

Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) is contracted via aerosol infection, typically affecting the lungs. Mycobacterium bovis bacillus Calmette-Guerin (BCG) is the only licensed vaccine and has variable efficacy in protecting against pulmonary TB. Additionally, chemotherapy is associated with low compliance contributing to development of multidrug-resistant (MDR) and extensively drug-resistant (XDR) Mtb. Thus, there is an urgent need for the design of more effective vaccines against TB. Experimental vaccines delivered through the mucosal route induce robust T helper type 17 (Th17)/ Interleukin (IL) -17 responses and provide superior protection against Mtb infection. Thus, the development of safe mucosal adjuvants for human use is critical. In this study, we demonstrate that nanoemulsion (NE)-based adjuvants when delivered intranasally along with Mtb specific immunodominant antigens (NE-TB vaccine) induce potent mucosal IL-17 T-cell responses. Additionally, the NE-TB vaccine confers significant protection against Mtb infection, and when delivered along with BCG, is associated with decreased disease severity. These findings strongly support the development of a NE-TB vaccine as a novel, safe and effective, first-of-kind IL-17 inducing mucosal vaccine for potential use in humans.
Keywords: Mucosal vaccines, nanoemulsion, IL-17 responses
Introduction
Mycobacterium tuberculosis (Mtb) latently infects one-third of the world's population, causing pulmonary tuberculosis in ∼9 million people and resulting in ∼1.4 million deaths each year [1]. The currently available TB vaccine, Mycobacterium bovis BCG (BCG), shows variable efficacy in protection against pulmonary tuberculosis. In addition, drug resistant Mtb strains have recently emerged. Thus, there is a great need for new TB vaccines [2]. TB vaccine development during the past decade has focused on targeting interferon-gamma (IFNγ) secretion from T helper 1 (Th1) cells to mediate early macrophage activation and bacterial killing [3]. A recombinant TB vaccine, MVA85A, was recently tested in human clinical trials. Despite inducing high levels of IFNγ production from T-cells [4, 5], this vaccine failed to protect against TB disease [6, 7]. These data highlight the importance of exploring new and more effective immune approaches to improve vaccine-induced immunity against TB.
Our recent work has demonstrated that T helper type 17 (Th17) cells, which produce the cytokine interleukin-17 (IL-17), are primary effector cells mediating vaccine-induced protection against Mtb [8-11]. Additionally, intranasal (I.N.) vaccines induce better mucosal immunity and confer superior protection against mucosal infectious diseases, including TB [12-15], when compared to systemic routes of immunization [16]. Importantly, we and others have recently demonstrated that mucosal vaccination of Mtb antigens in Heat Labile enterotoxin (HLT) [8] or cholera toxin [17] induced mucosal Th17 responses, which confer protection upon Mtb challenge. Our mechanistic studies demonstrated that IL-17 induced chemokines localize cytokine-producing T-cells near Mtb-infected macrophages, forming lymphoid follicles within granulomas to mediate Mtb control [11, 18]. In combination with DC transfer, a HLT-TB mucosal vaccine can provide superior near-sterilizing vaccine-induced protection against Mtb infection [8, 17, 19]. Despite the ability of these experimental mucosal adjuvants to induce protective Th17 responses and confer vaccine-induced protection, there are serious concerns regarding the safety of toxin subunits as mucosal adjuvants in human vaccines. For example, Bell's palsy has been observed following I.N. application of the vaccine Nasalflu, which contains E. coli HLT as an adjuvant [20]. Therefore, there is an urgent need to identify safe and effective mucosal TB vaccines that can induce lung mucosal IL-17 T-cell responses for use in humans.
Nanoemulsions (NE) are oil-in-water emulsions formulated with antigen. NE adjuvant was safe and well-tolerated in human volunteers when used as a flu vaccine, and elicited both systemic and mucosal immunity following a single I.N. vaccination [21]. When delivered with antigen in mice by I.N. route, the NE-based vaccine induces Th17 responses [22]. In the current study, we tested whether NE-adjuvanted TB vaccine will confer protection against Mtb infection, and provide any advantage when compared to use of BCG vaccination. Our results show that mucosal delivery of NE along with Mtb immunodominant antigens (NE-TB vaccine) can confer Mtb control, to levels similar to BCG vaccination in mice. Importantly, our data also show that NE-TB vaccination either given concurrently or sequentially as a boost to initial priming using BCG, can significantly limit TB disease in infected mice. Thus our findings provide the basis for development of a NE-TB vaccine as a novel, safe and effective, first-of-kind IL-17 inducing mucosal vaccine for potential use in humans.
Materials and methods
Mice
C57BL/6J (B6), (Jackson Laboratories, Bar Harbor, ME) mice were bred under specific pathogen-free conditions at the Washington University in St. Louis. Mice maintained were used at 6 to 8 weeks of age and sex matched for all experiments. All animal experiments were performed in accordance with National and Institutional guidelines for animal care under approved protocols.
Vaccination and Mtb infection
The antigen proteins, ESAT-6 and Ag85B, were provided by BEI Resources (Manassas, VA) obtained under National Institutes of Health [NIH] contract AI-75320. The NE-TB vaccine was prepared by simple mixing of ESAT-6 and Ag85B antigens (1.5-fold concentrated) in PBS together with 60% W805EC nanoemulsion (NE) mucosal adjuvant (NanoBio Corporation, Ann Arbor, MI) at a 2:1 (volume : volume) ratio. NE is produced by high-speed emulsification of highly-refined soybean oil together with cetyl pyridinium chloride, Tween 80 and ethanol in water [22]. The nanoemulsion droplets have an average diameter of 450 nm. The final mucosal vaccine formulation consisted of 20% NE + ESAT-6 (2083 μg/ml) protein or NE+ Ag85B (2083 μg/ml) protein, or a combination of both proteins with nanoemulsion (NE-TB vaccine) containing 20% NE + ESAT-6 (2083 μg/ml) + Ag85B (2083 μg/ml), such that I.N. administration of 12 ng/μL total volume provided an antigen dose of 25 μg of each protein (ESAT-6 / Ag85B) per animal as described below. As an adjuvant control, 20% NE also was prepared by mixing with PBS alone without addition of protein antigens.
Mycobacterium bovis Bacille Calmette Guerin (BCG Pasteur, Source: Trudeau Institute) and Mycobacterium tuberculosis strain HN878 (BEI Resources, Manassas, VA) were grown to mid-log phase in Proskauer Beck medium containing 0.05% Tween80 and frozen in 1 ml aliquots at -80°C. BCG-vaccinated mice received 1×106 colony forming units (CFU) BCG subcutaneously (S.C.) [23]. The I.N. NE-TB vaccinations were carried out using a sterile pipette tip applied to the nares, and the mice were administered 12μl (6μl/nare) of the NE formulation containing 25μg of antigen mixed with 20% NE. The NE-TB vaccine was delivered to 6-8-week-old B6 mice, three times at three week intervals, while mock-vaccinated mice received PBS as control. Some mice received S.C. BCG vaccination concurrently with NE-TB vaccine, or as a booster to BCG vaccination. Four weeks after the last booster immunization, mice were challenged by aerosol with a low dose (100 CFU) of Mtb strain HN878 (BEI Resources, Manassas, VA). Four weeks after challenge, unvaccinated and vaccinated mice were sacrificed by carbon dioxide (CO2) asphyxiation, and the lungs were aseptically excised and individually homogenized in physiological saline solution. Serial dilutions of lung homogenates were plated on 7H11 agar for CFU and counted after 3 weeks of incubation at 37°C as described before [24].
ELISpot assay
Antigen-specific IFNγ- and IL-17-producing cells in immunized lungs were detected by ELISpot assay as described [23]. Briefly, 2 weeks after the last immunization, single cell suspensions from lungs and spleens of immunized mice were seeded in antibody-coated plates at an initial density of 5×105 per well, n=4-5 individual mice were used per group. Each well represented cells from an organ from each mouse. Irradiated syngeneic spleen cells (20 Gy), IL-2 (final concentration of 10 U/ml) in the presence of ESAT-6 or Ag85B proteins (10 μg/ml) were added to the cultures of ESAT-6 vaccinated and Ag85B vaccinated mice. Cells isolated from BCG vaccinated mice were restimulated with Ag85B protein, while cells from vaccinated mice receiving NE alone, NE+ESAT-6, or NE+Ag85B were stimulated with both Ag85B and ESAT-6 proteins. After 18 h, the cells secreting IFNγ or IL-17 were detected using BCIP/NBT (Sigma, St.Louis, MO) according to the manufacturer's instructions. The frequency of responding cells was calculated using ImmunoSpot software (Cellular Technology Limited, Shaker Heights, OH), and applied to the number of cells per sample to generate the total number of responding cells per organ. We have previously shown that neither cells cultured in the absence of peptide nor cells from uninfected mice produce detectable spots in ELISpot assays [10, 25].
Evaluation of inflammatory lesions and formation of B cell follicles in vaccinated mice by bright field and fluorescent microscopy
Lungs from vaccinated and unvaccinated Mtb-infected mice were perfused with 10% neutral buffered formalin and embedded in paraffin. 5 μm paraffin lung sections were stained with hematoxylin and eosin, and percentage of area occupied by inflammatory cell infiltrates was calculated. Quantitation of inflammation was performed in a blinded fashion in individual upper right lobe of mice infected with Mtb (n=10 mice per experimental group), collected at day 30 after infection. Inflammation in the entire lobe was outlined with an automated tool of the Axiovision Rel 4.8 software of the Zeiss microscope, at 100X magnification (100× = 10× lens × 10× ocular). After calculating the total area occupied by inflammatory cells per lobe, the periphery of individual lobes was outlined with the Axiovision Rel 4.8 software to estimate their sizes. Area occupied by the lumen of big blood vessels, bronchi and bronchioles was subtracted from the total area of the lobe. Finally, the percentage of area covered by inflammation in individual lobes was calculated. Serial sections of 5 μm paraffin embedded lung tissues were also stained with primary antibodies specific for CD3 (Clone M-20, Santa Cruz Biotechnology, Dallas, TX) and biotinylated antibodies against CD45R/B220 (clone RA3-6B2, BD Biosciences, San Jose, CA). To visualize the B cell follicles and T-cells inside TB granulomas, we incubated lung sections with Alexa fluor 568 donkey anti-goat IgG (A11057, Thermo Fisher Scientific, Waltham, MA) and Alexa fluor 488 streptavidin (S11223, Thermo Fisher Scientific, Waltham, MA). After washing slides, they were mounted with prolong gold antifade with DAPI (P36931, Thermo Fisher Scientific, Waltham, MA) and representative pictures were taken with a Axioplan Zeiss microscope and recorded with a Hamamatsu camera. Morphometric analysis of B cell follicles was performed with the Axiovision Rel 4.8 software of the Zeiss Axioplan microscope.
Detection of cytokines in culture supernatants
Lung homogenates were assayed for multiple cytokines using Milliplex (EMD Millipore, Billerica, MA) according to recommended standard protocols.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism (La Jolla, CA, USA). For experiments with two groups, two-tailed student t-tests were performed. For two or more groups, a one-way ANOVA was used.
Results
NE-TB vaccine induces mucosal IL-17 T-cell responses in mice
Mucosal vaccines induce better mucosal immunity and confer superior protection against TB, when compared to systemic routes of immunization. Furthermore, mucosal route of vaccination induces Th17 responses in mice [8]. We thus determined whether I.N. delivery of NE along with Mtb immunodominant antigens could drive mucosal IL-17 and/or IFNγ responses in vaccinated mice. B6 mice were mucosally vaccinated and boosted twice with NE adjuvant formulated with either immunodominant Mtb antigen ESAT-6 or Ag85B whole protein. Two weeks after the last booster vaccination, we determined antigen-specific IFNγ and IL-17 CD4+ T-cell responses in lungs and spleens of vaccinated mice. Our results show that B6 mice mucosally vaccinated with ESAT-6 in NE or Ag85B in NE vaccine induced IL-17 mucosal T-cell responses in the lung, while neither subcutaneous BCG vaccination nor mucosal delivery of NE by itself induced Mtb-specific IL-17 lung T-cell responses (Figure. 1A). Both BCG vaccination and mucosal delivery of NE with Mtb antigens induced IL-17 T-cell responses in the spleen (Figure. 1B), when compared to NE alone without Mtb antigens. BCG vaccination induced IFNγ T-cell responses in both lung and the spleen. However, while ESAT-6 in NE adjuvant induced some IFNγ T-cell responses in the lung, Ag85B along with NE did not induce IFNγ T-cell responses in either lung or spleen (Figure. 1 C, D). Thus, our data show that mucosal vaccination of NE along with Mtb antigens induces both systemic and mucosal IL-17 T-cell responses in the lung, with lower induction of IFNγ T-cell responses in the lung.
Figure 1. NE-TB vaccine induced IL-17 mucosal responses.

(A-D) B6 mice were mucosally vaccinated I.N. three times with three week intervals with Ag85B or ESAT-6 protein (25 μg each) along with NE adjuvant (20%), or NE adjuvant alone (20%), or with BCG S.C (1×106 CFU). Two weeks after last booster, lungs and spleens were harvested and IL-17 and IFNγ responses were determined by antigen-driven ELISpot assays. The data points represent the means (±SD) of values from 4-5 mice. ***P< 0.0001, **P<0.01, *P<0.05 by one way ANOVA.
NE-TB vaccination confers vaccine-induced protection and decreases TB disease upon Mtb challenge
Our results show that NE adjuvant, in combination with Mtb immunodominant antigens, induces potent IL-17 mucosal responses, while ESAT-6 and not Ag85B, induces local IFNγ responses in the lungs. Thus, we next determined whether a NE-TB vaccine (NE formulated with both ESAT-6/Ag85B proteins) was protective in a mouse model of Mtb challenge, and whether NE-TB vaccine delivered as a booster to BCG vaccination improved protection upon Mtb challenge. Thus, we mucosally vaccinated B6 mice with NE-TB vaccine using different protocols: mice received I.N. vaccination with NE-TB followed by two I.N. boosts 3 weeks apart; (NE:NE), or mice received a single priming dose of BCG S.C. followed sequentially by two I.N. NE boosts; (BCG:NE), or mice received S.C. with BCG concurrently with simultaneous I.N. NE-TB as the priming immunization, followed by two I.N. NE-TB boosts alone, without BCG (BCG+NE:NE). Additionally, we included mice that received BCG vaccination S.C. alone (BCG). Mice receiving PBS were included as unvaccinated controls. All groups of mice were rested for four weeks, and then challenged with a low dose of aerosolized hypervirulent clinical strain, Mtb HN878. As expected, BCG vaccination resulted in significant protective efficacy upon Mtb challenge (Figure. 2A). NE-TB vaccine also resulted in significant protection when compared to Mtb CFU in unvaccinated Mtb challenged mice (Figure. 2A). Additionally, mucosal delivery of NE-TB vaccine either concurrently at the time of BCG vaccination or alone as a booster to initial BCG vaccination did not alter Mtb control in vaccinated mice (Figure. 2A). These results suggest that NE-TB mucosal vaccine is protective upon Mtb challenge and provides protection, similar to the gold standard vaccine, BCG.
Figure 2. NE-TB vaccination conferred vaccine-induced protection and decreased TB disease upon.

Mtb challenge. B6 mice either left unvaccinated (PBS group) or vaccinated S.C. with BCG(BCG group, 1×106 CFU). B6 mice were also vaccinated I.N. three times with three week intervals with NE-TB vaccine (Ag85B and ESAT-6 proteins (25 μg each) with 20% NE, (NE:NE group). B6 mice received BCG S.C. (1×106 CFU) and boosted sequentially with NE-TB vaccine twice (BCG:NE group) or vaccinated concurrently both BCG (S.C.) (1×106 CFU) and mucosally with NE-TB vaccine, followed by two boosts with NE-TB vaccine (BCG+NE:NE group). All groups of B6 mice were rested for 4 weeks after which mice were challenged with Mtb HN878 (100 CFU). (A) Mtb CFU was determined on 30 days post-infection. (B, C) Pulmonary inflammation was quantitated on formalin-fixed, paraffin-embedded lung sections from mice on H&E-stained sections. The data points represent the means (±SD) of values from 10 mice per group. ***P< 0.0001, **P<0.01, *P<0.05 by one way ANOVA.
Although prevention of infection and Mtb control are readouts of vaccine efficacy, alleviation of TB disease is another critical outcome for an effective vaccine response. Thus, we measured lung inflammation in the different groups of vaccinated Mtb-infected mice. We found that while unvaccinated Mtb-infected mice demonstrated increased inflammation, BCG vaccination moderately dampened lung inflammation in Mtb-infected mice (Figure. 2 B, C). Interestingly, NE-TB vaccination either by itself (NE: NE) or when carried out sequentially after a priming vaccination using BCG alone (BCG: NE), or when administered concurrently with BCG vaccination for the priming dose (BCG+NE: NE) resulted in decreased lung inflammation, when compared to unvaccinated Mtb-infected mice, or animals vaccinated using BCG alone (Figure. 2 B, C). These results suggest that mucosal immunization using the NE-TB vaccine limits disease inflammation and improves disease outcome in Mtb-infected mice, even beyond levels conferred by the gold standard BCG vaccine.
Decreased chemokine induction and improved B cell lymphoid follicle formation is associated with mucosal delivery of NE-TB vaccine in previously BCG vaccinated mice
Our results show that a novel mucosal NE-TB vaccine confers protection, and when used concurrently with BCG vaccination limits TB disease in Mtb-infected mice. Our recent published studies have described the generation of B cell follicle containing TB granulomas that allow for colocalization of T-cells near Mtb-infected macrophages for control of Mtb infection [18]. Thus, we measured the area occupied by B cell follicles within TB granulomas in the various groups of vaccinated and unvaccinated Mtb-infected mice. While, NE-TB vaccination resulted in formation of well-defined B cell follicles within TB granulomas (Figure. 3A), mice that received BCG vaccination along with concurrent mucosal NE-TB vaccine delivery resulted in the most effective formation of organized lymphoid structures within TB granulomas. Additionally, mice that received BCG vaccination along with mucosal NE-TB vaccine delivery also exhibited decreased induction of inflammatory chemokines including C-X-C motif chemokine (CXCL-9), CXCL-2 and C-C-motif ligand (CCL-5) (Figure. 3B-D). Thus, these results together demonstrate that not only does NE-TB vaccine induce effective Mtb control, but NE-TB vaccination alone, or given alongside BCG vaccination resulted in decreased TB disease and improved formation of protective granulomas in the lung.
Figure 3. Decreased chemokine induction and improved B cell lymphoid follicle formation is associated with mucosal delivery of NE-TB vaccine in previously BCG vaccinated mice.
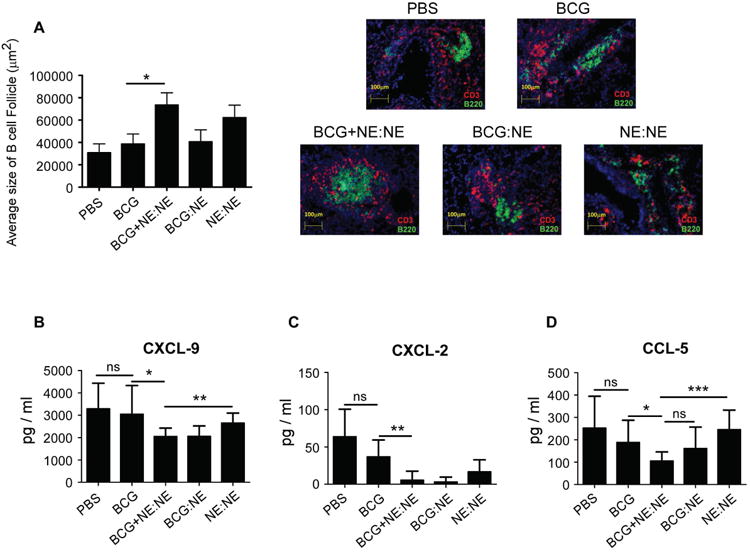
(A) B cell lymphoid follicle formation was determined by CD3 and B220 staining on formalin-fixed, paraffin-embedded sections by immunofluorescent staining. The total area occupied by B cell follicles per lobe was quantitated using the morphogenetic Axiovision Rel 4.8 software tool of the Zeiss Axioplan microscope. Representative images of B cell follicles from the different groups are shown, at a magnification of 20×. (B-D) Lung homogenates from mice groups in Figure 2 were used to measure chemokine levels by Milliplex assay. The data points represent the means (±SD) of values from 10 mice per group. (Student t-test) *P<0.05, **P<0.005, ***P<0.0005, ns- not significant.
Discussion
TB is a significant cause of global mortality and morbidity. However with the continuing specter of TB worldwide, an efficacious human vaccine is still unavailable. Despite the urgent need for the development of an effective human TB vaccine, BCG remains as the only licensed vaccine against TB over the past hundred years. Since the natural route of TB infection is through the mucosal surface, vaccines delivered through the mucosal route are known to provide superior protection against Mtb infection [14, 15]. However, there is no mucosal TB vaccine that is currently in clinical trials. Thus, in this study we demonstrate that NE adjuvant already tested to be safe in humans [22], when delivered with Mtb immunodominant antigens induces potent mucosal IL-17 T-cell responses and confers protective vaccine-induced immunity against Mtb infection. Importantly, this NE-based mucosal TB vaccine when used concurrently with BCG vaccination significantly limits TB disease severity, when compared to use of the BCG vaccination alone. Thus, our studies strongly support the future use of NE-TB vaccine as a first-of-kind mucosal vaccine for development into a human TB vaccine.
Substantial recent evidence demonstrates a pivotal role for Th17 cells and IL-17 in vaccine-mediated immunity against TB [8, 10, 17]. Several mucosally delivered adjuvants such as cholera toxin [26, 27], heat labile enterotoxin [8], Monophosporyl lipid A (MPL) along with chitosan [25], when delivered with Mtb antigens in experimental models have all been shown to induce potent lung Th17 responses and confer Mtb control. However, there are serious concerns regarding the safety of toxin subunits as mucosal adjuvants in human vaccines. So far, of the IL-17-inducing adjuvants, MPL [28], chitosan [29], and MPL along with chitosan [30] are the only adjuvants that have been proven safe for human mucosal immunization. NE-based vaccines are safe for human mucosal use as shown in phase I clinical trials [21], and thus stand out in being compatible for use in a human TB vaccine. With regard to choice of antigens, we combined the use of immunodominant antigens ESAT-6 and Ag85B, as both antigens when individually delivered in NE, induced potent IL-17-producing T-cells, possibly both Th17 and CD8+ T-cells in the lung. This is consistent with the known role for NE in activating mucosal DCs and epithelial cells to secrete Th17-inductive cytokines including IL-1β, IL-6 and TGFβ [31], and in driving mucosal Th17 responses [22]. However, NE delivered with ESAT-6 antigen additionally induced IFNγ T-cell responses in the lung. This is a surprising finding considering ESAT-6 protein has been shown to induce TGF-β and IL-6 in TLR-2, Myd88-dependent manner and induce Th17 responses [32]. Nonetheless, we combined use of Ag85B and ESAT-6 along with NE to drive both potent mucosal IL-17 T-cell responses, for testing protective efficacy in Mtb-infected mice. Our results demonstrating that use of NE-TB vaccine in mice confers protection upon Mtb challenge, further supports the development of IL-17-inducing vaccines for TB. However, our results here as well as published studies [8] demonstrate that while sub-unit vaccines delivered mucosally confer Mtb control, the level of protection induced by sub-unit vaccines is not as robust when compared to protection induced by use of a live attenuated TB vaccine, such as BCG.
A hallmark of pulmonary TB in both humans and experimental animals is the formation of granulomas containing Mtb-infected macrophages [33]. The role of IL-17 in mediating early protective immunity against Mtb HN878, is through IL-17 receptor (IL-17R) signaling in non-haemopoietic cells, to induce expression of the chemokine CXCL-13 [34]. Protective TB granulomas comprise of organized lymphoid structures that are mediated by the expression of CXCL-13 [35-38]. CXCL13 controls the formation of B cell follicles, T-cell localization, and the optimal activation of macrophages for Mtb control [35-37]. Our results show that use of NE-TB vaccine either by itself, or in combination with BCG reduces overall lung inflammation. Coincident with decreased TB disease, our results also demonstrate that use of NE-TB vaccine either by itself or in combination with BCG vaccination, induces effective formation of B cell follicles in the lungs of the Mtb-infected mice. It is possible that mucosal IL-17 T-cell responses induced by NE-TB vaccines in BCG vaccinated mice drive rapid lung CXCL-13 expression to induce early lymphoid structures, thus decreasing TB disease, expression of inflammatory chemokines, and mediating Mtb control. Therefore the use of NE-TB vaccine may be effective in not only providing protection against Mtb challenge but will also aid in lowering the level of inflammation in the lungs, and perhaps even limit TB reactivation in latently infected individuals. The current slate of TB vaccines are projected to replace BCG with an improved live vector-based vaccine, or as a boost in BCG primed hosts [1]. Thus, our data demonstrate that co-administration of the NE-TB mucosal vaccine along with BCG vaccination significantly decreases TB disease. These studies provide a strategy to improve the efficacy of BCG vaccination in humans. Further validation of the NE-TB vaccine in a non-human primate animal model is necessary to test the efficacy of the NE-TB vaccine candidate for use in humans for protection against Mtb infection and disease. Once approved, the NE-TB vaccine may be administered to humans as a mucosal booster in the later life of an infant after the intradermal BCG vaccination at birth.
In summary, we demonstrate that the NE-TB vaccine induces IL-17 T-cell responses that significantly protect against the hypervirulent clinical Mtb strain HN878, in mice. While conferring Mtb protection, the NE-TB vaccine in combination with BCG vaccination offers the added advantage of limiting TB disease. These findings support and enable the development of a NE-TB vaccine as a novel, safe and effective, IL-17-inducing mucosal vaccine for use in humans. Furthermore, Th17 vaccine responses are critical for protection against other significant pulmonary pathogens namely Streptococcus pneumoniae [39-42], Bordetella pertussis [41, 42], Klebsiella pneumoniae [43], and Pseudomonas aeruginosa [44]. Therefore the identification of novel, safe and potent IL-17-inducing adjuvants such as NE can be advanced towards the development of effective mucosal vaccines against several pulmonary diseases.
Acknowledgments
This work was supported by Washington University in St Louis, National Institutes of Health grants HL105427 and AI127172 to S.A.K. J.R.-M. was supported by funds of the Department of Medicine, University of Rochester. The authors thank Sarah Squires (WashU) for animal breeding.
Abbreviations
- Ag85B
Antigen 85B
- ANOVA
Analysis of variance
- BCG
Mycobacterium bovis bacillus Calmette-Guerin
- BCIP/NBT
5-Bromo-4-chloro-3-indolyl phosphate/ nitro blue tetrazolium chloride
- C
Centigrade
- C57BL/6J
B6
- CCL-5
Chemokine (C-C motif) ligand 5
- CD
cluster of differentiation
- CFU
colony forming unit
- CXCL-13
Chemokine (C-X-C motif) ligand 13
- CXCL-2
Chemokine (C-X-C motif) ligand 2
- CXCL-9
Chemokine (C-X-C motif) ligand 9
- DAPI
4′,6-diamidino-2-phenylindole
- DC
dendritic cells
- ESAT-6
early secreted antigenic target 6 kDa protein
- Gy
(gray) absorbed radiation dose
- HLT
heat labile enterotoxin
- I.N
intranasal
- IFNγ
interferon gamma
- IL
interleukin
- ml
milliliter
- MPL
monophosphoryl lipid A
- Mtb
Mycobacterium tuberculosis
- MVA85A
modified vaccinia Ankara 85A
- MyD88
Myeloid differentiation primary response gene 88
- NE
nanoemulsion
- nm
nanometer
- ns
not significant
- PBS
phosphate buffered saline
- S.C
subcutaneous
- S.D
standard deviation
- TB
tuberculosis
- TGF-β
Transforming growth factor beta
- Th1
T helper type 1
- Th17
T helper type 17
- TLR
toll-like receptor
- μg
microgram
- μl
microliter
- μm
micrometer
Footnotes
The authors have no conflicts to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xing Z, Jeyanathan M, Smaill F. New approaches to TB vaccination. Chest. 2014;146:804–12. doi: 10.1378/chest.14-0439. [DOI] [PubMed] [Google Scholar]
- 2.Lienhardt C, Fruth U, Greco M. The blueprint for vaccine research & development: walking the path for better TB vaccines. Tuberculosis (Edinb) 2012;92(1):S33–5. doi: 10.1016/S1472-9792(12)70011-2. [DOI] [PubMed] [Google Scholar]
- 3.Rook GA, Dheda K, Zumla A. Immune responses to tuberculosis in developing countries: implications for new vaccines. Nat Rev Immunol. 2005;5:661–7. doi: 10.1038/nri1666. [DOI] [PubMed] [Google Scholar]
- 4.Hawkridge T, Scriba TJ, Gelderbloem S, Smit E, Tameris M, Moyo S, et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J Infect Dis. 2008;198:544–52. doi: 10.1086/590185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tameris M, Geldenhuys H, Luabeya AK, Smit E, Hughes JE, Vermaak S, et al. The candidate TB vaccine, MVA85A, induces highly durable Th1 responses. PLoS One. 2014;9:e87340. doi: 10.1371/journal.pone.0087340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. 2013;381:1021–8. doi: 10.1016/S0140-6736(13)60177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ndiaye BP, Thienemann F, Ota M, Landry BS, Camara M, Dieye S, et al. Safety, immunogenicity, and efficacy of the candidate tuberculosis vaccine MVA85A in healthy adults infected with HIV-1: a randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2015;3:190–200. doi: 10.1016/S2213-2600(15)00037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, et al. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol. 2013;6:972–84. doi: 10.1038/mi.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths KL, Khader SA. Novel vaccine approaches for protection against intracellular pathogens. Curr Opin Immunol. 2014;28:58–63. doi: 10.1016/j.coi.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T-cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 11.Monin L, Griffiths KL, Slight S, Lin Y, Rangel-Moreno J, Khader SA. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal Immunol. 2015;8:1099–109. doi: 10.1038/mi.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Wang J, Zganiacz A, Xing Z. Single intranasal mucosal Mycobacterium bovis BCG vaccination confers improved protection compared to subcutaneous vaccination against pulmonary tuberculosis. Infect Immun. 2004;72:238–46. doi: 10.1128/IAI.72.1.238-246.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goonetilleke NP, McShane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J Immunol. 2003;171:1602–9. doi: 10.4049/jimmunol.171.3.1602. [DOI] [PubMed] [Google Scholar]
- 14.Santosuosso M, Zhang X, McCormick S, Wang J, Hitt M, Xing Z. Mechanisms of mucosal and parenteral tuberculosis vaccinations: adenoviral-based mucosal immunization preferentially elicits sustained accumulation of immune protective CD4 and CD8 T-cells within the airway lumen. J Immunol. 2005;174:7986–94. doi: 10.4049/jimmunol.174.12.7986. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Thorson L, Stokes RW, Santosuosso M, Huygen K, Zganiacz A, et al. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J Immunol. 2004;173:6357–65. doi: 10.4049/jimmunol.173.10.6357. [DOI] [PubMed] [Google Scholar]
- 16.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–58. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths KL, Stylianou E, Poyntz HC, Betts GJ, Fletcher HA, McShane H. Cholera toxin enhances vaccine-induced protection against Mycobacterium tuberculosis challenge in mice. PLoS One. 2013;8:e78312. doi: 10.1371/journal.pone.0078312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest. 2013;123:712–26. doi: 10.1172/JCI65728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffiths KL, Ahmed M, Das S, Gopal R, Horne W, Connell TD, et al. Targeting dendritic cells to accelerate T-cell activation overcomes a bottleneck in tuberculosis vaccine efficacy. Nat Commun. 2016;7:13894. doi: 10.1038/ncomms13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, Linder T, et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N Engl J Med. 2004;350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 21.Stanberry LR, Simon JK, Johnson C, Robinson PL, Morry J, Flack MR, et al. Safety and immunogenicity of a novel nanoemulsion mucosal adjuvant W805EC combined with approved seasonal influenza antigens. Vaccine. 2012;30:307–16. doi: 10.1016/j.vaccine.2011.10.094. [DOI] [PubMed] [Google Scholar]
- 22.Bielinska AU, Gerber M, Blanco LP, Makidon PE, Janczak KW, Beer M, et al. Induction of Th17 cellular immunity with a novel nanoemulsion adjuvant. Crit Rev Immunol. 2010;30:189–99. doi: 10.1615/critrevimmunol.v30.i2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gopal R, Lin Y, Obermajer N, Slight S, Nuthalapati N, Ahmed M, et al. IL-23-dependent IL-17 drives Th1-cell responses following Mycobacterium bovis BCG vaccination. Eur J Immunol. 2012;42:364–73. doi: 10.1002/eji.201141569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, et al. Antigen-specific T-cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–87. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed M, Jiao H, Domingo-Gonzalez R, Das S, Griffiths KL, Rangel-Moreno J, et al. Rationalized design of a mucosal vaccine protects against Mycobacterium tuberculosis challenge in mice. J Leukoc Biol. 2017 doi: 10.1189/jlb.4A0616-270R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Datta SK, Sabet M, Nguyen KP, Valdez PA, Gonzalez-Navajas JM, Islam S, et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc Natl Acad Sci U S A. 2010;107:10638–43. doi: 10.1073/pnas.1002348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JB, Jang JE, Song MK, Chang J. Intranasal delivery of cholera toxin induces th17-dominated T-cell response to bystander antigens. PLoS One. 2009;4:e5190. doi: 10.1371/journal.pone.0005190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter D, Reed SG. Role of adjuvants in modeling the immune response. Curr Opin HIV AIDS. 2010;5:409–13. doi: 10.1097/COH.0b013e32833d2cdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills KH, Cosgrove C, McNeela EA, Sexton A, Giemza R, Jabbal-Gill I, et al. Protective levels of diphtheria-neutralizing antibody induced in healthy volunteers by unilateral priming-boosting intranasal immunization associated with restricted ipsilateral mucosal secretory immunoglobulin a. Infect Immun. 2003;71:726–32. doi: 10.1128/IAI.71.2.726-732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Kamary SS, Pasetti MF, Mendelman PM, Frey SE, Bernstein DI, Treanor JJ, et al. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis. 2010;202:1649–58. doi: 10.1086/657087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makidon PE, Belyakov IM, Blanco LP, Janczak KW, Landers J, Bielinska AU, et al. Nanoemulsion mucosal adjuvant uniquely activates cytokine production by nasal ciliated epithelium and induces dendritic cell trafficking. Eur J Immunol. 2012;42:2073–86. doi: 10.1002/eji.201142346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatterjee S, Dwivedi VP, Singh Y, Siddiqui I, Sharma P, Van Kaer L, et al. Early secreted antigen ESAT-6 of Mycobacterium tuberculosis promotes protective T helper 17 cell responses in a toll-like receptor-2-dependent manner. PLoS Pathog. 2011;7:e1002378. doi: 10.1371/journal.ppat.1002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saunders BM, Cooper AM. Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol Cell Biol. 2000;78:334–41. doi: 10.1046/j.1440-1711.2000.00933.x. [DOI] [PubMed] [Google Scholar]
- 34.Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 2014;10:e1004099. doi: 10.1371/journal.ppat.1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, et al. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol. 2011;187:5402–7. doi: 10.4049/jimmunol.1101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khader SA, Rangel-Moreno J, Fountain JJ, Martino CA, Reiley WW, Pearl JE, et al. In a murine tuberculosis model, the absence of homeostatic chemokines delays granuloma formation and protective immunity. J Immunol. 2009;183:8004–14. doi: 10.4049/jimmunol.0901937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12:639–46. doi: 10.1038/ni.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci U S A. 2007;104:10577–82. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malley R, Srivastava A, Lipsitch M, Thompson CM, Watkins C, Tzianabos A, et al. Antibody-independent, interleukin-17A-mediated, cross-serotype immunity to pneumococci in mice immunized intranasally with the cell wall polysaccharide. Infect Immun. 2006;74:2187–95. doi: 10.1128/IAI.74.4.2187-2195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. 2009;119:1899–909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banus S, Stenger RM, Gremmer ER, Dormans JA, Mooi FR, Kimman TG, et al. The role of Toll-like receptor-4 in pertussis vaccine-induced immunity. BMC Immunol. 2008;9:21. doi: 10.1186/1471-2172-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T-cells. J Immunol. 2006;177:7980–9. doi: 10.4049/jimmunol.177.11.7980. [DOI] [PubMed] [Google Scholar]
- 43.Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, et al. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity. 2011;35:997–1009. doi: 10.1016/j.immuni.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Priebe GP, Walsh RL, Cederroth TA, Kamei A, Coutinho-Sledge YS, Goldberg JB, et al. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J Immunol. 2008;181:4965–75. doi: 10.4049/jimmunol.181.7.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]