

Abstract
1,25-dihydroxyvitamin D3 (1,25D) used to treat human acute myeloid leukemia (AML) cells induces features of normal monocytes, but the mechanisms underlying this response is not fully understood. We hypothesized that one or more microRNAs (miRNAs) known to control mouse hematopoiesis and lineage commitment might contribute to the ability of 1,25D to control the malignant phenotype. Here we report that 1,25D markedly induces expression of miR-32 in human myeloid leukemia cells, where it targets the 3′-UTR of the mRNA encoding the pro-apoptotic factor Bim to reduce its expression. RNAi-mediated suppression of the miRNA-processing enzymes Drosha and Dicer increased Bim levels, in support of the concept that Bim is under miRNA control in AML cells. Antisense-mediated suppression of miR-32 was sufficient to upregulate Bim expression in AML cells. Conversely, ectopic expression of miR-32 downregulated Bim expression and increased the differentiation response to 1,25D treatment in a manner that was associated with increased cell survival. The positive effects of miR-32 on cell survival were confirmed by evidence of increased cell death in AML cells pre-exposed to antisense miR-32 before treatment with arabinocytosine, a chemotherapeutic drug used to treat human AML. Together, our findings indicate that miR-32 blockade is sufficient to elevate Bim expression and sensitize AML cells to chemotherapy-induced apoptosis. Thus, agents which can inhibit miR-32 expression may offer clinical utility by enhancing therapeutic efficacy in human AML.
Keywords: MicroRNA-32, Vitamin D Differentiation, Bim, Acute Myeloid Leukemia, AraC, Apoptosis
Introduction
Acute myeloid leukemia (AML) is a hematological disease characterized by blocks at various stages of hematopoietic differentiation, which lead to uncontrolled cell proliferation and accumulation of immature myeloid cells in bone marrow and the peripheral blood. The disease has extremely poor prognosis, even with the available treatment regimens, which currently are based on the eradication of the malignant stem and myeloid precursor cells (blasts) by cytotoxic agents such as arabinocytosine (AraC). Unfortunately, toxicity of these drugs to the patients limits their dosage, and recurrences of the disease are frequent (1). Thus, effective other treatment modalities are urgently needed.
The differentiation blocks responsible for the disease can be overcome in cultured AML cells by supra-physiological concentrations of the active form of vitamin D, 1,25-dihydroxyvitamin D3 (1,25D) (2–4). The rationale for the ability of 1,25D to achieve this effect has been presented as due to an elevation of the levels of transcription factors (TFs), e.g. jun/AP1 and C/EBP beta, which permit the blasts to bypass the barrier presented by the leukemia-causing mutations, frequently by a switch from myeloid to monocytic differentiation lineage induced by these TFs (5–9). However, while all-trans retinoic acid (ATRA) has been successfully used in the clinic, induction of differentiation of AML blasts by 1,25D has so far not resulted in notable clinical success. The reasons for this may include the fact that in addition to pro-differentiation activity ATRA also promotes cell death, and is particularly effective when combined with arsenic trioxide (As2O3), a toxic agent (10, 11). Similarly, increased cytotoxicity is seen when, following AraC exposure, 1,25D is added to cultured AML cells (12, 13). It seems reasonable, therefore, to explore if changes in cell survival mechanisms that accompany 1,25D–induced differentiation can be modified to increase the therapeutic potential of 1,25D.
Using a miRNA microarray platform we observed that miR-32 was the most highly elevated miRNA in human leukemia 60 (HL60) cells treated with 1,25D. One of the predicted targets of miR-32 lies in the 3′ untranslated region (UTR) of BCL2L11 gene, which encodes the pro-apoptotic protein Bim (14, 15). Therefore, we investigated if the increased levels of miR-32 in 1,25D-treated AML cells can be validated by qRT-PCR, and if so, if miR-32 regulates the expression of Bim, and whether this is associated with changes in the ability of cytotoxic agents such as AraC to induce the death of AML cells.
Materials and Methods
Cell culture
HL60-G cells, derived from a patient with promyeloblastic leukemia (16), and U937 cells, obtained from ATCC (Manassas, VA) were cultured in suspension in RPMI-1640 medium supplemented with 10% bovine calf serum (Hyclone, Logan, UT) in a humidified atmosphere containing 5% CO2 at 37°C. For U937 cells concentration of 1,25D routinely used was 10 nM, as these cells are less sensitive to 1,25D than HL60 cells, which were treated with 1 nM 1,25D. Cells were passaged 2–3 times a week and were used in the exponential growth phase. Cells were routinely tested for mycoplasma by selective culture techniques. For all experiments the cells were resuspended in fresh medium and each experiment was repeated at least three times.
Isolation of mononuclear cells from peripherial blood and selection of monocytes
Peripheral blood samples were obtained from five healthy volunteers according to institutional IRB protocol. Mononuclear cells were isolated by using Histopaque-1077 (Sigma-Aldrich, St. Louis, MO), as previously described (17). Monocytes were positively selected with CD14 MicroBeads (Miltenyi Biotec Inc. Auburn, CA) as directed by manufacturer’s protocol. Homogeneity of CD14 positive cells was determined by using EPICS XL flow cytometer (Beckman Coulter, Miami, FL).
Chemicals and antibodies
1,25D was a kind gift from Dr. Milan Uskokovic (Bioxell, Nutley NJ). Antisense oligonucleotides against Drosha (Cat#L-016996-00) and Dicer (Cat#L-003483-00) were obtained from Thermo Scientific (Chicago, IL), hsa-miR-32 anti-miR miRNA inhibitor (ID: AM12716) and hsa-miR-32 pre-miR miRNA precursor (ID: PM12716) were obtained from Applied Biosystems (Foster City, CA). Complete protease inhibitor cocktail was purchased from Hoffmann-La Roche (Nutley, NJ). Antibodies were procured from Cell Signaling Technologies, Denver, MA (Bim, Bax, secondary anti-rabbit, and anti-mouse linked to HRP), and from Santa Cruz Biotechnology, Santa Cruz, CA (Crk-L and Calregulin).
MicroRNA target predictions and pathway analysis
Public web-based prediction sites under miRbase were used to identify miRNA 32 target binding sites in the 3′UTR of human gene transcripts (18). miRBase currently links miRNA-32 to targets predicted by microcosm Targets (19); microRNA.org (20); TargetScan (21) and Pictar-Vert (22), and aims to provide a more extensive target prediction aggregation service in the future. In addition, other target prediction online softwares (DIANA-microT, miRanda, PITA) were used. Targets of miRNAs which were differentially and significantly (>1.5-fold change and p < 0.05) expressed by 1,25D were subjected to Ingenuity Pathway Analysis (IPA) (Ingenuity® Systems) performed by uploading specific miRNA lists into miRNA array analysis program. The list of gene targets of miRNAs predicted by IPA was filtered to remove duplicates and genes with no annotation in IPA listed apoptosis pathways, resulting in a list of network-eligible genes associated with the apoptosis signaling pathway.
Transfection of siRNA against Drosha and Dicer, of antisense oligonucleotides against miR-32, and of miR-32 precursor
This was carried out using Endo-Porter delivery reagent from Gene Tools Inc. (Philomath, OR). Si-Drosha, si-Dicer (Thermo Scientific), anti-miR-32 inhibitor (a chemically modified, single stranded nucleic acid designed to specifically bind to and inhibit endogenous miRNA molecules), pre-miR-32 (Ambion, Austin, TX) and appropriate non-targeting control oligonucleotides were transfected at a final concentration of 20 nM for 48 h before exposure to other compounds.
RNA isolation and quantitative RT-PCR
Total RNA was extracted by using Trizol (Invitrogen, CA) according to manufacturer’s protocol and reverse transcribed for quantification by TaqMan microRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) as previously described (23). Mature miRNAs were quantitated using two-step TaqMan RT-PCR with TaqMan microRNA kit. MiR-32 expression level were normalized using U6 rRNA as an internal control (Applied Biosystems).
Quantitative RT-PCR for Bim was carried out using a FastStart DNA SYBR Green PCR kit (Roche Diagnostics, Indianapolis, IN) as described before (23). Fold changes of mRNA levels in target gene relative to the RNA polymerase II (RPII) control were calculated by relative quantification analysis. Primers used for Bim were: upstream 5′-AGTTCTGAGTGTGACCGAGAAGGT-3′, downstream 5′-TCCTGTCTTGTGGCTCTGTCTGT A-3′; for RP II, upstream 5′-GCACCACGTCCAATGACAT-3′, downstream 5′-GTGCGG CTGCTTCCATAA-3′. The quality of PCR products were monitored using post-PCR melting curve analysis.
Markers of monocytic differentiation
Aliquots of 1 × 106 cells were harvested, washed twice with phosphate buffered saline (PBS) and incubated for 45 min at room temperature with 0.5 μl MY4-RD-1 and 0.5 μl MO1-FITC antibodies (Beckman Coulter Inc, Brea CA) to analyze the expression of monocytic differentiation surface markers CD14 and CD11b, respectively. The cells were then washed three times with ice-cold PBS, resuspended in 1 ml PBS and analyzed using EPICS XL flow cytometer (Beckman Coulter). Isotypic mouse IgG1 was used to set threshold parameters.
Cell cycle distribution
The DNA content of the cells was determined as follows: 1 × 106 cells were harvested and washed twice with PBS, then fixed with 75% ethanol at −20°C for 24 h. Cells were then collected and resuspended in 1 ml of PBS with 10 μg/ml RNase (Sigma) and 10 μg/ml propidium iodide (Sigma) for 30 min at 37°C. PI stained cells were analyzed using EPICS XL flow cytometer. The resultant distribution of DNA content was gated and analyzed using the multicycle program to determine the proportion of cells in the sub-G1/G0 fraction, which represents the non-viable cells.
Annexin V and propidium iodide staining
AML cells were induced to apoptosis by 100 μM AraC (Sigma) for 24 h. Samples were collected, washed once with PBS and resuspended in the 10 mM HEPES/NaOH binding buffer, containing 0.14 M NaCl and 2.5 mM CaCl2, pH 7.5. Apoptotic cells were stained using an Annexin V-FITC Kit (Sigma). Cells were incubated with 50 μg/ml Annexin V and 20 μg/ml propidium iodide in the dark, at room temperature for 10 min, and immediately analyzed by flow cytometry (EPICS XL). Cells Annexin V-positive/PI-negative were considered as early apoptotic, cells doubly positive, as late apoptotic.
Western blotting
Western blotting was performed using whole cell extracts as described before (23). Briefly, membranes were incubated overnight with different primary antibodies, and then blotted with a HRP-linked secondary antibodies for 1 h. The protein bands were visualized using a chemiluminescence assay system (Pierce Biotechnology, Rockford, IL), each membrane was stripped and reprobed for internal control (Crk-L or calregulin). The optical density of each band was quantitated using ImageQuant 5.0 (Molecular Dynamics, Sunnyvale, CA).
Statistical methods
Each experiment was performed at least three times. The results were expressed relative to vehicle controls, or as percentages of the cell population (mean ± SE). Significance of the differences between mean values was assessed by a two-tailed Student’s t-test. All computations were performed with an IBM-compatible personal computer using Microsoft EXCEL.
Results
Upregulation of miR-32 by 1,25D in HL60 and U937 cells
The initial experiments were performed using a miRNA microarray platform (24) to identify the expression of miRNAs after exposure to 1,25D. Ingenuity Pathway analysis indicated that only miR-32 was differentially expressed in both HL60 and U937 1,25D-treated cells. As shown in Fig 1A, miR-32 was markedly (~11-fold) and highly significantly increased in HL60 cells, but the increase was less marked and below the level of statistical significance in U937 cells. However, validation of these results by quantitative real-time PCR, shown in Fig 1B, demonstrated that although the increase in miR-32 level was less marked in U937 than in HL60 cells, the increase was statistically significant in both cell lines. While Taq-Man results were less dramatic than those obtained by the microarray, they were clearly confirmatory.
Fig. 1. Expression levels of miR-32 are up-regulated by 1,25D in HL60 and U937 cells.
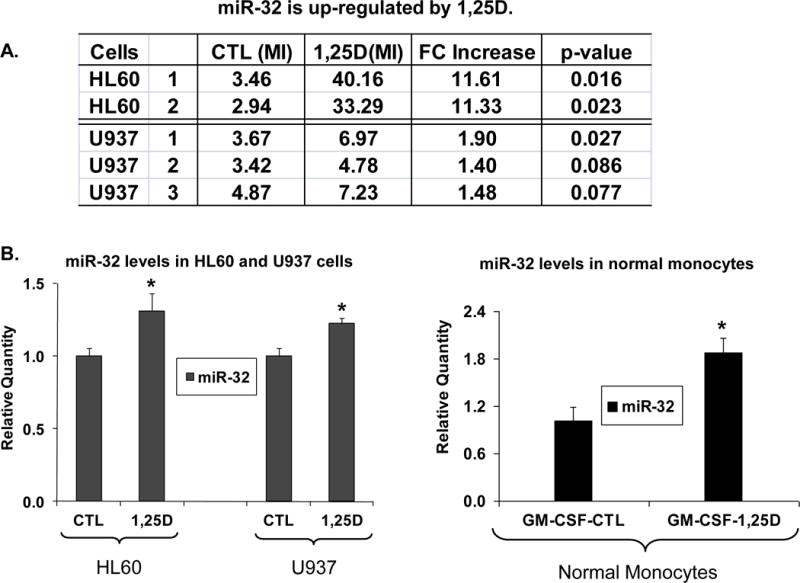
HL60 cells were exposed to 1 nM 1,25D for 48 h, and the less sensitive U937 cells to 10 nM 1,25D for 72 h, while normal monocytes to 100 nM 1,25D for 96 h. The miR-32 expression level was estimated using (A) miRNA microarray platform (24) and (B) TaqMan qRT-PCR assays. Asterisks show the statistically significant differences from the corresponding controls, p < 0.05; bars represent mean values +/− SE, n = 8–12 for cell lines, n=5 for monocytes. CTL=control, MI=median intensity of signal, FC=fold change from CTL, GM-CSF= granulocyte/macrophage colony stimulating factor (20 ng/ml), used to support survival/growth of monocytes in primary culture.
Treatment with 1,25D of monocytes isolated from healthy volunteers also significantly (p< 0.01) increased miR-32 levels.
The pro-apoptotic protein Bim is a potential target of miR-32 in AML cells
In silico studies reported in at least six public searches (Fig 2A) revealed that 3′UTR of BCL2L11 gene, which encodes the pro-apoptotic protein Bim (14, 15), has a potential miR-32 binding site at chromosome 2q13, which is conserved between human and murine genomes (Fig 2B). Direct regulation in AML cells of Bim protein expression by miR-32 was suggested by the marked decrease in Bim mRNA and Bim protein, its largest isoform known as Bim-EL (15), in both HL60 and U937 cells after exposure to 1,25D (Fig 2C). This is consistent with the report that in human prostate cancer cells LNCaP a luciferase reporter construct containing BCL2L11 3′UTR with the predicted miR-32 target sequence, showed translational inhibition by miR-32 (25). While cell type-restricted specificity of miRNA targets is well known, this suggests that Bim may be a general target of miR-32 in human cells.
Fig. 2. Bim has a potential miR-32 binding site and is down-regulated by 1,25D in HL60 and U937 cells.
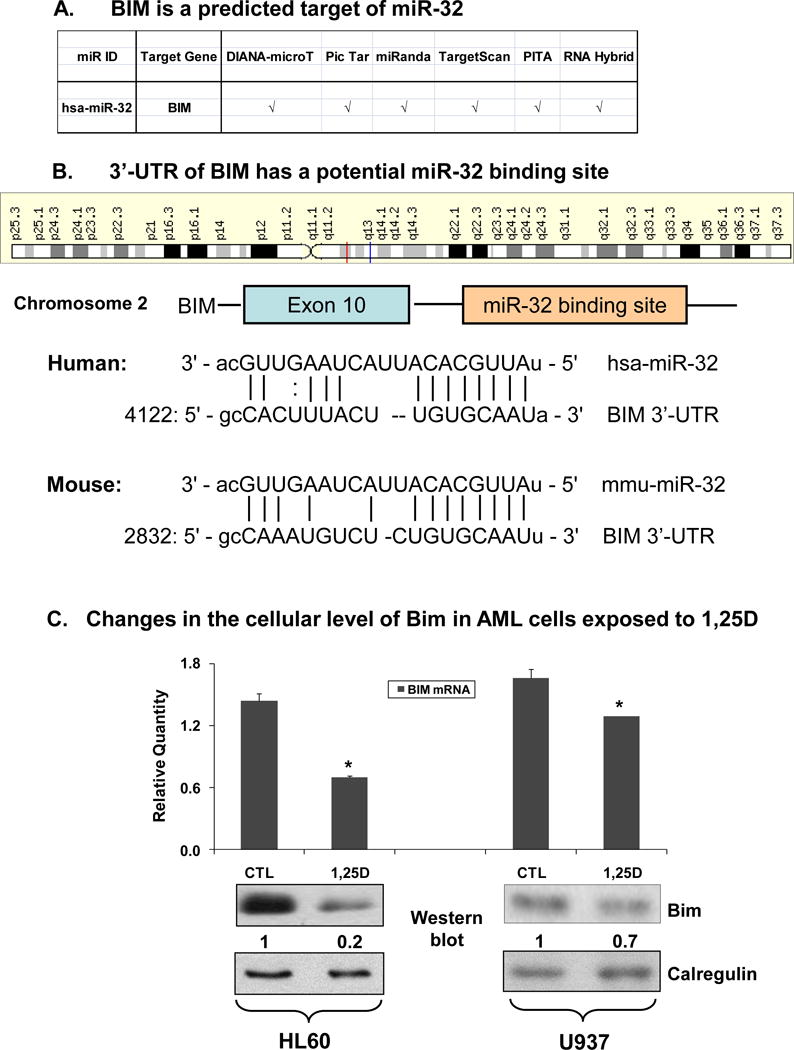
(A) Six public web sites examined all predict miR-32 target sites in the 3′-UTR of human Bim gene. (B) Schematic representation of the potential miR-32 binding sites within the 3′-UTR of human and mouse Bim genes. (C) Bim mRNA and protein levels are decreased in AML cells exposed to 1,25D for 48 h. HL60 cells were exposed to 1 nM 1,25D, while the less 1,25D-sensitive U937 cells were exposed to 10 nM 1,25D. The levels of Bim mRNA were determined by SYBRGreen qRT-PCR. The asterisks show statistically significant differences from the corresponding controls (CTL). p < 0.05; bars represent mean values +/− SE, n = 4. The levels of Bim protein were determined by Western blotting and representative blots of four experiments are shown here with signal densities indicated below each panel. Calregulin was used as a loading control.
Expression of Bim is regulated by miRNAs
To confirm that the above predictions apply to AML cells, we first tested if an interference with miRNA processing affects Bim protein expression in these cells. To accomplish this we reduced the levels of the RNases which process miRNA precursors, the RNase III Drosha, which processes miRNAs in the nucleus, and the cytosolic RNase III, Dicer (26–29). As shown in Fig 3A, transfection of silencing constructs of Dicer and Drosha effectively abrogated 1,25D-induced up-regulation of miR-32 expression, though the ambient levels of miR-32 were not sufficiently reduced to detect statistical significance. As expected, the changes in Bim protein levels were reciprocally affected by the reduction in miRNA processing enzymes, with decreased Bim levels noted after 1,25D exposure, which were abrogated and actually increased by Dicer and Drosha (Fig 3B). The increase in Bim levels when miRNA processing is reduced corroborates that Bim expression is regulated by a miRNA or several miRNAs.
Fig. 3. The expression of Bim protein is regulated by miRNA.
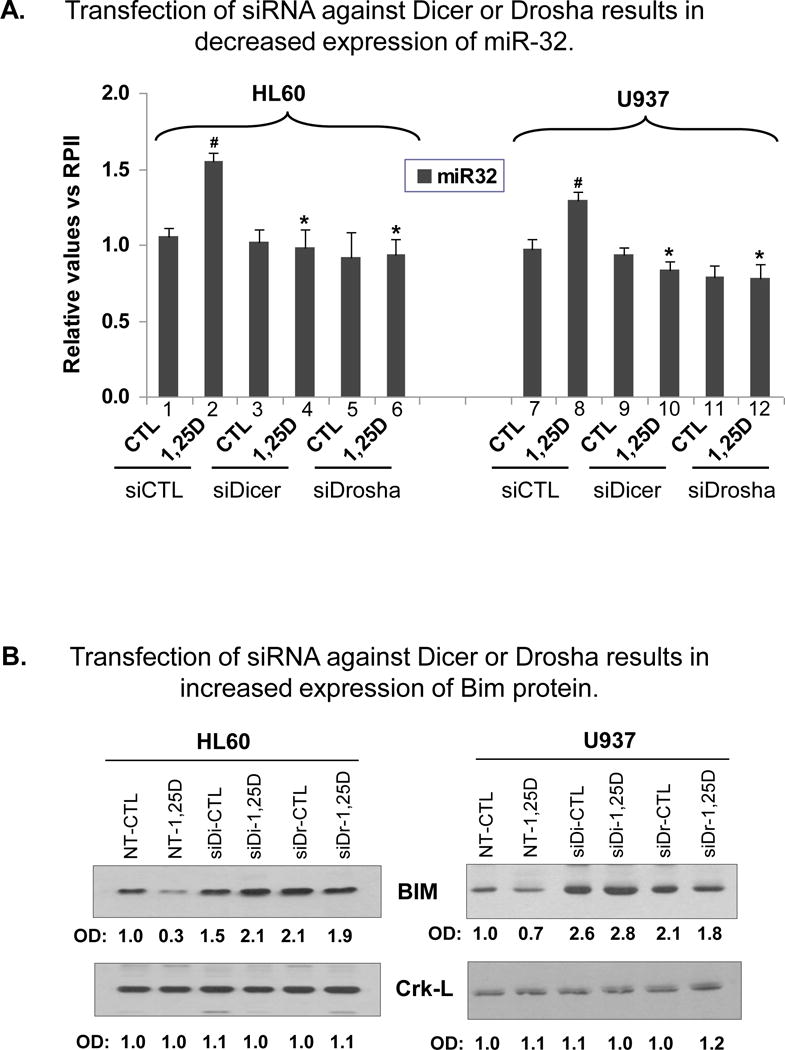
Transfection of siRNA against miRNA processing enzymes Drosha or Dicer results in: (A) abrogation of 1,25D-induced increase in the expression of miR-32, and (B) increased expression of Bim protein in HL60 and U937 cells. siRNA (20 nM) against Drosha or Dicer, or the non-targeting control (denoted siCTL in panels A, or NT-CTL in panels B) oligonucleotides were transfected into the cells for 48 h, then the cells were exposed to 1 nM (HL60) or 10 nM (U937) 1,25D for further 24 h. The level of miR-32 was determined by TaqMan qRT-PCR. The # symbol indicates the statistically significant differences in 1,25D-treated cells (lanes 2 and 8) from the vehicle-treated cells (lanes 1 and 7). The asterisks (*) show significant differences from the corresponding siCTL group, apparent only in 1,25D-treated groups. p < 0.05; bars represent mean values +/− SE, n = 4. The levels of Bim protein and the loading control Crk-L were determined by Western blotting. Band densities are shown below each panel, which are representative blots of three experiments. NT, siCTL = Non-targeting siRNA control, siDi= siRNA against Dicer; siDr= siRNA against Drosha . The asterisks (*) show significant differences from the corresponding siCTL group, apparent only in 1,25D-treated groups. p < 0.05; bars represent mean values +/− SE, n = 4. The levels of Bim protein and the loading control Crk-L were determined by Western blotting. Band densities are shown below each panel, which are representative blots of four experiments. NT, siCTL = Non-targeting siRNA control, siDi= siRNA against Dicer; siDr= siRNA against Drosha.
Precursor and antisense miR-32 regulate Bim protein levels
To establish that miR-32 is at least one of the miRNAs which affected Bim protein expression when the processing of miRNAs was inhibited, we used antisense miR-32 oligonucleotides (anti-miR-32) and precursor miR-32 (pre-miR-32) to modulate cellular levels of miR-32. Under the conditions employed, anti-miR-32 produced a significant decrease in the cellular levels of endogenous miR-32 (Fig 4A, bars 3–4) with corresponding increases in Bim mRNA (Fig 4B, bars 3–4) and protein (Fig 4C). Conversely, pre-miR-32 resulted in significant increases in cellular levels of miR-32 (Fig 4A, bars 7–8), while Bim mRNA (Fig 4B, bars 7–8) and protein (Fig 4D) levels decreased. This indicates that Bim is a target of miR-32 in AML cells, and that 1,25D-induced down-regulation of Bim can be reversed by anti-miR-32. In contrast, the levels of Bax, another pro-apoptotic protein, are essentially unaltered by the manipulation of miR-32 cellular levels (Fig 4, C and D).
Fig. 4. Modulation of miR-32 expression by antisense oligonucleotides or by miR-32 precursor results in reciprocal changes in the expression of Bim mRNA and protein in AML cells.
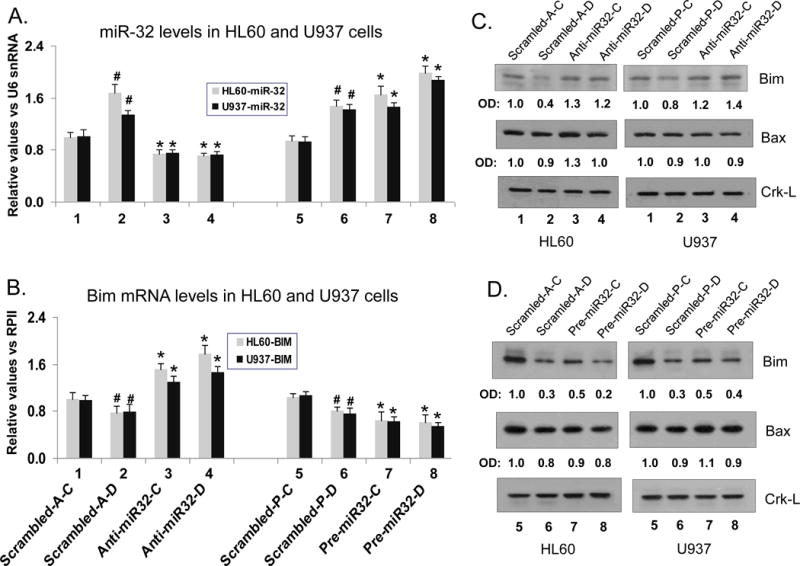
(A) HL60 and U937 cells were transfected with 20 nM antisense oligonucleotides against miR-32 (lanes 3 and 4) or 20 nM pre-miR-32 (lanes 7 and 8) for 48 h, then exposed to 1 nM and 10 nM 1,25D, respectively for further 48 h. MiR-32 levels were determined by TaqMan qRT-PCR. The # symbols indicate the statistically significant differences in 1,25D-treated cells (lane 2 or lane 6) from the corresponding vehicle-treated cells (lanes 1 and 5). The asterisks (*) show the statistically significant differences from the corresponding (scrambled) controls transfected with non-targeting scrambled oligos (scrambled-A, lane 1 vs anti-miR-32 lane 3; or scrambled-P lane 5 vs pre-miR-32,lane 7). A comparison is also shown for 1,25D-treated cells lane 4 vs lane 2, and lane 8 vs lane 6). p < 0.05; bars represent mean values +/− SE, n = 4. (B) Bim mRNA levels were determined by SYBRGreen qRT-PCR in the experiments described in (A) above. (C) Levels of Bim and Bax proteins after transfection of anti-miR-32, and (D) pre-miR-32, as determined by Western blotting. Crk-L was used as a loading and transfer control. OD ratio of each band versus CrkL is shown below each panel, which is representative of three experiments.
Precursor miR-32 promotes 1,25D-induced AML cell differentiation while antisense miR-32 inhibits differentiation
It has been observed that the processes associated with cell maturation and acquisition of function include cell increased cell survival capacity (30–32). We therefore determined the effect on 1,25D-induced differentiation of the modulation of cellular miR-32 levels by anti-miR-32 or pre-miR-32, and found that anti-miR-32 inhibited cell differentiation (Fig 5A), while pre-miR-32 enhanced differentiation (Fig 5B). Thus, the changes in the expression of cell surface markers of the monocytic phenotype CD11b and CD14 (Fig 5) paralleled the cellular levels of miR-32 shown in Fig 4A. The enhancement of monocytic differentiation by pre-miR-32 was also shown by the increased expression of the cytoplasmic esterase NSE, another marker of monocytic phenotype (data not shown). Thus, perhaps indirectly, miR-32 has a minor but clear role in monocytic differentiation.
Fig. 5. Modulation of miR-32 expression by antisense miR-32 and precursor miR-32 influences 1,25D-induced AML cell differentiation.
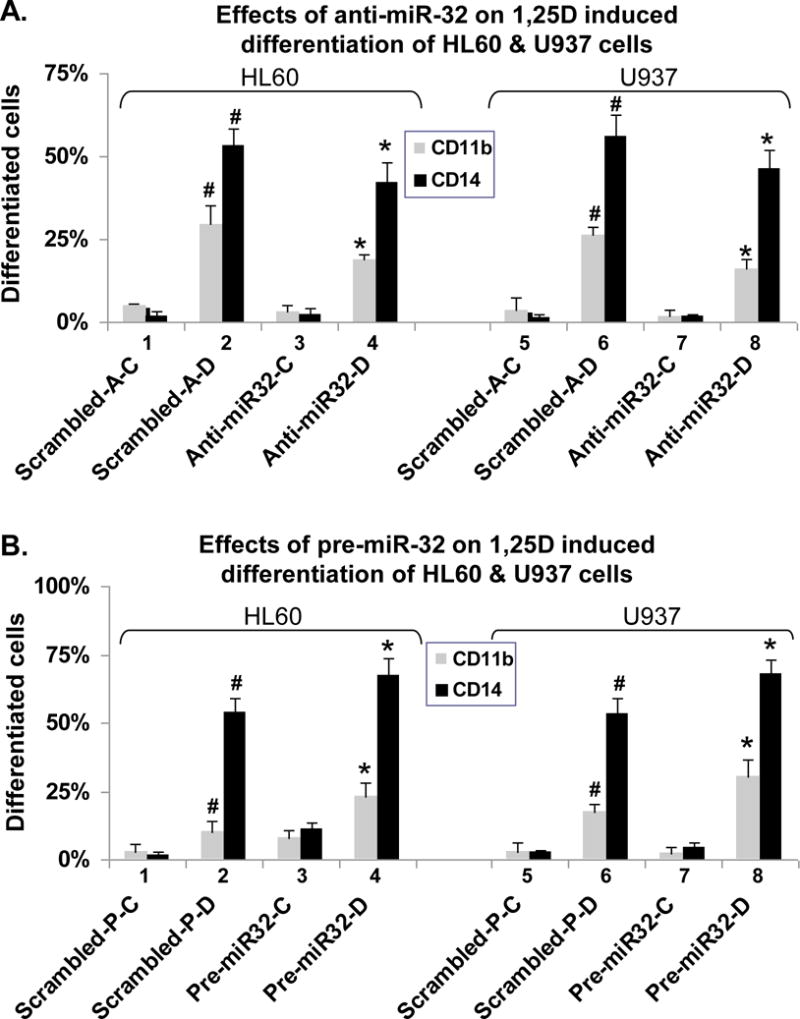
(A) Effects of anti-miR-32 on 1,25D induced differentiation of HL60 and U937 cells. (B) Effects of pre-miR-32 on 1,25D-induced differentiation. The conditions and symbols are described in the legend to Fig 4 and the determination of differentiation markers in “Materials and Methods”.
Inhibition of miR-32 expression by anti-miR-32 increases the toxicity of AraC to AML cells
The observed effects of miR-32 on the expression of Bim protein suggest that a reduction of cellular levels of miR-32 should make AML cells more susceptible to the therapeutic agents used to treat this disease, generally AraC. This was tested by pre-incubating AML cells for 48 h with anti-miR-32, then treating the cells with 100 uM AraC for 24 h, and determining Trypan Blue permeability, an indication of necrosis, and the estimation of apoptosis by Annexin V, as well as by the sub G1/G0 fraction obtained from flow cytometric measurement of cell cycle distribution. All three sets of determinations showed that anti-miR-32 increases cell death induced by AraC (Table 1A, groups 3 and 4, 7 and 8) and abrogates the previously reported anti-apoptotic effect of 1,25D (31).The protective effect of 1,25D is seen here for HL60 cells (Table 1A, group 1 vs group 2), although the complexity of the transfection syst-em does not make this apparent in U937 cells. Anti-miR-32 also enhanced apoptosis when doxorubicin or daunomycin were used as the toxic agents (data not shown).
Table 1. Inhibition of miR-32 expression by antisense oligonucleotides increases the toxicity of AraC to HL60 and U937 cells.
HL60 and U937 cells were transfected with 20 nM antisense oligonucleotides against miR-32 for 48 h, and then exposed to 1 nM (HL60) or 10 nM (U937) 1,25D for further 48 h. Then the cells were exposed to 100 μM AraC for further 24 h. To detect apoptosis cells were stained using 50 μg/ml Annexin V and 20 μg/ml propidium iodide and analyzed by flow cytometry. Annexin V-positive/PI-negative cells were considered as early apoptotic, cells doubly positive, as late apoptotic. Bold type shows the statistically significant differences from the corresponding scrambled controls. p < 0.05; mean values +/− SE are shown, n = 4. C= vehicle control; D=1,25D-treated; A=anti-miR32; P=pre-miR32.
| A: Anti-miR-32 increases toxicity of AraC to HL60 and U937 cells | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Groups | Cells | Treatment | Viability Mean±SD (%) |
Early-Apts Mean±SD (%) |
Late-Apts Mean±SD (%) |
Total-Apts Mean±SD (%) |
SubG1 Mean±SD (%) |
| 1 | HL60 | Scrambled-A-C | 79.3 ± 6.8 | 9.6 ± 2.9 | 22.3 ± 6.4 | 31.9 ± 6.5 | 9.9 ± 4.4 |
| 2 | HL60 | Scrambled-A-D | 75.3 ± 1.0.5 | 7.6 ± 1.7 | 16.7 ± 7.7 | 24.3 ± 6.6 | 6.0 ± 2.1 |
| 3 | HL60 | Anti-miR32-C | 65.3 ± 8.3 | 9.7 ± 1.3 | 31.9 ± 7.9 | 41.6 ± 6.5 | 16.6 ± 4.7 |
| 4 | HL60 | Anti-miR32-D | 63.0 ± 10.3 | 10.9 ± 2.4 | 31.3 ± 7.7 | 42.2 ± 9.5 | 13.2 ± 3.5 |
|
| |||||||
| 5 | U937 | Scrambled-A-C | 70.3 ± 5.0 | 10.4 ± 1.2 | 33.3 ± 6.6 | 43.7 ± 7.0 | 8.6 ± 1.5 |
| 6 | U937 | Scrambled-A-D | 71.7 ± 4.2 | 9.5 ± 1.7 | 32.1 ± 3.6 | 41.6 ± 4.1 | 7.6 ± 3.1 |
| 7 | U937 | Anti-miR32-C | 61.0 ± 5.5 | 10.5 ± 2.1 | 45.1 ± 6.9 | 55.6 ± 3.1 | 11.4 ± 2.1 |
| 8 | U937 | Anti-miR32-D | 63.3 ± 3.8 | 9.1 ± 2.4 | 40.7 ± 7.0 | 49.8 ± 4.2 | 12.5 ± 2.7 |
| B: Pre-miR-32 reduces toxicity of AraC to HL60 and U937 cells | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Groups | Cells | Treatment | Viability Mean±SD(%) |
Early-Apts Mean±SD(%) |
Late-Apts Mean±SD(%) |
Total-Apts Mean±SD(%) |
SubG1 Mean±SD(%) |
| 1 | HL60 | Scrambled-P-C | 77.0 ± 11.5 | 9.0 ± 2.2 | 27.3 ± 4.8 | 36.3 ± 6.7 | 8.4 ± 1.6 |
| 2 | HL60 | Scrambled-P-D | 79.0 ± 10.5 | 7.0 ± 1.6 | 19.9 ± 4.9 | 26.9 ± 7.8 | 6.4 ± 1.8 |
| 3 | HL60 | Pre-miR32-C | 79.5 ± 11.5 | 7.0 ± 1.2 | 14.9 ± 3.7 | 21.9 ± 5.5 | 4.7 ± 1.6 |
| 4 | HL60 | Pre-miR32-D | 80.5 ± 10.5 | 7.1 ± 2.1 | 13.8 ± 4.2 | 20.9 ± 6.1 | 4.0 ± 1.5 |
|
| |||||||
| 5 | U937 | Scrambled-P-C | 61.0 ± 5.1 | 15.3 ± 1.2 | 36.9 ± 6.3 | 52.2 ± 7.2 | 8.6 ± 1.9 |
| 6 | U937 | Scrambled-P-D | 62.3 ± 7.2 | 12.5 ± 1.8 | 43.2 ± 6.0 | 55.7 ± 7.5 | 7.8 ± 2.3 |
| 7 | U937 | Pre-miR32-C | 67.5 ± 6.0 | 6.6 ± 1.0 | 35.5 ± 5.9 | 42.1 ± 7.1 | 5.3 ± 1.5 |
| 8 | U937 | Pre-miR32-D | 68.0 ± 6.1 | 12.4 ± 1.9 | 27.6 ± 5.7 | 40.0 ± 7.3 | 4.9 ± 1.4 |
The complementary approach, the use of pre-miR-32, to show the protective effect of miR-32 on AML cell survival, confirmed the protective effect (Table 1B). In the experiments where pre-miR-32 was transfected the changes in cell survival were less marked than those obtained with anti-miR-32, with AraC-induced necrosis not being significantly affected, and only early apoptosis detectable in U937 cells not treated with 1,25D (Table 1B). This is perhaps seen because other components of cell survival network play a greater role than Bim when pre-miR-32 reduces Bim levels, while increased Bim levels in cells treated with anti-miR-32 have a more dominant effect on cell survival. Taken together, the two approaches clearly show that the response of AML cells to therapeutic cytotoxic agents may be increased by lowering cellular miR-32 levels. This raises the possibility that the combination of differentiation (by 1,25D) with cytotoxic (AraC) therapy may be further enhanced by agents or conditions which lower cellular miR32 levels, such as anti-miR32.
Discussion
The studies reported here provide several mechanistic insights, and may also have a translational significance. They reveal a novel aspect of the mechanism of vitamin D action, have a bearing on the specificity of miRNA targets, and enhance the understanding of AML cell survival mechanisms. The latter may point the way to increasing the effectiveness of AraC-based induction of disease-free patient survival.
Previous work in our and other laboratories has shown that 1,25D-induced differentiation of human AML cells is accompanied by increased cell survival capacity, which is likely to be multifactorial. These include activation of the ERK an AKT pathways (33–36), though the specific details regarding the sequence of molecular events are few. One possibility is that the hKSR2 gene, which is directly up-regulated by 1,25D (37), provides an upstream platform for activation of MAPK pathways, which then signal pro-survival events, as has been shown by knock-down of hKSR2 expression (31). The pro-survival events include up-regulation of the anti-apoptotic Mcl-1 (30), and altered Bcl-2/Bad ratios (31). Here, we document a role for miR-32 in the pro-survival events, but its relationship to the other signalling members of apoptosis/survival network remains to be elucidated.
The exquisite cell context specificity of miRNA targets is well known, and any given mRNA may be targeted by different miRNAs in different cell types or even cell subtypes. (e.g. (38, 39)). It is therefore important to note that in both HL60 and U937 cells, which belong to differrent FAB subtypes, miR-32 targets Bim (Fig 4). Also, recent studies in the Croce laboratory have shown that in two subtypes of cultured human prostate cancer cells miR-32 inhibits the expression of Bim by a 3′UTR-mediated mechanism (25). Taken together with results reported here, this suggests that the miR-32-Bim relationship is of wide significance in human cells. This contrasts with the targeting of p27Kip1 by miR-181 previously reported in AML cells (23, 40), whereas in a variety of solid tumors p27Kip1 is targeted by miR-221/222 (41–43). These examples show that miRNA targets need to be established one cell type at a time.
Currently, there are only a few published reports of the ability of 1,25D to regulate key cellular functions through modulation of miRNA expression. Previously, we showed that an exposure of human AML HL60 and U937 cells to 1,25D results in down-regulation of the miR-181 family, with the most prominent effect on miR-181a (23). This was associated with the up-regulation of p27Kip1 expression, and contributed to 1,25D-induced cell cycle arrest. More recently, miR-24 was reported to be up-regulated by 1,25D and related to diminished cell proliferation (44), and a role for miRNA in cell cycle control by 1,25D was described in cultured non-malignant RWPE-1 human prostate epithelial cells (45). In RWPE-1 cells 1,25D up-regulates the DNA helicase MCM7 gene in which the miR-106 is embedded in intron 13 (46, 47), which then targets p21/CDKN1A and contributes to cell cycle arrest. Thus, the current report provides a new aspect of the accumulating evidence that miRNAs participate in the critical aspects of vitamin D regulation of the essential cellular processes, such as those controlling the cell cycle, cell survival, and cell differentiation.
Selective up-regulation of miR-32 in at least some human cancers including prostate carcinoma and multiple myeloma (25, 48) appears to play a role in malignant transformation by providing survival advantage to cells with high expression of miR-32 and reduced levels of Bim. A similar mechanism described here appears to aid the phagocytic function of monocytes, which produces intracellular stress as a consequence of generation of ROS needed to dispose of phagocytised material. But since increased survival capacity is a disadvantage when malignant cells are to be eradicated by cytotoxic agents, consideration should be given to the design of differentiation therapy regimens in which compounds or conditions which reduce the expression of miR-32 or increase the levels of Bim (49, 50), are administered along with the therapeutic agents.
Acknowledgments
The study was supported by the NIH Grant RO1-CA-044722-21 (to GPS), and by the Polish Science Foundation (to EG).
Abbreviations
- miRNA
microRNA
- 1,25D
1,25-dihydroxyvitamin D3
- UTR
untranslated region
- Bim
Bcl-2-interacting mediator of the cell death, Bcl-2 like protein 11, Bim-EL
- Anti-miR-32
antisense oligonucleotides to miR-32
- pre-miR-32
precursor miR-32
- AraC
arabinocytosine
References
- 1.Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol. 2009;37:649–658. doi: 10.1016/j.exphem.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Miyaura C, Abe E, Kuribayashi T, Tanaka H, Konno K, Nishii Y, Suda T. 1 alpha,25-Dihydroxyvitamin D3 induces differentiation of human myeloid leukemia cells. Biochem Biophys Res Commun. 1981;102:937–943. doi: 10.1016/0006-291x(81)91628-4. [DOI] [PubMed] [Google Scholar]
- 3.Koeffler HP. Induction of differentiation of human acute myelogenous leukemia cells: therapeutic implications. Blood. 1983;62:709–721. [PubMed] [Google Scholar]
- 4.Studzinski GP, Bhandal AK, Brelvi ZS. A system for monocytic differentiation of leukemic cells HL 60 by a short exposure to 1,25-dihydroxycholecalciferol. Proc Soc Exp Biol Med. 1985;179:288–295. doi: 10.3181/00379727-179-42098. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Studzinski GP. Inhibition of p38MAP kinase potentiates the JNK/SAPK pathway and AP-1 activity in monocytic but not in macrophage or granulocytic differentiation of HL60 cells. J Cell Biochem. 2001;82:68–77. doi: 10.1002/jcb.1141. [DOI] [PubMed] [Google Scholar]
- 6.Wang Q, Wang X, Studzinski GP. Jun N-terminal kinase pathway enhances signaling of monocytic differentiation of human leukemia cells induced by 1,25-dihydroxyvitamin D3. J Cell Biochem. 2003;89:1087–1101. doi: 10.1002/jcb.10595. [DOI] [PubMed] [Google Scholar]
- 7.Pan Z, Hetherington CJ, Zhang DE. CCAAT/enhancer-binding protein activates the CD14 promoter and mediates transforming growth factor beta signaling in monocyte development. J Biol Chem. 1999;274:23242–23248. doi: 10.1074/jbc.274.33.23242. [DOI] [PubMed] [Google Scholar]
- 8.Studzinski GP, Wang X, Ji Y, Wang Q, Zhang Y, Kutner A, Harrison JS. The rationale for deltanoids in therapy for myeloid leukemia: role of KSR-MAPK-C/EBP pathway. J Steroid Biochem Mol Biol. 2005;97:47–55. doi: 10.1016/j.jsbmb.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcinkowska E, Garay E, Gocek E, Chrobak A, Wang X, Studzinski GP. Regulation of C/EBPbeta isoforms by MAPK pathways in HL60 cells induced to differentiate by 1,25-dihydroxyvitamin D3. Exp Cell Res. 2006;312:2054–2065. doi: 10.1016/j.yexcr.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, Han ZG, Ni JH, Shi GY, Jia PM, Liu MM, He KL, Niu C, Ma J, Zhang P, Zhang TD, Paul P, Naoe T, Kitamura K, Miller W, Waxman S, Wang ZY, de The H, Chen SJ, Chen Z. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- 11.Shen Y, Shen ZX, Yan H, Chen J, Zeng XY, Li JM, Li XS, Wu W, Xiong SM, Zhao WL, Tang W, Wu F, Liu YF, Niu C, Wang ZY, Chen SJ, Chen Z. Studies on the clinical efficacy and pharmacokinetics of low-dose arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia: a comparison with conventional dosage. Leukemia. 2001;15:735–741. doi: 10.1038/sj.leu.2402106. [DOI] [PubMed] [Google Scholar]
- 12.Studzinski GP, Bhandal AK, Brelvi ZS. Potentiation by 1-alpha,25-dihydroxyvitamin D3 of cytotoxicity to HL-60 cells produced by cytarabine and hydroxyurea. J Natl Cancer Inst. 1986;76:641–648. doi: 10.1093/jnci/76.4.641. [DOI] [PubMed] [Google Scholar]
- 13.Studzinski GP, Reddy KB, Hill HZ, Bhandal AK. Potentiation of 1-beta-D-arabinofuranosylcytosine cytotoxicity to HL-60 cells by 1,25-dihydroxyvitamin D3 correlates with reduced rate of maturation of DNA replication intermediates. Cancer Res. 1991;51:3451–3455. [PubMed] [Google Scholar]
- 14.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM. Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm Genome. 2001;12:163–168. doi: 10.1007/s003350010242. [DOI] [PubMed] [Google Scholar]
- 16.Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, Gallo R. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood. 1979;54:713–733. [PubMed] [Google Scholar]
- 17.Gocek E, Kielbinski M, Baurska H, Haus O, Kutner A, Marcinkowska E. Different susceptibilities to 1,25-dihydroxyvitamin D3-induced differentiation of AML cells carrying various mutations. Leuk Res. 2010;34:649–657. doi: 10.1016/j.leukres.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. Rna. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 22.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Gocek E, Liu CG, Studzinski GP. MicroRNAs181 regulate the expression of p27Kip1 in human myeloid leukemia cells induced to differentiate by 1,25-dihydroxyvitamin D3. Cell Cycle. 2009;8:736–741. doi: 10.4161/cc.8.5.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A, Wang H, Sun H, Volinia S, Alder H, Calin GA, Liu CG, Andreeff M, Croce CM. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene. 2007;26:4148–4157. doi: 10.1038/sj.onc.1210186. [DOI] [PubMed] [Google Scholar]
- 25.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, Yfantis HG, Stephens RM, Croce CM. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. Embo J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. Embo J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 29.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Studzinski GP. Antiapoptotic action of 1,25-dihydroxyvitamin D3 is associated with increased mitochondrial MCL-1 and RAF-1 proteins and reduced release of cytochrome c. Exp Cell Res. 1997;235:210–217. doi: 10.1006/excr.1997.3667. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Patel R, Studzinski GP. hKSR-2, a vitamin D-regulated gene, inhibits apoptosis in arabinocytosine-treated HL60 leukemia cells. Mol Cancer Ther. 2008;7:2798–2806. doi: 10.1158/1535-7163.MCT-08-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lombardero M, Kovacs K, Scheithauer BW. Erythropoietin: a hormone with multiple functions. Pathobiology. 2011;78:41–53. doi: 10.1159/000322975. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Studzinski GP. Activation of extracellular signal-regulated kinases (ERKs) defines the first phase of 1,25-dihydroxyvitamin D3-induced differentiation of HL60 cells. J Cell Biochem. 2001;80:471–482. doi: 10.1002/1097-4644(20010315)80:4<471::aid-jcb1001>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 34.Marcinkowska E, Kutner A. Side-chain modified vitamin D analogs require activation of both PI 3-K and Erk1,2 signal transduction pathways to induce differentiation of human promyelocytic leukemia cells. Acta Biochim Pol. 2002;49:393–406. [PubMed] [Google Scholar]
- 35.Zhang Y, Zhang J, Studzinski GP. AKT pathway is activated by 1, 25-dihydroxyvitamin D3 and participates in its anti-apoptotic effect and cell cycle control in differentiating HL60 cells. Cell Cycle. 2006;5:447–451. doi: 10.4161/cc.5.4.2467. [DOI] [PubMed] [Google Scholar]
- 36.Gocek E, Kielbinski M, Marcinkowska E. Activation of intracellular signaling pathways is necessary for an increase in VDR expression and its nuclear translocation. FEBS Lett. 2007;581:1751–1757. doi: 10.1016/j.febslet.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Wang TT, White JH, Studzinski GP. Expression of human kinase suppressor of Ras 2 (hKSR-2) gene in HL60 leukemia cells is directly upregulated by 1,25-dihydroxyvitamin D3 and is required for optimal cell differentiation. Exp Cell Res. 2007;313:3034–3045. doi: 10.1016/j.yexcr.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inomata M, Tagawa H, Guo YM, Kameoka Y, Takahashi N, Sawada K. MicroRNA-17-92 down-regulates expression of distinct targets in different B-cell lymphoma subtypes. Blood. 2009;113:396–402. doi: 10.1182/blood-2008-07-163907. [DOI] [PubMed] [Google Scholar]
- 39.Debernardi S, Skoulakis S, Molloy G, Chaplin T, Dixon-McIver A, Young BD. MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia. 2007;21:912–916. doi: 10.1038/sj.leu.2404605. [DOI] [PubMed] [Google Scholar]
- 40.Cuesta R, Martinez-Sanchez A, Gebauer F. miR-181a regulates cap-dependent translation of p27Kip1 mRNA in myeloid cells. Mol Cell Biol. 2009;29:2841–2851. doi: 10.1128/MCB.01971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM, Stein GS. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68:2773–2780. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillies JK, Lorimer IA. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle. 2007;6:2005–2009. doi: 10.4161/cc.6.16.4526. [DOI] [PubMed] [Google Scholar]
- 43.Visone R, Russo L, Pallante P, De Martino I, Ferraro A, Leone V, Borbone E, Petrocca F, Alder H, Croce CM, Fusco A. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer. 2007;14:791–798. doi: 10.1677/ERC-07-0129. [DOI] [PubMed] [Google Scholar]
- 44.Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O’Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O, Becker KG, Gorospe M, Hide W, Lieberman J. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Mol Cell. 2009;35:610–625. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thorne JL, Maguire O, Doig CL, Battaglia S, Fehr L, Sucheston LE, Heinaniemi M, O’Neill LP, McCabe CJ, Turner BM, Carlberg C, Campbell MJ. Epigenetic control of a VDR-governed feed-forward loop that regulates p21Waf1/Cip1 expression and function in non-malignant prostate cells. Nucleic Acids Res. 2011;39:2045–2056. doi: 10.1093/nar/gkq875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saramaki A, Banwell CM, Campbell MJ, Carlberg C. Regulation of the human p21Waf1/Cip1 gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006;34:543–554. doi: 10.1093/nar/gkj460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, Kobayashi SV, Lim L, Burchard J, Jackson AL, Linsley PS, Cleary MA. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–2174. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, Munker R, Volinia S, Boccadoro M, Garzon R, Palumbo A, Aqeilan RI, Croce CM. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–238. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Rahmani M, Anderson A, Habibi JR, Crabtree TR, Mayo M, Harada H, Ferreira-Gonzalez A, Dent P, Grant S. The BH3-only protein Bim plays a critical role in leukemia cell death triggered by concomitant inhibition of the pI3K/Akt and MEK/ERK1/2 pathways. Blood. 2009;114:4507–4516. doi: 10.1182/blood-2008-09-177881. [DOI] [PMC free article] [PubMed] [Google Scholar]