

Abstract
Natural genetic promoters are regulated by multiple cis and trans regulatory factors. For quantitative studies of these promoters, the concentration of only a single factor is typically varied to obtain dose response or transfer function of the promoters with respect to the factor. Such design of experiments has limited our ability to understand quantitative, combinatorial interactions between multiple regulatory factors at promoters. The limitation is primarily due to the intractable number of experimental combinations that arise from multifactorial design of experiments. To overcome this major limitation, we integrate impact printing and cell-free systems to enable multi-dimensional studies of genetic promoters. We first present a gradient printing system which comprises parallel piezoelectric cantilever beams as a scalable actuator array to generate droplets with tunable volumes in the range of 100pL – 10nL, which facilitates highly accurate direct dilutions in the range of 1 – 10,000 fold in a 1μL drop. Next, we apply this technology to study interactions between three regulatory factors at a synthetic genetic promoter. Finally, a mathematical model of gene regulatory modules is established using the multi-parametric and multi-dimensional data. Our work creates a new frontier in the use of cell-free systems and droplet printing for multi-dimensional studies of synthetic genetic constructs.
Graphical Abstract
A multi-parametric gradient generation system has been established for multi-dimensional, high-throughput, and low-consumption quantitative analysis of a synthetic genetic module.
Introduction
Studies of synthetic genetic promoters in cell-free systems are becoming increasingly important for high-throughput expression screening, high yield protein synthesis, and computational modeling of gene circuits.1, 2 For instance, mathematical models have been established to predict the dependence of protein expression levels on template DNA concentration and experimental timing in cell-free systems.3 Promoter constructs have been designed and tested using cell-free systems, providing a valuable tool for rapidly prototyping DNA regulatory elements for synthetic biology.4–6 Cell-free systems have been miniaturized using compartmentation either without gradient or with one-dimensional gradient of stimuli.7–10 In general, these studies have focused on one-dimensional study of individual regulatory factor. Such focus contradicts the common understanding that genetic promoters are regulated by complex interactions between multiple factors with a wide range of concentrations. 11, 12 Recent advances in precision medicine, regulatory genomics and synthetic biology have demanded for high-throughput and high-efficiency ways to facilitate screening and optimization of transcription factors for promoter regulation.3, 13 Therefore, the technology for multi-parametric studies of genetic promoters is important for understanding molecular interactions at the promoters,14 establishing new gene control modules,13 and optimizing the system for high-throughput protein synthesis.15
A fundamental requirement for multifactorial studies of genetic promoters is the generation of concentration gradient for each regulatory factor across multiple log folds.16 However, the classic procedures to establish concentration gradients lack efficiency, modularity, and throughput. In most biological laboratories, serial dilutions employing manual micropipettes and robotic dispensing systems are still considered the gold standard.17 This conventional method is time-consuming, wastes reagents, and introduces substantial potential for cross-contamination. Importantly, potential human and systematic errors based on the classic methods can elevate to a significant level (30%) when the sample volume is below 1 μL.18–20 The latest advancements in microfluidics and lab-on-a-chip systems have enabled gradient generation by using microfluidic architectures to diffuse or mix.21 The former method implements diffusive channels with the control of different flow rates of individual reactants, 22, 23 while the mixing approach requires the separation and mixing of liquid volumes in segregated reaction chambers.17 Both methods permit concentration gradients to be maintained over an extended period of time, on the order of hours.24 Recent introduction to droplet microfluidics provides an alternative approach to generate gradient, in which multi-component aqueous droplets can be physically compartmentalized from samples merged from different flow channels at different flow rates. Droplet microfluidics allow for high-throughput gradient generation and analysis with extremely low sample consumption (pico-liter to nano-liter reactant volumes), effectively enabling this technology to adapt to copious uses in biological assaying, including protein crystallization concentration screening25 and enzyme inhibition assays.26
However, current droplet microfluidic devices present several technical limitations when dealing with multi-parametric gradient generation. First, current devices use a fixed design and thus are only useful in specific applications with fixed dimensions and a limited number of inputs.27 Since microfluidic operations require fine tuning of the fluidic dynamics in the microchannel, multi-parametric experiments can be complicated to implement and operate with current devices. In fact, current microfluidic multi-parametric concentration gradient generation designs only allow for 10-fold concentration gradient in two-dimensional gradient generation.15, 28 To this end, our past work has addressed the aforementioned limitations by adapting simple microfluidic impact printing cartridges fabricated by standard soft lithography to a plug-and-play actuation mechanism to produce droplet sequences on demand. Uniquely, the reversibly pluggable microfluidic cartridge allows for experimental customization with high adaptability and scalable multiplexability; for instance, one can implement various microfluidic functionalities, such as mixing and pumping, into the cartridge prior to droplet ejection.29 It is worth noting that unlike the continuous and digital droplet microfluidics, the microfluidic impact printing platform enables independent volume, content, and spatial controls of each individual droplet, and thus, the printed droplet containers can be individually manipulated, positioned, and analyzed.30 The full automated operation of the microfluidic impact printing system has also reduced the minimal analytical volume to less than 1 μL. Compared with previous arts of impact printing31, our proposed platform is more suitable for biological experiments with low-viscosity fluids (< 20cP), high frequency (1 kHz), easy expansion of channel numbers, as well as disposable cartridges.
As multi-parametric analyses is demanded by study of complex biological processes, our work aims to resolve the throughput, accuracy and multiplexibility issues in conventional synthetic genetic promoter characterization methods. We first report a multiplexable gradient generation system derived from the aforementioned microfluidic impact printing concept, from which quantitative multi-parametric analyses can be performed on complex biological processes in a high-efficiency and high-throughput fashion. This newly established analytical system utilizes parallel piezoelectric cantilevers as external drivers for droplet generation, which facilitate linear scalability, high frequency (1000 Hz), and tunable droplet volumes (100pL to 10nL). Using this scalable droplet gradient generation platform, we have realized high-throughput wide-range concentration gradient generation of multiple fluorescent proteins within micro-liter drops with the following features: 1) efficient, wide range volume control to meet the dilution requirements, 2) scalable fluidic inputs to tailor for multi-parametric analyses, 3) a wide range of fluidic adaptability, and 4) a minimal dead volume to mitigate waste of precious bio samples. Specifically, this platform has been applied to study multi-parametric interactions of multiple transcription/translation regulation factors at a synthetic genetic promoter, from which a mathematical model of gene regulatory modules has been established. In summary, our work presents an efficient tool for automating quantitative multi-parametric analyses of gene regulation.
Design
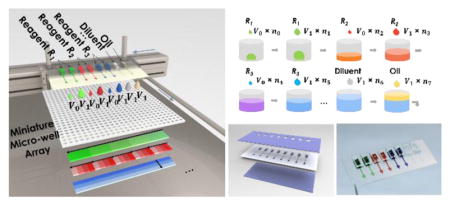
As illustrated in Figure 1, biological reagents (e.g., R1 – R3) will be loaded in parallel into a multi-channel microfluidic printing cartridge with tunable dispensing volume (from 100pL – 10nL), along with diluent and oil. The printing cartridge will be inserted into the printer head assembly consisting of individually addressable piezoelectric actuators. From the cartridge, desired volume of each reagent can be printed into the targeted micro-wells with high speed and digital accuracy, followed by printing of complimentary volume diluent to achieve desired concentrations and oil to prevent evaporation. To facilitate the dilution speed while maintaining volume accuracy, we have adapted a two-volume printing strategy, e.g., we providing two standard droplet volumes. With two different nozzle sizes of 60μm and 120μm, two standard droplets (volume of V0 and V1) can be generated, respectively, with V1 > 5V0. The desired drop numbers can be calculated accordingly. Such a two-volume printing strategy significantly increases generation speeds of any mixing profiles. For instance, when 1, 100, and 10,000-fold dilution in 1μL drop is needed, with V0 equals to 0.1nL and V1 equals to 10nL, one can print one hundred drops of 10nL in Well #1, one drop of 10nL in Well #2, and one of 0.1nL in Well #3, which reduces the printing time by 100 fold, compared with conventional single volume strategy. Eventually, a miniaturized micro-well array combining multiplexed concentration gradients can be generated in a high-throughput manner, of which the individual gradients of R1, R2 and R3 are also illustrated (in Figure 1a), respectively, for the purpose of quantitative multi-parametric analyses. Importantly, the gradient profiles can be designed according to the demand as linear, logarithmic, or exponential. With accurate pico-liter volume control, scalable fluidic inputs, and high-efficient gradient generation strategy, the proposed MIP system would enable high-throughput quantitative assessments for multi-dimensional and multi-parametric biological interactions in massive droplet reactors.
Figure 1.

a) Design of the microfluidic multi-parametric gradient generation system. Multiple reagents (R1, R2, and R3) can be loaded into parallel microfluidic channels of a cartridge with tunable volume resolution. Overlapped gradient patterns of different reagents in the same region of a multi-well array are illustrated. b) Illustration of sequential printing of three reagents into a single well, using a two-volume strategy. nk (k=1, 2…7) represents number of droplets in each step, which can be calculated based on the desired dispensing volume and standard droplet sizes. In Figure 1b, total volume of R1 is (V0n0 + V1n1), thus concentration of R1 is (V0n0 + V1n1)/ΣVknk).
Methods
Gradient printing system
A multiplexed, high-speed, scalable, and compact gradient printing system has been established for multi-parametric analysis, based on the principle of microfluidic impact printing.32 The microfluidic impact printing technology operates by striking on the membrane of a separated microfluidic cartridge to squeeze liquid outside nozzle (illustrated in Figure S1a).
Compared with our previous version of microfluidic impact printing using piezo discs32, this newly established multiplexable gradient generation system removed the need of mechanical levers by adopting parallel piezoelectric cantilevers as external drivers for droplet generation. Piezoelectric cantilevers (T226-A4-103X, Piezo Systems, Inc.) were chosen for their large stroke at relatively low driving voltage, linear scalability and improved frequency (1,000 Hz), while maintaining electronically tunable vibration amplitude. As a result, these new actuators occupied 12-times less space per channel and allowed for multiplexibility in a compact palm-size printer head. Polymethyl methacrylate (PMMA) and aluminum sheets (McMaster-Carr) were machined by a Kern Micro 24 laser cutter and a CNC milling machine respectively to fabricate the printer head. Piezoelectric cantilevers were inserted into the slots in the printer head during assembling. Metal pins (Keystone Electronics) were attached to the end of the PZT actuators for striking the deformable actuation membrane.
The microfluidic multi-parametric gradient generation system used a separable cartridge independent from the actuator, reducing time for washing and risks of contamination for analyzing multiple reagents. 22, 23 A disposable, multi-channel microfluidic cartridge was designed, with minimal reagent costs, as illustrated in Figure 2a. For cartridge fabrication, two 100μm-thickness hydrophilic polyester (PET) films (3M™ 9984 Diagnostic Microfluidic Surfactant Free Fluid Transport film) were cut by a CO2 laser machine (Universal Laser Systems, VersaLaser 2.30) and used as a top elastic layer connecting to reservoirs and a bottom nozzle layer with small holes. Top and bottom layers were hydrophilic, making reagent loading easier during injection loading. A 150μm-thickness polyester film with double-sided adhesive film (3M 467 200MP) was cut by the CO2 laser machine as a middle channel layer. Three layers were assembled together under a mask aligner. A photograph of an 8-channel cartridge is shown in Figure 2b. The polymeric cartridge can hold up to 8-channels in a 2cm × 4cm area. The distance between adjacent channels was 5mm.
Figure 2.

a). Illustration of a 3-layer cartridge. b) Photograph of a transparent 8-channel cartridge filled with color dyes.
The control system (Figure S1b) comprised a customized printing control software that enabled tuning of driving signals for each PZT cantilevers, an Arduino microcontroller unit (MCU) integrated with a multi-channel switch circuit board, a custom made high voltage amplifier (HVA), a custom made multi- channel printer head as a housing of PZT cantilevers and cartridges, a disposable multi-channel microfluidic cartridge, and a 3-axis motorized stage with a controller (BBD202, Thorlabs). We managed to use a common amplifier for all channels though a channel selector, thus cutting down the cost to scale up channel numbers. The MCU generated waveforms through DAC port and relayed waveforms to the HVA for signal amplification. Channel selection was controlled by digital I/O outputs of the channel selector. Number of channels can be easily altered through the graphic interface in the software. Channel switching was fully automated with high position accuracy and repeatability. The printer head was amounted on the precision travelling stages (LTS300, DDS220, Thorlabs) with travelling speeds up to 300 mm/s and positional accuracy <3.0 μm. In our experiment, the location misalignment in X and Y directions on a planar surface were both smaller than 25 μm with 2.15mm printing distance.
To make the multi-well containers for the biological experiments, 25 × 25 through-holes with 1.2mm diameter were laser cut in a PMMA sheet (1.59 mm thickness), with 1.6mm center-to-center intervals, which was chemically bonded with a 0.2mm-thick PMMA bottom layer, as shown in Figure S2 in supplemental materials. Chemical bonding was performed with 150μL acetonitrile. The multi-well containers were placed under fume hood overnight to remove acetonitrile residues. Both cartridges and containers were sterilized under a UV lamp before use.
Prior to our experiment, parameters including number of reagents, gradient pattern of each reagent (linear, logarithmic, or exponential), and calibrated volume resolution data were input into the software. Then the software translated these into commands for sequential channel selection, volume switching, assigning number of droplets (n0~7) and stage travelling. All the following printing procedures were performed automatically.
Reagents
Fluorescent proteins TagBFP, GFP and mCherry were cloned downstream a T7 promoter in plasmid pET15b (Novagen), with a 6x His-tag at the C-end. Plasmids were transformed into E. coli BL21 (DE3)-pLysS cells (Novagen) and clones were selected in LB-agar plates with carbenicillin and chloramphenicol. Expression of each fluorescent protein was induced by addition of 0.5 mM IPTG to cells in exponential growth phase cultured in LB media. Bacteria were lysed by sonication and His-tagged fluorescent proteins were purified using HisTrap FF columns (GE Healthcare). Protein concentrations were determined using Protein Assay 660 nm (Thermo Scientific).
The in-vitro transcription-translation reaction mix was prepared by mixing 2.2x reaction buffer, 1.3x protein mix, and RNase-free water at a ratio of 1:0.58:0.25. The 2.2x reaction buffer contained the following components: amino acid mix 121 mM (each amino acid 6.05 mM), tRNA (Roche) 118.8 UA260/mL, ATP 8.25 mM, GTP 5.5 mM, CTP 2.75 mM, UTP 2.75 mM, creatine phosphate 110 mM, folinic acid 60 μg/mL, HEPES-KOH 7.6 110 mM, potassium glutamate 770 mM, magnesium acetate 36 mM, spermidine 2.2 mM, DTT 11 mM, BSA 1.1 mg/mL, creatine kinase (Roche) 178.2 μg/ml, myokinase (Sigma Aldrich) 110 μg/mL, diphosphonucleotide kinase (Sigma Aldrich) 9 μg/mL, T7 RNAP (New England Biolabs) 440 U/μl, and RNAse inhibitor (New England Biolabs) 0.9 U/μl. The 1.3x protein mix was prepared by mixing purified translation machinery (TraMOS) and ribosomes (New England Biolabs) with 18 μg/μL TraMOS and 3 μM ribosomes. TraMOS was provided by Tan lab. EsaR was purified as described previously and prepared at stock concentration of 50.8 μM. AHL (3-oxo-hexanoyl-homoserine-lactone, 3OC6HSL, Sigma-Aldrich) was prepared at a stock concentration of 1 mM. Plasmid Ngo1, coding for GFP under control of a hybrid synthetic promoter T7/Esa box, was prepared using a Qiaprep midi kit (Qiagen), and a stock solution was prepared at 134 nM. In each well, 453nL out of a 550nL final volume was allotted to the transcription-translation reaction mix, which left 97nL volume capacity for concentration manipulation of regulating factors. During printing experiment, ambient humidity is controlled around 70% to reduce evaporation. Every reaction was generated in 3 replicates. 300μL low-viscosity Vapor-Lock PCR oil (981611, Qiagen) was printed to prevent evaporation before the aligning with the next well.
Calibration and imaging
Liquids were calibrated in our system prior to experiments to set up the resolutions accordingly. In reality, the minimal and maximal volumes of droplets generated from each liquid depended on its viscosity and surface tension. Prior to array printing, droplet volumes generated by each channel were calibrated by printing into 2mm-thick silicone oil (Sigma-Aldrich) and measuring the diameters of suspended droplets under a bright filed microscope (EVOS XL, Life Technologies). Tuning of volumes for droplets can be performed with modulation of voltage amplitude, pulse width, and nozzle diameters.32 We can set up resolutions with at least 50-time volume differences.
A Dino-Lite digital microscope (Pro AM4113T) was used to visually check the alignment. Images of a multi-well array were automatically taken and stitched by Nikon Eclipse Ti-E Inverted Microscopes in multipoint mode. Filter sets used were GFP (480nm excitation/535nm emission), DAPI (360nm excitation/460nm emission) and Texas Red (560nm excitation/630nm emission) filter cubes from Nikon. Different exposure times (5ms, 50ms, 300ms, and 2s) were used in the fluorescent protein dilution experiment to cover wide range of concentration. Intensities of fluorescent intensities were analyzed by an ImageJ Macro script. To better visualize concentration gradient generated in a wide concentration range, the fluorescent intensity data were normalized by exposure times and combined.
Results
Wide-range gradient generation
Fluorescent proteins are commonly used to quantify translation/transcription yield and efficiency.33 To illustrate that this droplet printer can be used for both controlling the concentration of input molecules and measuring the reporters of gene expression, we first demonstrated a 1–10,000 fold dilution of 3 fluorescent proteins individually. Due to the fact that this platform can generate volume as low as 100pL,32 a 10,000-fold dilution can be implemented within a 1μL drop. The coefficient of variation (CV) of droplet volume measured from 72 droplets (5nL) was 3.5% on our platform, compared with 4.3% using piezo stack actuated inkjet dispenser.29
Prior to printing, droplet volumes of fluorescent proteins were calibrated, based on which droplet numbers were calculated. In order to efficiently achieve a 10,000-fold concentration gradient with a binary dilution profile, the droplet volume of GFP was designed to vary by 64-fold, i.e., the volume of fluorescent protein droplets was set to be altered from 0.1 nL to 6.4 nL. Particularly, we used one nozzle with an inner diameter of 60μm with a droplet volume tunable from 0.1 to 3.5nL by changing the driving voltage (120V–200V) and pulse width (30μs–500μs). Adding another nozzle with an inner diameter of 120μm, it can produce a droplet volume up to 7.5nL.32 Therefore, we have selected a three-volume setting to facilitate the printing process: 0.1 nL, 0.8 nL and 6.4 nL, as in Table 1.
Table 1.
The three-volume setting for generating droplets of 0.1 nL, 0.8 nL and 6.4 nL
| Channel 1 | Channel 2 | ||
|---|---|---|---|
|
| |||
| Setting 1 | Setting 2 | ||
|
| |||
| Nozzle Diameter | 60μm | 60μm | 120μm |
| Voltage | 120V | 120V | 200V |
| Pulse Width | 30μs | 500μs | 500μs |
|
| |||
| Volume | 0.1 nL | 0.8 nL | 6.4 nL |
As shown in Table 2, the specific droplet numbers, based on the 3 standard volumes, have been calculated to achieve the desired concentrations. As a result, a dilution profile spanning 4 orders of magnitude (10,000-fold) can be established in a high-throughput fashion. That means it requires less than 200 droplets to generate each concentration, which can be completed within one second of printing, given the maximum printing speed of 400Hz. All three fluorescent proteins have been calibrated in the same way.
Table 2.
Assignment of drop number from 3 channels for a 10,000-fold GFP concentration gradient generation experiment from a stock concentration of C0.
| Drop Number | ||||
|---|---|---|---|---|
|
| ||||
| Channel 1 (C0) | Channel 2 (C0) | Channel 3 (Diluent) | ||
|
| ||||
| Diluted Concentration | 0.1 nL per drop | 0.8 nL per drop | 6.4 nL per drop | 6.4 nL per drop |
|
| ||||
| C0 | 8 | 1 | 156 | 0 |
| 2−1 C0 | 8 | 0 | 78 | 78 |
| 2−2 C0 | 4 | 0 | 39 | 117 |
| 2−3 C0 | 2 | 4 | 19 | 137 |
| 2−4 C0 | 1 | 6 | 9 | 146 |
| 2−5 C0 | 0 | 7 | 4 | 151 |
| 2−6 C0 | 4 | 3 | 2 | 154 |
| 2−7 C0 | 6 | 1 | 1 | 155 |
| 2−8 C0 | 7 | 4 | 0 | 156 |
| 2−9 C0 | 4 | 2 | 0 | 156 |
| 2−10 C0 | 2 | 1 | 0 | 156 |
| 2−11 C0 | 5 | 0 | 0 | 156 |
| 2−12 C0 | 2 | 0 | 0 | 156 |
| 2−13 C0 | 1 | 0 | 0 | 156 |
Before printing, 10 μL from each of TagBFP (116 μM), mCherry (23 μM) and GFP (44 μM) solutions were individually loaded into the sample channel. Two additional channels were loaded with 50μL PBS buffer and 15μL PCR oil. Through the control of droplet size and droplet numbers, we sequentially printed different amount of fluorescent proteins and PBS buffer solutions into custom-made PMMA microplates, leading to an ending volume of 1.3μL in each well, from which a wide-range concentration gradient of each individual fluorescent protein was generated. In our design, TagBFP was diluted from 116 μM to 6.9 nM (×214 fold), mCherry was diluted from 23 μM to 1.4 nM (×214 fold), and GFP was diluted from 44 μM to 2.7 nM (×214 fold), with 15 target concentrations (including control) and 3 replicates for each. As summarized in Figure 3, the microfluidic droplet generation system was capable to generate 10,000-fold concentration gradient profile. Noticeably, among the three fluorescent proteins, TagBFP had weakest signals (compared with that of mCherry and GFP), which was not detectable at the concentration range lower than 227nM. While, mCherry and GFP exhibited stronger signal intensities and continuous changes from the most diluted to the most concentrated, a 10,000-fold change.
Figure 3.

Wide-range fluorescent dilution curves of a) GFP, b) mCherry, and c) TagBFP. Fluorescent intensities were normalized by exposure times. On each figure, original one-dimensional dilution of each individual protein was shown in blue dots. All fitting curves had R2 >0.95. Data calculated from subsequent multi-parametric gradient generation section are illustrated by orange dots.
Multi-parametric gradient generation
The next demonstration of the microfluidic multi-parametric gradient generation system included combinatorial multi-parametric dilution by combining three fluorescent proteins in one digitally printed microarray, from which we can easily achieve a wide range of concentration profiles of multiple dilutes (up to 10,000-fold).
Using a single cartridge for all fluorescent proteins, each loaded into an individual channel, we created 5 concentrations of GFP (34 μM, 3.4 μM, 340 nM, 34 nM, and 3.4 nM), 4 concentrations of mCherry (1.8 μM, 180 nM, 18 nM, 1.8 nM), and 3 of TagBFP (17.9 μM, 1.79 μM, and 179 nM) in a 5 × 4 × 3 combinatorial micro-well array, as illustrated in Figure 4a. The overall volume of each well was kept at 1.3μL. Pictures taken through different optical filters (DAPI, TexasRed and GFP) were shown in Figures 4b–d, respectively, whereas, the final multi-parametric fluorescent patterns with all three components was combined in Figure 4e. To validate the accuracy of multi-parametric dilution, mean signal intensities of each individual fluorescent protein, were compared with previous calibration data of each individual protein. As shown in Figure 3, the data from multi-parametric dilution (highlighted as orange dots) were closely matched with the original calibration curve for GFP and mCherry proteins in the range above 34 nM and 18 nM. However, for the lower range of GFP, mCherry, and TagBFP solutions, the measured concentration from the multi-parametric dilution deviated from the predicted value of the individual dilution. This was likely attributed to the optical interference of fluorescent proteins with strong signals (GFP and mCherry) in the bandpass filters used for other fluorescent proteins, especially for TagBFP which presented lowest signals. In addition, GFP exhibited better sensitivity to concentration variation compared with mCherry because the slope of the fitting curve for GFP (Figure 3a) is greater than that for mCherry (Figure 3b). As a result, GFP fluorescent protein was considered most sensitive and used as a reliable indicator of expression level in the following multi-parametric characterization of transcription/translation regulation in a synthetic cell-free system, while the fitting curve generated in Figure 3a was used for back calculation of GFP expression level in the biological experiments.
Figure 4.

Multi-dimensional dilution using fluorescence proteins. a). Illustration of the combinatorial three-dimensional dilution in one array where TagBFP, mCherry, and GFP are diluted by 100 fold, 1000 fold and 10,000 fold, respectively. Microscopic picture taken with b) TagBFP filter, c) TexasRed filter, and d) GFP filter. e). Overlapped figure of b)–d).
Biological application: multi-parametric characterization of transcription/translation regulation in a synthetic cell-free system
Based on the established gradient printer, we demonstrated multi-parametric gradient generation for multi-factorial characterization of transcription regulation processes in a miniaturized synthetic cell-free system.34 A schematic of the transcription and translation process in a synthetic cell-free system is illustrated in Figure 5a. A transcription factor, EsaR, represses the transcription by binding to a specific sequence of DNA and reducing the rate of gene transcription.35 The repression function of EsaR is inhibited by acyl-homoserine lactone (AHL).4 Here, we studied the molecular interaction of GFP-encoding plasmid, EsaR, and AHL by implementing combinatorial large ratio dilution of the three molecules in a synthetic cell-free system and used the level of GFP expression as an indicator of transcription efficiency. The limit of concentration gradient depended on the value of total volume divided by volume resolution, thus the maximal dilution was limited within 1,000-fold, which still outperforms current automation technologies.3, 8, 9, 15 As described in the Figure 1b, for each reaction well, Plasmid/EsaR/AHL droplets were first printed at desired volume, followed by printing of RNase free water as a diluent to reach 97nL, and adding both 453nL transcription-translation reaction mix and 300nL PCR oil at the end before moving to the next well.
Figure 5.

a) Schematic of the transcription/translation process in a cell-free system, illustrating the interactions of plasmids, repressor EsaR,
One-dimensional and two-dimensional studies
To establish the miniaturized reaction system, we first examined the impact of plasmid concentration on gene expression (Figure 5b). In the experiment for one parameter, we tested ten plasmid concentrations ranging from 0.01 nM to 10 nM (1,000 fold) with 3 technical replicates on the same array. Fluorescence generated by expressed GFP was measured after 4-hour incubation at 37°C. We observed that the GFP expression level increased monotonically with plasmid concentration (Figure 5b). A linear increasing range existed from 1nM to 6nM. Above 6nM, the fluorescent signal started to saturate in our optical detector. Below 1nM, expression level was low and the relationship was no longer linear.
Next, we measured the EsaR concentration that will modulate transcriptional response. We performed a two-dimensional concentration characterization with 600-fold variation of plasmid concentration along with 10-fold variation of EsaR concentration. 55 reactions with 3 replicates were conducted on one multi-well array, followed by 4-hour incubation. As shown in Figure 5c, only plasmid concentration above 0.72nM would generate a transcriptional response to EsaR in our synthetic cell-free system. We further plotted the data as fluorescent intensity against EsaR/plasmid concentration ratio (Figure 5d). Using the microfluidic multi-parametric gradient generation system, a 10,000-fold change of EsaR/plasmid ratio has been achieved, showing a monotonically decreasing relationship with fluorescent intensity. As expected, higher ratios (more EsaR dimers per DNA molecule) led to higher inhibition of the transcription process. The inhibition efficacy saturated at the ratio of 300. Furthermore, if EsaR/plasmid ratio was lower than 20, EsaR did not inhibit gene expression in our system. This 2-dimensional analysis established function of EsaR in cell-free systems.
Three-dimensional characterization
To obtain a biophysical model of the genetic module (Fig. 6a), a three-dimensional characterization experiment was conducted with 16-fold variation of plasmids (0.8nM–12.7nM), 64-fold variation of EsaR (0.08μM to 5.4μM), and 128-fold variation of AHL (0.098μM to 12.5μM) in a single array. We chose 3 plasmid concentrations (12.7nM, 3.2nM, 0.8nM) corresponding to different regions of interest on Figure 5b (saturation, linear, low). 170 reactions have been conducted on one multi-well array with 3 repeats (510 reactions in total). Approximately 1,000 fold change of AHL/EsaR ratio was achieved using the printer. As expected, the results indicated that GFP intensity increased with higher promoter copy number, decreased with higher EsaR concentration, and increased with higher AHL concentration. In addition, the multi-dimensional data were used to fit a biophysical model (Eq. 1) using the nonlinear fitting function in Matlab.
Figure 6.

Combinatorial variation of three regulatory factors generated high-throughput data for fitting of a biophysical model. a) A schematic of the genetic module. (b) Higher EsaR concentration led to less GFP intensity. Higher copy number of the promoter led to higher GFP intensity. (c) Higher AHL concentration led to higher GFP intensity. The color bars indicated GFP concentrations in μM.
| [Eq. 1] |
The model structure was formulated based on the assumption that EsaR dimer forms a complex with DNA to inhibit GFP transcription and that AHL binds to EsaR to inhibit its repression function (Figure 6a). The results demonstrated the ability of the cell-free array in generating multi-parameteric and multidimensional data that can be used to identify mathematical model of gene regulatory modules.
Conclusions
A multiplexed, scalable, and compact microfluidic multi-parametric gradient generation system has been established and demonstrated for wide-range, multi-parametric, low-consumption, and high-throughput quantitative analysis of a synthetic genetic module.31 Modular design of the cartridge and control system enabled easy extension of channel numbers, which facilitated multi-parametric parallel study of complex biomolecular reaction systems. The system generated wide-range concentration gradient across 10,000-fold of fluorescent proteins (including GFP, mCherry, and TagBFP) and their mixtures. Finally, this system was used to measure multi-parametric interactions in a synthetic cell-free system, which yielded a quantitative model for the interaction between a promoter, a transcription factor, and a co-activator.
The other main advantage of our method, compared with continuous microfluidic methods, is the reduction of sample consumption, due to the on-demand printing nature. 31, 32,36, 37 Since the microfluidic multi-parametric gradient generation system was essentially a drop-on-demand printing system, the calibration step usually consumed only 5 droplets (less than 100nL) but worked for an arbitrary number of target concentrations, significantly reducing the waste of reagents. In addition, due to the microfluidic channel design and hydrophilic coating, the minimal loading volume into a single channel was as low as 2μL, with a dead volume as low as 0.25μL at the tip of cartridge (illustrated in Figure S3 in supplemental materials), both significantly reduced compared with previous nano-liter dispensing technologies38–42. The non-contact and disposable nature of the impact printing cartridge also has great potential in reducing cross-contamination during experiment and cutting down maintenance costs. As a reference for future bio-printing applications, the proposed platform also reduces several shortcomings of fluid handling at small scales that plague all this kind of printing technologies: evaporation, minimum droplet size limitations, droplet size variability, and clogging.43 Evaporation issue is reduced by increasing speed, printing a cover layer of oil and control of ambient humidity. Minimal droplet size is pushed down to 0.1nL by manipulating nozzle diameters, driven waveform and liquid viscosity. The CV of droplet volume measured was as low as 3.5% using this piezoelectric and microfluidic impact printing configuration. Thanks to the disposable property of cartridges, channel clogging is no longer a critical issue, minimizing the waste of time for instrument maintenance.
For synthetic biology, the platform demonstrates, for the first time, a modular and rapid ex vivo method that enables multi-parametric studies of synthetic genetic modules. This platform resolves a critical technological challenge in the field of synthetic biology by allowing for the generation of high-throughput and high-content information about complex promoters that are regulated by multiple cellular factors. In addition, our platform enables high throughput, quantitative analysis of potential therapeutics ex vivo prior to their use in vivo. For instance, the design of effective CRIPSR/Cas systems with low off-target effects currently requires extensive tests using mammalian cells, which are time-consuming and expensive. The platform may be used to speed up the experimentation and validation of CRISPR/Cas ex vivo by simultaneously testing interactions between different CRISPR/Cas designs with multiple DNA targets. In addition, dynamics of synthetic genetic modules are generally known to change in different host environments. Along this line, the platform may be used to identify the host factors that govern context-dependent functions of synthetic genetic modules in a high-throughput manner. These potential applications are made possible due to the modularity of the platform that can be adapted to different studies of genetic promoters.
Supplementary Material
Acknowledgments
This research work has been supported in part by the National Science Foundation Awards ECCS-0846502 and DBI-1256193 to TP. JF acknowledges a traineeship support from National Institutes of Environmental Health Sciences of NIH Superfund Research Program (No. P42ES004699). BL acknowledges a grant support from National Natural Science Foundation of China (No. 51675505). CT acknowledges a grant support from the Branco-Weiss Fellowship. Authors would like to acknowledge Ray Lin for his assistance in software programming and circuit board design and Samantha Kennedy for her assistance in mechanical printer-head designs.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Harris DC, Jewett MC. Curr Opin Biotech. 2012;23:672–678. doi: 10.1016/j.copbio.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villarreal F, Tan C. Frontiers of Chemical Science and Engineering. 2017;11:58–65. [Google Scholar]
- 3.Stogbauer T, Windhager L, Zimmer R, Radler JO. Integrative Biology. 2012;4:494–501. doi: 10.1039/c2ib00102k. [DOI] [PubMed] [Google Scholar]
- 4.Chappell J, Jensen K, Freemont PS. Nucleic Acids Res. 2013;41:3471–3481. doi: 10.1093/nar/gkt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garamella J, Marshall R, Rustad M, Noireaux V. Acs Synth Biol. 2016;5:344–355. doi: 10.1021/acssynbio.5b00296. [DOI] [PubMed] [Google Scholar]
- 6.Noireaux V, Bar-Ziv R, Godefroy J, Salman H, Libchaber A. Phys Biol. 2005;2:P1–P8. doi: 10.1088/1478-3975/2/3/P01. [DOI] [PubMed] [Google Scholar]
- 7.Dittrich PS, Manz A. Nat Rev Drug Discov. 2006;5:210–218. doi: 10.1038/nrd1985. [DOI] [PubMed] [Google Scholar]
- 8.Dittrich PS, Jahnz M, Schwille P. Chembiochem. 2005;6:811-+. doi: 10.1002/cbic.200400321. [DOI] [PubMed] [Google Scholar]
- 9.Mei Q, Fredrickson CK, Simon A, Khnouf R, Fan ZH. Biotechnol Progr. 2007;23:1305–1311. doi: 10.1021/bp070133p. [DOI] [PubMed] [Google Scholar]
- 10.Gulati S, Rouilly V, Niu XZ, Chappell J, Kitney RI, Edel JB, Freemont PS, Demello AJ. J R Soc Interface. 2009:6. doi: 10.1098/rsif.2009.0083.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li GW, Xie XS. Nature. 2011;475:308–315. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharp PA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Tan C, Marguet P, You LC. Nat Chem Biol. 2009;5:842–848. doi: 10.1038/nchembio.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeger-Lotem E, Sattath S, Kashtan N, Itzkovitz S, Milo R, Pinter RY, Alon U, Margalit H. P Natl Acad Sci USA. 2004;101:5934–5939. doi: 10.1073/pnas.0306752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Angenendt P, Nyarsik L, Szaflarski W, Glokler J, Nierhaus KH, Lehrach H, Cahill DJ, Lueking A. Anal Chem. 2004;76:1844–1849. doi: 10.1021/ac035114i. [DOI] [PubMed] [Google Scholar]
- 16.Sun K, Wang ZX, Jiang XY. Lab Chip. 2008;8:1536–1543. doi: 10.1039/b806140h. [DOI] [PubMed] [Google Scholar]
- 17.Fan JZ, Li BQ, Xing SY, Pan TR. Lab Chip. 2015;15:2670–2679. doi: 10.1039/c5lc00432b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedges AJ. Int J Food Microbiol. 2002;76:207–214. doi: 10.1016/s0168-1605(02)00022-3. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi Y, Matsuda R. Anal Sci. 1994;10:881–888. [Google Scholar]
- 20.Grgicak CM, Urban ZM, Cotton RW. J Forensic Sci. 2010;55:1331–1339. doi: 10.1111/j.1556-4029.2010.01460.x. [DOI] [PubMed] [Google Scholar]
- 21.Lee K, Kim C, Kim Y, Jung K, Ahn B, Kang JY, Oh KW. Biomed Microdevices. 2010;12:297–309. doi: 10.1007/s10544-009-9385-6. [DOI] [PubMed] [Google Scholar]
- 22.Jeon NL, Dertinger SKW, Chiu DT, Choi IS, Stroock AD, Whitesides GM. Langmuir. 2000;16:8311–8316. [Google Scholar]
- 23.Lee K, Kim C, Ahn B, Panchapakesan R, Full AR, Nordee L, Kang JY, Oh KW. Lab Chip. 2009;9:709–717. doi: 10.1039/b813582g. [DOI] [PubMed] [Google Scholar]
- 24.Seidi A, Kaji H, Annabi N, Ostrovidov S, Ramalingam M, Khademhosseini A. Biomicrofluidics. 2011:5. doi: 10.1063/1.3580756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng B, Roach LS, Ismagilov RF. J Am Chem Soc. 2003;125:11170–11171. doi: 10.1021/ja037166v. [DOI] [PubMed] [Google Scholar]
- 26.Cai LF, Zhu Y, Du GS, Fang Q. Anal Chem. 2012;84:446–452. doi: 10.1021/ac2029198. [DOI] [PubMed] [Google Scholar]
- 27.Leung K, Zahn H, Leaver T, Konwar KM, Hanson NW, Page AP, Lo CC, Chain PS, Hallam SJ, Hansen CL. P Natl Acad Sci USA. 2012;109:7665–7670. doi: 10.1073/pnas.1106752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu SW, Xu BY, Xu JJ, Chen HY. Biomicrofluidics. 2013;7:64116. doi: 10.1063/1.4847815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bsoul A, Pan S, Cretu E, Stoeber B, Walus K. Lab Chip. 2016;16:3351–3361. doi: 10.1039/c6lc00636a. [DOI] [PubMed] [Google Scholar]
- 30.Teh SY, Lin R, Hung LH, Lee AP. Lab Chip. 2008;8:198–220. doi: 10.1039/b715524g. [DOI] [PubMed] [Google Scholar]
- 31.La D, Ciardella RL, Babiarz AJ, Bouras CE. Google Patents. 1994. [Google Scholar]
- 32.Li B, Fan J, Li J, Chu J, Pan T. Biomicrofluidics. 2015;9:054101. doi: 10.1063/1.4928298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Biophys J. 1997;73:2782–2790. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodgman CE, Jewett MC. Metab Eng. 2012;14:261–269. doi: 10.1016/j.ymben.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schu DJ, Carlier AL, Jamison KP, von Bodman S, Stevens AM. J Bacteriol. 2009;191:7402–7409. doi: 10.1128/JB.00994-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsieh YL, Ho TY, Chakrabarty K. Ieee T Comput Aid D. 2012;31:1656–1669. [Google Scholar]
- 37.Huang JD, Liu CH, Lin HS. Ieee T Comput Aid D. 2013;32:1484–1494. [Google Scholar]
- 38.Smith KP, Kirby JE. J Clin Microbiol. 2016;54:2288–2293. doi: 10.1128/JCM.00932-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose D. Drug Discov Today. 1999;4:411–419. doi: 10.1016/s1359-6446(99)01388-4. [DOI] [PubMed] [Google Scholar]
- 40.Kong FW, Yuan L, Zheng YF, Chen WD. Jala-J Lab Autom. 2012;17:169–185. doi: 10.1177/2211068211435302. [DOI] [PubMed] [Google Scholar]
- 41.Dutka F, Opalski AS, Garstecki P. Lab Chip. 2016;16:2044–2049. doi: 10.1039/c6lc00265j. [DOI] [PubMed] [Google Scholar]
- 42.Vaughn BS, Tracey PJ, Trevitt AJ. Rsc Adv. 2016;6:60215–60222. [Google Scholar]
- 43.Oosterbroek E, Van den Berg A. Lab-on-a-chip: miniaturized systems for (bio) chemical analysis and synthesis. Elsevier; 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.