenth the averaged spectral hit numbers for the BirA*Ras-expressing samples were designated as background proteins. A list of these background proteins is available on request.
After background protein subtraction, our list of biotinylated proteins (available on request) included 748 members: 440 were found in the G12D sample, 410 in WT, 447 in S17N, and 383 in C185S/G12D. We initially examined these hits with respect to the numbers of proteins that were biotinylated by different combinations of the BirA* variants, and compared the numbers to what would be predicted if the biotinylation patterns were independent. Interestingly, 122 proteins were biotinylated by all four BirA* variants, higher than predicted by random assortment. We also observed that the number of proteins biotinylated only by the C185S/G12D variant (116 proteins) was much higher than random assortment prediction. Similarly, we observed a higher than random assortment predicted number of proteins (133 proteins) that were biotinylated by the three farnesylation-positive variants (G12D, WT, S17N), but not by the C185S/G12D protein. These differences reflect the localization differences observed in our fluorescence and subcellular fractionation experiments.
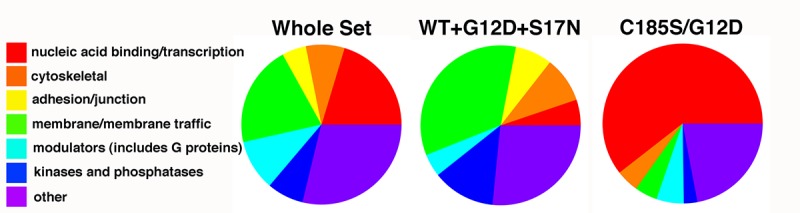
When biotinylated proteins from transfected cells were classified according to protein class ontology, several groups were observed (Figure 7). These included cytoskeletal, adhesion/junction, membrane/membrane traffic, modulator, kinase/phosphatase, as well as nucleic acid binding/ transcription and other proteins. Not surprisingly, the subset of proteins biotinylated by the G12D, WT and S17N, but not by C185/G12D was enriched for membrane and membrane traffic proteins (Figure 7). In contrast, proteins solely biotinylated by the C185/G12D variant were markedly enriched in nucleic acid binding and transcription proteins, and these results are discussed in more detail in the Conclusions section.
Figure 7. Classification of biotinylated proteins in transfected HEK293T cells. Proteins from the whole set of biotinylated proteins (748 total), from the set of proteins that were biotinylated all variants except the double mutant (WT+G12D+S17N; 133 total), or from the proteins that were biotinylated only by the C185S/G12D BirA*Ras variant (116 total) were classified according to the Panther classification system based on protein class ontology. Note that relative to the whole set, the WT+G12D+S17N set is enriched for membrane/membrane traffic proteins, while the C185S/G12D set is enriched for nucleic acid binding/transcription proteins.
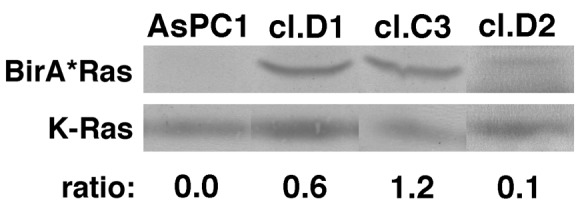
To extend our observations, we examined K-Ras protein interactions in human AsPC-1 pancreatic cancer cells, that express the G12D version of K-Ras (56). For these studies, we transduced AsPC-1 cells with the G12D BirA*Ras variant via lentivirus vector infection (see Methods). Individual AsPC-1 clones were isolated by limiting dilution, screened for BirA* expression and rescreened to compare transgene BirA*Ras and endogenous K-Ras expression levels. On this basis, we picked three clones, clones C3, D1 and D2 for analysis. As shown in Figure 8, the three clones respectively express G12D BirA*Ras proteins at roughly 0.6, 1.2, and 0.1 times the endogenous G12D K-Ras expression levels.
Figure 8. BirA*Ras levels in AsPC-1 clones. K-Ras and BirA*Ras protein levels in the AsPC-1 parent cell lines and clones D1, C3, and D2 were determined by immunoblotting with a K-Ras antibody, and densitometric quantitation of protein levels. Ratios indicate the levels of transgene expression relative to endogenous K-Ras expression, and were 0.6, 1.2, and 0.1 respectively for the D1, C3, and D2 clones.
Proteins biotinylated by BirA*Ras proteins in AsPC-1 clone C3, D1, and D2 cells were identified by LC-MS/MS sequencing as described above. Proteins were designated as background hits if the normalized spectral hits for the C3 plus D1 clones were less than five, or less than ten times the number of spectral hits for the parental AsPC-1 cell line: a list of these background proteins is available on request. After background subtraction, fifty-six putative BirA*Ras interacting partners were identified (Table I). These proteins were characterized by high normalized spectral count numbers in AsPC-1 clones C3 and/or D1, and zero or low values for the parental AsPC-1 cell line, and clone D2, which expressed very low levels of BirA*Ras, relative to K-Ras (Figure 8). The majority of biotinylated proteins in AsPC-1 cells were identified as putative K-Ras-interacting partners in transfected HEK293T cells. Noteworthy abundant AsPC-1 hits included the giant, putative tumor suppressor protein AHNAK (67), but also multiple cell surface signaling proteins, including EGFR, integrin β1, mucin 13, ephrin and its receptor, and CD44. These results are discussed below.
Discussion
We have modified the promiscuous biotin ligase tagging system (42-4,85-88) to analyze the protein interaction networks of the G12D, WT, S17N, and C185S/G12D K-Ras proteins. As shown, our chimeric BirA*Ras proteins were expressed as expected, localized as predicted, and their localization patterns matched the patterns of the proteins they biotinylated (Figures 1-4). K-Ras proteins also have been reported to oligomerize (17,20,89-91), and we observed that WT, G12D, and S17N BirA*Ras proteins biotinylated WT pAMCherry1-KRas proteins in trans (Figure 5). These results are of interest in the context of data that show both GDP-bound and GTP-bound K-Ras proteins oligomerize in cells (20). The C185S/G12D protein did not biotinylate WT pAMCherry1-KRas proteins (Figure 5), presumably because it did not localize with the target protein, or because oligomerization elements in the C-terminal K-Ras HVR were impaired (17,20). We also examined BirA*Ras protein interactions with the Raf-1 effector protein in transfected cells (Figure 6). As expected, the constitutively active G12D BirA*Ras protein interacted with Raf-1 as indicated by the strong biotinylated WT Raf-1 signals in co-transfected samples. Also as expected, the inactive S17N BirA*Ras protein failed to biotinylate Raf-1, while WT BirA*Ras biotinylated Raf-1 at lower levels than the G12D protein. Interestingly, the C185S/G12D BirA*Ras protein yielded higher levels of Raf-1 biotinylation than did the WT Ras variant. Given that the C185S/G12D protein is expected to be in the GTP-bound form (5,10-11,84), it is perhaps reasonable to expect K-Ras/C-Raf binding in a co-transfection system, as was observed for G12V/C186S H-Ras binding to Raf (84). However, because C185S/G12D BirA*Ras failed to induce focus formation (Figure 4), these results support the notion that K-Ras triggering of the RAF-MEK-ERK cascade not only requires Ras-Raf binding, but requires that the binding occur in the proper subcellular membrane location.
Our sequencing results of transfected cells identified 748 proteins that were biotinylated by at least one of our BirA*Ras variants. Biotinylated proteins appeared to be enriched for cytoskeletal, adhesion/junction, membrane/ membrane traffic, modulator, kinase/phosphatase, nucleic acid binding/transcription and other proteins (Figure 7). Relative to mock-transfected cells, 122 proteins were biotinylated by all four BirA*Ras variants. These included 100 proteins that were identified at roughly equal levels in all four BirA*Ras samples, but also 22 that were identified at least three-fold higher levels in the WT, G12D, and S17N BirA*Ras samples than in the C185S/G12D BirA*Ras sample. Of the 100 proteins that were identified at roughly equal levels in all BirA*Ras samples, fourteen (protein ID: 41, AFAD, AHNK, CD2AP, CDV3, IF4B, IF4G1, IF5, FLNA, LYRIC, PERQ2, SYEP, TPR, XRN1) were previously observed to have been biotinylated by the BirA*-tagged myristoylated HIV-1 structural (Gag) protein that assembles at the PMs of infected cells (44): these proteins presumably are involved in common pathways of Gag and K-Ras protein expression, regulation, or trafficking. Concerning the 22 proteins that were biotinylated by all four BirA*Ras variants but were identified at reduced levels in the C185S/G12D sample, these were similar to the 133 proteins that were biotinylated only by the WT, G12D and S17N variants, in that they were enriched for membrane and membrane traffic proteins (Figure 7): these results suggest that the localizations of K-Ras-GTP and GDP are at least partially overlapping. In contrast, proteins that were biotinylated only by the C185S/G12D variant were greatly enriched for nucleic acid binding and transcription proteins (Figure 7). This observation may simply represent the anomalous localization of the mutant K-Ras protein in the nucleus as a consequence of its potentially karyophilic, lysine-rich C-terminal sequence. However, H-Ras has been shown to localize to cell nuclei in a cell cycle-dependent fashion (93). Moreover, p19 H-Ras has been shown to bind to both the tumor suppressor protein p73β and its repressor, MDM2, enhancing p73β signaling (94). While current data concerning K-Ras nuclear localization are unclear (95-6), it would seem worthwhile to examine its potential nuclear activities.
Perhaps the most interesting associations of K-Ras in transfected cells are those with surface signalling proteins, kinases and ligases, and G proteins, GEFs, and GAPs. Over forty surface signaling proteins were biotinylated by at least one of the BirA*Ras variants. The vast majority of these were identified at higher levels with the G12D, WT, and S17N variants than with the C185S/G12D variant. Also of note is that the EGFR was biotinylated by all the membrane-associated BirA*Ras variants, as were other RTKs, including the Ephrin type-A receptor 2, and Plexin. Integrin β1 similarly interacted with BirA*Ras proteins, consistent with its role as a downstream Ras modulator (68-9). Other notable interactions with surface molecules included component 2 of the progesterone receptor (70), the putative tumor suppressors Robo1 (roundabout homolog 1; (71)) and nectin-2 (Poliovirus receptor-related receptor 2; (72)). Over fifty kinases or phosphatases were biotinylated by at least one of the BirA*Ras proteins. Our co-transfection experiments indicated that activated BirA*Ras proteins biotinylated Raf-1 whereas the S17N variant did not (Figure 6), and thus it was not surprising that biotinylated Raf-1, A-Raf and B-Raf proteins were detected by LC-MS/MS from cells expressing the activated BirA*Ras proteins. A significant number of kinases and phosphatases were biotinylated at higher levels for the membrane-targeted BirA*Ras variants than for the C185S/G12D mutant, reflecting the localization preferences of these enzymes (73). Along with surface signalling proteins, kinases, and phosphatases, G proteins and their activators and exchange factors also were biotinylated by BirA*Ras proteins. K-Ras itself was readily biotinylated as a domain of the chimeric BirA*Ras proteins (Figure 1) and served as a cell sample biotinylation control. Another GTPase identified at roughly 10% the levels of K-Ras, was Septin-9, a breast cancer marker involved in cytokinesis (74). We also detected a number of GTPase-activating proteins, including the Ras GAPs, GAPD1 and nGAP (RASAL2), and GAPs for Rho, Rac, and Ral proteins (DOCK7, RHG21, SLIT-ROBO, RGPA1, RHG01). Conspicuously absent from our list is the major K-Ras GEF, SOS (1-9), which is expressed in HEK293T cells (75). We currently do not know the reason for the lack of SOS biotinylation by any of our BirA*Ras proteins, but we do note that the Abl interactor 1 protein, which forms a complex with EPS8 and SOS (76) was detected in all our BirA*Ras LC-MS/MS biotinylation spectra. We also note that Raf biotinylation levels, while observed, were lower than what might have been expected, possibly due to endogenous Raf expression levels and/or the relative accessibility of target Raf lysine residues.
To extend our observations, we decided to examine proteins that interact with the K-Ras in pancreatic cancer cells. To do so, clones that expressed G12D BirA*Ras proteins at similar levels to the AsPC-1 endogenous G12D K-Ras protein were selected. These clones, C3, D1 and D2 respectively expressed BirA*Ras proteins at 1.2-, 0.6- and 0.1-times the levels of the endogenous K-Ras proteins (Figure 8), and were subjected to LC-MS/MS sequence analysis of biotinylated proteins. As shown in Table I, a total of fifty-six host cell proteins were biotinylated preferentially in the C3 and D1 AsPC-1 cell clones. Of these, the majority, but not all proteins were biotinylated by at least one BirA*Ras variant in transfected HEK293T cells. This supports the notion that K-Ras interacts with a set of common proteins across multiple cell types, but also with certain partners in a cell type-dependent context. In AsPC-1 cells, the protein with the highest spectral count is the giant (600-700 kDa) AHNAK protein, which was biotinylated by BirA*Ras proteins in HEK293T cells, and also by a BirA* tagged HIV-1 structural protein (44). One probable reason AHNAK and its sister protein AHNAK2 were identified in our study is that they both present large biotinylation target sizes. However, both are plasma membrane-associated, and have been observed to be up-regulated in certain cancers, including pancreatic cancers (77).
As is evident from Table I, the majority of BirA*Ras-biotinylated proteins in AsPC-1 cells were cytoskeletal proteins or plasma membrane proteins. Biotinylated cytoskeletal proteins included protein 4.1, the band 4.1-like protein, adducins, filamin A, myosin 9, and cortactin; and likely reflect the localization of K-Ras proteins at the cytoskeleton-cell surface interface. Plasma membrane proteins included several transporters (AAAT, AT1A1, AT2B1, NHRF1), but predominantly signaling and cell juncture/adhesion molecules. Not surprisingly, the EGFR was a major biotinylated protein, but we also observed significant biotinylation of the RTK ephrin A2, as well as the ephrin B1 ligand, both of which have been associated with a variety of cancers (78). Interestingly, a significant number of cell juncture/adhesion proteins were biotinylated, including mucin 13; integrins β1, β5, and α6, CD44, catenin δ, desmoglein, the coxsackie virus and adenovirus receptor, and the 4F2 cell-surface antigen heavy chain (Table I). All of these have been identified as potential cancer biomarkers (79-83,92), but their specific interactions with K-Ras and contributions to the progression of pancreatic cancers have yet to be thoroughly elucidated. We anticipate that analysis of these and other K-Ras interactions in pancreatic cancer cells, and in other cancers will be worthy of further scrutiny.
Acknowledgements
EB was supported by OHSU School of Medicine Research Core Initiative funding and by a grant from the OHSU Brenden-Colson Center for Pancreatic Care. PS was supported by NIH grants 1 R01 DK090309-01 A1 and 1 R21 CA191392-01. XN was supported by NCI 5U54CA143836, and research in the Nan lab on Ras biology is currently supported by startup funds from OHSU, the Damon Runyon Cancer Research Foundation, and the Brenden-Colson center for Pancreatic Care. We are grateful to Isabel Cylinder, Emily Platt, and Rachel Sloan for assistance and advice at the beginning of this project. Mass spectrometry was performed through the OHSU Proteomics Shared Resource, and we are particularly grateful to Larry David, John Klimek, and Ashok Reddy for their invaluable help.
References
- 1.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins MA, Pasca di Magliano M. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front Physiol. 2014;4:407–414. doi: 10.3389/fphys.2013.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, Roberts B, Zhang T, Chenard M, Haines B, Andersen J, Nagashima K, Paweletz C, Lynch B, Feldman I, Dai H, Huang P, Watters J. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010;3:26. doi: 10.1186/1755-8794-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773:1177–1195. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Jancik S, Drabek J, Radzioch D, Hajduch M. Clinical relevance of KRAS in human cancers. J Biomed Biotech. 2007;2007:150960. doi: 10.1155/2010/150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, Feiler HS, Ko AH, Olshen AB, Danenberg KL, Tempero MA, Spellman PT, Hanahan D, Gray JW. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldfinger LE. Choose your own path: specificity in Ras GTPase signaling. Mol Biosyst. 2008;4:293–299. doi: 10.1039/b716887j. [DOI] [PubMed] [Google Scholar]
- 9.Raaijmakers JH, Bos JL. Specificity in Ras and Rap signaling. J Biol Chem. 2009;284:10995–10999. doi: 10.1074/jbc.R800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smit V, Boot A, Smits A, Fleuren G, Cornelisse C, Bos J. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucl Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris V, Lucas F, Overman M, Eng C, Morelli M, Jiang Z, Luthra R, Meric-Bernstam F, Maru D, Scheet P, Kopetz S, Vilar E. Clinicopathologic characteristics and gene expression analyses of non-KRAS 12/13, RAS-mutated metastatic colorectal cancer. Ann Oncol. 2014;25:2008–2014. doi: 10.1093/annonc/mdu252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nassar N, Singh K, Garcia-Diaz M. Structure of the dominant negative S17N mutant of Ras. Biochemistry. 2010;49:1970–1974. doi: 10.1021/bi9020742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rocks O, Peyker A, Bastiaens PI. Spatio-temporal segregation of Ras signals: one ship, three anchors, many harbors. Curr Opin Cell Biol. 2006;18:351–357. doi: 10.1016/j.ceb.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Barcelo C, Paco N, Beckett A, Alvarez-Moya B, Garrido E, Gelabert M, Tebar F, Jaumot M, Prior I, Agell N. Oncogenic K-Ras segregates at spatially distinct plasma membrane signaling platforms according to its phosphorylation status. J Cell Sci. 2013;126:4553–4559. doi: 10.1242/jcs.123737. [DOI] [PubMed] [Google Scholar]
- 16.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann H. Small molecule inhibition of KRAS-PDEdelta interaction impairs ongogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 17.Lin WC, Iversen L, Tu HL, Rhodes C, Christensen S, Iwig J, Hansen SD, Huang WY, Groves JT. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci USA. 2014;111:2996–3001. doi: 10.1073/pnas.1321155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 19.Hancock J, Paterson H, Marshall C. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–139. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- 20.Nan X, Tamgüney TM, Collisson EA, Lin LJ, Pitt C, Galeas J, Lewis S, Gray JW, McCormick F, Chu S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc Natl Acad Sci USA. 2015;112:7996–8001. doi: 10.1073/pnas.1509123112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy. Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 25.Rebocho A, Marais R. ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene. 2013;32:3207–3212. doi: 10.1038/onc.2012.330. [DOI] [PubMed] [Google Scholar]
- 26.Nan X, Collisson EA, Lewis S, Huang J, Tamgüney TM, Liphardt JT, McCormick F, Gray JW, Chu S. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci USA. 2013;110:18519–18524. doi: 10.1073/pnas.1318188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Takahashi M, Stork PJ. Ras-mutant cancer cells display B-Raf binding to Ras that activates extracellular signal-regulated kinase and is inhibited by protein kinase A phosphorylation. J Biol Chem. 2013;288:27646–27657. doi: 10.1074/jbc.M113.463067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor S, Shaw AS. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fabian JR, Vojtek AB, Cooper JA, Morrison DK. A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc Natl Acad Sci USA. 1994;91:5982–5986. doi: 10.1073/pnas.91.13.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giehl K, Skripczynski B, Mansard A, Menke A, Gierschik P. Growth factor-dependent activation of the Ras-Raf-MEK-MAPK pathway in the human pancreatic carcinoma cell line PANC-1 carrying activated K-ras: implications for cell proliferation and cell migration. Oncogene. 2000;19:2930–2942. doi: 10.1038/sj.onc.1203612. [DOI] [PubMed] [Google Scholar]
- 31.Przybylskia M, Kozlowskaa A, Pietkiewicza P, Lutkowskaa A, Lianeria M, Jagodzinskia P. Increased CXCR4 expression in AsPC1 pancreatic carcinoma cells with RNA interference-mediated knockdown of DNMT1 and DNMT3B. Biomedicine and Pharmacotherapy. 2010;64:254–258. doi: 10.1016/j.biopha.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Kovacevic Z, Chikhani S, Lui G, Sivagurunathan S, Richardson D. The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits PI3K and Ras signaling pathways. Antioxid Redox Signal. 2013;18:874–887. doi: 10.1089/ars.2011.4273. [DOI] [PubMed] [Google Scholar]
- 33.Serra-Pages C, Kedersha NL, Fazikas L, Medley QG, Debant A, Streuli M. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J. 1995;14:2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch D, Winata S, Lyons R, Hughes W, Lehrbach G, Wasinger V, Corthals G, Cordwell S, Daly RJ. A Cortactin-CD2-associated Protein (CD2AP) Complex Provides a Novel Link between Epidermal Growth Factor Receptor Endocytosis and the Actin Cytoskeleton. J Biol Chem. 2003;278:21805–21813. doi: 10.1074/jbc.M211407200. [DOI] [PubMed] [Google Scholar]
- 35.Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, Raddaoui E, Almomen AK, Al-Muhsen S, Geha RS, Alkuraya FS. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immun. 2012;130:481–488. doi: 10.1016/j.jaci.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei P, Zhang N, Xu Y, Li X, Shi D, Wang Y, Li D, Cai S. TPX2 is a novel prognostic marker for the growth and metastasis of colon cancer. J Trans Med. 2013;11:313–325. doi: 10.1186/1479-5876-11-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bischoff F, Klebe C, Kretschmer J, Wittinghofer A, Ponstingl H. RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc Natl Acad Sci USA. 1994;91:2587–2591. doi: 10.1073/pnas.91.7.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu A, Tebar F, Alvarez-Moya B, Lopez-Alcala C, Calvo M, Enrich C, Agell N, Nakamura T, Matsuda M, Bachs O. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J Cell Biol. 2009;184:863–879. doi: 10.1083/jcb.200807186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dow L, Elsum I, King C, Kinross K, Richardson H, Humbert P. Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene. 2008;27:5988–6001. doi: 10.1038/onc.2008.219. [DOI] [PubMed] [Google Scholar]
- 40.Pearson H, Perez-Mancera P, Dow L, Ryan A, Tennstedt P, Bogani D, Elsum I, Greenfield A, Tuveson DA, Simon R, Humbert PO. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Investigation. 2011;121:4257–4267. doi: 10.1172/JCI58509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O’Donnell L, Reguly T, Nixon J, Ramage L, Winter A, Sellam A, Chang C, Hirschman J, Theesfeld C, Rust J, Livstone MS, Dolinski K, Tyers M. The BioGRID interaction database: 2015 update. Nucleic Acids Research 1 (Database) 2015:470–478. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roux K, Kim D, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim D, Birendra K, Zhu W, Motamedchaboki K, Doye V, Roux K. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci USA. 2014;111:2453–2461. doi: 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie C, Cylinder I, Platt EJ, Barklis E. Analysis of HIV-1 Gag protein interactions via biotin ligase tagging. J Virol. 2015;89:3988–4001. doi: 10.1128/JVI.03584-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu B, Chao JA, Singer RH. Fluorescence fluctuation spectroscopy enables quantitative imaging of single mRNAs in living cells. Biophys J. 2012;102:2936–2944. doi: 10.1016/j.bpj.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alting-Mees MA, Short JM. pBluescript II: gene mapping vectors. Nucleic Acids Res. 1989;17:9494. doi: 10.1093/nar/17.22.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.López CS, Sloan R, Cylinder I, Kozak SL, Kabat D, Barklis E. RRE-dependent HIV-1 Env RNA effects on Gag protein expression, assembly and release. Virology. 2014;462-463:126–134. doi: 10.1016/j.virol.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zufferey R, Nagy D, Mandel R, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 49.Noviello C, López C, Kukull B, McNett H, Still A, Eccles J, Sloan R, Barklis E. Second-site compensatory mutations of HIV-1 capsid mutations. J Virol. 2011;85:4730–4738. doi: 10.1128/JVI.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carey KD, Watson RT, Pessin JE, Stork PJ. The requirement of specific membrane domains for Raf-1 phosphorylation and activation. J Biol Chem. 2003;278:3185–3196. doi: 10.1074/jbc.M207014200. [DOI] [PubMed] [Google Scholar]
- 51.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein–Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- 54.Guerrero S, Casanova I, Farre L, Mazo A, Capella G, Mangues R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60:6750–6756. [PubMed] [Google Scholar]
- 55.Cheng CM, Li H, Gasman S, Huang J, Schiff R, Chang EC. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol. 2011;31:983–997. doi: 10.1128/MCB.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deer E, Gonzalez-Hernandez J, Coursen J, Shea J, Ngatia J, Scaife C, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen M, Jelinek L, Jones RS, Stegeman-Olsen J, Barklis E. Assembly and composition of intracellular particles formed by Moloney murine leukemia virus. J Virol. 1993;67:5163–5174. doi: 10.1128/jvi.67.9.5163-5174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A. 2010;77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 60.Shevchenko A, Tomas H, Havli J, Olsen J, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 61.Wilmarth P, Riviere M, David LL. Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. J Ocul Biol Dis Infor. 2009;2:223–234. doi: 10.1007/s12177-009-9042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keller A, Nesvizhskii A, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 63.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res (Database issue) 2012;41:D377–386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ingaramo M, Beckett D. Selectivity in post-translational biotin addition to five 885 human carboxylases. J Biol Chem. 2012;287:1813–1822. doi: 10.1074/jbc.M111.275982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abu-Elheiga L, Brinkley W, Zhong L, Chirala S, Woldegiorgis G, Wakil S. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci USA. 2000;97:1444–1449. doi: 10.1073/pnas.97.4.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Svistunova DM, Musinova YR, Polyakov VY, Sheval EV. A Simple Method for the Immunocytochemical Detection of Proteins Inside Nuclear Structures That Are Inaccessible to Specific Antibodies. J Histochem Cytochem. 2012;60:152–158. doi: 10.1369/0022155411429704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee I, Sohn M, Lim H, Yoon S, Oh H, Shin S, Shin J, Oh SH, Kim J, Lee DK, Noh DY, Bae DS, Seong JK, Bae YS. Ahnak functions as a tumor suppressor via modulation of TGFβ/Smad signalling pathway. Oncogene. 2014;33:4675–4684. doi: 10.1038/onc.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kinbara K, Goldfinger LE, Hansen M, Chou FL, Ginsberg MH. Ras GTPases: integrins’ friends or foes. Nat Rev Mol Cell Biol. 2003;4:767–776. doi: 10.1038/nrm1229. [DOI] [PubMed] [Google Scholar]
- 69.Desgrosellier J, Cheresh D. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu L, Wang J, Zhao L, Nilsen J, McClure K, Wong K, Brinton RD. Progesterone increases rat neural progenitor cell cycle gene expression and proliferation via extracellularly regulated kinase and progesterone receptor membrane components 1 and 2. Endocrinology. 2009;150:3186–3196. doi: 10.1210/en.2008-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parray A, Siddique HR, Kuriger JK, Mishra SK, Rhim JS, Nelson HH, Aburatani H, Konety BR, Koochekpour S, Saleem M. ROBO1, a tumor suppressor and critical molecular barrier for localized tumor cells to acquire invasive phenotype: study in African-American and Caucasian prostate cancer models. Int J Cancer. 2014;135:2493–2506. doi: 10.1002/ijc.28919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boles KS, Barchet W, Diacovo T, Cella M, Colonna M. The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood. 2005;106:779–786. doi: 10.1182/blood-2005-02-0817. [DOI] [PubMed] [Google Scholar]
- 73.Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies. Blood. 2000;96:1655–1669. [PubMed] [Google Scholar]
- 74.Connolly D, Yang Z, Castaldi M, Simmons N, Oktay MH, Coniglio S, Fazzari MJ, Verdier-Pinard P, Montagna C. Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Res. 2011;13:R76. doi: 10.1186/bcr2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burkard TR, Planyavsky M, Kaupe I, Breitwieser FP, Bürckstümmer T, Bennett KL, Superti-Furfga G, Colinge J. Initial characterization of the human central proteome. BMC Syst Biol. 2011;5:17. doi: 10.1186/1752-0509-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan P-D, Goff SP. Abl interactor 1 binds to sos and inhibits epidermal growth factor- and v-Abl-induced activation of extracellular signal-regulated kinases. Mol Cell Biol. 2000;20:7591–7601. doi: 10.1128/mcb.20.20.7591-7601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goonesekere A, Wang X, Ludwig L, Guda C. A meta analysis of pancreatic microarray datasets yield new targets as cancer genes and biomarkers. Plos One. 2014 doi: 10.1371/journal.pone.0093046. http://dx.doi.org/10.1371/journal.pone.0093046. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Surawska H, Ma C, Salgia R. The role of ephrins and Eph receptors in cancer. Cytokine & Growth Factor Reviews. 2004;15:419–433. doi: 10.1016/j.cytogfr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 79.Maher D, Gupta B, Nagata S, Jaggi M, Chauhan S. Mucin 13: structure, function, and potential roles in cancer pathogenesis. Mol Cancer Res. 2011;9:531–537. doi: 10.1158/1541-7786.MCR-10-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheng Z, Assfag V, Shi X, Lin S, Xia J, Yang P, Huser N, Shen F. Effect of p120 catenin silencing on biological behaviors of PANC-1 cells. J Huazhong Univ Sci Technolog Med Sci. 2012;32:707–712. doi: 10.1007/s11596-012-1022-6. [DOI] [PubMed] [Google Scholar]
- 81.Ormanns S, Altendorf-Hofmann A, Jackstadt R, Horst D, Assmann G, Zhao Y, Bruns C, Kirchner T, Knosel T. Desmogleins as prognostic biomarkers in resected pancreatic ductal adenocarcinoma. Br J Cancer. 2015;113:1460–1466. doi: 10.1038/bjc.2015.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vindrieux D, Le Corre L, Hsie J-T, Metivier R, Escobar P, Caicedo A, Brigitte M, Lazennec G. Coxsackie and adenovirus receptor is a target and a mediator of estrogen action in breast cancer. Endocrine-Related Cancer. 2011;18:311–321. doi: 10.1530/ERC-10-0230. [DOI] [PubMed] [Google Scholar]
- 83.Sun C, Zargham R, Shao Q, Gui X, Marcus V, Lazaris A, Salman A, Metrakos P, Qu X, Gao Z. Association of CD98, integrin b1, integrin b3 and Fak with the progression and liver metastases of colorectal cancer. Pathol Res Pract. 2014;210:668–674. doi: 10.1016/j.prp.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 84.Miyake M, Mizutani S, Koide H, Kaziro Y. Unfarnesylated transforming Ras mutant inhibits the Ras-signaling pathway by forming a stable Ras-Raf complex in the cytosol. FEBS Letters. 1996;378:15–18. doi: 10.1016/0014-5793(95)01416-0. [DOI] [PubMed] [Google Scholar]
- 85.Mehus AA, Anderson RH, Roux KJ. BioID identification of lamin-associated proteins. Methods Enzymol. 2016;569:3–22. doi: 10.1016/bs.mie.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell. 2016;27:1188–1196. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim DI, Jensen SC, Roux KJ. Identifying protein-protein associations at the nuclear envelope with BioID. Methods Mol Biol. 2016;1411:133–146. doi: 10.1007/978-1-4939-3530-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haugsten EM, Sorensen V, Kunova Bosakova M, de Souza GA, Krejci P, Wiedlocha A, Wesche J. Proximity labeling reveals molecular determinants of FGFR4 endosomal transport. J Proteome Res. 2016;15:3841–3855. doi: 10.1021/acs.jproteome.6b00652. [DOI] [PubMed] [Google Scholar]
- 89.Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R. Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem J. 2016;473:1719–1732. doi: 10.1042/BCJ20160031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prakash P, Sayyed-Ahmad A, Cho KJ, Dolino DM, Chen W, Li H, Grant BJ, Hancock JF, Gorfe AA. Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci Rep. 2017;7:40109. doi: 10.1038/srep40109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, Hancock JF. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell. 2017;168:239–251. doi: 10.1016/j.cell.2016.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi T, Niepel M, McDermott JE, Gao Y, Nicora CD, Chrisler WB, Markillie LM, Petyuk VA, Smith RD, Rodland KD, Sorger PK, Qian WJ, Wiley HS. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci Signal. 2016;9(436):rs6. doi: 10.1126/scisignal.aaf0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Contente S, Yeh TJA, Friedman R. H-Ras localizes to cell nuclei and varies with the cell cycle. Genes Cancer. 2011;2:166–172. doi: 10.1177/1947601911405042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jeong MH, Bae J, Kim WH, Yoo SM, Kim JW, Song PI, Choi KH. p19ras interacts with and activates p73 by involving the MDM2 protein. J Biol Chem. 2006;281:8707–8715. doi: 10.1074/jbc.M513853200. [DOI] [PubMed] [Google Scholar]
- 95.Fuentes-Calvo I, Blazquez-Medela AM, Santos E, Lopz-Novoa JM, Martinez-Salgado C. Analysis of K-Ras nuclear expression in fibroblasts and mesangial cells. PLoS ONE. 2010;5:E8703. doi: 10.1371/journal.pone.0008703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang CJ, Huang SH, Chien CC, Lee HHC, Yang SH, Chang CC, Lee CL. Impact of microsatellite status on chemotherapy for colorectal cancer patients with KRAS or BRAF mutation. Oncology Let. 2016;12:4427–4434. doi: 10.3892/ol.2016.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]