


Abstract
Human MutY homolog (hMYH), an adenine DNA glycosylase, can effectively remove misincorporated adenines opposite template G or 8-oxoG bases, thereby preventing G:C→T:A transversions. Human cell extracts possess the adenine DNA glycosylase activity of hMYH and can form protein–DNA complexes with both A/G and A/8-oxoG mismatches. hMYH in cell extracts was shown to be the primary binding protein for A/G- and A/8-oxoG-containing DNA substrates by UV cross-linking. However, recombinant hMYH expressed in bacteria has much weaker glycosylase and substrate-binding activities towards A/G mismatches than native hMYH. Moreover, the protein–DNA complex of bacterially expressed hMYH migrates much faster than that of native hMYH in a non-denaturing polyacrylamide gel. Dephosphorylation of native hMYH reduces the glycosylase activity on A/G more extensively than on A/8-oxoG mismatches but does not alter the gel mobility of the protein–DNA complex. Our results suggest that hMYH in human cell extracts may be associated with other factors in the protein–DNA complex to account for its slower mobility in the gel. hMYH and apurinic/apyrimidinic endonuclease (hAPE1) co-migrate with the protein–DNA complex formed by the extracts and A/8-oxoG-containing DNA.
INTRODUCTION
Cellular DNA damage induced by reactive oxygen species (ROS) has been implicated in causing genetic instability, aging and cancer (1). Among the various DNA adducts induced by environmental carcinogens and endogenous oxidative phosphorylations that produce ROS, 7,8-dihydro-8-oxo-deoxyguanine (8-oxoG) is one of the most abundant and deleterious mutagenic oxidative lesions in human cells (2). The major toxicity of 8-oxoG is its high frequency of pairing with adenine during DNA replication that can result in G:C→T:A transversion mutations (3).
Dramatic progress has been made recently in understanding DNA repair mechanisms, including reducing 8-oxoG in Escherichia coli and higher organisms. It has been proposed that MutT, MutM and MutY of E.coli are the major lines of cellular defense against 8-oxoG lesions (4,5). In eukaryotes the repair mechanisms analogous to the E.coli MutT-, MutM- and MutY-dependent pathways have been identified. Human MutT homolog (hMTH1) hydrolyzes 8-oxo-dGTP to 8-oxo-dGMP and pyrophosphate, similarly to E.coli MutT (6,7). Removal of the oxidized dGTP precursors for DNA polymerases reduces the 8-oxoG content in DNA. Human 8-oxoG glycosylase (hOGG1), a functional homolog of E.coli MutM, can efficiently remove 8-oxoG lesions opposite cytosine but very poorly when opposite adenine (8–11). Human MutY homolog (hMYH) has been characterized in nuclear extracts (12,13) and can cross-react with antibodies against E.coli MutY (14). The adenine DNA glycosylase activity of mammalian MYH can effectively remove adenines misincorporated opposite 8-oxoG or G following DNA replication (12,14), thus eliminating G:C→T:A transversions. Mismatch repair carried out by MutS and MutL homologs (MSH and MLH) may be involved in the repair of oxidative DNA damage. The MSH2/MSH6 heterodimer (MutSα) of Sacchromyces cerevisiae has been shown to bind to A/8-oxoG-containing DNA and to be involved in the repair of A/8-oxoG mismatches (15). Because S.cerevisiae does not contain MutY and MutT homologs, the role of MutSα remains to be investigated in other organisms.
hMYH shares sequence homology and functional similarity with E.coli MutY. A cDNA of the human mutY gene (hMYH) has been reported with an open reading frame encoding a 535 residue, 59 kDa protein (16). Takao et al. have shown that there are two types of hMYH proteins: a mitochondrial form (Type 1, residues 1–535) and a nuclear form (Type 2, residues 15–535) (17). However, Tsai-Wu et al. showed that Type 1 is localized to the nuclei (18). Ohtsubo et al. also showed that multiple forms of hMYH are located in the nuclei and mitochondria (13). These controversial results remain to be resolved. The hMYH protein from the cloned cDNA has been expressed in an in vitro transcription–translation system (17) and in E.coli (18,19) and partially characterized. The expressed recombinant hMYH has adenine DNA glycosylase activity on A/8-oxoG-containing DNA substrates but very weak activity on A/G-containing DNA substrates (17–19). However, the hMYH protein expressed in a baculovirus system has efficient binding and adenine DNA glycosylase activities on both A/G- and A/8-oxoG-containing DNA substrates at low salt (1–50 mM) concentrations (20). In this paper we compare the substrate specificities of native and recombinant hMYH and investigate the underlining mechanisms for their differential substrate specificities. Because the protein–DNA complex of bacterially expressed recombinant hMYH migrates much faster than that of native hMYH in a non-denaturing polyacrylamide gel, we suspect that hMYH in human cell extracts may be associated with other factors. We demonstrate here that hMYH and human apurinic/apyrimidinic endonuclease (hAPE1) co-migrate in the protein–DNA complex formed by the extracts and A/8-oxoG-containing DNA. Human MYH, not MutSα, is the major protein in human cell extracts recognizing A/G and A/8-oxoG mismatches and consequently repairs the mismatches.
MATERIALS AND METHODS
Preparation of human cell extracts
H2009, a human non-small cell lung cancer (NSCLC) cell line, was kindly supplied by Dr Herbert K.Oie (National Cancer Institute and National Naval Medical Center, Bethesda, MD). H2009 cells were grown to late log phase in RPMI 1640 medium (Life Technologies, Rockville, MD) supplemented with 10% fetal bovine serum (HyClone, Logan, UT). The cell pellet from three T-75 flasks (∼3 × 107 cells) was resuspended in 0.5 ml of buffer containing 50 mM potassium phosphate, pH 7.4, 50 mM KCl, 1 mM dithiothreitol, 0.1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 10% glycerol. An equal volume of 0.1 mm glass beads was added to the cell suspension and cells were disrupted by vigorous vortexing for 10 s at 4°C and cooled on ice for 20 s. Vortexing was repeated 10 times. The mixture was then centrifuged at 12 000 g for 15 min. The supernatant was aliquoted and stored at –80°C. Human HeLa S3 cells, grown in Joklik’s minimal essential medium with 5% newborn calf serum, 2 g/l NaHCO3, 1% non-essential amino acids, 4 mM l-glutamine and 1% penicillin/streptomycin, were purchased from The Cell Culture Center (Minneapolis, MN). HeLa nuclear extracts were prepared as described (12,21). The protein concentration was determined by Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Construction and expression of recombinant hMYH
The hMYH cDNA, ∼1.6 kb in length, was amplified from a HeLa cDNA library (Invitrogen, Carlsbad, CA) by PCR using gene-specific primers Chang 262 (5′-GCTATCATTAATATGACACCGCTCGTCTCCCGC-3′) and Chang 227 (5′-GCTAACCTAGGTCACTGGGCTGCACTGTTGAG-3′) and the Expand High Fidelity PCR system (Boheringer Mannheim, Indianapolis, IN). The AsnI–BamHI fragment of the PCR product was subcloned into the pET11a vector at the cloning sites of NdeI and BamHI. The second codon, ACA, of the hMYH coding sequence was silently replaced with ACC to increase the efficiency of translation, because ACA is a rare codon in E.coli. The sequence of the hMYH gene was confirmed by DNA sequencing and was as published (16). The recombinant hMYH protein was expressed under control of a T7 promoter. The expression host, GBE943 [lacIp4000(LacIq)lacZp4008(LacL8)srlC300::Tn10λ–IN(rrD-rrnE)1micA68::Tn10Kan], harboring the λDE3 lysogen was constructed according to the manufacturer’s procedures (Invitrogen). Escherichia coli strain GBE943/DE3 harboring an expression plasmid containing the hMYH cDNA was grown in LB broth containing 50 µg/ml ampicillin at 37°C. Expression of the protein was induced at an A600 of 0.6 by addition to the culture of isopropyl-1-thio-β-d-galactopyranoside to a final concentration of 0.4 mM at 20°C. The cells were harvested 10 h later.
Purification of recombinant hMYH
Recombinant hMYH was purified according to the purification procedure detailed for recombinant Schizosaccharomyces pombe MYH (SpMYH) expressed in E.coli (22). Cells (21 g) were resuspended in 60 ml of buffer T (50 mM Tris–HCl, pH 7.6, 0.1 mM EDTA, 0.5 mM dithiothreitol and 0.1 mM PMSF) and disrupted with 0.1 mm glass beads in a bead beater (Biospec Products, Bartlesville, OK). The cell debris was removed by centrifugation and the supernatant was treated with 5% streptomycin sulfate to remove nucleic acids. After stirring for 45 min the solution was centrifuged and the supernatant was collected as Fraction I. Ammonium sulfate was added to a concentration of 55% (w/v) and the protein was precipitated for 4 h. After centrifugation the protein pellet was resuspended in 25 ml of buffer T and dialyzed twice against 2 l of buffer T for 4 h each. The protein sample was diluted 2-fold with buffer A (20 mM potassium phosphate, pH 7.4, 0.1 mM EDTA, 0.5 mM dithiothreitol and 0.1 mM PMSF) containing 0.05 M KCl as Fraction II and was loaded onto a 30 ml phosphocellulose column, which had been equilibrated with buffer A containing 0.05 M KCl. After washing with 60 ml of buffer A containing 0.05 M KCl, proteins were eluted with a linear gradient of KCl (0.05–0.6 M) in buffer A. Fractions that had hMYH adenine glycosylase activity on a DNA substrate containing an A/8-oxoG mismatch were pooled as Fraction III. Fraction III was then loaded onto a 20 ml hydroxyapatite column equilibrated with buffer B (0.01 M potassium phosphate, pH 7.4, 10 mM KCl, 0.1 mM EDTA, 0.5 mM dithiothreitol and 0.1 mM PMSF). The flow-through and the early elution fractions were pooled and dialyzed against 1 l of buffer A containing 0.05 M KCl and 10% (v/v) glycerol for 2 h to yield Fraction IV. Fraction IV was then loaded onto a 5 ml heparin–agarose column equilibrated with buffer A containing 0.05 M KCl and 10% glycerol. Proteins were eluted with a linear gradient of KCl (0.05–0.6 M) in buffer A with 10% glycerol. Fractions containing hMYH adenine glycosylase activity were pooled (Fraction V), divided into small aliquots and stored at –80°C.
Western blotting
Cell extracts (10 µg protein) and partially purified recombinant hMYH (230 ng protein in 14 µl) were mixed with 3.5 µl of 5-fold SDS loading dye (155 mM Tris–HCl, pH 6.0, 25% v/v glycerol, 5% SDS, 0.5 mg/ml bromophenol blue and 5% v/v β-mercaptoethanol), fractionated by 10% SDS–PAGE and transferred to a nitrocellulose membrane. In some experiments extracts were allowed to react with DNA substrates (see below), fractionated on an 8% native polyacrylamide gel and transferred to a nitrocellulose membrane. The membranes were allowed to react with affinity purified antibodies against E.coli MutY, hMYH peptides, hAPE1 (C-20; Santa Cruz Biotechnology, Santa Cruz, CA), hPCNA (Ab-1; Calbiochem-Novabiochem, Darmstadt, Germany) and hMSH2 (Ab-2; Calbiochem-Novabiochem). Antibodies against E.coli MutY were raised in rabbits and purified essentially as described previously (14). The α344 and α516 hMYH peptide antibodies against residues 344–361 (FPRKASRKPPREESSATC) and residues 516–535 (CDNFFRSHISTDAHSLNSAA) of hMYH were raised in rabbits and purified by peptide affinity chromatography as described (23). Western blotting was detected with the Enhanced Chemiluminescence (ECL) analysis system (Amersham International) according to the manufacturer’s protocol.
Protein dephosphorylation
Human MYH protein in cell extracts was dephosphorylated with shrimp alkaline phosphatase (SAP) (Boehringer Manheim). Cell extracts (10 µg protein) were treated with 2 U SAP at 25°C for 10 min, after which proteins were fractionated by 10% SDS–PAGE followed by western blotting with the affinity purified antibodies against E.coli MutY. In addition, cell extracts (25 µg protein) were treated with 5 U SAP at 25°C for 10 min while one-fifth of the treated sample (5 µg protein from cell extracts) was used for hMYH binding and glycosylase assays as described below.
Preparation of oligonucleotide substrates
The 19mer oligonucleotide substrates containing the normal base or 8-oxoG were synthesized and purified as previously described (24). The sequences were Chang-68, 5′-CCGAGGAATTAGCCTTCTG-3′, Chang-69, 5′-GCAGAAGGCGAATTCCTCG-3′ and Chang-179, 5′-GCAGAAGGCOAATTCCTCG-3′, where O represents 8-oxoG and the mismatched bases are underlined. Heteroduplexes containing A/G or A/8-oxoG mismatches were constructed by annealing Chang-68 with Chang-69 or Chang-179, respectively. One picomole of annealed duplexes was labeled at the 3′-end of the upper ‘A’ strand as described by Lu et al. (25) and Lu (26). The resulting DNA duplexes were 20 bp in length and were purified by passing them through G-25 Quick-Spin columns (Boehringer Mannheim, Mannheim, Germany).
Assay of hMYH glycosylase activity
The hMYH glycosylase reaction mixture (20 µl) contained 5 µg extract protein or 82 ng partially purified recombinant hMYH, 1.8 fmol DNA substrate containing an A/8-oxoG mismatch labeled at the 3′-end of the A-containing strand, 75 µg/ml BSA, 10 mM Tris–HCl, pH 7.4, 0.5 mM dithiothreitol, 2.25 mM EDTA, 5 µM ZnCl2 and 1.45% glycerol. The reactions were performed at 37°C for 1 h. Unless specified, samples after reactions were lyophilized to dryness, resuspended in 3 µl of formamide dye (90% formamide, 10 mM EDTA, 0.1% xylene cyanol and 0.1% bromophenol blue), heated at 90°C for 3 min, loaded onto 14% polyacrylamide/8.3 M urea sequencing gels and analyzed by autoradiography.
Assay of protein–DNA binding complexes
The reaction mixture for hMYH binding activity on the A/8-oxoG-mismatched DNA substrate was similar to that for the glycosylase assay, except that 20 mM NaCl and 1 µg/ml poly(dI·dC) were added to the reactions. The reactions were carried out at 37°C for 30 min and were supplemented with 1.5 µl of 50% glycerol. The protein–DNA complexes were then resolved on an 8% polyacrylamide gel in 50 mM Tris–borate buffer. The gel was dried and autoradiographed.
Large-scale binding reactions were performed with 40 µg H2009 cell extract or 0.8 µg recombinant hMYH protein and 10 pmol A/8-oxoG-containing DNA substrate in a 50 µl reaction according to the procedure described above, except that 2 µg/ml poly(dI·dC) was used. After electrophoresis the complex was transferred to a nitrocellulose membrane and subjected to western blotting analysis. Alternatively, the gel slices containing the protein–DNA complexes were excised, crushed and soaked in 0.5 ml of 50% phosphate-buffered saline (PBS) solution with 1 mM dithiothreitol and 0.1 mM PMSF overnight at 4°C. The gel pieces were removed with a Spin-X centrifuge filter unit (Costar, Cambridge, MA) and washed with 0.2 ml of 50% PBS solution twice. The filtrate was lyophilized to dryness, dissolved in 28 µl of distilled water and mixed with 7 µl of the 5-fold SDS loading dye. The sample was boiled and fractionated by 10% SDS–PAGE followed by western blotting analysis with the antibody against hMYH peptide (α516).
UV cross-linking and immunoprecipitation
H2009 cell extracts (20 µg protein) were incubated with 18 fmol 3′-end-labeled DNA substrate containing an A/8-oxoG or A/G mismatch in a 20 µl reaction according to the binding assay as described above. After incubating at 37°C for 30 min the reactions were irradiated with a Stratalink UV Crosslinker 1800 (Stratagene, La Jolla, CA) under 254 nm UV light bulbs (8 W) with a distance of 5 cm from the bottom of the 1.5 ml microcentrifuge tube to the UV light bulb. Samples were supplemented with 5 µl of 5-fold SDS loading buffer, boiled for 5 min and then resolved by 10% SDS–PAGE. For immunoprecipitation, 40 µl of protein G–agarose (50% in slurry; Life Technologies, Rockville, MD) was washed twice with 0.5 ml of PBS buffer and incubated with affinity purified hMYH peptide antibody α344 (1 µg/ml) or α516 (2 µg/ml) in 0.1 ml of PBS buffer at 4°C for 2 h with gentle rocking. The beads were blocked with 8% bovine serum albumin at room temperature for 30 min followed by washing with 0.5 ml of PBS buffer three times. The UV irradiated sample was added to the pretreated protein G–agarose bound or not with antibodies and incubated at 4°C for 2 h. The mixtures were washed twice with 800 µl of PBS buffer and the pellets collected after centrifugation at 2500 g for 5 min. SDS sample buffer (40 µl) was added to the pellets, which were then resolved by 10% SDS–PAGE. The gel was dried and autoradiographed.
RESULTS
Native hMYH and recombinant hMYH have different gel mobilities
The 535 residue hMYH protein was expressed in E.coli and partially purified as described in Materials and Methods. This form (Type 1) of hMYH has been shown by Takao et al. to be transported into the mitochondria (17,27), but has been shown by Tsai-Wu et al. to be localized to the nuclei (18). The expression levels of recombinant hMYH from hMYH cDNA cloned into the pET11a expression vector were not very high, which was also observed by other laboratories with different expression systems or fusion protein constructs (17–19), probably because its sequence contains many rare codons for E.coli or it is insoluble and toxic to the bacteria. However, we were able to partially purify hMYH for characterization. The final hMYH protein preparation was analyzed by 10% SDS–PAGE followed by silver staining (Fig. 1A, lane 1) and by western blotting with affinity purified antibodies against E.coli MutY (Fig. 1A, lane 2) and against the hMYH peptide (α516) (Fig. 1A, lane 3). The partially purified hMYH preparation contained other proteins with molecular masses in the range 30–45 kDa, however, a band migrating at ∼60 kDa reacted with both of the antibodies against hMYH peptide (α516) and E.coli MutY.
Figure 1.
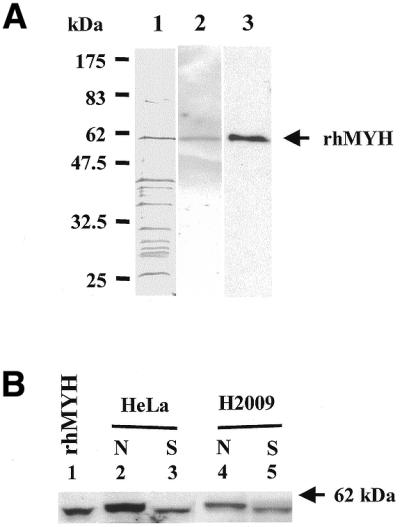
(A) Partially purified recombinant hMYH from E.coli. The partially purified recombinant hMYH (230 ng protein in 14 µl of Fraction V) was analyzed by 10% SDS–PAGE followed by silver staining (lane 1) and by western blotting with affinity purified antibodies against E.coli MutY (lane 2) and against the hMYH peptide (α516) (lane 3). The arrow indicates the position of the recombinant hMYH (rhMYH). (B) Native hMYH is phosphorylated in human cell extracts. Human HeLa nuclear extracts (lanes 2 and 3) and NSCLC H2009 total cell extracts (lanes 4 and 5) (10 µg protein per lane) were separated by SDS–PAGE and hMYH was detected by western blotting with antibodies against E.coli MutY. Samples in lanes 3 and 5 were treated with SAP. N indicates samples without SAP treatment and S indicates SAP-treated samples. Lane 1, the partially purified recombinant hMYH expressed in E.coli (14 µl of Fraction V). The arrow indicates the 62 kDa molecular mass standard (New England Biolabs prestained markers).
Native hMYH, with a molecular mass of ∼60 kDa, was detected in both nuclear extracts of human HeLa cells and total cell extracts of human H2009 cells by western blotting with hMYH antibody (α516) (Fig. 1B, lanes 2 and 4, respectively). The native form of hMYH migrated slightly slower than bacterially expressed recombinant hMYH (Fig. 1B, compare lane 1 with lanes 2 and 4). We show that native hMYH might be modified in human cells because the mobility of native hMYH (Fig. 1B, lanes 2 and 4) shifted to the same position as recombinant hMYH after treatment with SAP (Fig. 1B, lanes 3 and 5).
Native hMYH and recombinant hMYH exhibit differential adenine DNA glycosylase activities on A/G and A/8-oxoG mismatches
To test whether recombinant hMYH expressed in E.coli is active, we assayed its DNA glycosylase activity on A/G- or A/8-oxoG-containing DNA substrates. As shown in Figure 2A, recombinant hMYH has strong glycosylase activity on an A/8-oxoG mismatch but very weak activity on an A/G mismatch (Fig. 2A, compare lane 8 with lane 4). A weak A/G glycosylase activity of recombinant hMYH was also observed in previous reports (17,19). However, H2009 cell extracts exhibited comparable DNA glycosylase activities on both A/G- and A/8-oxoG-containing DNA substrates (Fig. 2A, lanes 3 and 7, respectively), as did purified E.coli MutY protein (Fig. 2A, lanes 2 and 6, respectively). This result is consistent with findings observed for native MYH from human Jurkat cells (13) and calf thymus (14).
Figure 2.

(A) Glycosylase activities of native hMYH (H), recombinant hMYH (R) and E.coli MutY (Y). DNA substrates containing A/G (lanes 1–4) or A/8-oxoG (lanes 5–8) were incubated with their respective enzymes (as marked at the top of each lane) at 37°C for 1 h. Reactions in lanes 3 and 7 contained 5 µg H2009 cell extract protein, those in lanes 4 and 8 contained 82 ng of recombinant hMYH and those in lanes 2 and 6 contained 72 fmol purified MutY. Lanes 1 and 5 were DNA substrates alone. (B) Dephosphorylation of native hMYH reduced its glycosylase activity. Cell extracts of H2009 (5 µg protein) were applied to the hMYH glycosylase assay with DNA substrates containing an A/G (lanes 1–3) or A/8-oxoG (lanes 4–6) mismatch. Extracts used in lanes 1 and 4 were not treated with SAP, those used in lanes 2 and 5 were treated with SAP and those in lanes 3 and 6 were incubated with SAP buffer without SAP. All the DNA samples were fractionated on a 14% polyacrylamide/8.3 M urea sequencing gel. Arrows mark the intact DNA substrate (I) and the nicked product (N).
Because bacterially expressed recombinant hMYH lacks the proper protein modifications whereas native hMYH in human cell extracts has these, protein modifications may contribute to the substrate discrepancy between native hMYH and recombinant hMYH. To test whether phosphorylation of hMYH has any effect on glycosylase activity, we assayed native hMYH glycosylase activity after it had been treated with SAP. About 40% of A/G- and A/8-oxoG-containing DNA substrates were cleaved by the untreated cell extracts (Fig. 2B, lanes 1, 3, 4 and 6), however, only 0.8 and 10% of A/G- and A/8-oxoG-containing DNA, respectively, were cleaved by the SAP-treated extracts (Fig. 2B, lanes 2 and 5). The extent of reduction in hMYH glycosylase activity on an A/G mismatch by phosphatase treatment was greater than that on an A/8-oxoG mismatch. In control reactions, E.coli MutY glycosylase activity was not affected by SAP treatment (data not shown) and the buffer used in the SAP treatment had no effect on hMYH glycosylase activity (Fig. 2B, lanes 3 and 6). These findings suggest that protein phosphorylation does affect hMYH glycosylase activity.
Native hMYH and recombinant hMYH have differential binding affinities for A/G and A/8-oxoG mismatches
Recombinant hMYH could form a protein–DNA complex with the DNA substrate containing an A/8-oxoG mismatch (Fig. 3A, lane 8), however, binding to an A/G mismatch substrate was very weak (Fig. 3A, lane 4). This recombinant hMYH binding specificity is similar to a previous report on recombinant hMYH expressed in an in vitro transcription–translation system (17).
Figure 3.
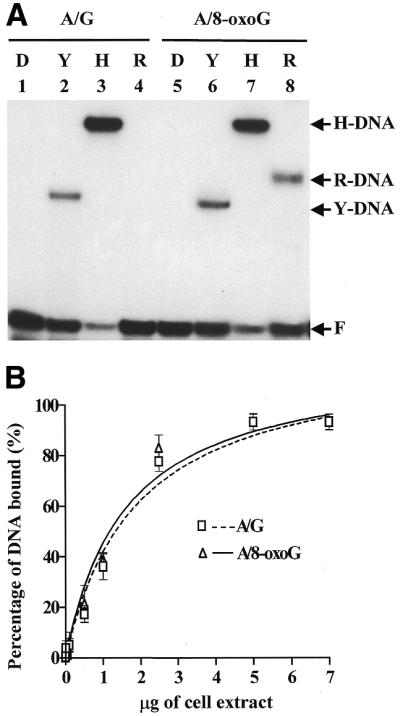
(A) DNA-binding activities of native hMYH (H), recombinant hMYH (R) and E.coli MutY (Y). DNA substrates containing an A/G (lanes 1–4) or A/8-oxoG (lanes 5–8) were incubated with the respective enzymes (as marked at the top of each lane) and fractionated on non-denaturing gels. No protein was added to the reactions in lanes 1 and 5. Lanes 2 and 6, reactions performed with 72 fmol purified MutY; lanes 3 and 7, reactions with 5 µg H2009 cell extract protein; lanes 4 and 8, reactions with 82 ng recombinant hMYH (Fraction V). Arrows indicate the positions of the native hMYH–DNA complex (H-DNA), recombinant hMYH–DNA complex (R-DNA), MutY–DNA complex (Y-DNA) and free DNA substrate (F). (B) Binding affinities of cell extracts on DNA substrates containing A/G and A/8-oxoG mismatches. Different amounts of the cell extracts were incubated with 1.8 fmol DNA substrates containing A/G (marked by squares and a dotted line) or A/8-oxoG (marked by triangles and a solid line) mismatch in the binding assay. The percentages of DNA bound were analyzed with a PhosphorImager and plotted against the amount of added cell extract of H2009. Data were obtained from three independent experiments.
As shown in Figure 3A, H2009 cell extracts could effectively bind to DNA substrates containing A/G or A/8-oxoG mismatches (lanes 3 and 7, respectively). Figure 3B further demonstrates that the extent of binding of H2009 cell extracts with substrates containing A/G or A/8-oxoG mismatches was dependent upon the amount of cell extract. Moreover, Figure 3B demonstrates that hMYH in human cell extracts had similar binding affinities for A/G and A/8-oxoG mismatches. Therefore, native hMYH in human cell extracts has a differential affinity for an A/G mismatch from recombinant hMYH. Dephosphorylation of hMYH in cell extracts had no effect on hMYH binding affinity for both A/G and A/8-oxoG mismatches (data not shown) although it could affect hMYH glycosylase activity (Fig. 2B). To our surprise, the protein–DNA complex (H-DNA complex) formed by the cell extracts (Fig. 3A, lane 7) migrated much slower than that formed by recombinant hMYH and A/8-oxoG-containing DNA (R-DNA complex) (Fig. 3A, lane 8).
Because recombinant hMYH is not pure and cell extracts contain other mismatch-binding proteins, it is possible that the R-DNA and H-DNA complexes may not be derived from hMYH itself. Other repair enzymes, such as the hMSH2/hMSH6 heterodimer, may bind to A/8-oxoG-containing DNA, as do the yeast homologs (15). To determine whether hMYH is present in the binding complexes with an A/8-oxoG-containing DNA in lanes 7 and 8 of Figure 3A, gel slices were excised from the lanes containing unlabeled DNA substrate (Fig. 4A, lanes 2 and 4) by alignment with complex bands containing labeled DNA substrates (Fig. 4A, lanes 1 and 3). Proteins were eluted from the gel slices and resolved by 10% SDS–PAGE. Western blotting with antibody against hMYH peptide (α516) detected hMYH in the eluted samples (Fig. 4B, lanes 1 and 2). Consistent with the result in Figure 1B, Figure 4B also shows that recombinant hMYH (Fig. 4B, lane 2) migrated slightly faster on SDS–PAGE than native hMYH (Fig. 4B, lane 1). Thus, hMYH is present in both the R-DNA and H-DNA complexes.
Figure 4.

hMYH is present in the protein–DNA complexes. (A) Gel retardation assay. Lanes 1 and 3, DNA binding assay performed with H2009 cell extracts or recombinant hMYH and 32P-labeled A/8-oxoG-containing DNA as described in lanes 7 and 8 of Figure 3A, respectively. The large-scale binding reactions were performed with 40 µg H2009 cell extract (lane 2) or 0.8 µg recombinant hMYH (lane 4) and 10 pmol unlabeled A/8-oxoG-containing DNA substrate in 50 µl reactions. Gel slices were excised from lanes 2 and 4 (boxed) by alignment with the complexes in lanes 1 and 3, presumably containing the H-DNA and R-DNA complexes, respectively. Arrows indicate the positions of the free DNA substrate (F), native hMYH–DNA complex (H-DNA) and recombinant hMYH–DNA complex (R-DNA). (B) Western blotting analysis of eluted protein–DNA complexes. Protein–DNA complexes were eluted from the gel pieces in lanes 2 and 4 of (A) and fractionated by 10% SDS–PAGE followed by western blotting with antibody against hMYH (α516). Lane 1 indicates native hMYH and lane 2 is recombinant hMYH. Molecular mass standards (New England Biolabs prestained markers) were run in a parallel lane. Arrows mark the positions of native hMYH and recombinant hMYH (rhMYH).
hMYH is the major protein recognizing A/8-oxoG mismatches in human cell extracts
Further experiments were designed to determine whether the A/8-oxoG-containing DNA substrate was primarily bound by hMYH. The complexes formed by extracts of H2009 cells and a labeled 20 bp DNA substrate containing an A/8-oxoG mismatch were cross-linked by UV irradiation and resolved by 10% SDS–PAGE. To evaluate the method, we analyzed purified E.coli MutY (∼39 kDa) with a 20 bp DNA substrate containing an A/8-oxoG mismatch and detected one complex with a size of 52 kDa after 5 min UV irradiation (data not shown). No cross-linked product was detected with human cell extracts and an A/8-oxoG-containing DNA when UV irradiation was omitted (Fig. 5, lane 1), implying there was no protein covalently bound to the DNA substrate under binding conditions. The cross-linked protein–DNA complex could be detected after 0.5 min UV irradiation and its amount increased as exposure time to UV irradiation increased (Fig. 5A, lanes 2–7). The major UV cross-linked protein–DNA product had a molecular mass of ∼73 kDa, which is consistent with the sum of the molecular masses of hMYH protein (∼60 kDa) and the double-stranded 20 bp DNA (∼13 kDa). With a longer exposure time to UV irradiation two minor complexes with molecular masses of ∼75 and ∼60 kDa (>2 min UV, Fig. 5A, lanes 5–7) and another complex with a molecular mass of ∼180 kDa (>40 min UV, data not shown) were also detected. The formation of 32P-labeled protein–DNA complexes could be titrated out by adding a 100-fold excess of unlabeled DNA substrate containing an A/8-oxoG mismatch (Fig. 5A, lane 8) and were also abolished by proteinase K treatment (Fig. 5A, lane 9), indicating that the cross-linked complexes were composed of proteins and DNA. Similarly, native hMYH could be cross-linked with A/G-containing DNA substrate by 5 min UV irradiation (Fig. 5A, lane 11).
Figure 5.
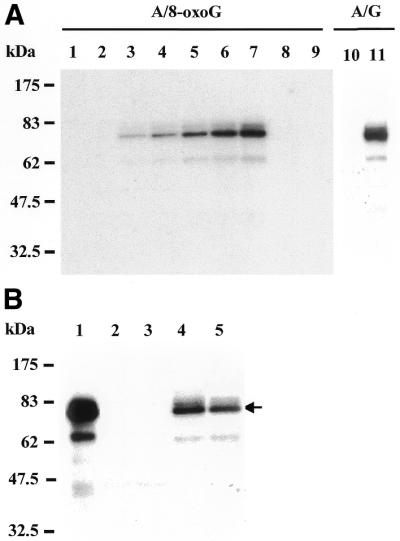
hMYH is the primary protein recognizing A/8-oxoG and A/G mismatches in human cell extracts. (A) UV cross-linking of human H2009 cell extracts with DNA containing A/8-oxoG (lanes 1–9) or A/G (lanes 10 and 11). Lanes 1–7, time course of UV irradiation. Lane 1, the reaction sample with A/8-oxoG-containing DNA was not UV irradiated. Lanes 2–7, samples were irradiated with UV light for 0.17, 0.5, 1, 2, 3 and 5 min, respectively. Lane 8, as lane 7 but 100-fold unlabeled A/8-oxoG-containing DNA substrate was added. Lane 9, as lane 7 but the sample was treated with 3 µg proteinase K for 10 min at room temperature. Lane 10, the reaction sample with A/G-containing DNA was not UV irradiated. Lane 11, the extract with A/G-containing DNA was irradiated with UV light for 5 min. Lanes 10 and 11 are from a non-concurrent experiment. The results are not intended to compare the intensity of hMYH cross-links to A/G- and A/8-oxoG-containing DNA substrates. Molecular mass standards (New England Biolabs prestained markers) were run in a parallel lane. (B) Human MYH is cross-linked to the A/8-oxoG-containing DNA. The UV cross-linked protein–DNA complexes were immunoprecipitated by antibodies against hMYH peptides. Lane 1, the reaction sample was irradiated with UV light for 5 min and was similar to lane 7 in (A) but without the immunoprecipitation procedures. Lane 2, immunoprecipitate with α344 antibodies from an unirradiated sample. Lane 3, immunoprecipitate from a 5 min UV irradiated sample but with no antibody added. Lane 4, immunoprecipitate with α344 antibodies from a 5 min UV irradiated sample. Lane 5, immunoprecipitate with α516 antibodies from a 5 min UV irradiated sample. The arrow marks the position of the major protein–DNA complex. Molecular mass standards (New England Biolabs prestained markers) were run in a parallel lane.
To confirm that hMYH is indeed the major protein binding to DNA substrates containing an A/8-oxoG mismatch the UV cross-linked protein–DNA complexes were immunoprecipitated by antibodies against human hMYH peptides (α344 or α516). As shown in Figure 5B, the 73 kDa protein–DNA complex could be immunoprecipitated by hMYH antibodies α344 (Fig. 5B, lane 4) and α516 (Fig. 5B, lane 5). The 73 kDa binding complex was not found in the immunoprecipitants from reactions without either UV irradiation or added antibodies (Fig. 5B, lanes 2 and 3, respectively). Taken together, our results indicate that hMYH is not only present in the H-DNA binding complex in Figure 3A (lane 7) but also is the primary protein recognizing the 20 bp DNA substrate containing an A/8-oxoG mismatch.
Determining putative hMYH-interacting proteins in the protein–DNA binding complex
Two possibilities may explain the mobility differences between the two complexes of A/8-oxoG-containing DNA with native and recombinant hMYH. First, native hMYH from cell extracts, but not recombinant hMYH from bacteria, may be post-translationally modified. We have ruled out an effect of phosphorylation because the complex of phosphatase-treated native hMYH and DNA migrated to the same position as the untreated protein (data not shown). The effects of other types of modification require further testing.
Second, the H-DNA complex may be derived from hMYH complexed with its associated protein(s). Recently we showed that hMYH physically interacts with human AP endonuclease I (hAPE1), proliferating cell nuclear antigen (PCNA) and replication protein A (RPA) by co-immunoprecipitation and affinity binding to glutathione S-transferase (GST)–hMYH fusion protein (28). To investigate whether these hMYH-interacting proteins are present in the H-DNA complex we performed a gel shift assay with cell extracts and an A/8-oxoG-containing DNA, transferred the complex to a nitrocellulose membrane and then applied western blotting with antibodies against hMYH (α516), hAPE1, PCNA and hMSH2 (Fig. 6, lanes 2–5, respectively). A similar binding reaction with labeled DNA substrates was run concurrently and an autoradiogram is shown in lane 1 of Figure 6. The western blots and the autoradiogram were aligned with Rainbow protein markers (Amersham Pharmacia Biotech). As shown in Figure 6, hMYH protein detected by western blotting (lane 2) co-migrated with the H-DNA complex (lane 1) on a non-denaturing polyacrylamide gel. The majority of hAPE1 also co-migrated with the H-DNA complex (Fig. 6, lane 3). Another weak hAPE1 band that migrated faster than the H-DNA complex may be derived from free hAPE1, hAPE1–DNA or hAPE1 complexed with other proteins. However, PCNA and hMSH2 migrated faster or slower, respectively, than did the H-DNA complex (Fig. 6, lanes 4 and 5). Our results suggest that the H-DNA complex contains hMYH and hAPE1 but not PCNA and hMSH2. Thus, hAPE1 may interact directly with hMYH in the base excision repair process and contribute to the slower migration of the protein–DNA complex formed by human cell extracts.
Figure 6.
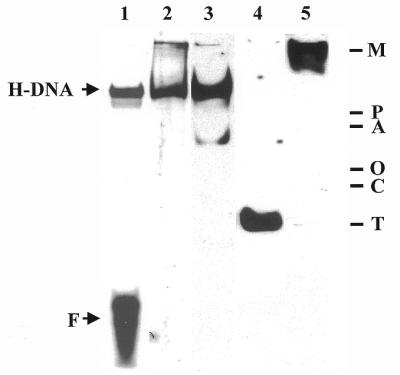
Western blotting of proteins fractionated on a native gel. Lane 1 is the DNA binding reaction with H2009 cell extract and labeled A/8-oxoG-containing DNA and is from an autoradiogram. Samples in lanes 2–5 were then transferred to a nitrocellulose membrane and subjected to western blotting analyses using antibodies against hMYH (α516, lane 2), hAPE1 (lane 3), PCNA (lane 4) and hMSH2 (lane 5). Arrows indicate the positions of the native hMYH–DNA complex (H-DNA) and free DNA (F). The Rainbow protein markers used to align the western blots with the autoradiogram are marked: M, 220 kDa myosin; P, 184 kDa phosphorylase; A, 66 kDa albumin; O, 45 kDa ovalbumin; C, 30 kDa carbonic anhydrase; T, 20 kDa trypsin inhibitor.
DISCUSSION
In this paper we compare recombinant hMYH expressed in E.coli with native hMYH. Our results on the Type 1 form (residues 1–535) of recombinant hMYH are consistent with previous findings on this protein expressed in E.coli and by an in vitro transcription–translation system (17–19). According to Takao et al. (17) Type 1 hMYH is transported into the mitochondria, however, Tsai-Wu et al. (18) showed that Type 1 is localized in the nuclei. We have identified A/G- and A/8-oxoG-specific adenine glycosylase activities in human and calf nuclear extracts (12,14) as well as in calf mitochondria (29). Both nuclear extracts and total cell extracts contain one major form of hMYH with a molecular mass of ∼60 kDa (Fig. 1B), suggesting that the nuclear form of hMYH is dominant in human cells. As shown in Figure 5A, in the cross-linking experiments the 73 kDa hMYH–DNA complex is the major form and its molecular mass is consistent with the sum of the molecular masses of hMYH protein (∼60 kDa) and the double-stranded 20 bp DNA (∼13 kDa).
Recombinant and native hMYH differ in several respects. First, native hMYH protein in human cell extracts, but not bacterially expressed hMYH, is phosphorylated. As shown in Figures 1B and 4B, native hMYH migrated slightly slower than recombinant hMYH on SDS–PAGE. Dephosphorylation of hMYH protein from human cell extracts by phosphatases shifted migration of native hMYH on SDS–PAGE to the same position as recombinant hMYH (Fig. 1B). Consistent with this result, we found that bacterially expressed hMYH could be phosphorylated with [γ-32P]ATP in vitro by several protein kinases (Y.Gu and A.Lu, unpublished data). Secondly, recombinant hMYH expressed in bacteria has much weaker glycosylase and substrate-binding activities on an A/G mismatch than does native hMYH. SAP-treated extracts had only 2 and 25% of the hMYH glycosylase activity on A/G and A/8-oxoG mismatches, respectively, of the untreated extracts. However, dephosphorylation of native hMYH protein did not alter its DNA-binding activity or gel mobility of the protein–DNA complex. Therefore, protein phosphorylation may modulate hMYH glycosylase activity and may, in part, account for the preferential reduction in A/G glycosylase activity of bacterially expressed hMYH. Recently Shinmura et al. (20) reported that hMYH expressed in a baculovirus system had efficient binding and glycosylase activities on an A/G-containing DNA at low salt (1–50 mM) concentrations, the same low salt concentrations as used in our hMYH assay reactions. This suggests that hMYH expressed in the baculovirus system may be phosphorylated. When bacterially expressed hMYH was phosphorylated by protein kinase C its binding and glycosylase activities were not significantly enhanced (data not shown). This may be because of the low efficiency of in vitro phosphorylation or because protein kinase C is not the in vivo kinase of hMYH.
Thirdly, the protein–DNA complex of recombinant hMYH migrates much faster than that of native hMYH in a non-denaturing polyacrylamide gel. We have ruled out an effect of phosphorylation, but other types of modification are not excluded. Our results also eliminate the possibility that the protein–DNA complex (H-DNA complex) formed with cell extracts may not be derived from hMYH itself. Figures 4 and 5 demonstrate that hMYH is indeed in the H-DNA complex. Our data support the possibility that the H-DNA complex may contain hMYH and its associated protein(s). Recently we detected physical interactions of hMYH with hAPE1, PCNA and RPA by co-immunoprecipitation and affinity binding to GST–hMYH fusion protein (28). Our results suggest that the H-DNA complex may contain hMYH and hAPE1 but not PCNA and hMSH2. Parker et al. observed that the interaction of PCNA and hMYH was weaker than that of hAPE1 and hMYH (28). The weak interaction between PCNA and hMYH may not be detected under the stringent conditions used in the gel retardation assay. Another possibility is that the interaction of PCNA and hMYH may be weakened in the presence of mismatch-containing DNA.
hAPE1 cleaves the phosphodiester bond on the 3′-side of an AP site to generate a 3′-OH group for DNA polymerases. The presence of hMYH and hAPE1 in the H-DNA complex suggests that upon removal of the mismatched adenines hAPE1 may be recruited to the complex to carry out the next step of DNA repair, so that the cytotoxic and mutagenic AP sites can be completely processed. Our results are consistent with a recent report by Yang et al. that hAPE1 can stimulate mouse MYH glycosylase activity by enhancing MYH–DNA complex formation (30). However, Yang et al. did not observe a MYH–APE1–DNA tertiary complex (30). It is possible that the H-DNA complex may contain other proteins besides hMYH and hAPE1. hAPE1 can also enhance activities of other glycosylases, such as thymine-DNA and uracil-DNA glycosylases (31–33). In contrast to hMYH, direct interactions of hAPE1 with these glycosylases have not been demonstrated and hAPE1 promotes the dissociation of these glycosylases from AP sites.
This study also demonstrates, for the first time, that hMYH is the major protein in human cell extracts recognizing A/G and A/8-oxoG mismatches by UV cross-linking. In addition to the major band formed by hMYH, some other minor bands were observed at longer times of UV irradiation (Fig. 5A). These complexes may be derived from different forms or degraded products of hMYH as well as other proteins. Saccharomyces cerevisiae MSH2/MSH6 heterodimer (MutSα) has been shown to bind to A/8-oxoG mismatches and to be involved in repairing 8-oxoG damage (15). Human MSH6 (hMSH6) has also been shown to cross-link to T/G mismatches upon UV irradiation (34). If the hMSH2/hMSH6 heterodimer functions like the yeast homologs and can bind to an A/8-oxoG mismatch, the UV cross-linked complex of MSH6 with a 20 bp DNA would be expected to have a molecular mass of ∼175 kDa. After 40 min UV irradiation one complex with a molecular mass of ∼180 kDa appears (data not shown), presumably an hMSH6–DNA complex. Consistent with the notion that hMutSα is not the major repair protein for A/8-oxoG mismatches, hMSH2 is not found in the protein–DNA complex on the non-denaturing polyacrylamide gel (Fig. 6). A homolog of E.coli MutY in S.pombe (SpMYH) has been characterized as an adenine DNA glycosylase (22). Schizosaccharomyces pombe SpMYHΔ strains have increased mutation rates (D.Chang, Y.Gu and A.Lu, unpublished data). These results provide supporting evidence that MutY homologs are the major enzymes repairing A/8-oxoG mismatches in S.pombe. However, our conclusion does not minimize the importance of MSH2/MSH6-dependent repair in S.cerevisiae, because S.cerevisiae does not contain a MutY-like protein. Rather, our finding that hMYH is the key protein recognizing and repairing A/8-oxoG mismatches in human cell extracts emphasizes the biological significance of hMYH in defending against the mutagenic effects of 8-oxo lesions.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dr Herbert K.Oie of the National Cancer Institute and National Naval Medical Center, Bethesda, MD, for providing the H2009 NSCLC cell line. We also thank the Cell Culture Center (Minneapolis, MN) for growing the HeLa cells. This work was supported by grant CA78391 from the National Cancer Institute, NIH.
References
- 1.Ames B.N. (1983) Dietary carcinogens and anticarcinogens: oxygen radicals and degenerative diseases. Science, 221, 1256–1264. [DOI] [PubMed] [Google Scholar]
- 2.Ames B.N. (1989) Endogenous oxidative DNA damage, aging and cancer. Free Radic. Res. Commun., 7, 121–128. [DOI] [PubMed] [Google Scholar]
- 3.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 4.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 5.Tchou J. and Grollman,A. (1993) Repair of DNA containing the oxidatively-damaged base, 8-oxoguanine. Mutat. Res., 299, 277–287. [DOI] [PubMed] [Google Scholar]
- 6.Furuichi M., Yoshida,M.C., Oda,H., Tajiri,T., Nakabeppu,Y., Tsuzuki,T. and Sekiguchi,M. (1994) Genomic structure and chromosome location of the human mutT homologue gene MTH1 encoding 8-oxo-dGTPase for prevention of A:T to C:G transversion. Genomics, 24, 485–490. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto K., Toyokuni,S., Kim,W., Ogawa,O., Kakehi,Y., Arao,S., Hiai,H. and Yoshida,O. (1996) Overexpression of human mutT homologue gene messenger RNA in renal-cell carcinoma: evidence of persistent oxidative stress in cancer. Int. J. Cancer, 65, 437–441. [DOI] [PubMed] [Google Scholar]
- 8.Aburatani H., Hippo,Y., Ishida,T., Takashima,R., Matsuba,C., Kodama,T., Takao,M., Yasui,A., Yamamoto,K. and Asano,M. (1997) Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res., 57, 2151–2156. [PubMed] [Google Scholar]
- 9.Arai K., Morishita,K., Shinmura,K., Kohno,T., Kim,S., Nohmi,T., Taniwaki,M., Ohwada,S. and Yokota,J. (1997) Cloning of a human homolog of the yeast Ogg1 gene that is involved in the repair of oxidative DNA damage. Oncogene, 14, 2857–2861. [DOI] [PubMed] [Google Scholar]
- 10.Bessho T., Tano,K., Kasai,H., Ohtsuka,E. and Nishimura,S. (1993) Evidence for two DNA repair enzymes for 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) in human cells. J. Biol. Chem., 268, 19416–19421. [PubMed] [Google Scholar]
- 11.Radicella J.P., Dherin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomycescerevisiae. Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeh Y., Chang,D., Masin,J. and Lu,A. (1991) Two nicking enzyme systems specific for mismatch-containing DNA in nuclear extracts from human cells. J. Biol. Chem., 266, 6480–6484. [PubMed] [Google Scholar]
- 13.Ohtsubo T., Nishioka,K., Imaiso,Y., Iwai,S., Shimokawa,H., Oda,H., Fujiwara,T. and Nakabeppu,Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res., 28, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGoldrick J.P., Yeh,Y.C., Solomon,M., Essigmann,J.M. and Lu,A.L. (1995) Characterization of a mammalian homolog of the Escherichiacoli MutY mismatch repair protein. Mol. Cell. Biol., 15, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ni T.T., Marsischky,G.T. and Kolodner,R.D. (1999) MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell, 4, 439–444. [DOI] [PubMed] [Google Scholar]
- 16.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.H., Wei,Y.F. and Miller,J.H. (1996) Cloning and sequencing a human homolog (hMYH) of the EscherichiacolimutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol ., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takao M., Zhang,Q.M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsia-Wu J.J., Su,H.T., Wu,Y.L., Hsu,S.M. and Wu,C.H.H. (2000) Nuclear localization of the human mutY homologue hMYH. J. Cell. Biochem., 77, 666–677. [PubMed] [Google Scholar]
- 19.Slupska M.M., Luther,W.M., Chiang,J.H., Yang,H. and Miller,J.H. (1999) Functional expression of hMYH, a human homolog of the EscherichiacoliMutY protein. J. Bacteriol ., 181, 6210–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinmura K., Yamaguchi,S., Saitoh,T., Takeuchi-Sasaki,M., Kim,S.R., Nohmi,T. and Yokota,J. (2000) Adenine excisional repair function of MYH protein on the adenine:8-hydroxyguanine base pair in double-stranded DNA. Nucleic Acids Res., 28, 4912–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu A.L. and Fawcett,W.P. (1998) Characterization of the recombinant MutY homolog, an adenine DNA glycosylase, from yeast Schizosaccharomycespombe. J. Biol. Chem., 273, 25098–25105. [DOI] [PubMed] [Google Scholar]
- 23.Fuller S.A., Takahashi,M. and Hurrell,J.G.R. (1995) Purification of monoclonal antibodies. In Ausubel,F.M., Brent,R., Kingston,R., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (eds) Current Protocols In Molecular Biology. John Wiley & Sons, New York, Vol. 2, pp. 11.11.2–11.11.4.
- 24.Lu A.L., Yuen,D.S. and Cillo,J. (1996) Catalytic mechanism and DNA substrate recognition of Escherichiacoli MutY protein. J. Biol. Chem., 271, 24138–24143. [DOI] [PubMed] [Google Scholar]
- 25.Lu A.L., Tsai-Wu,J.J. and Cillo,J. (1995) DNA determinants and substrate specificities of Escherichia coli MutY. J. Biol. Chem., 270, 23582–23588. [DOI] [PubMed] [Google Scholar]
- 26.Lu A.L. (2000) Repair of A/G and A/8-oxoG mismatches by MutY adenine DNA glycosylase. In Vaughan,P. (ed.) DNA Repair Protocols. Humana Press, Totowa, NJ, Vol. 152, pp. 3–16. [DOI] [PubMed]
- 27.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker A., Gu,Y., Mahoney,W., Lee,S.-H., Singh,K.K. and Lu,A.L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long-patch DNA base excision repair. J. Biol. Chem., 276, 5547–5555. [DOI] [PubMed] [Google Scholar]
- 29.Parker A., Gu,Y. and Lu,A.L. (2000) Purification and characterization of a mammalian homolog of Escherichia coli MutY mismatch repair protein from calf liver mitochondria. Nucleic Acids Res., 28, 3206–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H., Clendenin,W.M., Wong,D., Demple,B., Slupska,M.M., Chiang,J.H. and Miller,J.H. (2001) Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res., 29, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bharati S., Krokan,H.E., Kristiansen,L., Otterlei,M. and Slupphaug,G. (1998) Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res., 26, 4953–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parikh S.S., Mol,C.D., Slupphaug,G., Bharati,S., Krokan,H.E. and Tainer,J.A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J., 17, 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waters T., Gallinari,P., Jiricny,J. and Swann,P. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem., 274, 67–74. [DOI] [PubMed] [Google Scholar]
- 34.Iaccarino I., Marra,G., Palombo,F. and Jiricny,J. (1998) hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSα. EMBO J., 17, 2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]