

Abstract
Background/Aim: Activin and its antagonist follistatin (FST) have been implicated in several solid tumours. This study investigated the role of FST in breast cancer. Materials and Methods: FST expression was examined using reverse transcription polymerase chain reaction (RT-PCR), real-time quantitative polymerase chain reaction (qPCR) and immunohistochemistry in a cohort of breast cancer samples. Expression was correlated to pathological and prognostic parameters in our patient cohort. FST was overexpressed in MCF-7 cells and assays for growth and invasion were performed. Results: FST is expressed in breast tissue, in the cytoplasm of mammary epithelial cells. Expression was decreased in breast cancer tissue in comparison to normal mammary tissue. Over-expression of FST in vitro led to significantly increased growth rate and reduced invasion. Higher FST associates with lower-grade tumours and better survival. Conclusion: Our results suggest a role for FST as a suppressor of invasion and metastasis in breast cancer.
Keywords: Follistatin, breast cancer, survival
Follistatin (FST) is a secreted extracellular regulatory protein that binds activin and, with less affinity, related TGFβ superfamily members such as bone morphogenetic proteins (BMPs), preventing access to their receptors (1,2). Originally thought to act on the pituitary to regulate FSH release, FST has subsequently been noted in many other tissues, in particular co-localising with activin resulting in autocrine and paracrine cellular regulation (1,3). As an antagonist of activin, FST has diverse regulatory roles in embryogenesis, tissue differentiation and repair, gonadal function and inflammatory and immune processes (3,4).
Three major FST isoforms can be produced, namely FST288 (from splice variant mRNA precursor FST317), FST315 (from splice variant mRNA precursor FST344) and a third FST isoform, FST303, produced from the post-translational truncation of the FST315 C-terminus (5). These three main FST isoforms can also be glycosylated to yield six further FST isoforms (6). FST288 and FST315 are differentially expressed in human tissues, but FST315 is the predominant isoform, whilst the FST288 isoform accounts for less than 5% of the encoded mRNA (6,7) .
The FST/activin interaction has been implicated in tumour proliferation, angiogenesis and metastasis in several solid tumours (8-11). Activin has primarily been seen as an inhibitor of cellular proliferation, although certain cell populations seem to be stimulated by activin. Thus, the overall result of activin/FST action in cancer may be both context and cell type specific (3,6).
In the breast, FST is expressed in the normal mammary gland and in different breast proliferative diseases, with additional experimental evidence suggesting that FST can modulate the breast cancer cell cycle (3,12-14). However, its role in breast cancer growth and metastasis is far from clear. In this study we demonstrated a clinical correlation between FST expression and survival in breast cancer, and that overexpression of FST in vitro reduces invasion of breast cancer cells.
Patients and Methods
Cell lines and patient tissue samples. Human breast cell lines MCF-7,MDA MB 231, ZR-751, BT 549 and BT-20 were obtained from and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal calf serum and antibiotics (PAA Laboratories Ltd., Somerset, UK). Cells were incubated at 37˚C in 5% CO2 at 95% humidity.
Breast cancer tissue and normal breast tissue samples were collected during surgery at the University Hospital of Wales. In total, 93 tumour samples and 30 normal breast tissue samples were obtained from patients who were enrolled in the study. The tissues obtained during surgery were snap-frozen in liquid nitrogen and stored at −80˚C. The presence of tumour cells was verified by a pathologist (Anthony Douglas-Jones). Background tissues were confirmed to be free of tumour deposits. The median follow-up time was 120 months. All human specimens and data were obtained according to a protocol reviewed and approved by the local ethical committee, and all patients signed an informed consent form.
Immunohistochemistry and antibodies. Frozen sections of breast tumours and background breast tissue were cut at a thickness of 6 μm. Sections were air-dried and fixed in a mixture of 50% acetone and 50% methanol before a rehydration using Tris-buffered saline (TBS) buffer for 20 min. The sections were then incubated for 20 min in a 0.6% bovine serum albumin TBS and probed with FST antibody (1:100 dilution, sc-23553, Santa Cruz Biotechnology, TX, USA) for 1 h at room temperature. Following three washes with TBS, sections were incubated for 30 min with a secondary biotinylated antibody (1:1,000, Multi Link Swine anti-goat/ mouse/rabbit immunoglobulin; Dako Inc., Ely, Cambridgeshire, UK). Avidin/Biotin Complex (Vector Laboratories Ltd, Peterborough, UK) was then applied before a subsequent incubation with the chromogen 3, 3’-diaminobenzidine (Vector Laboratories Ltd.) in the dark for 5 min, and were then counterstained in Gill’s haematoxylin.
Reverse transcription, PCR and real-time PCR. RNA was isolated using the TRI Reagent from Sigma (USA) according to the manufacturer’s instructions. RNA quantification was carried out using a spectrophotometer. RNA (0.5 μg) was reverse transcribed into cDNA using a standard reverse transcription kit. The quality of generated cDNA was verified using GAPDH primers (Table I). Reaction conditions started with an initial denaturation of 5 min at 94˚C followed by 35 cycles of 10 sec at 94˚C, 30 sec at 55˚C for annealing and 30 sec at 72˚C, with a final elongation of 72˚C for 10 min. Amplified products were then separated on a 1% agarose gel and stained using Sybr safe DNA gel stain (Thermo Scientific UK Ltd., Loughborough, UK).
Table I. Primers used for PCR and QPCR.
Transcript levels of FST in the breast tissue specimens were determined using real-time PCR as reported previously (15). An additional sequence (5’-actgaacctgaccgtaca-3’) was added to the antisense primer which is complementary to the universal Z probe (Intergen Inc., Oxford, UK). The reaction was carried out using iCycler iQ™ (Bio-Rad, Hemel Hempstead, Hertfordshire, UK), which is equipped with an optical unit that allows real-time detection of 96 reactions. The sequences of the primers used in the current study are listed in Table I.
FST over-expression model. Full-length FST344 was amplified from a cDNA library of normal human prostate tissue and was then cloned into a mammalian expression plasmid vector (TOPO TA pEF/His, Invitrogen Inc., Paisley, UK). The constructed FST expression plasmid vector was verified using DNA sequencing. For the overexpression experiments, MCF-7 cells were transfected with the constructed FST expression plasmids or an empty pEF plasmid respectively. Transfected cells were selected using 5 μg/ml blasticidin for a period up to two weeks. Selected cells were subject to verification using real time PCR before further use in the experiments.
Cell growth assay. The growth of the breast cancer cell lines was assessed using a colorimetric-based method. 3×103 cells were seeded per well on a 96-well plate. Cells were fixed on day one, three, and five in 4% formaldehyde. Following staining with 0.5% crystal violet, the crystal violet was extracted using 10% acetic acid and the absorbance was read at a wave length of 540 nm. Growth rate was calculated using a formula, Growth rate (%)=absorbance (Day 3 or Day 5)/absorbance (Day1) ×100.
Cell invasion assay. Cell invasion assays were performed using inserts with 8-μm pores coated with 50 μg Matrigel (BD Biosciences, San Jose, CA, USA). Cells (2×105) were added to transwell inserts on top of the artificial basement membranes, and into 24-well plate as control. After three days incubation inserts were fixed with formalin and stained with crystal violet. The Matrigel and cells remained in the inserts were removed using a cotton swap. Crystal violet stained invaded cells were quantified by extraction of stain using 10% acetic acid. The invasion was measured using invasion rate, i.e. absorbance of invaded cells/ absorbance of corresponding control ×100 (%).
Statistical analysis. Analysis of the data was performed using SigmaPlot 11 (Systat Software, San Jose, CA, USA). Medians were compared using the Mann-Whitney U-test for non-normally distributed data, while means were compared using the two-sample t-test for normally distributed data. Kaplan Meier survival curves were produced from online expression databases KMplot (16) and Gene Expression Miner (17). Gene Expression Miner also used publically available expression data to produce a boxplot correlation of FST mRNA level with tumour grade.
Results
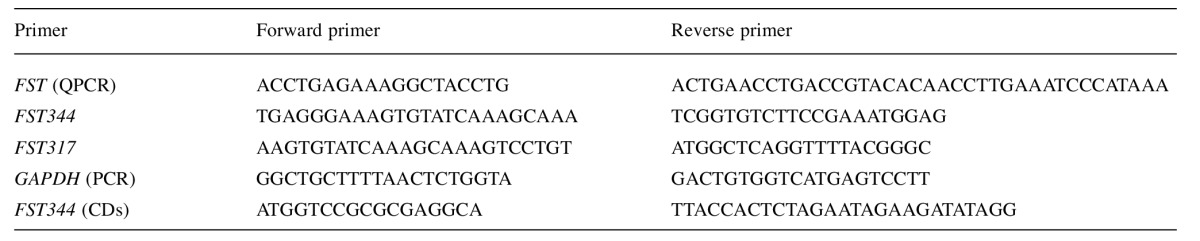
FST is expressed in a variety of normal tissues, cancer tissues and human breast cancer cell lines. As one might expect for a protein originally related to gonadal function and development, FST was expressed in the ovary. In our data it was also seen at a lower level in normal breast and colon. It was not expressed in bone, liver or skin (Figure 1A). This correlates well with gene expression data from the recent review by Shi et al. (6).
Figure 1. Expression of follistatin in human tissues and breast cancer cell lines. (A) The expression of follistatin transcripts in different human tissues using RT-PCR. L is DNA ladder. 1-4 are placenta. 5-7 are ovary. 8 and 9 are normal breast tissues. 10 is omentum. 11 is liver. 12 and 13 are skin. 14 is colon. 15 is bone. 16 and 17 are breast cancer tissue. 18 and 19 are prostate cancer tissues. 20 is negative control. (B) Follistatin transcript splicing variants (FST344, NM_013409.2 and FST317, NM_006350.3) were detected in five breast cancer cell lines, i.e. MCF-7, MDAMB-231, ZR-751, BT-549 and Bt-20, using RT-PCR.
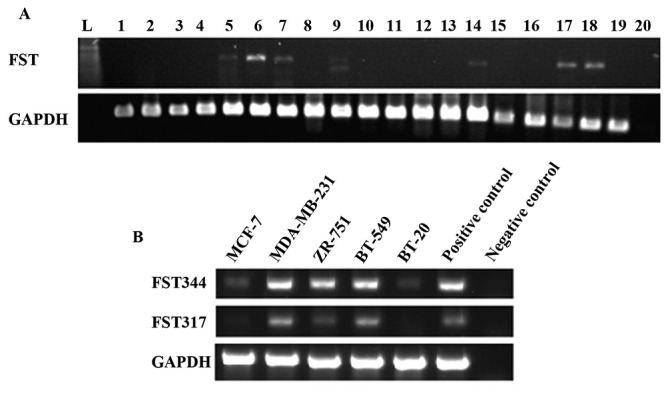
In human breast cell lines, both FST317 and FST344 were seen in MDA-MB-231 and BT-549 cells. Lower expression levels of both were seen in MCF-7 and BT-20 lines. In ZR-751 cells FST344 was more strongly expressed than FST317 (Figure 1B).
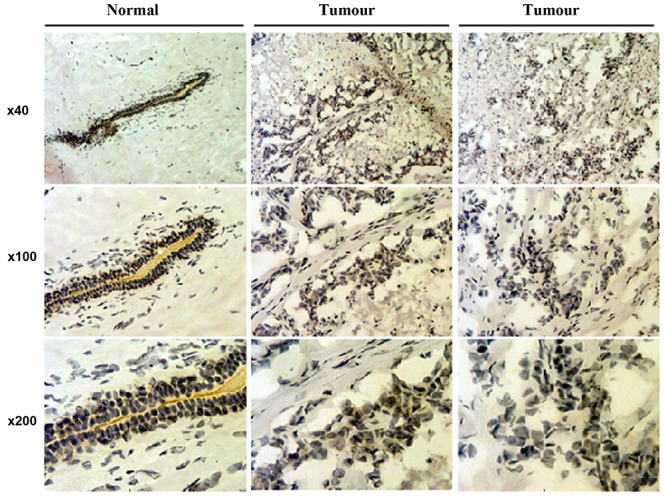
Follistatin is seen in the cytoplasm and nucleus of mammary epithelium and is reduced in invasive breast carcinoma. Immunohistochemical staining was shown to be more intense for FST in the cytoplasm of normal mammary epithelial cells, as well as the nucleus. As the cells become more invasive, through ductal carcinoma in situ (DCIS) to invasive carcinoma, the staining reduces in intensity (Figure 2). This is in agreement with other studies (12,18), one of which noted a decrease in FST transcripts on qPCR of invasive ductal carcinoma tissue samples compared to normal tissue.
Figure 2. Immunochemical staining of follistatin in human breast carcinomas, ductal carcinoma in situ (middle) and invasive ductal carcinoma (right) in comparison to normal breast tissues (left).
Overexpression of follistatin in MCF-7 cells increases proliferation. The constructed FST expression plasmid was successful at producing a 3.5-fold increase in FST expression in the transfected MCF-7 cells (Figure 3A). Our cellular growth assay (n=3) demonstrates a significant increase in proliferation in MCF-7 with FST overexpression compared to control MCF-7 pEF cells (Figure 3).
Figure 3. FST overexpression model and functional assays. (A) FST344 expression plasmid produced a 3.5-fold increase in FST expression compared to pEF control plasmid in the transfected MCF-7 cells. (B) Invasion assay demonstrating a significantly reduced invasion rate in FST-overexpressing MCF-7 cells compared to control (p<0.001). (C) Percentage growth rate at day three of the assay, with significant increase in growth rate in FST overexpressing MCF-7 cells compared to control (p<0.0001). (D) This is maintained at day five of growth assay (p<0.0001).
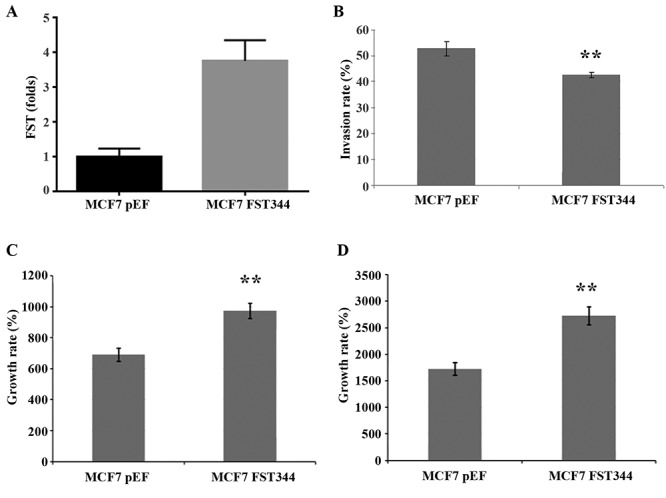
Overexpression of follistatin in MCF-7 cells reduces invasion. Invasion assay (n=3) showed that in comparison to pEF control, MCF-7 cells overexpressing FST showed significantly decreased invasion (Figure 3).
Low follistatin expression in the clinical cohort is associated with higher histological grade and poorer survival. The use of publically-available expression databases with clinical parameters, such as KMplot allow the collation of expression data to give a larger cohort of patients. This data shows that high FST expression in breast cancer tumours is associated with better relapse free survival (Figure 4A). There was no correlation between survival and FST expression in the KMplot analyses of subgroups of patients according to receptor status or lymph node positivity, which correlates with our cohort regarding lymph node status and FST expression (Table II). High FST expression was significantly correlated with better overall survival at 5 years (Figure 4B, p=0.007), but became less statistically significant over 10 and 20 years survival (Figure 4C and D, p=0.06 both). This may reflect the smaller numbers of patients in the cohort over a longer follow up period, but raises an interesting possibility that low FST marks more aggressive disease.
Figure 4. Correlation of FST expression to patient survival. (A) Relapse-free survival is better in tumours with higher FST expression (p<0.001). (B) Overall patient survival over 5 years follow-up shows significantly better survival in those with higher FST expression (p=0.007). (C) Overall patient survival over 10 years and (D) 20 years follow-up do not reach statistical significance in terms of survival difference (p=0.06), but does show a similar trend regarding better survival with higher FST expression.
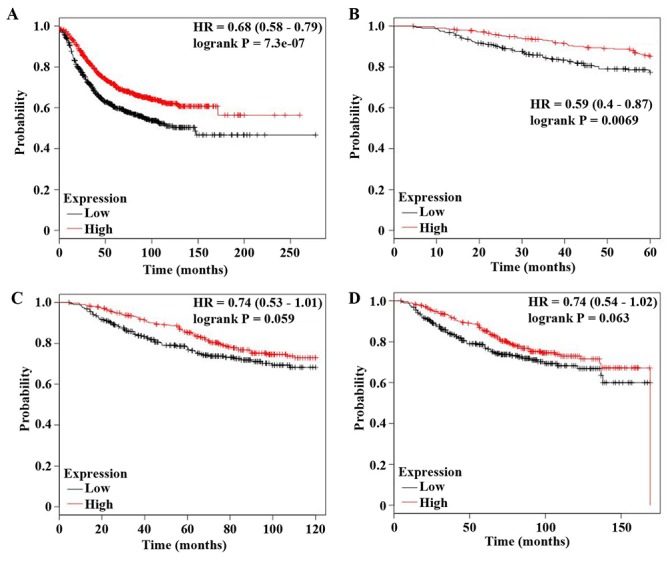
Table II. Tumour cohort FST expression levels.
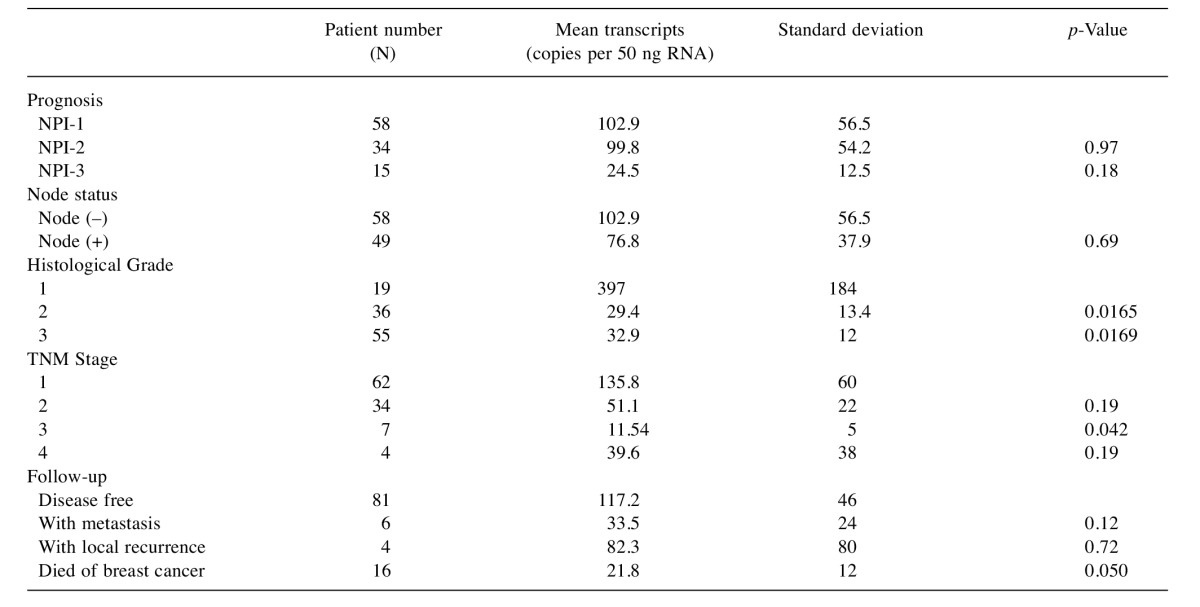
NPI, Nottingham prognostic index.
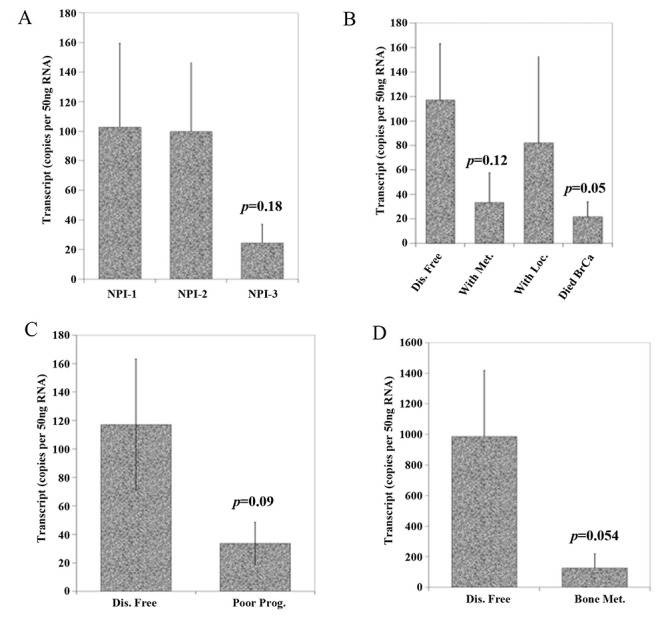
Furthermore, the qPCR transcript levels in our patient cohort showed that FST expression was decreased in undifferentiated and more aggressive higher grade tumours (Table II and Figure 5A). Geneminer data also support the finding (Figure 5B). Our cohort also showed that those with poor prognostic features such as high TNM stage and high Nottingham prognostic index score had lower FST transcript levels (Figures 5C and Figure 6), however, this did not consistently reach significance. Interestingly, low FST transcript levels appeared to be associated with death from breast cancer and bone metastases (p=0.05 both).
Figure 5. Expression of FST is associated with tumour grade and TNM staging. A) FST qPCR transcript levels from our clinical cohort according to patient’s histological tumour grade. The X axis represents tumour grade, a reflection of how differentiated and therefore how ‘aggressive’ it’s potential for invasion. Grade 1 is well differentiated, Grade 2 is moderately differentiated, whilst Grade 3 is poorly differentiated tumour. Those with grade 2 and 3 tumours had lower FST levels than grade 1 (p=0.0165 and 0.0169 respectively). B) Gene expression miner uses publicly available patient expression data from patient breast cancer tissue. Low FST expression correlated with higher grade, poorly differentiated breast tumours (p<0.0001), similar to our cohort in panel A. C) FST qPCR transcript levels according to TNM staging. Those tumours that are TNM stage 1 (i.e. smaller size, with less nodal and distant metastases) had higher FST expression than TNM 2, 3 and 4 (p=0.19, 0.042 and 0.19 respectively).

Figure 6. A) FST qPCR transcript levels according to patient’s Nottingham Prognostic Index level. Those with lower FST has a poorer prognostic score, but this does not reach significance (p=0.18). B) FST qPCR transcript levels according to disease outcomes showing those that died from their disease have a lower FST expression compared to those remaining disease free (p=0.05). C) and D) FST expression transcripts were higher in those remaining disease-free compared to those with poor prognosis (all though this did not reach significance p=0.09) and those with bone metastases (p=0.054).
Discussion
Activin and BMPs are both TGFβ superfamily members, that employ different combinations of serine-threonine kinase receptors that when phosphorylated, result in activation of canonical Smad 2/3 and Smad 1/5/8 intracellular signalling pathways respectively (19-21). Non-canonical Akt/PI3K, MAPK/ERK and Wnt/β-catenin pathways can also be activated, dependant on the combination of receptors recruited. Consequently, TGFβ, activin, and BMPs engage in a complex signalling network, resulting in the transcription of target genes with vital influences on cell growth, differentiation, proliferation and survival. It is not surprising therefore, that these proteins are implicated in tumorigenesis (19-22).
FST is a known transcription target of activin signalling (23) and acts as a negative feedback regulator, binding activin with high affinity to prevent its interaction with activin receptors and inhibiting its biological cellular functions (4,6). Due to this regulatory influence, follistatin has also been implicated in tumourigenesis in certain solid tumours (6). Some studies have examined the role of activin in breast cancer, however, very little is known regarding the influence of follistatin in breast cancer.
Our results show variation in FST expression, with MDA-MB-231, ZR-751 and BT-549 cells expressing FST at a higher level than MCF-7 and BT-20 cells. It is possible that differential FST expression amongst the cell lines reflects the heterogeneous nature of breast cancer and its molecular subtypes. The most clinically-relevant subtypes are those breast cancers that express oestrogen receptor (ER), progesterone receptor (PR) or human epidermal growth factor receptor 2 (HER2), as this determines treatment options and prognosis. Prior studies provided evidence for an interaction between downstream activin signalling pathway components, and ER signalling in breast cancer cells (24,25). One study (25) found activin and oestrogen signalling in breast cancer cells can be mutually repressive at a transcriptional level. Kalkhoven et al. (26) established that the presence of ER in breast cancer cells influences whether these cells are growth inhibited by activin. They determined ER-positive cell lines were growth inhibited by treatment with activin, with ZR-751 cells being the most sensitive, whereas the ER-negative cell lines were resistant to the growth-inhibitory potential of activin. In BT-20 cells this resistance to activin signalling was thought to be related to low activin receptor levels, however, this was not the case in MDA-MB-231 cells, in which activin binding to receptors was detected despite their resistance to the anti-proliferative effect of activin, suggesting the effect is downstream of the activin receptors. Conversely, others have found that activin treatment inhibits the growth of both ER positive and negative cell lines (27). This study also demonstrated that the growth inhibition of activin was abrogated to a greater degree by the addition of follistatin treatment in ER negative cells, compared to ER positive cells. They show MDA-MB-231 cells actually secrete more follistatin than MCF-7 cells and suggest that this auto and paracrine signalling may be the reason that MDA-MB-231 cells are more resistant to activin effects on cellular growth. Follistatin has also been reported as being more highly expressed in ER negative tumours (13).
In our data FST is highly expressed in MDA-MB-231, BT-549 and ZR-751 cells, and less so in MCF-7 and BT-20 cells, which may reflect the different levels of activin pathway activity between the cell lines, different degrees of dysregulation in the activin pathways or crosstalk with other signalling pathways such as hormone receptors, that can alter FST transcription. This added layer of complexity in relation to follistatin and activin signalling in breast cancer certainly warrants more detailed exploration, particularly in relation to loss of hormone receptor expression, treatment resistance and progression of disease.
Activin is well-documented as slowing-down cell proliferation in breast cancer studies (26). Burdette et al. observed that activin treatment arrested T47D breast cancer cells in the G0-G1 cell cycle. This effect was abrogated either by using the type I receptor inhibitor SB431542, or an adenoviral dominant negative Smad3, or treatment with follistatin. When they treated the cells with follistatin alone, cells proliferated and accumulated in S phase, when compared to untreated cells (28). This is certainly supported by our data, which shows that overexpressing FST in MCF-7 breast cancer cells increases proliferation and growth. FST has also been seen at increased levels of expression in benign proliferative breast disorders such as fibroadenoma (12).
Interestingly, in a study that used a SCID murine model inoculated with FST288 expressing R30C mammary cells, the FST-expressing tumours were smaller in volume compared to control, or those expressing activin. This seemed to be as a result of increased apoptosis in the FST tumours (3). So, although FST may result in increased growth and proliferation of breast cancer cells, the rate of cellular death may also increase, such that in vivo, the tumours are smaller. On the other hand, BMP-promoted angiogenesis may also be involved, but is yet to be investigated in this context.
Our results are the first report that FST decreases invasiveness in breast cancer cells. Thus, these findings can only be indirectly supported by other studies such as Bashir et al. (18), who treated MCF-7 cells with activin, and found an increase in epithelial to mesenchymal transition (EMT) and decreased E-cadherin expression, implying a tendency to greater invasiveness. Both activin and TGFβ have been seen to promote EMT (18,29). In vivo, a murine model has shown MCF-7 cells overexpressing activin establish tumours with more mesenchymal transition features compared to control tumours (18). We might, thus, expect higher FST levels to antagonise this effect of activin and reduce EMT and invasive properties. Follistatin also directly binds to TGF-β3 and completely blocks TGF-β3-induced EMT of the normal murine mammary gland (NMuMG) epithelial cell line in vitro (29). This inhibition of EMT by FST is reflected in our clinical data, that low FST expression correlates with higher grade tumours, which typically display more aggressive and invasive clinical behaviour and are histologically less well differentiated. In our cohort FST protein was decreased in invasive breast cancer compared to normal tissue, supported by Bashir et al., who found qPCR transcript levels of FST were significantly lower in breast tumours (18) which may indicate reduction in FST expression as invasive characteristics develop. Conversely, publically available mRNA expression EST data (see Shi et al. (6)) suggests an increased expression of FST in breast cancer as compared to normal mammary tissue and Bloise et al. report no significant difference on semi-quantitative scoring of FST expression in normal mammary tissue versus in situ and invasive carcinoma (12). Of course this variability can be accounted for by differing size and tumour characteristics of the patient cohorts and the different experimental methods utilised. Mange et al. (30) found relative secretion of FST was increased in tumourigenic MCF10.DCIS and MCF10.CA1d cells compared to non tumourigenic MCF10A cells. They then determined, using enzyme-linked immunosorbant assay (ELISA) that serum concentration of FST was higher in invasive breast cancer patients compared to healthy matched controls. The breast cancer patients were predominantly histological grade one and two, ER positive, and of small size, suggesting FST serum levels increase in early breast cancers with better prognostic features compared to healthy controls, but does not allow us to conclude what the serum levels in those with more advanced disease may be.
We have correlated higher tumour FST expression with better survival, particularly in the first 5 years of follow-up, suggesting that FST may have a protective role in regards to progression and metastasis of breast cancer that is worthy of further exploration. The increase in cellular proliferation and decreased invasiveness in breast cancer cells in relation to higher expression of FST that we have demonstrated suggests a dual role in tumourigenesis for FST. A balance between proliferation and differentiation is coordinated by TGFβ ligands and their antagonists. This is well recognised in TGFβ family members, such as TGFβ itself which, similarly to activin, is known to have inhibitory effects on cellular proliferation in several cancers, but is also associated with EMT, invasion and metastasis in aggressive and progressive disease (19,22).
The most frequent metastatic site in breast cancer is the bone, resulting in osteolytic lesions that cause significant morbidity and reduce survival (31). As an antagonist of BMPs, which are regulators of bone development and implicated in development of bone metastases in several solid tumours (20), FST may also have a role to play in metastasis to bone specifically. Our clinical cohort demonstrated a significantly lower expression of FST in primary tumours that metastasised to bone.
Overexpression of another BMP antagonist Noggin in breast cancer cells, is associated with the potential to develop osteolytic lesions in bone (32). And when looking at serum markers in breast cancer patients (33), activin levels were reported to be significantly increased in breast cancer compared with age-matched control donors, those with bone metastases having significantly higher levels than patients without.
Kang et al. (34) found follistatin overexpression to be part of a bone metastasis gene signature in a subpopulation of MDA-MD-231 cells, which included overexpression of CXCR4, MMP1, FGF5, CTGF, and IL-11. This bone metastasis signature differed from that of adrenal metastasis gene signature. However, Kang et al. demonstrated only one potential bone metastasis gene signature-there may well be several others in which overexpression of FST is not utilised. Overexpression of FST may also emerge later in the course of the disease, and thus may not be reflected in the expression profile of the patient’s primary tumour. Nevertheless, these initial investigations raise significant interest in the role of follistatin specifically in bone metastases.
Taken together, we suggest that FST has varying potential roles in breast cancer development, progression, invasion and metastasis that are worthy of note and further elucidation. From our data, FST over-expression appears to promote in vitro proliferation and reduce invasiveness. Higher FST expressing tumors are associated with improved patient survival, and FST may thus prove a potential prognostic and therapeutic target in breast cancer.
Acknowledgements
The Authors would like to thank the Department of Surgery at the University Hospital of Wales, and Dr. Anthony Douglas-Jones. The Authors also wish to thank Cancer Research Wales for the support to the study.
References
- 1.Tsuchida K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H, Inokuchi K. Activin signaling as an emerging target for therapeutic interventions. Cell Commun Signal. 2009;7:15. doi: 10.1186/1478-811X-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nolan K, Thompson TB. Bmp and bmp regulation: Structure and function. In: Bone morphogenetic proteins: Systems biology regulators. Vukicevic S and Sampath KT (eds.) Springer International Publishing. 2017:pp. 73–111. [Google Scholar]
- 3.Krneta J, Kroll J, Alves F, Prahst C, Sananbenesi F, Dullin C, Kimmina S, Phillips DJ, Augustin HG. Dissociation of angiogenesis and tumorigenesis in follistatin- and activin-expressing tumors. Cancer Res. 2006;66(11):5686–5695. doi: 10.1158/0008-5472.CAN-05-3821. [DOI] [PubMed] [Google Scholar]
- 4.Harrison CA, Gray PC, Vale WW, Robertson DM. Antagonists of activin signaling: Mechanisms and potential biological applications. Trends Endocrinol Metab. 2005;16(2):73–78. doi: 10.1016/j.tem.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Sugino K, Kurosawa N, Nakamura T, Takio K, Shimasaki S, Ling N, Titani K, Sugino H. Molecular heterogeneity of follistatin, an activin-binding protein. Higher affinity of the carboxyl-terminal truncated forms for heparan sulfate proteoglycans on the ovarian granulosa cell. J Biol Chem. 1993;268(21):15579–15587. [PubMed] [Google Scholar]
- 6.Shi L, Resaul J, Owen S, Ye L, Jiang WG. Clinical and therapeutic implications of follistatin in solid tumours. Cancer Genomics Proteomics. 2016;13(6):425–435. doi: 10.21873/cgp.20005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneyer AL, Wang Q, Sidis Y, Sluss PM. Differential distribution of follistatin isoforms: Application of a new fs315-specific immunoassay. J Clin Endocrinol Metab. 2004;89(10):5067–5075. doi: 10.1210/jc.2004-0162. [DOI] [PubMed] [Google Scholar]
- 8.Ogino H, Yano S, Kakiuchi S, Muguruma H, Ikuta K, Hanibuchi M, Uehara H, Tsuchida K, Sugino H, Sone S. Follistatin suppresses the production of experimental multiple-organ metastasis by small cell lung cancer cells in natural killer cell-depleted scid mice. Clin Cancer Res. 2008;14(3):660–667. doi: 10.1158/1078-0432.CCR-07-1221. [DOI] [PubMed] [Google Scholar]
- 9.Deli A, Kreidl E, Santifaller S, Trotter B, Seir K, Berger W, Schulte-Hermann R, Rodgarkia-Dara C, Grusch M. Activins and activin antagonists in hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1699–1709. doi: 10.3748/wjg.14.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razanajaona D, Joguet S, Ay AS, Treilleux I, Goddard-Leon S, Bartholin L, Rimokh R. Silencing of flrg, an antagonist of activin, inhibits human breast tumor cell growth. Cancer Res. 2007;67(15):7223–7229. doi: 10.1158/0008-5472.CAN-07-0805. [DOI] [PubMed] [Google Scholar]
- 11.Reis FM, Luisi S, Carneiro MM, Cobellis L, Federico M, Camargos AF, Petraglia F. Activin, inhibin and the human breast. Mol Cell Endocrinol. 2004;225(1-2):77–82. doi: 10.1016/j.mce.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Bloise E, Couto HL, Massai L, Ciarmela P, Mencarelli M, Borges LE, Muscettola M, Grasso G, Amaral VF, Cassali GD, Petraglia F, Reis FM. Differential expression of follistatin and flrg in human breast proliferative disorders. BMC Cancer. 2009;9:320. doi: 10.1186/1471-2407-9-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couto HL, Dela Cruz C, Buzelin MA, Toppa NH, Wainstein AJ, Reis FM. Follistatin expression in human invasive breast tumors: Pathologic and clinical associations. Appl Immunohistochem Mol Morphol. 2016 doi: 10.1097/PAI.0000000000000385. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14.Sengupta D, Bhargava DK, Dixit A, Sahoo BS, Biswas S, Biswas G, Mishra SK. Errbeta signalling through fst and bcas2 inhibits cellular proliferation in breast cancer cells. Br J Cancer. 2014;110(8):2144–2158. doi: 10.1038/bjc.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye L, Bokobza S, Li J, Moazzam M, Chen J, Mansel RE, Jiang WG. Bone morphogenetic protein-10 (bmp-10) inhibits aggressiveness of breast cancer cells and correlates with poor prognosis in breast cancer. Cancer science. 2010;101(10):2137–2144. doi: 10.1111/j.1349-7006.2010.01648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szasz AM, Lanczky A, Nagy A, Forster S, Hark K, Green JE, Boussioutas A, Busuttil R, Szabo A, Gyorffy B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7(31):49322–49333. doi: 10.18632/oncotarget.10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jezequel P, Campone M, Gouraud W, Guerin-Charbonnel C, Leux C, Ricolleau G, Campion L. Bc-genexminer: An easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat. 2012;131(3):765–775. doi: 10.1007/s10549-011-1457-7. [DOI] [PubMed] [Google Scholar]
- 18.Bashir M DS, Mukherjee G, Kondaiah P. Activin-a signaling promotes epithelial–mesenchymal transition, invasion, and metastatic growth of breast cancer. Breast Cancer. 2015;1(15007):13. doi: 10.1038/npjbcancer.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loomans HA, Andl CD. Intertwining of activin a and tgfbeta signaling: Dual roles in cancer progression and cancer cell invasion. Cancers (Basel) 2014;7(1):70–91. doi: 10.3390/cancers7010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antsiferova M, Werner S. The bright and the dark sides of activin in wound healing and cancer. J Cell Sci. 2012;125(Pt 17):3929–3937. doi: 10.1242/jcs.094789. [DOI] [PubMed] [Google Scholar]
- 21.Davis H, Raja E, Miyazono K, Tsubakihara Y, Moustakas A. Mechanisms of action of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev. 2016;27:81–92. doi: 10.1016/j.cytogfr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Lebrun JJ. The dual role of tgfbeta in human cancer: From tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;2012:381428. doi: 10.5402/2012/381428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartholin L, Maguer-Satta V, Hayette S, Martel S, Gadoux M, Corbo L, Magaud JP, Rimokh R. Transcription activation of flrg and follistatin by activin a, through smad proteins, participates in a negative feedback loop to modulate activin a function. Oncogene. 2002;21(14):2227–2235. doi: 10.1038/sj.onc.1205294. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, Liu Z, Lu C, Mao Z, Cao X. Smad4 as a transcription corepressor for estrogen receptor alpha. J Biol Chem. 2003;278(17):15192–15200. doi: 10.1074/jbc.M212332200. [DOI] [PubMed] [Google Scholar]
- 25.Burdette JE, Woodruff TK. Activin and estrogen crosstalk regulates transcription in human breast cancer cells. Endocrine-related Cancer. 2007;14(3):679–689. doi: 10.1677/ERC-07-0054. [DOI] [PubMed] [Google Scholar]
- 26.Kalkhoven E, Roelen BA, de Winter JP, Mummery CL, van den Eijnden-van Raaij AJ, van der Saag PT, van der Burg B. Resistance to transforming growth factor beta and activin due to reduced receptor expression in human breast tumor cell lines. Cell Growth Differ. 1995;6(9):1151–1161. [PubMed] [Google Scholar]
- 27.Wilson C, Ottewell P, Coleman RE, Holen I. The differential anti-tumour effects of zoledronic acid in breast cancer – evidence for a role of the activin signaling pathway. BMC Cancer. 2015;15:55. doi: 10.1186/s12885-015-1066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin a mediates growth inhibition and cell cycle arrest through smads in human breast cancer cells. Cancer Res. 2005;65(17):7968–7975. doi: 10.1158/0008-5472.CAN-04-3553. [DOI] [PubMed] [Google Scholar]
- 29.Nogai H, Rosowski M, Grun J, Rietz A, Debus N, Schmidt G, Lauster C, Janitz M, Vortkamp A, Lauster R. Follistatin antagonizes transforming growth factor-beta3-induced epithelial-mesenchymal transition in vitro: Implications for murine palatal development supported by microarray analysis. Differentiation. 2008;76(4):404–416. doi: 10.1111/j.1432-0436.2007.00223.x. [DOI] [PubMed] [Google Scholar]
- 30.Mange A, Dimitrakopoulos L, Soosaipillai A, Coopman P, Diamandis EP, Solassol J. An integrated cell line-based discovery strategy identified follistatin and kallikrein 6 as serum biomarker candidates of breast carcinoma. J Proteomics. 2016;142:114–121. doi: 10.1016/j.jprot.2016.04.050. [DOI] [PubMed] [Google Scholar]
- 31.Yardley DA. Pharmacologic management of bone-related complications and bone metastases in postmenopausal women with hormone receptor-positive breast cancer. Breast Cancer. 2016;8:73–82. doi: 10.2147/BCTT.S97963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarragona M, Pavlovic M, Arnal-Estape A, Urosevic J, Morales M, Guiu M, Planet E, Gonzalez-Suarez E, Gomis RR. Identification of nog as a specific breast cancer bone metastasis-supporting gene. J Biol Chem. 2012;287(25):21346–21355. doi: 10.1074/jbc.M112.355834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leto G, Incorvaia L, Badalamenti G, Tumminello FM, Gebbia N, Flandina C, Crescimanno M, Rini G. Activin a circulating levels in patients with bone metastasis from breast or prostate cancer. Clin Exp Metastasis. 2006;23(2):117–122. doi: 10.1007/s10585-006-9010-5. [DOI] [PubMed] [Google Scholar]
- 34.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]