

Abstract
Background
In hypertrophy, progressive loss of function due to impaired diastolic compliance correlates with advancing cardiac fibrosis. It has been shown that endothelial cells contribute to this process through endothelial-to-mesenchymal transition (EndMT) due to inductive signals such as TGF-β. Vascular endothelial growth factor (VEGF) has proven effective in preserving systolic function and delaying the onset of failure. In this study, we hypothesize that VEGF inhibits EndMT and prevents cardiac fibrosis, thereby preserving diastolic function.
Methods
Newborn rabbits underwent banding of the descending aorta. At 4 and 6wks, hypertrophied animals (H) were treated with intrapericardial VEGF protein (T) and compared to controls (C; n=6/groups). Weekly transthoracic echocardiography measured peak systolic stress. At 7wks, diastolic stiffness was determined through pressure-volume curves, fibrosis by Masson’s Trichrome (MT) and hydroxyproline assay (HP), EndMT by immunohistochemistry, and activation of TGF-β and SMAD2/3 by qRT-PCR.
Results
Peak systolic stress was preserved during the entire observation period and diastolic compliance was maintained in T (H:20±1 versus T:11±3 and C:12±2, p<0.05). Collagen was significantly higher in H (MT: H:3.1±0.9 versus T:1.8±0.6, and HP: H:2.8±0.6 versus T:1.4±0.4; p<0.05). Fluorescent immunostaining showed active EndMT in H, but significantly less in T, which was directly associated with a significant increase in TGF-β/SMAD-2 mRNA expression.
Conclusions
EndMT contributes to cardiac fibrosis in hypertrophied hearts. VEGF treatment inhibits EndMT and prevents the deposition of collagen that leads to myocardial stiffness through TGF-β/SMAD-dependent activation. This presents a therapeutic opportunity to prevent diastolic failure and preserve cardiac function in pressure-loaded hearts.
Keywords: hypertrophy, vascular endothelial growth factor, fibrosis, endothelial-to-mesenchymal transition
In response to increased pressure loading, the myocardium adjusts by altering myocardial structure and function. This series of complex cellular and molecular changes ultimately leads to the progression of compensated hypertrophy to heart failure. Cardiomyocytes are central to the contractile function of the myocardium but the cardiac interstitium and its fibrillar collagen matrix also play a critical role in cardiac performance (1). Morphological changes involve hypertrophy and apoptosis of cardiomyocytes, but the destruction of the dynamic balance within the extracellular matrix, affecting vascularization, and collagen synthesis and degradation is equally as important (2,3,4). During the compensated stage of hypertrophy, these changes permit the parallel increase in cardiomyocyte growth and collagen deposition. In contrast, later, excess deposition of collagen occurs as a result of combined effects of loss of cardiomyocytes with increased extracellular matrix serving to replace necrosed or apoptotic cardiomyocytes, change in growth factor profile, creating an anti-angiogenic environment leading to rarefaction of the microvasculature and resulting cardiomyocyte loss, and mechanical forces, with activation of transforming growth factor-β (TGF-β) (2,5,6). Increased collagen content of the heart (fibrosis) is tightly linked to TGF-β signaling, thus, making TGF-β a target for heart failure treatment (7,8).
The TGF-β family is a class of structurally related proteins which regulates cell growth and apoptosis, cellular proliferation and migration, as well as extracellular matrix remodeling (9). SMAD proteins are the intracellular signaling pathway proteins of the TGF-β superfamily. In addition to TGF-β, fibrosis is often associated with other growth factors such as vascular endothelial growth factor (VEGF). VEGF down-regulation accompanied by up-regulation of endogenous anti-angiogenic proteins has been found to result in increased organ fibrosis, whereas VEGF up-regulation has anti-fibrotic effects (10). As we have previously shown, microvascular rarefaction, as a result of up-regulated levels of anti-angiogenic in combination with lack of increase of pro-angiogenic stimuli in the hypertrophied myocardium, is a key component for the development of heart failure (4,11). Microvascular rarefaction has been attributed to loss of endothelial cells through a process called endothelial-to-mesenchymal transformation (EndMT) which has been shown to significantly increase the pool of fibroblasts (12). Promoting fibroblast growth increases extracellular collagen, and decreases ventricular compliance (8), causing a shift of the pressure-volume curve upwards and to the left, as a higher pressure is needed to fill the ventricle to the same volume (13). Impaired diastolic contractile function is a direct result of increased fibrosis (14) and is increasingly recognized as an important component of heart failure (6). VEGF’s intricate participation in the regulation of normal development and its ability to modify pathologic processes in the heart are evidence of its therapeutic potential for cardiac dysfunction which we further examine in this study.
We have already shown that exogenous administration of VEGF is effective in preserving systolic function and delaying the onset of failure by improving cardiac perfusion, preventing apoptosis of cardiomyocytes, and degrading extracellular matrix through the activation of matrix metalloproteinases (2,3). In this study, we hypothesize that exogenous administration of VEGF prevents cardiac fibrosis through inhibition of EndMT and, thereby preserves diastolic compliance of the hypertrophied left ventricle.
Material and Methods
Animal Model
We established a pressure-overload hypertrophy model through banding of the descending thoracic aorta in ten-day-old New Zealand White rabbits (Millbrook Farms, Concord, MA). For all surgical procedures, anesthesia was induced with intramuscular ketamine (50–60mg/kg I.M.) and xylazine (2–5mg/kg I.M.), and maintained with isoflurane (0.5–2%) via face mask. Animals were monitored for anesthesia depth through assessing ear pinch, blink reflex and respiratory rate. Bupivacaine 0.25% (<3mg/kg) was injected locally and for postoperative pain medication, buprenorphine (0.1mg/kg I.M.) immediately after surgery once and followed by keteroloac (1mg/kg I.M.) was provided for the first 72 hours to minimize discomfort from the surgical procedure. Animals were examined daily for signs of pain for the first week postoperatively. At four and six weeks old, animals received 2μg/kg rhVEGF165 (R&D Systems, Minneapolis, MN) through intrapericardial administration under the same anesthetic and analgesic management as described above. Details about this model have been previously published by our group (2,3). Unsedated, slightly restrained animals were followed by weekly transthoracic echocardiography using a Philips 7500 equipped with a S12 transducer. Two-dimensional cross-sectional images and M-mode of the LV were obtained for measurements of LV peak systolic stress as we have previously described in more detail (15). The study endpoint was chosen based on survival data from our previous studies using this animal model (2).
Animal Care
All animals received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH Publication No. 86–23, revised 1996). The protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Boston Children’s Hospital.
Determination of Fibrosis
De-paraffinized tissue sections obtained from untreated hypertrophied hearts (n=7), VEGF-treated hypertrophied hearts (n=7), and sham-operated controls (n=7) were stained with Masson’s Trichrome which results in fibrotic (collagen-enriched) areas appearing blue and cellular elements appearing red. Slides were visualized using a microscope with a Nikon 10x objective and a 10x eyepiece. Collagen content was measured through morphometric differentiation of blue collagen fibers and red cellular components on 15 random fields of vision (LV free wall and intraventricular septum) per each part of the tissue section using imaging system software (AxioVision, Zeiss, Germany). Papillary muscles were excluded from the measurement of fibrosis. Percentage area of fibrosis was calculated by dividing the sum of the fibrotic areas of all sections by that of the total tissue areas and used as the index for the degree of fibrosis as described by Tanaka et al (16).
Secondly, total collagen content was measured through determination of hydroxyproline concentration (17). In brief, myocardial tissue was homogenized and solid material was precipitated from the tissue homogenate with 10% trichloroacetic acid by centrifugation at 5000g for 30min at 4°C. The pellet was mixed with 12N hydrochloric acid and boiled at 110°C until samples were dry. The samples were re-suspended in 1.4% chloramine T in 0.5M sodium acetate/10% isopropanol and incubated for 20min at room temperature. Serial dilutions of trans-4-hydroxy-L-proline standard were prepared starting at 0.5mg/ml. Ehrlich’s solution (1.0 M p-dimethylaminobenzaldehyde in 70% isopropanol/30% perchloric acid) was added to sample (or standard), mixed, and incubated at 65°C for 15min. After samples returned to room temperature, the optical density of each sample and standard was measured at 550 nm and the concentration was calculated from the hydroxyproline standard curve and normalized to mg protein. All materials were obtained from Sigma-Aldrich, St. Louis, MO.
Soluble and insoluble (cross-linked) collagen was isolated by pepsin digestion following a protocol by Miller et al. (18) and subsequently hydroxyproline content was measured.
Determination of Diastolic Stiffness
At study end-point (7 weeks of age), animals were administered heparin (500 U/kg intravenously) and euthanized with an overdose of ketamine (100 mg/kg IV) and xylazine (10 mg/kg IV). The hearts were excised and, after aortic cannulation, were perfused in a non-working, non-recirculating Langendorff method, as described previously in more detail (19). Left ventricular end-diastolic compliance was determined with an intracavitary fluid-filled latex balloon connected to a catheter-tipped micromanometer (Millar Instruments, Houston, Texas) inserted through the left atrial appendage. After 15 minutes of stabilization, the heart was relaxed through a 2-minute infusion of normothermic cardioplegic solution (50mL) of modified Krebs-Henseleit buffer containing a final concentration of 22.5 mmol/L KCl. Pressure-volume curves were recorded in untreated hypertrophied hearts (n=7), VEGF treated hypertrophied hearts (n=7), and age-matched, sham-operated control hearts (n=7) by blotting gradually increasing balloon volumes against measured end-diastolic pressures. These curves were obtained to determine passive stiffness of the myocardium.
Determination of Endothelial-to-Mesenchymal Transition (EndMT)
Determination of EndMT was performed through immunohistochemical staining of frozen sections with antibodies directed against CD-31, an endothelial cell marker (Dako, Carpinteria, CA), and fibroblast-specific protein-1 (FSP-1; Dako, Carpinteria, CA) and α-smooth muscle actin, fibroblast markers (αSMA; Sigma-Aldrich, St. Louis, MS). Phospho-SMAD2/3 (Santa Cruz, Dallas, TX) was used for detection of activation of EndMT in CD-31 positive endothelial cells when co-localized with respective nuclei. Secondary immunoreagents conjugated to the red-fluorescent Alexa-647™ or green-fluorescent Alexa-488™ fluorophores were used and nuclei were stained with blue fluorescent DAPI nucleic acid stain (Thermofisher, Cambridge, MA). Double-stained cells or nuclei, respectively, were indicative of endothelial cells undergoing EndMT.
Determination of TGF-β Signaling Pathway Activation
Total RNA was prepared using Trizol reagent (Sigma-Aldrich, St. Louis, MS). 100ng/rxn of RNA was transcribed and a One-Step PCR for TGF-β, SMAD2 and SMAD3 using a B-R 1-Step SYBR Green qRT-PCR Kit (Quanta Biosciences, Gaithersburg, MD). Primers were obtained from Eurofins MWG Operon (Huntsville, AL). Primer sequences were as follows: TGF-β - forward 5′-tgaagttgcagcctatgcac-3′ and reverse 5′-tggctgtaccacagcaagag-3′; SMAD2 - forward 5′-ggaatttgctgctcttctgg-3′ and reverse 5′-ctgccttcggtattctgctc-3′; SMAD3 – forward 5′-ccctggctacctgagtgaag-3′ and reverse 5′-ggctcgcagtaggtaactgg-3′. Conditions for quantitative PCR reaction were a primary incubation period at 48°C for 1 hour and a secondary incubation period at 94°C for 10 min, followed by 40 cycles of 94°C for 15 sec, 53.4°C for 30 sec and 72°C for 45 sec using a Stratagene Mx3000P RT-PCR machine (Agilent Technologies, Santa Clara, CA).
Statistical Analysis
Data were analyzed using SPSS software package (version 16.0, SPSS Inc., Chicago, IL). One-way ANOVA was used for comparison among and between groups, or Kruskal-Wallis test if normality was not passed, followed by Bonferroni’s or Dunn’s post-hoc analysis where appropriate. Probability values of ≤0.05 were regarded statistically significant.
Results
Peak Systolic Stress by Echocardiography
Normalization of wall stress as adaptive mechanism was preserved in VEGF-treated hearts (Figure 1A). In contrast, untreated hypertrophied hearts failed to compensate and progressed to failure (p<0.05; n=7/group). At study endpoint, heart weight to body weight ratios (g/kg) were significantly higher in the untreated hypertrophied and VEGF-treated hearts compared to controls (C: 4.23±0.2; H: 7.97±0.3; T: 7.65±0.4; p<0.001).
Figure 1.

Peak systolic stress and diastolic compliance: (A) Peak systolic stress was determined by weekly echocardiography over the entire observation period. Normalization of wall stress as adaptive mechanism to compensate for increased pressure-loading is preserved in VEGF-treated hypertrophied hearts compared to untreated hearts (n=7/group; *p<0.05 versus VEGF-treated and control). (B) Pressure volume curves were obtained on arrested isolated perfused hearts. Passive stiffness of the myocardium, determined in arrested hearts, is significantly higher in untreated hypertrophied hearts corresponding with a higher amount of collagen deposition in the extracellular matrix (n=7/group; *p<0.05 versus VEGF-treated and control).
Passive Diastolic Compliance
As indicated in Figure 1B, the intracavitary pressure curve for increasing volumes in arrested hearts was shifted to the left in the untreated hypertrophy group compared to VEGF-treated and control hearts, indicating a lack of ventricular compliance secondary to myocardial stiffness (H:20±1mmHg versus T:11±3mmHg and C:12±2mmHg; p<0.05).
Degree of Fibrosis
Total collagen as well as soluble and insoluble collagen, which was determined by hydroxyproline assay (Figure 2), were consistently elevated in the untreated hypertrophy group while remained at normal levels in VEGF-treated hearts. These results were supported by analysis of Masson’s Trichrome staining which also showed significantly lower collagen deposition in VEGF-treated hearts (Figure 3).
Figure 2.

Myocardial fibrosis determined by hydroxyproline assay: (A) Total amount of collagen is significantly increased in untreated hypertrophied hearts compared to VEGF-treated and control hearts (n=7/group; *p<0.05). (B) Soluble collagen is significantly increased in untreated hypertrophied hearts compared to VEGF-treated and control hearts (n=7/group; *p<0.05). (C) Cross-linked collagen is significantly increased in untreated hypertrophied hearts compared to VEGF-treated and control hearts (n=7/group; *p<0.05).
Figure 3.

Histological assessment of fibrosis: (A) As indicated by the representative histological sections stained with Masson’s Trichrome, collagen deposition (blue) is significantly higher in untreated hypertrophied hearts rather than VEGF-treated and control hearts (n=7/group; *p=0.001). (B) Total amount of collagen was analyzed by light microscopy and summary of data is depicted here (n=7/group; *p<0.05 versus VEGF-treated and control).
Activation of EndMT
EndMT was investigated through fluorescent immunohiostochemistry using labeled antibodies directed against CD31, an endothelial marker, and FSP-1 or αSMA, fibroblast markers whereof FSP-1 and αSMA indicated the same staining pattern. Co-expression of markers in an individual cell is considered an indicator of active transformation to a mesenchymal phenotype. As indicated in Figure 4A and B, fluorescent immunostaining showed the presence of cells undergoing EndMT in the untreated hypertrophy group (only FSP-1 data shown) and significantly less endothelial cells undergoing EndMT were found in VEGF-treated hypertrophied hearts (p=0.0001) and no EndMT was present in control hearts (data not shown). PhosphoSMAD2/3 was also used to determine activation of EndMT in endothelial cells which showed that significantly more nuclei were positive in the untreated hypertrophied hearts (Figure 4C and D).
Figure 4.

Endothelial-to-mesenchymal transition (EndMT): (A) Representative immunohistochemical sections of EndMT in untreated hypertrophied hearts and VEGF treated hypertrophied hearts are shown. EndMT is identified by double-labeling of cells with CD31, an endothelial cell marker in red, and FSP-1, a fibroblast marker in green. (B) Data summary shows cells undergoing EndMT (cells merged for CD31 and FSP-1) expressed per total number of CD31 positive cells. There are significantly less endothelial cells undergoing EndMT in VEGF treated hypertrophied hearts comparable to control hearts compared to untreated hypertrophied hearts (n=4/group; p=0.001 VEGF-treated and control versus untreated hypertrophy). (C) Representative immunohistochemical sections of EndMT in untreated hypertrophied hearts and VEGF treated hypertrophied hearts are shown. EndMT is identified by double-labeling nuclei of CD31 positive cells in red with pSMAD2/3 in green (indicated by white arrows). (D) Data summary shows cells undergoing EndMT (CD31 positive cells with merged nuclei) expressed per total number of CD31 positive cells. These results support the extent of EndMT (n=4/group; p=0.001).
The involvement of TGF-β and its down-stream mediators, SMAD2 and SMAD3, was quantified through real-time PCR. Untreated hypertrophied hearts had a significant increase in TGF-β and SMAD-2 mRNA expression compared to VEGF-treated hearts, suggesting VEGF’s role in inhibiting the main signaling pathway of EndMT induction (Data expressed as mean ±SD in Figure 5).
Figure 5.
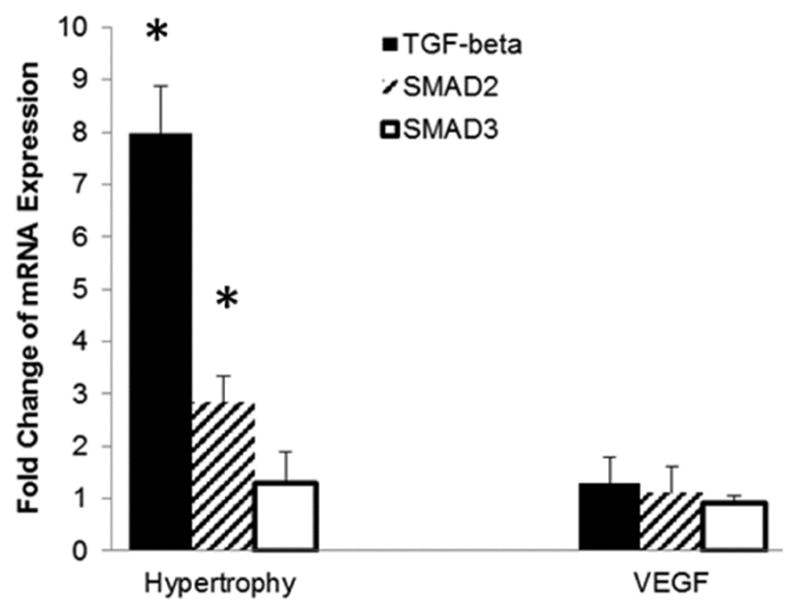
TGF-β pathway activation via qRT-PCR: TGF-β and SMAD2 are significantly up-regulated in untreated hypertrophied hearts which coincides with EndMT. Data are expressed as fold-change from control (n=6/group; *p=0.01).
Comment
In this study we confirm that EndMT contributes to cardiac fibrosis in pressure-overload hypertrophied rabbit hearts. As we have already reported, exogenous VEGF is effective as therapeutic strategy to preserve cardiomyocyte viability (2,3). At the same time we now identify VEGF as an inhibitor of fibroblastic transformation of endothelial cells. Decreasing the amount of collagen producing fibroblasts, prevents excessive deposition of collagen which reduces myocardial diastolic stiffness and heart failure. VEGF-induced effects on EndMT are mediated through down-regulation of TGF-β and SMAD-2. This presents a therapeutic opportunity to halt the progression of diastolic failure and preserve cardiac function in pressure-loaded hearts.
Under physiologic conditions, a network of collagen fibers serves a number of functions important to the overall performance of the heart. It provides supportive scaffolding for cardiomyocytes, myofibrils, and the vasculature, critical to maintaining the structural integrity and overall geometry of the heart. The extracellular matrix tethers individual myocytes and myofibrils together enabling transduction of forces generated by cardiac contraction. In addition, interstitial collagen provides tensile strength to the myocardium and is an important determinant of diastolic stiffness. However, in cases of excessive collagen deposition, cardiac fibrosis develops, restricting the delivery of nutrients to cardiomyocytes which we and others have reported (19). At the same time, fibrosis alters the mechanical properties of the myocardium (e.g. impaired relaxation) through tissue stiffness which restricts ventricular filling with significant hemodynamic consequences (2,20).
As indicated by our results, the changes to the extracellular matrix during pressure-overload-induced hypertrophy affect both the quantity with more collagen, and the composition of the matrix with more collagen fiber cross-linking. However, the main event is the increase in collagen producing cells. It has been shown that endothelial cells contribute to this process through a phenomenon called endothelial-to-mesenchymal transition (EndMT) in which cells change their phenotype due to inductive signals such as TGF-β. In this process, cells lose their polarity and cell-cell junctions to acquire invasive and migratory properties; this is accompanied by a loss of endothelial-specific markers like CD-31 and a gain of mesenchymal markers such as FSP-1 or α-smooth muscle actin (21,22). Up to 40% of fibroblasts are derived through this process (23). Promoting fibroblast growth increases extracellular collagen and decreases ventricular compliance (6). Although EndMT was initially discovered as a component of normal heart development (24), it has now been related to pathogenesis of diseases characterized by fibrosis in organs such as the heart (12), eye (25), lung (26), kidney (27), and intestine (28), and even cancer (21). Molecular and fibroblast lineage analyses in pressure-overload (12), myocardial infarction (29), and diabetes mellitus (30) have elucidated that EndMT is responsible for this process with TGF-β signaling as main mediator (22).
TGF-β is a well-known member of a superfamily of growth factors that includes bone morphogenetic proteins and activins. It has a vital role in processes ranging from embryonic development to growth, homeostasis, and tissue repair (31). The essential role of TGF-β in cardiac hypertrophy and fibrogenesis was elucidated in TGF-β knock-out mice (32). These results are supported by our data which indicate that TGF-β plays a significant role in fibrosis of pressure-overload hypertrophy. TGF-β exerts its function through a complex of serine/threonine kinase transmembrane receptors and a variety of second messengers with regulatory and inhibitory properties (22), but SMADs were discovered as the direct effectors of TGF-β signaling (33). SMAD2 and SMAD3 are phosphorylated following TGF-β receptor binding and translocate to the nucleus to activate target genes and stimulate transcription. The dominant role of SMAD2/3 in TGF-β signaling is underlined by studies on overexpression of SMAD2 and SMAD3 which resulted in maximal TGF-β-induced stimulation of collagen gene transcription (34), and in a study of cardiac fibrosis where SMAD2/3 were found to be the major mediators of TGF-β signaling in endothelial cells (35). In our study, TGF-β/SMAD2 up-regulation in untreated hypertrophied hearts is directly associated with an increase of fibrosis supporting the central role of TGF-β/SMAD signaling in the regulation of fibrosis as it has been previously suggested (8).
VEGF is known for its effect on endothelial cells to induce angiogenesis, a process of new capillaries developing from the pre-existing vasculature. We have previously shown that angiogenesis can be stimulated by exogenous VEGF administration in pathological situations with lack of adaptive capillary growth such as hypertrophy. As a consequence of capillary growth, oxygen and nutritional supply to hypertrophied myocardial tissue is restored. Furthermore, cardiomyocyte apoptosis is prevented indirectly through better perfusion but also directly through VEGF’s interaction with cardiomyocytes (2). As in previous studies published by our group, VEGF was administered into the pericardial sac utilizing the pericardial space as drug reservoir for extended exposure of the heart to the protein. This mode of administration for a protein drug is not new and has been shown to be advantageous over intracoronary and intravenous delivery with regard to myocardial deposition and retention (36). It is unclear however, how a pro-angiogenic protein directly affects the entire myocardium as we and others have shown and not only the epicardial surface (2,36). Potential explanations include modification of the microenvironment due to increased vasodilation and vascular permeability (37). Some of VEGF’s actions are mediated through the activation of matrix metalloproteinases and subsequent extracellular matrix degradation which potentially also release matrix bound growth factors to promote angiogenesis (3). Ultimately, VEGF has been shown to stimulate recruitment and homing of stem cells resulting in paracrine effects through a large number of cytokines including VEGF (38,39). Taken together, several of these localized actions of VEGF can lead to generalized effects on the myocardium which we observed in this study.
In a more recent study, VEGF gene therapy was able to prevent angiotensin II-induced diastolic dysfunction through the proliferation of endothelial cells and dilation of pre-existing capillaries in the affected myocardium (40). Incidentally, the authors also report a trend for decreasing fibrotic area which suggests supplementary mechanisms to VEGF’s contribution to the interruption of disease progression to diastolic heart failure. Our data support this observation that VEGF administration results in significantly reduced myocardial fibrosis. Furthermore, we could show that VEGF’s anti-fibrotic properties are due to the down-regulation of TGF-β and SMAD2 which resulted in the inhibition of EndMT. VEGF’s effects on the preservation of the endothelial cell phenotype reduced the amount of collagen-producing fibroblasts. As an added benefit, overall quality of collagen is also improved with less cross-linked collagen present following VEGF treatment. Mechanisms through which pressure-overload hypertrophy increases myocardial collagen cross-linking or VEGF prevents it, have not been determined in this study but are of interest for future investigations. Taken together, it further emphasizes the key role of VEGF in organ homeostasis under physiological and pathological conditions.
In conclusion, our study shows that VEGF treatment prevents fibrosis in progressive hypertrophy through inhibition of TGF-β mediated activation of EndMT. VEGF preserves the endothelial phenotype. Besides an overall decrease in collagen, less cross-linked collagen is present in VEGF-treated hypertrophied hearts. Our data clearly support the conclusion that VEGF preserves diastolic compliance, and maintains peak systolic stress normal, and thus maintains contractile function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pelouch V, Dixon IM, Golfman L, Beamish RE, Dhalla NS. Role of extracellular matrix proteins in heart function. Mol Cell Biochem. 1993;129(2):101–20. doi: 10.1007/BF00926359. [DOI] [PubMed] [Google Scholar]
- 2.Friehs I, Barillas R, Vasilyev NV, Roy N, McGowan FX, del Nido PJ. Vascular endothelial growth factor prevents apoptosis and preserves contractile function in hypertrophied infant heart. Circulation. 2006;114(1 Suppl):I290–5. doi: 10.1161/CIRCULATIONAHA.105.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friehs I, Margossian RE, Moran AM, Cao-Danh H, Moses MA, del Nido PJ. Vascular endothelial growth factor delays onset of failure in pressure-overload hypertrophy through matrix metalloproteinase activation and angiogenesis. Basic Res Cardiol. 2006;101(3):204–13. doi: 10.1007/s00395-005-0581-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez B, Gonzalez A, Querejeta R, Larman M, Diez J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol. 2006;48:89–96. doi: 10.1016/j.jacc.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 5.Kaza E, Ablasser K, Poutias D, et al. Up-regulation of soluble vascular endothelial growth factor receptor-1 prevents angiogenesis in hypertrophied myocardium. Cardiovasc Res. 2011;89(2):410–8. doi: 10.1093/cvr/cvq321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sequra AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Failure. 2014;19(2):173–85. doi: 10.1007/s10741-012-9365-4. [DOI] [PubMed] [Google Scholar]
- 7.Van Wamel AJ, Ruwhof C, van der Valk-Kokshoorn LJ, Schrier PI, van der Laarse A. Mechanical stimuli on the myocardium release TGF-β stretch-induced paracrine hypertrophic stimuli increase TGF-beta1 expression in cardiomyocytes. Mol Cell Biochem. 2002;236(1–2):147–53. doi: 10.1023/a:1016138813353. [DOI] [PubMed] [Google Scholar]
- 8.Kuwahara F, Kai H, Tokuda K, et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–5. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 9.Massague J, Gomis RR. The logic of TGF beta signaling. FEBS Lett. 2006;580:2811–20. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 10.Zen K, Okigaki M, Hosokawa Y, et al. Myocardium-targeted delivery of endothelial progenitor cells by ultrasound-mediated microbubble destruction improves cardiac function via an angiogenic response. J Mol Cell Cardiol. 2006;40(6):799–809. doi: 10.1016/j.yjmcc.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Nikolova A, Ablasser K, Wyler von Ballmoos MC, et al. Endogenous angiogenesis inhibitors prevent adaptive capillary growth in left ventricular pressure overload hypertrophy. Ann Thorac Surg. 2012;94(5):1509–17. doi: 10.1016/j.athoracsur.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 13.Lalande S, Johnson BD. Diastolic dysfunction: a link between hypertension and heart failure. Drugs Today (Barc) 2008;44(7):503–13. doi: 10.1358/dot.2008.44.7.1221662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hein S, Arnon E, Kostin S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart - structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 15.Moran AM, Friehs I, Takeuchi K, et al. Noninvasive serial evaluation of myocardial mechanics in pressure overload hypertrophy of rabbit myocardium. Herz. 2003;28(1):52–62. doi: 10.1007/s00059-003-2392-0. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka M, Fujiwara H, Onodera T, Wu DJ, Hamashima Y, Kawai C. Quantitative analysis of myocardial fibrosis in normal, hypertensive hearts, and hypertrophic cardiomyopathy. Br Heart J. 1986;55:575–81. doi: 10.1136/hrt.55.6.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stegemann H, Stalder K. Determination of hydroxyproline. Clin Chim Acta. 1967;18(2):267–73. doi: 10.1016/0009-8981(67)90167-2. [DOI] [PubMed] [Google Scholar]
- 18.Miller EJ, Rhodes RK. Preparation and characterization of the different types of collagen. Methods Enzymol. 1982;82(Pt A):33–64. doi: 10.1016/0076-6879(82)82059-4. [DOI] [PubMed] [Google Scholar]
- 19.Friehs I, Cao-Danh H, Nathan M, McGowan FX, del Nido PJ. Impaired insulin-signaling in hypertrophied hearts contributes to ischemic injury. Biochem Biophys Res Com. 2005;331:15–22. doi: 10.1016/j.bbrc.2005.03.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OH. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation. 1995;91:161–70. doi: 10.1161/01.cir.91.1.161. [DOI] [PubMed] [Google Scholar]
- 21.Lin F, Wang N, Zhang T-C. The role of endothelial–mesenchymal transition in development and pathological process. IUBMB Life. 2012;64(9):717–23. doi: 10.1002/iub.1059. [DOI] [PubMed] [Google Scholar]
- 22.Van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012;347:177–86. doi: 10.1007/s00441-011-1222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107(11):1304–12. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markwald RR, Fitzharris TP, Smith WN. Structural analysis of endocardial cytodifferentiation. Dev Biol. 1975;42:160–80. doi: 10.1016/0012-1606(75)90321-8. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Dong F, Jia Y, et al. Notch Signal regulates corneal endothelial-to-mesenchymal transition. Am J Pathol. 2013;183(3):786–95. doi: 10.1016/j.ajpath.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 26.Arciniegas E, Neves CY, Carrillo LM, Zambrano EA, Ramírez R. Endothelial–mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium. 2005;12:193–200. doi: 10.1080/10623320500227283. [DOI] [PubMed] [Google Scholar]
- 27.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–87. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rieder F, Kessler SP, West GA, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–73. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech. 2011 doi: 10.1242/dmm.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Widyantoro B, Emoto N, Nakayama K, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–18. doi: 10.1161/CIRCULATIONAHA.110.938217. [DOI] [PubMed] [Google Scholar]
- 31.Massague J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–91. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 32.Schultz Jel J, Witt SA, Glascock BJ, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–96. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh AK, Quaggin SE, Vaughan DE. Molecular basis of organ fibrosis: potential therapeutic approaches. Exp Biol Med (Maywood) 2013;238(5):461–81. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 34.Zhang W, Ou J, Inagaki Y, Greenwel P, Ramirez F. Synergistic cooperation between Sp1 and SMAD3/SMAD4 mediates transforming growth factor beta1 stimulation of alpha 2(I)-collagen (COL1A2) transcription. J Biol Chem. 2000;275:39237–45. doi: 10.1074/jbc.M003339200. [DOI] [PubMed] [Google Scholar]
- 35.Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 36.Laham RJ, Rezaee M, Post M, Xu X, Sellke FW. Intrapericardial administration of basic Fibroblast Growth Factor: Myocardial and tissue distribution and comparison with intracoronary and intravenous administration. Catheter Cardiovasc Interv. 2003;58:375–81. doi: 10.1002/ccd.10378. [DOI] [PubMed] [Google Scholar]
- 37.Fukumura D, Gohongi T, Kadambi A, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc NatlAcad Sci USA. 2001;98:2604–9. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang JM, Wang JN, Zhang L, et al. VEGF/SDF-1 promotes cardiac stem cell mobilization and myocardial repair in the infarcted heart. Cardiovasc Res. 2011;91:402–11. doi: 10.1093/cvr/cvr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signalling and therapy. Circ Res. 2008;103:1204–1219. doi: 10.1161/CIRCRESAHA.108.176826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serpi R, Tolonen AM, Huusko J, et al. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc Res. 2011;89(1):204–13. doi: 10.1093/cvr/cvq267. [DOI] [PubMed] [Google Scholar]