

Abstract
Multidrug resistance against the existing antibiotics is one of the most challenging threats across the globe. Antimicrobial peptides (AMPs), in this regard, are considered to be one of the effective alternatives that can overcome bacterial resistance. MSI-594, a 24-residue linear alpha-helical cationic AMP, has been shown to function via carpet mechanism to disrupt the bacterial membrane systems. To better understand the role of lipid composition on the function of MSI-594, in the present study, eight different model membrane systems have been studied using accelerated molecular dynamics (aMD) simulation. The simulated results are helpful in discriminating the particular effects of cationic MSI-594 against zwitterionic POPC, anionic POPG and POPS, and neutral POPE lipid moieties. Additionally, the effects of various heterogeneous POPC/POPG (7:3), POPC/POPS (7:3), and POPG/POPE (1:3 and 3:1) bilayer systems on the dynamic interaction of MSI-594 have also been investigated. The effect on the lipid bilayer due to interaction with the peptide is characterized by lipid acyl-chain order, membrane thickness, as well as acyl-chain dynamics. Our simulation results show that the lipid composition affects the membrane interaction of MSI-594 suggesting that membrane selectivity is crucial to its mechanism of action. The resullts reported in this study are helpful to obtain accurate atomistic-level information governing MSI-594 and its membrane disruptive antimicrobial mechanism of action, as well as to design next generation potent antimicrobial peptides.
Graphical Abstract
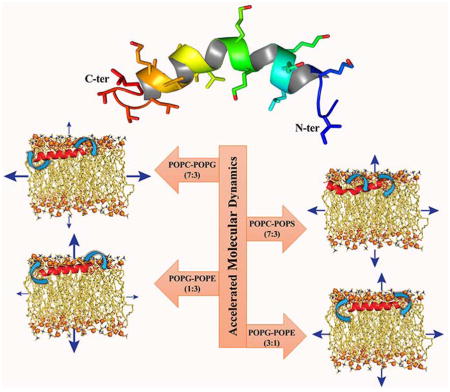
Introduction
The evolution of multidrug-resistant bacteria is one of the most challenging issues in the medical sciences that render the modern antibiotics ineffective at a global level. Additionally, the scarcity of antibiotics that can combat infectious diseases is also acknowledged publicly with increased incidence of untreatable infections. As an alternative, the use of antimicrobial peptides (AMPs) as therapeutic agents could potentially bridge the gap between small molecules and natural antibiotics. Therefore, AMPs can cover the immediate need for potential alternatives for effective treatment of infections. AMPs are small peptide fragments that are present in organisms as part of their natural defence mechanism. The mechanism of action of antibiotics includes their interaction with different protein receptors, whereas AMPs mainly interact with the membrane lipids and are responsible for causing disruption in membrane integrity.1 The lipid bilayer is the fundamental constituent of all natural cell membranes. It has been hypothesized that the mode of action of antimicrobial peptides possesses minimal possibility of triggering the evolution of new resistance, as changing the cell membrane composition and topology is an energetically expensive event.2 Notably, AMPs have the ability to incisively interact with the lipid components in the bacterial membrane with various established models like barrel-stave, toroidal pore, and carpet model.3, 4 Both hydrophobic and cationic residues are present in typical AMPs, which supports the interaction with the inner cell membrane architecture, and further leads to disruption of the cell membrane.5
MSI-5946 is an amphipathic α-helical peptide comprising of 24 amino acids that was originally designed and synthesized by Genaera Corporation. It is a hybrid of MSI-78, an analogue of magainin-2 and melittin. Magainin is a naturally occurring AMP found in the African clawed frog,7 and melittin is a haemolytic peptide from bee venom8 that stimulates phospholipase A2. These peptide fragments are mainly rich in lysine residues, which facilitate the cell lysing mechanism. Typically, different physicochemical properties such as the net charge,9 hydrophobic moments,10 helical content,11, 12 and the angle delimited by the polar/apolar faces,13 play a significant role in the interaction of AMPs with the membrane. These properties are mainly taken into consideration in the design of antimicrobial peptides for the purpose of improving their efficacy. For example, according to Hodges et al.,14 an increase in hydrophobicity may lead to a gain in the haemolytic activity of the peptide.
MSI-594 has been previously reported as a potent antimicrobial peptide with a higher degree of membrane interaction.15 Through solid-state nuclear magnetic resonance (NMR) spectroscopy,16 differential scanning calorimetry (DSC),17 and 31P NMR spectroscopy,18 it was elucidated that MSI-594 adopts α-helical conformation and aligns itself into a parallel orientation in the lipid bilayer. For a better understanding of its interaction with Gram-negative bacteria, NMR spectroscopy was carried out in the presence of lipopolysaccharide (LPS) micelles.15 It is worth mentioning that LPS is the major component of the outer membrane of Gram-negative bacteria. Interestingly, MSI-594 adopts a helical hairpin or helix-loop-helix conformation in LPS micelles. It was determined that the hydrophobic hub, consisting of Phe5, Ile2, Ile13, Leu17, and Leu20, was mainly responsible for the conformational stability of peptide, thereby causing perturbation to the bacterial outer membrane.16 It has also been reported that there exists a close contact between the LPS and the aromatic ring of Phe5 and the side-chain methyl groups of Ile and Leu residues, which governs the interaction phenomenon. Owing to the contribution of these amino acids, MSI-594 interacts with the lipid tails in the hydrophobic core of the bilayer and perturbs the membrane integrity. Importantly, experiments with these inferences have shown that MSI-594 employs a detergent-type mechanism for membrane fragmentation followed by cell death.6
Since the inception of AMPs, research efforts have focused on biophysical studies that can elucidate the mechanism of interaction between the antimicrobial peptide and bacterial membrane. As the AMPs specifically interact with lipid components of the membrane, it becomes inevitable to understand the exact mechanism behind the interaction at the atomic level. However, despite the efforts made to date, structural insights into the antimicrobial activity of MSI-594 are still far from being sufficient. In an attempt to gain better insights, we have explored in this study the interaction of MSI-594 with different lipid systems to trace their probable influence towards membrane fluidity. We also have attempted to elucidate the dynamic behavior of each lipid membrane. Notably, we have modelled zwitterionic POPC, anionic POPG and POPS, and neutral POPE lipid moieties in various ratios to mimic biological membrane systems. We explicate the molecular mechanism behind the interaction between MSI-594 and different membrane systems by emulating the Gram-negative, Gram-positive, and mammalian membrane (Table 1) through accelerated molecular dynamics (aMD) simulations. Our study elucidates that the helical structure of the MSI-594 peptide plays a significant role in translocating through the LPS layer in Gram-negative bacteria.19 As this AMP acts by membrane-lytic activity, there is a high probability of disrupting the host cell membrane as well. Hence, studying the interaction of MSI-594 with the particular lipid species is imperative, as the lipid composition is not the same for eukaryotic and prokaryotic cells. Thus, we have focused on mimicking the bacterial membrane along with the mammalian cell membrane to compare the lipid-peptide interactions. We have also carried out MD simulations for each lipid bilayer without the peptide as a control for a comparative analysis. Furthermore, with this information, the next generation antimicrobial peptides can be designed to be less effective toward mammalian membrane, while affecting the bacterial membrane with higher specificity.
Table 1.
Details of bilayer membrane systems employed for accelerated molecular dynamics (aMD) simulation.
| Systems | No. of Lipids | Total No. of Atoms | System mimic | Reference | |
|---|---|---|---|---|---|
| POPC | 41 | 13168 | Outer leaflet of plasma membrane | 24 | |
| POPG | 68 | 20176 | Anionic membrane system | Control | |
| POPE | 47 | 13894 | Neutral membrane system. | Control | |
| POPS | 46 | 13430 | Inner leaflet of plasma membrane | 24 | |
| POPC-POPG (7:3) | 50 | POPC: 35 | 14068 | Inner leaflet of bacterial membrane | 26, 34 |
| POPG: 15 | |||||
| POPC-POPS (7:3) | 50 | POPC: 35 | 14041 | Bacterial membrane. | 26 |
| POPS: 15 | |||||
| POPG-POPE (1:3) | 48 | POPG: 12 | 12687 | Gram-negative | 27, 35 |
| POPE: 36 | |||||
| POPG-POPE (3:1) | 48 | POPG: 36 | 12918 | Gram-positive | 27, 36 |
| POPE: 12 | |||||
Experimental
Peptide and Model Membrane Selection
The lysine-rich 24-amino acid antimicrobial MSI-594 peptide (GIGKFLKKAKKGIGAVLKVLTTG), which is derived from magainin and melittin, has been adopted for computational studies.20 MSI-594 is a cationic antimicrobial peptide showing a high degree of activity against bacteria, fungi, and viruses. Herpes simplex virus plaque forming units get significantly reduced by the application of this peptide.21 MSI-594 adopts a helical hairpin structure in LPS environment,22 and Phe5 residue of MSI-594 plays an important role in inter-helical interactions.23
We have constructed a total of eight lipid bilayer systems for our theoretical study. Zwitterionic 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids were used to model the outer leaflet of the mammalian membrane.24 Anionic 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS) lipids were also used to model the inner leaflet of the mammalian membrane, as the negatively charged phosphatidylserine (PS) lipid molecule contributes almost 96% of the lipid composition in the inner leaflet of mammalian cell membranes.25 A mixture with a 7:3 molar ratio of zwitterionic POPC to anionic 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) lipids mimicking the bacterial inner membrane26 and a combination with a 7:3 molar ratio of zwitterionic POPC to anionic POPS lipids were used to model the bacterial membranes.26 To model the membranes of the Gram-positive bacterial membrane, such as Bacillus subtilis, we simulated a 3:1 ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), while a 1:3 ratio of POPG/POPE was used to model the inner membranes of Gram-negative bacteria, such as Escherichia coli.27
Construction of Membrane Systems
Membrane Builder module of CHARMM-GUI28 server was utilized for the preparation of peptide-embedded membrane system in the present work. The coordinates of MSI-594 were adopted from the work of Porcelli et al.23 The initial orientation of the peptide in the membrane system was analyzed based on OPM server prediction.29 Orientations of different peptides in membrane models using OPM server have been explicitly described in previous studies.30 The membrane normal is assumed to be parallel to the Z-axis, and its center is located at Z = 0. The initial positioning of the peptide in the membrane system was made along the Z-axis by a 16 Å translation. The membrane geometry was fixed to 20 Å and 40 Å along the X-axis and Y-axis, respectively. Water thickness of 20 Å is maintained on both sides of lipid bilayer leaflet in rectangular box type arrangement for all systems. System neutralisation was dictated with the addition of counter sodium ions (Na+) or chloride ions (Cl−), in which the positional arrangement was determined based on the Monte Carlo method. The input geometry for the heterogeneous membrane system was specified with lipid moieties as per the membrane system ratio in both upper and lower leaflets. The system temperature was maintained at 300 K in all model membranes. The input files generated were further processed in NAMD31 for energy minimization and production run. All simulations were performed using Charmm36 force field.32
Computational Methodology
The three-dimensional structure of MSI-594 utilized for the molecular dynamics simulation was determined in detergent micelles using solution state NMR spectroscopy and its orientation was determined in lipid bilayers using solid-state NMR spectroscopy.23 The atomic-resolution three-dimensional structure determined in a lipid environment is well suited for the simulation study. In particular, we have evaluated the dynamicity of different lipid bilayers in the presence of antimicrobial peptide through eight independent 50 ns long accelerated molecular dynamics (aMD) simulations using NAMD program with the CHARMM36 force field. Initially, we have carried out eight 10 ns unbiased all-atom simulations to calculate the average potential energy threshold and the inverse strength boost factor for the dihedral energy, which is required to execute the accelerated simulation. Detailed values of potential energy threshold and dihedral boost used for each system are given in Supporting Information Table S1. The simulation study also highlights the change in peptide kinetics in different lipid environments. For simulating water, the TIP3P model has been used.33 Details of the eight systems are given in Table 1. Of note, the membrane geometry has been kept constant in all the membrane systems by constructing the models with fixed lengths of 20 Å and 40 Å along the x- and y-axes, respectively. The peptide has been placed in the bilayer using replacement method, which also affects the number of lipids in each system owing to the difference among the individual lipid moieties. The odd numbers of lipid moieties are thus attributed to this methodological implication in system setup. Furthermore, it is also noteworthy to mention that the primary objective of the study is to shed light on the difference in nature of the interaction between peptide and model membrane systems. Thus, the representative models are framed to explore such biophysical details.
The isobaric-isothermal (NPT) and periodic boundary conditions were maintained throughout all aMD simulations. Integration time step was fixed to 2 fs for all simulations. Langevin dynamics and Nose-Hoover-Langevin piston were used to retain the temperature at 300 K and the pressure at 1 bar, respectively.37 The cut-off value was set at 10 Å for the calculation of all short-range interactions, while long-range interactions were measured using particle mesh Ewald method.38 RATTLE39 and SETTLE40 algorithms were used to restrain all bond lengths that involve hydrogen atoms.
Accelerated Molecular Dynamics (aMD)
We have used accelerated molecular dynamics (aMD) for all simulations in this study. Accelerated molecular dynamics (aMD) helps to improve sampling by decreasing the energy barrier between two high-energy states, and thereby renders robust conformational sampling with comparatively less CPU time than that of conventional molecular dynamics protocols. Also, aMD provides a boost of potential energy to the system when the potential energy reaches the threshold level. The relation between the modified potential and the real potential is defined according to Equations (1) and (2):
| (1) |
Where V*(r) is the modified potential and ΔV(r) is the boost potential.
| (2) |
Analysis
In the present study, 50 ns all-atom aMD simulation was carried out for eight different model membrane systems in the presence of MSI-594. The simulation data mainly elucidates the dynamics of MSI-594 in heterogeneous membrane systems, which was also compared to previously published reports as well as to control homogeneous membrane systems. To analyse the membrane dynamics in the presence of MSI-594, we have used the membrane plugin analysis tool MEMPLUG in VMD (http://www.ks.uiuc.edu/Research/vmd/).
Area per Lipid
The area per lipid tool analyses the membrane under consideration and calculates the total area per lipid as well as the area per lipid moieties in the modelled membrane systems. This tool examines the membrane as single continuous plane and does not take membrane pore or any other significant empty spaces under consideration. The simulation trajectory assumes to be aligned to the Z-axis. Residue names are defined according to the lipid species present in the membrane. In particular, one lipid, as well as a triad of lipid species, can be calculated simultaneously. In bilayer simulation, both leaflets are analysed and denoted as “0” and “1” for upper and bottom leaflets, respectively. Both leaflets are selected, and the average value is plotted. In our study, 50 frames are analysed per iteration. At constant temperature, the fluctuations in area per lipid are associated with isothermal area compressibility modulus, KA, according to Equation (3):
| (3) |
where kB is Boltzmann’s constant, σ2 is the variance associated with AL, NL is the number of lipids, and the angle brackets denote time and ensemble averages.
Membrane Thickness
The membrane thickness tool measures the distance between two phosphate atoms of two lipid moieties in opposite leaflets considering the middle point between them. The central point is normalised based on the mass density of the selected atom along the normal of the membrane. Variation in membrane thickness gives insights into the membrane perturbation in the presence of the antimicrobial peptide.
SCD Order Parameter
The order parameter analysed the mobility of the C-H bond of the aliphatic lipid tails based on the bond orientation. The value of order parameter (-SCD) reflects on the membrane fluidity and is calculated using Equation (4):
| (4) |
Where θ is the angle between the C-H bond and the bilayer normal. Atom names are selected for the calculations of order parameter for that particular lipid. As the lipids under consideration have different chain assignments, therefore the specific lipid chains are selected for the order parameter calculation. Leaflets are designated as “0” and “1” for an upper and bottom leaflet, respectively.
Root-Mean-Squared Fluctuation (RMSF)
The root-mean-square fluctuation (RMSF) is vital to delineate the local changes that occur in the protein chain due to the change in atomic mobility of its residues. The RMSF for residue i of a protein can be represented according to Equation (5):
| (5) |
Where T is the trajectory time, tref is the reference time, ri is the position of the residue, r′ is the position of atoms in residue i after superposition on the reference, and the average of the square distance of atoms in the residue are designated by the angle brackets.
Results and Discussion
Dynamics of Membrane Models in the presence of MSI-594
The primary interest of this study is based on the motivational work of previous studies that have suggested that the MSI variants exhibit fragmentation of membrane in a detergent-like mechanism.6 In the present study, we focus on the changes in membrane integrity and the conformational changes in the antimicrobial MSI-594 peptide using all-atom accelerated MD simulation studies. The initial stage of the simulation provides information about the changes in orientation of the lipid moieties in the presence of the peptide. The conformational sampling of MSI-594 was processed using conventional MD simulation method for a period of 10 ns, which in turn provides potential energy term of the system (Supporting Information Table S1). Following this, accelerated MD simulations for a period of 50 ns, which are known to provide an accelerated sampling of conformations,41 were performed.
Area per lipid and the membrane thickness are two important parameters that provide structural insights of membrane perturbation with reference to the absence and presence of MSI-594. In this context, we have found that the area per lipid for different membrane model systems increases in the presence of the antimicrobial peptide MSI-594, which is attributed to the steric hindrance caused by the presence of MSI-594 in the bilayer, and the extent is indicative of the influence of this AMP in various bilayer systems (Supporting Information Table S3). It has been previously observed, both experimentally and theoretically, that the area per lipid is much lower for homogeneous lipid bilayer systems in the absence of any peptide.42, 43 Experimental studies have suggested an average value of 63.6 Å for the area per lipid of POPC/POPG (7:3) bilayer.44 Our control simulation in the absence of peptide also shows an average value of 62.74 ± 1.20 Å. However, our simulations show that this value increases up to 72.96 ± 2.67 Å in the presence of MSI-594, indicating that MSI-594 substantially facilitates the dynamicity of individual lipid moieties in this heterogeneous system (Supporting Information Table S3). Comparing POPC and POPG systems, POPC shows relative larger area per lipid with a value of 74.34 ± 2.14 Å compared to 73.72 ± 2.18 Å for POPG. Likewise, for the POPC/POPS (7:3) system, the increase in area per lipid value was up to 71.05 ± 1.89 Å (Supporting Information Table S3). This result shows more perturbation in the membrane in the presence of MSI-594, which is responsible for more fluctuations in lipid acyl chain. In the POPG/POPE (3:1) system, the area per lipid value of 65.22 ± 1.99 Å and 71.65 ± 1.81 Å was obtained in the absence and presence of MSI-594, respectively (Supporting Information Table S3). In contrast, the area per lipid was found to be 67.84 ± 1.72 Å in the case of POPG/POPE (1:3), which indicates that this system is relatively less perturbed compared to POPG/POPE (3:1). Note that POPG/POPE (3:1) and POPG/POPE (1:3) represent the Gram-positive and Gram-negative bacterial membranes, respectively. This behaviour of MSI-594, which shows relative perturbations in different bacterial membranes, also correlates well with previously reported studies.9, 45, 46
To better understand the behaviour of MSI-594 in membrane systems, we have also investigated homogeneous lipid bilayer systems as a control. In single lipid bilayer systems, the area per lipid also changes in the presence of MSI-594, which reflects that the change in area per lipid is maximum in the case of anionic POPG. The reported area per lipid was 56.1 Å under experimental conditions, while it increases up to 73.72 Å in the presence of MSI-594.44 In the case of POPC, POPE, and POPS, the change in area per lipid value is in the range of 5–10 Å. The values reported for POPC, POPE and POPS are 68.4 Å, 64.8 Å and 56 Å, respectively,44 while our simulation infers that the area per lipid increases up to 74.34 Å, 66.92 Å, and 68.71 Å, respectively in the presence of MSI-594 (Supporting Information Figure S2). A graphical plot indicating the fluctuation of area per lipid in the presence of MSI-594 is shown in Figure 1. The changes in the area per lipid throughout the simulation time span are mainly observed for POPG/POPE (1:3) and POPG/POPE (3:1) (Figure 1C, 1D). Importantly, when POPG is present in a lower concentration within the heterogeneous lipid bilayer, the fluctuations in simulation time course is higher. Note that lipid electrostatics and head group size are contributing factors in bilayer property. Generally, area per lipid decreases when acyl chain length of the lipid increases, indicating an increase of van der Waals attraction at a fixed temperature. However, area per lipid differs among the phosphatidylcholine (PC) series and phosphatidylethanolamines (PE) at the same temperature as the head group methylation plays a greater role than acyl chain length.47 The zwitterionic lipid (phosphatidylcholine) has a comparatively larger head group with three methyl groups attached to the N atom. This is further ascribed to the fact that larger head groups tend to stabilise the membrane integrity mediated through electrostatic interaction. According to the results reported by Ramamoorthy and co-workers,48 the cationic AMP interacts more with the anionic lipid POPG compared to zwitterionic or neutral lipid moieties. This is also evident from our simulation, as the interaction between anionic POPG and MSI-594 leads to higher degree of fluctuation of average area per lipid in the bilayer. Overall, these changes in the average area per lipid shed light on the increased permeability of the membrane, which is attributed to the presence of the antimicrobial peptide.
Figure 1.

Area per lipid is estimated for heterogeneous bilayer systems. The average value (black line) is also plotted against simulation time. (A) POPC/POPG 7:3, (B) POPC/POPS 7:3, (C) POPG/POPE 1:3 and (D) POPG/POPE 3:1. POPC, POPS, POPG and POPE are represented by red, purple, green and blue colour, respectively.
Membrane Deformation Due to the Presence of MSI-594
The orientation and mobility of the C-H bond can be measured by characterising the lipid order.49 Biological membranes do not consistently remain in a single homogeneous fluid phase, as they are extremely heterogeneous and display phase transition.50 The parameters such as “lateral” and “orientational” order, and more specifically the trans-gauche isomerization that occurs in the liquid state of the membrane, tend to affect the conformational order of the membrane. Notably, the bilayers display phase transition between the gel-like liquid crystalline state and the highly disordered liquid state.50 These two different phases affect the fluidity and permeability of the membrane, thereby affecting the membrane functions. Thus, order parameter helps to gain insights into the state of the system, which correlates with its function.51 The substantial distortion in lipid bilayer induced by the peptide has a significant impact in its biological function.52
In the completely ordered state, the acyl chain of lipid moieties is aligned at a right angle with the bilayer (modelled system before equilibration state) and have an all-atom extended configuration with -SCD = 0.5. However, upon the successive progression of the simulation, the value becomes -SCD = 0 in the completely disordered state, which will be governed by molecular mechanics in the all-atom simulation.53 Therefore, the higher value of -SCD shows enhanced lipid order. The order parameter also varies depending on the amount of chain deflection during simulation.54 With a view to gain similar acyl chain deflection, SCD values have been calculated for all the lipid species present in different membrane models (Figure 2). The order parameter values show a decaying pattern in the presence of MSI-594. The upper portion of the lipid chain, attached to the phosphate head group, shows an ordered orientation according to the -SCD values of the carbon atoms present in that region. On the other hand, the relative order of head group size follows the order POPC>POPG>POPS> POPE.55
Figure 2.

Lipid order parameter (-SCD) is measured on the basis of POPC, POPS, POPG and POPE lipid fragments in (A) POPC/POPG 7:3, (B) POPC/POPS 7:3, (C) POPG/POPE 1:3 and (D) POPG-POPE 3:1 heterogeneous systems. POPC, POPS, POPG and POPE are represented with red, purple, green and blue respectively.
Likewise, the lower part exhibits a greater extent of disorientation owing to its –SCD (Figure 2). As shown in the figure, the carbon atoms close to the head group of POPG show more ordered orientation in a PG/PE 3:1 mixed bilayer compared to the control POPG bilayer (Figure 2C and 2D; Supporting Information Figure S2). Notably, the anionic lipid (phosphatidylglycerol) also has a larger head group compared to neutral lipid (POPE), owing to the presence of the glycerol group. On the contrary, POPG shows similarly ordered state in PC/PG 7:3 bilayer systems relative to the control system (Figure 2A). Also, the neutral lipid (phosphatidylethanolamine) has only three hydrogens attached to its N atom resulting in the formation of a comparatively smaller head group. This fact aligns with the observation that infers the carbon atoms close to the phosphate head groups in POPG remain ordered in the presence of POPE lipid, thereby rendering more stability to the bilayer. In contrast, the presence of POPC does not impart any change in the order parameter of POPG. It has been previously reported that higher concentration of the lipid moieties with a larger head group brings instability into the bilayer due to electrostatic interactions.56 In contrast, POPE shows a more disordered orientation when present in POPG/POPE (3:1) heterogeneous system. The carbon atoms close to the phosphate head groups of POPE become more disoriented, owing to the presence of POPG lipid, as evident from –SCD values obtained from the POPE control system (Supporting Information Figure S2).
The C10, C16 and C18 carbon atoms show more flexibility in POPS, POPG and POPE acyl chains, respectively. The flexibility of acyl chain of POPC lipid moieties remains almost similar in both the control and mixed bilayer systems. Notably, POPS show a slightly elevated level of ordered state in the presence of POPC in heterogeneous systems. The carbon numbers C9–C11, C16 and C18 are more fluctuating in the case of zwitterionic POPC moieties.
MSI-594 perturbs Membrane Thickness
The composition and distribution of lipid moieties are critical in biological membranes. The presence of hydrophobic tail (acyl chain length), a hydrophilic head group of lipid, and extent of unsaturation in acyl chain are the key players in determining the membrane perturbation in the presence of AMPs.57 This dynamicity of the membrane can be well characterized by evaluating the membrane thickness over the course of the simulation. This interaction between the hydrophobic lipid tail and the protein leads to an alteration in the membrane structure, which results in a vacillating membrane thickness.
The computed membrane thickness varies in accordance with the modification of lipid composition of the studied model systems (Supporting Information Table S3). The heterogeneous bilayer composed of POPG/POPE (1:3) shows a 38.11 Å average membrane thickness in the presence of MSI-594, which is the largest among the heterogeneous membrane model systems under consideration (Figure 3C and Supporting Information Table S3). According to our simulation study, the estimated membrane thickness of POPG/POPE (1:3) is 41.65 Å in the absence of any peptide fragment.20 On the other hand, the PG/PE 3:1 membrane shows a 36.11 Å average thickness over the course of simulation in the presence of MSI-594 (Supporting Information Table S3). Further, it is noteworthy to mention that Gram-negative bacterial membrane is modelled by PG/PE 1:3 lipid composition. Thus, this observation supports the experimentally proved antimicrobial activity of MSI-594.58, 59 Similarly, in the other two heterogeneous bilayer systems, PC/PG 7:3 and PC/PS 7:3, membrane thickness was found to be 36.04 Å and 37.01 Å, respectively (Figure 3A, B), which indicates no significant difference between the two systems.
Figure 3.

Average membrane thickness is computed with respect to simulation time for (A) POPC/POPG 7:3, (B) POPC/POPS 7:3, (C) POPG/POPE 1:3 and (D) POPG/POPE 3:1 bilayer systems.
A significant difference in membrane thickness is observed upon the alteration of anionic lipid POPG to POPS in the model systems (Figure 3B). This result supports the lipid-specific action in the membrane-lytic activity of MSI-594. In homogeneous POPC membrane model, previously reported as well as our simulation study shows the membrane thickness 39.46 Å in the absence of any peptide.20 In our case, the estimated average membrane thickness for the control POPC homogeneous system appears to be 36.48 Å in the presence of MSI-594, which correlates with previous reports. More importantly, POPC is one of the essential lipid moieties present in the outer leaflet of the mammalian membrane. Hence, the comparative lower activity of MSI-594 in POPC bilayer suggests that MSI-594 shows selective activity against bacterial membrane systems. The average calculated membrane thickness for POPG, POPE, and POPS single lipid bilayer systems is 36.25 Å, 38.31 Å and 37.99 Å, respectively (Supporting Information Table S3 and Figure S3). Based on these results, it can be recognized that MSI-594 is more active in POPE and POPS bilayer systems compared to POPG. In other words, the interaction between cationic AMP and anionic POPG might be responsible for stabilizing the membrane integrity. The plots in Figure 3 show that the fluctuation in membrane thickness was reflective up to 11 ns, following which the thickness appears to be converged in the remaining simulation time. Similar convergence is also reflective in the final 10 ns timescales for other systems (Supporting Information Figure S3).
Behaviour of MSI-594 in Different Lipid Environments
The interaction between the AMP and the lipid molecules is crucial to study the dynamicity of the peptide within the membrane. Additionally, understanding the mechanism responsible for the interaction may lead to better insights into the application of MSI-594 as an antimicrobial drug. To characterize the interaction between MSI-594 and the lipid moieties, we have calculated the center of mass (CoM) keeping Gly12 as the key residue. According to a previous report by Porcelli et al.,23 it was highlighted that Gly12-Gly14 segment of MSI-594 is responsible for additional dynamicity in the zwitterionic DPC micelle, thereby increasing the overall flexibility. In order to calculate the CoM, we have focused on the distance-drift between the center of the bilayer membrane and Cα of Gly12 and Cα of the residue that has acquired a maximum bend in the course of simulation time. The increased CoM distance is indicative of peptide drift towards the interface between the aqueous solvent and the phosphate head groups. This observation further renders the hydrophilic or hydrophobic interaction between the peptide and membrane moieties. In other words, the above observation divides the total interaction energy into two parts, viz. electrostatic and van der Waals interactions.
Figure 4 shows the probability histograms for the CoM distances. The CoMs measured for the Cα of Gly12 residue in POPC/POPG (7:3) and POPC/POPS (7:3) heterogeneous bilayers appear to be 34.5 Å and 37 Å, respectively. In both the POPC/POPG (7:3) and POPC/POPS (7:3) bi-layers the maximum bend is observed in Gly15. The COM calculated for the Gly15 residue has got a maximum count for 37 Å and 39 Å in POPC/POPG and POPC/POPS system, respectively. On the other hand, for the homogeneous bilayer systems (POPC, POPG, and POPS), the CoMs calculated for Gly12 residue are 10, 39, and 39 Å, respectively (Supporting Information Figure S4). Notably, a significant change in CoM calculated for Lys8 is observed in POPC homogeneous system, which is near 43 Å. This marked difference between POPC (zwitterionic) and PG/PS (anionic) could be attributed to the physical nature of lipid moieties, which also governs the pattern of interaction between MSI-594 and corresponding lipid moieties. The relative decrease in CoM for heterogeneous bilayers is indicative of membrane stability that is achieved by mixing different lipid moieties. Also, the contribution of the electrostatic interaction mediated through phosphate head groups is contributing to these stability criteria. The marked difference be-tween POPC/POPG (7:3) and POPC/POPS (7:3) further indicates that the hydrophilic interaction mediated by phosphate head groups and dynamic water molecules dominates in the case of POPC/POPS compared to POPC/POPG (Figure 4A, B). With regard to homogeneous systems like zwitterionic system (POPC), the interaction is driven mainly by the hydrophobic interaction between MSI-594 and acyl chain. The other three control bilayers POPE, POPG and POPS, show maximum conformations at 41 Å, 39 Å and 39 Å distances, respectively, which dictates significant hydrophilic interaction between MSI-594 and phosphate head groups as well as contribution from dynamic water molecules. The CoM for POPG/POPE (1:3 and 3:1) shows values of 36.5 Å and 35 Å, thereby dictating nearly similar interaction pattern (Figure 4C, D). In the case of POPG-POPE (1:3) and POPG-POPE (3:1) systems, which mimic Gram-negative and Gram-positive bacterial membrane, respectively, the maximum bend angle is observed for Lys7 residue. The calculated CoM for Lys7 residue for both the system shows maximum count in 40 Å and 39 Å respectively.
Figure 4.

The maximum number of MSI-594 configurations present at a particular distance from the center of bilayer is plotted by histogram analysis and representative snapshots are given from simulation trajectory for (A) POPC/POPG 7:3, (B) POPC/POPS 7:3, (C) POPG/POPE 1:3 and (D) POPG/POPE 3:1 bilayer systems. [Blue represents the distance between center of bilayer and the residue with maximum bend angle; Red represents the distance between center of bilayer and Gly12 residue.]
The calculated residue-wise root mean square fluctuations (RMSF) of MSI-594 in the presence of membrane moieties are shown in Figure 5. The significance of this data is that it gives the amplitude of atomic fluctuation of the Cα carbon atoms that corresponds to relative dynamicity of peptide segment in conjunction to the corresponding inter-action phenomenon. In particular, the highest degree of variation is observed for POPC/POPG (7:3) compared to other heterogeneous systems, which suggests that relative dynamicity of MSI-594 is greater in this system (Figure 5A). Residue-wise, the fluctuations are more significant in C-terminal (C-ter) and N-terminal (N-ter) residues compared to the residues present in the central region of MSI-594. This observation is consistent in all model systems (Figure 5A). The dynamicity of MSI-594 is found to be relatively rigid in POPG/POPE (1:3), which corresponds to Gram-negative bacterial membrane systems. This result correlates well with the results of area per lipid (Figure 1), which indicates that membrane perturbation induced by MSI-594 in POPG/POPE (1:3) is less compared to Gram-positive bacterial membrane mimicking system POPG/POPE (3:1). A close trajectory analysis reveals that the relative dynamicity of positively charged amino acids is found to be significant, as indicated in Figure 5B–E and Figure S5 (Supporting Information).
Figure 5.
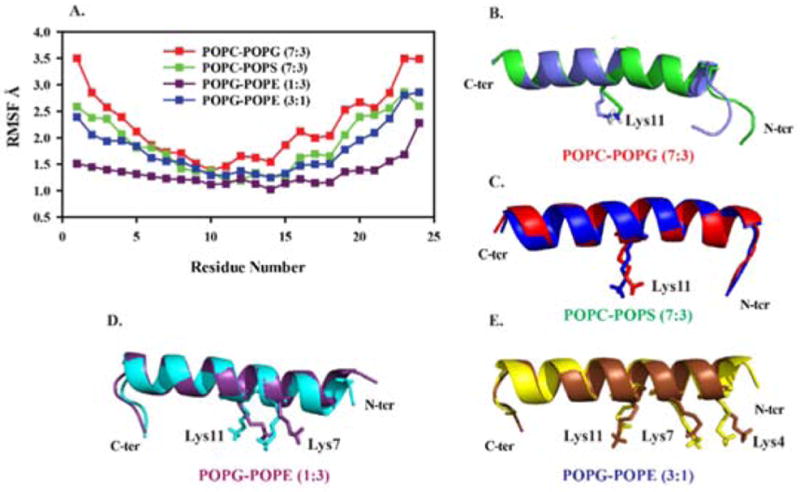
(A) RMSF plots for the Cα carbon atoms of MSI-594 in heterogeneous lipid environments and (B–E) superimposition of MSI-594 conformations from starting and end-point simulation timescale. The side chain of residues that shows significant difference are highlighted in stick representations. Figures were generated using PyMol visualization tool.
Helix Bending
To obtain further understanding of the secondary structure of the MSI-594, detailed analysis for the structural and positional changes of the residues have been calculated. The structural bending or distortion in the helical structure of transient ensemble of MSI-594 in the presence of membrane models was analyzed using Bendix program.60 This program evaluates the structure of the helix based on local structural changes. Figure 6 shows the maximum bend angle observed in MSI-594 that accounts for the effect of different lipid environments. In POPC/POPS (7:3) heterogeneous system, helix bend angle of 8.8° is observed formed by residues Gly14, Leu17, and Leu20. This suggests that the structural stability is mediated mainly via hydrophobic interactions. In POPC/POPG (7:3) the bend angle is 14.6° and the residues involved are Gly14, Leu17, and Leu20, which is indicative of hydrophobic association. Notably, these residues also take part in bend formation in POPS control system (Supporting Information Figure S6). Comparing the helix bending of POPG/POPE membrane systems, the helix bend is observed to be 13.8° in both 1:3 and 3:1 molar-ratio systems. Importantly, the residues involved in both systems are Lys4, Lys7, and Lys10, which are the N-terminal region of MSI-594, responsible for inducing the helix bend. Based on these observations, the electro-static interactions tend to stabilize the interaction between MSI-594 and lipid moieties in 3:1 and 1:3 POPG/POPE systems.
Figure 6.
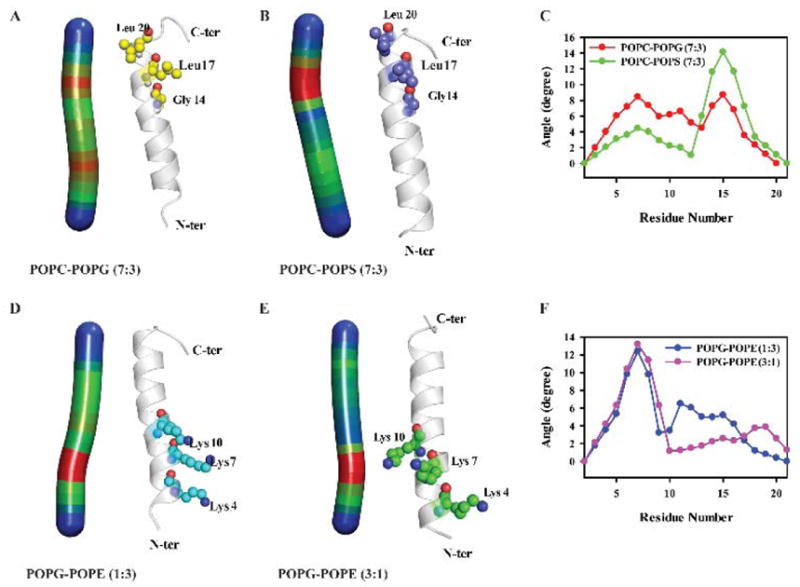
Heat map for the helix bend angle and the residues involved along with the degree of angle plot against the residue (E, F) are shown for (A) POPC/POPG 7:3, (B) POPC/POPS 7:3, (D) POPG/POPE 1:3 and (E) POPG/POPE 3:1 heterogeneous lipid systems.
Conclusions
The present study elucidates the atomistic information for the mechanism of action of MSI-594 in different membrane model systems. Notably, the use of dual boost accelerated molecular dynamics simulation helps in obtaining enhanced conformational sampling that in turn reveals the driving force responsible for the AMP-membrane interaction. The study comprises of heterogeneous membrane systems like POPC/POPG (7:3) and POPC/POPS (7:3) that correspond to previously reported experimental studies, as well as POPG/POPE (1:3), POPG/POPE (3:1) that mimic Gram-negative and Gram-positive bacterial outer membrane, respectively. The results obtained are helpful in differentiating the level of activity of MSI-594 in the presence of different lipid species in the membrane models. In particular, we have observed that MSI-594 exhibit significant perturbation in a homogeneous zwitterionic POPC bilayer and least disruption in a neutral POPE bilayer system, as reflected by the area per lipid calculations (Supporting information Figure S7). Similarly, it was also observed that anionic POPG renders maximum fluctuations when present in lower concentrations, such as in POPG/POPE (1:3) and POPC/POPG (7:3) bilayers (Figure 7). The selectivity of MSI-594 is found to be against bacterial membrane systems, as reflective of lower activity in POPC system. Our simulations also dictate that in POPC/POPS (7:3) system, the interaction is hydrophilic in nature, whereas the interaction is found to be hydrophobic against POPC system. Overall, this information is helpful in dictating the mechanism of action and membrane selectivity of MSI-594, as well as that it may lead to the design of MSI variants as active AMPs.
Figure 7.

Structural models indicating the relative dynamicity of MSI-594 (in green acyl chain) and membrane system (in yellow acyl chain). The structural drift for C-ter, G-I-G middle section, and N-ter is shown with a curved arrow, which corresponds to relative dynamicity of MSI-594 from aMD. The membrane thickness (vertical arrows) and the average change in area (horizontal arrows) reflect the perturbation of membrane system. The thickness of arrow is indicative of their extent of perturbation. The model summarizes perturbation effect over (A) POPC/POPG 7:3, (B) POPC/POPS 7:3, (C) POPG/POPE 1:3, and (D) POPG/POPE 3:1 systems due to the presence of MSI-594.
Supplementary Material
Acknowledgments
This study was partly supported by Bose Institute (Plan Project-II (to AB)) and partly by NIH (to AR). SM and RKK are grateful to UGC and CSIR, Govt. of India for junior and senior research fellowships, respectively.
Footnotes
Electronic Supplementary Information (ESI) available.
Dedication
This paper is dedicated to Professor Dr. Thomas Peters, University of Lübeck, Germany, for his 60th birthday.
Notes and references
- 1.Bond PJ, Khalid S. Protein Pept Lett. 2010;17:1313–1327. doi: 10.2174/0929866511009011313. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen LT, Haney EF, Vogel HJ. Trends Biotechnol. 2011;29:464–472. doi: 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Mihajlovic M, Lazaridis T. Biochim Biophys Acta. 2010;1798:1485–1493. doi: 10.1016/j.bbamem.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sengupta D, Leontiadou H, Mark AE, Marrink SJ. Biochim Biophys Acta. 2008;1778:2308–2317. doi: 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Shai Y. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 6.Lee DK, Bhunia A, Kotler SA, Ramamoorthy A. Biochemistry. 2015;54:1897–1907. doi: 10.1021/bi501418m. [DOI] [PubMed] [Google Scholar]
- 7.Zasloff M, Martin B, Chen HC. Proc Natl Acad Sci U S A. 1988;85:910–913. doi: 10.1073/pnas.85.3.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dempsey CE. Biochim Biophys Acta. 1990;1031:143–161. doi: 10.1016/0304-4157(90)90006-x. [DOI] [PubMed] [Google Scholar]
- 9.Maloy WL, Kari UP. Biopolymers. 1995;37:105–122. doi: 10.1002/bip.360370206. [DOI] [PubMed] [Google Scholar]
- 10.Wieprecht T, Dathe M, Krause E, Beyermann M, Maloy WL, MacDonald DL, Bienert M. FEBS Lett. 1997;417:135–140. doi: 10.1016/s0014-5793(97)01266-0. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzaki K, Sugishita K, Fujii N, Miyajima K. Biochemistry. 1995;34:3423–3429. doi: 10.1021/bi00010a034. [DOI] [PubMed] [Google Scholar]
- 12.Dathe M, Schümann M, Wieprecht T, Winkler A, Beyermann M, Krause E, Matsuzaki K, Murase O, Bienert M. Biochemistry. 1996;35:12612–12622. doi: 10.1021/bi960835f. [DOI] [PubMed] [Google Scholar]
- 13.Uematsu N, Matsuzaki K. Biophys J. 2000;79:2075–2083. doi: 10.1016/S0006-3495(00)76455-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. Antimicrob Agents Chemother. 2007;51:1398–1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soblosky L, Ramamoorthy A, Chen Z. Chem Phys Lipids. 2015;187:20–33. doi: 10.1016/j.chemphyslip.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramamoorthy A, Thennarasu S, Lee DK, Tan A, Maloy L. Biophys J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spink CH. Methods Cell Biol. 2008;84:115–141. doi: 10.1016/S0091-679X(07)84005-2. [DOI] [PubMed] [Google Scholar]
- 18.Yang P, Ramamoorthy A, Chen Z. Langmuir. 2011;27:7760–7767. doi: 10.1021/la201048t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domadia PN, Bhunia A, Ramamoorthy A, Bhattacharjya S. J Am Chem Soc. 2010;132:18417–18428. doi: 10.1021/ja1083255. [DOI] [PubMed] [Google Scholar]
- 20.Murzyn K, Róg T, Pasenkiewicz-Gierula M. Biophys J. 2005;88:1091–1103. doi: 10.1529/biophysj.104.048835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egal M, Conrad M, MacDonald DL, Maloy WL, Motley M, Genco CA. Int J Antimicrob Agents. 1999;13:57–60. doi: 10.1016/s0924-8579(99)00094-1. [DOI] [PubMed] [Google Scholar]
- 22.Bhunia A, Ramamoorthy A, Bhattacharjya S. Chemistry. 2009;15:2036–2040. doi: 10.1002/chem.200802635. [DOI] [PubMed] [Google Scholar]
- 23.Porcelli F, Buck-Koehntop BA, Thennarasu S, Ramamoorthy A, Veglia G. Biochemistry. 2006;45:5793–5799. doi: 10.1021/bi0601813. [DOI] [PubMed] [Google Scholar]
- 24.Loose M, Schwille P. J Struct Biol. 2009;168:143–151. doi: 10.1016/j.jsb.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 25.Zachowski A. Biochem J. 1993;294(Pt 1):1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramamoorthy A, Thennarasu S, Tan A, Gottipati K, Sreekumar S, Heyl DL, An FY, Shelburne CE. Biochemistry. 2006;45:6529–6540. doi: 10.1021/bi052629q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Jung SW, Cho AE. Langmuir. 2016;32:1782–1790. doi: 10.1021/acs.langmuir.5b04113. [DOI] [PubMed] [Google Scholar]
- 28.Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, Im W. J Comput Chem. 2014;35:1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. Nucleic Acids Res. 2012;40:D370–376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomize AL, Pogozheva ID. Methods Mol Biol. 2013;1063:125–142. doi: 10.1007/978-1-62703-583-5_7. [DOI] [PubMed] [Google Scholar]
- 31.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang J, MacKerell AD. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983:79. [Google Scholar]
- 34.Cheng JT, Hale JD, Elliott M, Hancock RE, Straus SK. Biochim Biophys Acta. 2011;1808:622–633. doi: 10.1016/j.bbamem.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 35.Amos ST, Vermeer LS, Ferguson PM, Kozlowska J, Davy M, Bui TT, Drake AF, Lorenz CD, Mason AJ. Sci Rep. 2016;6:37639. doi: 10.1038/srep37639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong C, Tieleman DP, Wang Y. Langmuir. 2014;30:11993–12001. doi: 10.1021/la502363b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.FSE, Yuhong Z, PRW, BBR The Journal of Chemical Physics. 1995:103. [Google Scholar]
- 38.Pedersen LG. World J Biol Chem. 2011;2:35–38. doi: 10.4331/wjbc.v2.i2.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.AHC Journal of Computational physics. 1983 Oct;52:24–34. [Google Scholar]
- 40.Shuichi M, KPA Journal of Computational Chemistry. 1992 Oct;13:952–962. [Google Scholar]
- 41.Hamelberg D, Mongan J, McCammon JA. J Chem Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 42.Appelt C, Eisenmenger F, Kühne R, Schmieder P, Söderhäll JA. Biophys J. 2005;89:2296–2306. doi: 10.1529/biophysj.105.063040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez Cascales JJ, Garro A, Porasso RD, Enriz RD. Phys Chem Chem Phys. 2014;16:21694–21705. doi: 10.1039/c4cp02537g. [DOI] [PubMed] [Google Scholar]
- 44.Gullingsrud J, Schulten K. Biophys J. 2004;86:3496–3509. doi: 10.1529/biophysj.103.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamb HM, Wiseman LR. Drugs. 1998;56:1047–1052. doi: 10.2165/00003495-199856060-00011. discussion 1053–1044. [DOI] [PubMed] [Google Scholar]
- 46.Gottler LM, Ramamoorthy A. Biochim Biophys Acta. 2009;1788:1680–1686. doi: 10.1016/j.bbamem.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrache HI, Dodd SW, Brown MF. Biophys J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee DK, Brender JR, Sciacca MF, Krishnamoorthy J, Yu C, Ramamoorthy A. Biochemistry. 2013;52:3254–3263. doi: 10.1021/bi400087n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vermeer LS, de Groot BL, Réat V, Milon A, Czaplicki J. Eur Biophys J. 2007;36:919–931. doi: 10.1007/s00249-007-0192-9. [DOI] [PubMed] [Google Scholar]
- 50.Chapman D, Urbina J. J Biol Chem. 1974;249:2512–2521. [PubMed] [Google Scholar]
- 51.Balali-Mood K, Harroun TA, Bradshaw JP. Eur Phys J E Soft Matter. 2003;12(Suppl 1):S135–140. doi: 10.1140/epjed/e2003-01-031-3. [DOI] [PubMed] [Google Scholar]
- 52.Kinnun JJ, Mallikarjunaiah KJ, Petrache HI, Brown MF. Biochim Biophys Acta. 2015;1848:246–259. doi: 10.1016/j.bbamem.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bera S, Korshavn KJ, Kar RK, Lim MH, Ramamoorthy A, Bhunia A. Phys Chem Chem Phys. 2016;18:16890–16901. doi: 10.1039/c6cp02023b. [DOI] [PubMed] [Google Scholar]
- 54.Petersen NO, Chan SI. Biochemistry. 1977;16:2657–2667. doi: 10.1021/bi00631a012. [DOI] [PubMed] [Google Scholar]
- 55.Dickey A, Faller R. Biophys J. 2008;95:2636–2646. doi: 10.1529/biophysj.107.128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shoemaker SD, Vanderlick TK. Biophys J. 2002;83:2007–2014. doi: 10.1016/S0006-3495(02)73962-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lohner K, Blondelle SE. Comb Chem High Throughput Screen. 2005;8:241–256. doi: 10.2174/1386207053764576. [DOI] [PubMed] [Google Scholar]
- 58.Sun Y, Shang D. Mediators Inflamm. 2015;2015:167572. doi: 10.1155/2015/167572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pulido D, Nogués MV, Boix E, Torrent M. J Innate Immun. 2012;4:327–336. doi: 10.1159/000336713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dahl AC, Chavent M, Sansom MS. Bioinformatics. 2012;28:2193–2194. doi: 10.1093/bioinformatics/bts357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.