

Abstract
Novel elongated and shortened derivatives of the peptidomimetic furin inhibitor phenylacteyl-Arg-Val-Arg-4-amidinobenzylamide were synthesized. The most potent compounds, e.g., Nα(carbamidoyl)Arg-Arg-Val-Arg-4-amidinobenzylamide (Ki = 6.2 pM), contain additional basic residues at the N-terminus and inhibit furin in the low picomolar range. Furthermore, to decrease the molecular weight of this inhibitor type, compounds lacking the P5-moiety were prepared. The best inhibitors of this series, 5-(guanidino)-valeroyl-Val-Arg-4-amidinobenzylamide and its P3 tert.leucine analogue, displayed Ki values of 2.50 nM and 1.26 nM, respectively. Selected inhibitors, together with our previously described 4-amidinobenzylamide derivatives as references, were tested in cell culture for their activity against furin-dependent infectious pathogens. The propagation of the alphaviruses Semliki Forest virus and chikungunya was strongly inhibited in the presence of selected derivatives. Moreover, a significant protective effect of the inhibitors against diphtheria toxin was observed. These results confirm that the inhibition of furin should represent a promising approach for a short-term treatment of acute infectious diseases.
Keywords: furin, inhibitors, alphavirus, chikungunya virus, Semliki forest virus, diphtheria toxin
TOC image
Elongated and shortened inhibitors of the proprotein convertase furin, based on the lead structure phenylacetyl-Arg-Val-Arg-4-amidinobenzylamide, have been developed. The best compounds inhibit the propagation of the alphaviruses chikungunya and Semliki forest virus in the micromolar range. Furthermore, the activation of diphtheria toxin was inhibited in the presence of selected compounds.
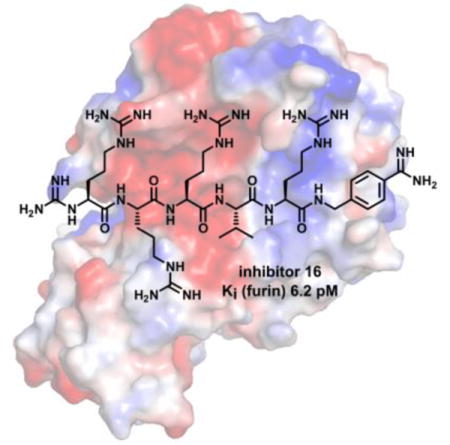
Introduction
The type-I transmembrane protein furin belongs to the family of proprotein convertases (PCs) and contains a Ca2+-dependent subtilisin-like serine protease domain. Furin is ubiquitously distributed in human tissues and preferentially cleaves its substrates after the multibasic motif Arg-Xaa-(Lys/Arg)-Arg↓Xaa.[1] Furin activates numerous precursors of hormones, enzymes and other proteins. Prominent cellular substrates are the precursor to insulin-like growth factor, albumin and the transforming growth factor β1.[2] In addition to its physiological functions, furin also contributes to numerous diseases such as cancer, metabolic disorders, osteoarthritis, cardiovascular diseases or neurodegenerative disorders.[3–5] Importantly, cellular furin can be hijacked by a number of pathogenic viruses and bacteria. Furin activates the fusogenic surface glycoproteins of several viruses including those from highly pathogenic influenza viruses, measles, ebola, and flaviviruses like Dengue and West Nile virus.[6] Furin also cleaves and thereby activates several bacterial toxin precursors, including anthrax, diphtheria or shiga toxins.[1,4,5] Although a furin knockout in mice leads to embryonic death at day 11, a short-term application of furin inhibitors in severe infectious diseases could represent a potential treatment option,[1] because no obvious adverse effects were observed in adult mice with a liver-specific conditional furin knockout, suggesting some redundancy between furin-like PCs.[7] In initial studies with living rodents, a protective effect of the furin inhibitor hexa-D-arginine against Pseudomonas aeruginosa exotoxin A and anthrax toxin was observed.[8] Several furin inhibitors have been published in the past, including engineered proteins, oligopeptides and small molecule inhibitors.[5,9,10]
We have previously developed various substrate-analogue furin inhibitors based on the lead structure phenylacetyl-Arg-Val-Arg-4-amidinobenzylamide (inhibitor 1, Figure 1).[11,12] Further optimization by incorporation of basic P5 groups in combination with tert.leucine (Tle) in P3 position has provided furin inhibitors with strongly improved potency, such as compounds 2 and 3 (Figure 1).[13–15] The analysis of their crystal structures in complex with human furin revealed numerous polar interactions contributing to their low picomolar affinity.[13,14] The best compounds produced a strong antiviral effect against infections with influenza virus and the paramyxovirus canine distemper virus in cell culture and also inhibited the activation of toxins from Bacillus anthracis, Shigella dysenteriae and Corynebacterium diphtheria.[11,13,16]
Figure 1.
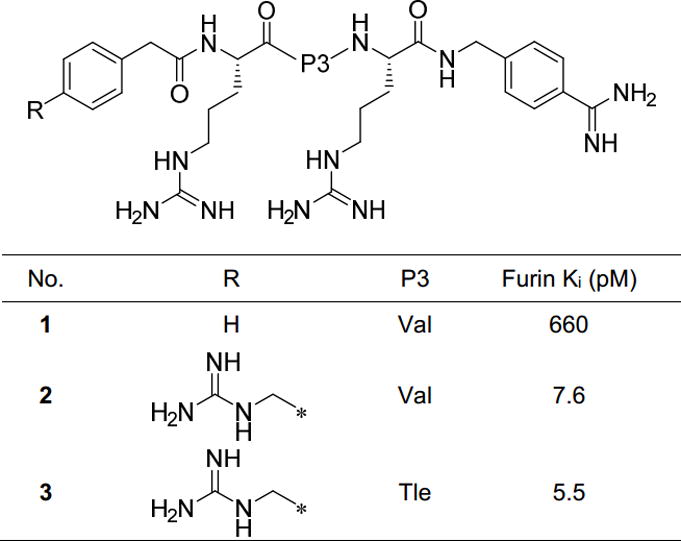
Known 4-Amba derived furin inhibitors 1–3 used as reference compounds.[13,16]
Numerous PC substrates[2] and inhibitors[17,18] possess additional basic residues in their P5-P8 region contributing to their recognition by furin. For this reason, a series of new N-terminally elongated inhibitors was prepared which contain a constant P4-P1 Arg-Xxx-Arg-4-Amba segment (where Xxx stands for valine or tert.leucine). In contrast, to decrease the molecular weight of this inhibitor type, additional compounds lacking the P5-moiety were synthesized. A significant antiviral and antibacterial effect was found in cell culture assays for selected inhibitors, as described below.
Results
Design and activity of the synthesized compounds
The inhibitors were prepared using a combination of solution and solid phase peptide synthesis, as described previously.[11] The side chain-protected peptide segment beginning from the P2 residue was synthesized on 2-chlorotrityl chloride resin according to a standard Fmoc-protocol with HBTU as the coupling reagent. This segment was cleaved from the resin under mild acidic conditions and coupled with 4-amidinobenzylamine · 2 HCl in solution. After strong acidic deprotection, the inhibitors were purified by preparative HPLC and lyophilized.
Various modifications of this synthesis were performed; the sequences of the peptides prepared are summarized in Table 1. The P4-P1 Arg-Val-Arg-4-Amba segment was elongated with further arginines in L- and/or D-configuration. Compounds 4 and 5 possess the basic structure Phac-(Arg)x-Val-Arg-4-Amba, where × is 2 (4) or 3 (5). In analogues 6 and 7, one Arg residue in position P5 or P6 was replaced by Ala in order to examine the importance of a basic residue in these positions.
Table 1.
Analytical characterization of the inhibitors and their potency against furin. Abbreviations of unusual residues are shown below the table.
| No. | Sequence | HPLC (min) | MS calc. | MS found (M+H)+ | Furin Ki (pM)[c] | R2[d] |
|---|---|---|---|---|---|---|
| 4 | Phac-Arg-Arg-Val-Arg-4-Amba | 23.1 | 834.51 | 835.33 | 56.3 ± 7.7[b] | 0.996 |
| 5 | Phac-Arg-Arg-Arg-Val-Arg-4-Amba | 22.5 | 990.61 | 496.27[a] | 13.8 ± 4.5[b] | 0.993 |
| 6 | Phac-Arg-Ala-Arg-Val-Arg-4-Amba | 21.6 | 905.55 | 906.54 | 45.9 ± 12.5[b] | 0.998 |
| 7 | Phac-Ala-Arg-Arg-Val-Arg-4-Amba | 23.8 | 905.55 | 453.73[a] | 49.1 ± 19.2[b] | 0.997 |
|
| ||||||
| 8 | H-Arg-Arg-Arg-Val-Arg-4-Amba | 15.9 | 872.57 | 437.29[a] | 33.7 ± 1.3[b] | 0.987 |
| 9 | H-DArg-Arg-Arg-Val-Arg-4-Amba | 16.3 | 872.57 | 873.43 | 28.3 ± 9.7[b] | 0.991 |
| 10 | H-DArg-DArg-Arg-Val-Arg-4-Amba | 15.5 | 872.57 | 873.53 | 51.0 ± 6.7[b] | 0.988 |
| 11 | H-DArg-DArg-DArg-Arg-Val-Arg-4-Amba | 16.5 | 1028.67 | 1029.66 | 75.4 ± 10.6[b] | 0.987 |
| 12 | H-DArg-DArg-DArg-DArg-Arg-Val-Arg-4-Amba | 17.1 | 1184.77 | 593.53[a] | 94.3 ± 24.3[b] | 0.979 |
| 13 | H-DArg-Arg-Val-Arg-4-Amba | 15.8 | 716.47 | 359.37[a] | 108 ± 61[b] | 0.967 |
| 14 | H-Arg-Arg-Val-Arg-4-Amba | 15.6 | 716.47 | 359.37[a] | 110 ± 10[b] | 0.974 |
| 15 | H-DArg-Arg-Tle-Arg-4-Amba | 17.1 | 730.48 | 366.39[a] | 37.8 ± 19.5[b] | 0.981 |
| 16 | N(Ca)Arg-Arg-Arg-Val-Arg-4-Amba | 17.0 | 914.59 | 458.38[a] | 6.2 ± 1.0[b] | 0.993 |
| 17 | N(Ca)Arg-Ala-Arg-Val-Arg-4-Amba | 16.8 | 829.53 | 415.87[a] | 11.9 ± 3.4[b] | 0.996 |
|
| ||||||
| 18 | Ac-Leu-Leu-Leu-Leu-Arg-Val-Lys-4-Amba | 38.1 | 1026.71 | 1027.80 | 491 ± 70 | |
| 19 | Ac-DLeu-Leu-Leu-Leu-Arg-Val-Lys-4-Amba | 40.1 | 1026.71 | 514.74[a] | 449 ± 34 | |
|
| ||||||
| 20 | Ac-Leu-Leu-Leu-Leu-Arg-Tle-Lys-4-Amba | 38.7 | 1040.72 | 1041.95 | 193 ± 33 | |
| 21 | Ac-Leu-Leu-Leu-Leu-Arg-Val-Arg-4-Amba | 38.6 | 1054.71 | 1055.8 | 164 ± 21[b] | 0.996 |
| 22 | Ac-Leu-Leu-Leu-Leu-Arg-Tle-Arg-4-Amba | 39.5 | 1068.73 | 1069.8 | 78.6 ± 8.4[b] | 0.998 |
|
| ||||||
| 23 | 3-AMe-Phac-Val-Arg-4-Amba | 19.3 | 551.33 | 552.40 | 48 200 ± 13 600 | |
| 24 | 4-AMe-Phac-Val-Arg-4-Amba | 18.6 | 551.33 | 552.30 | 37 800 ± 12 800 | |
| 25 | 3-GMe-Phac-Val-Arg-4-Amba | 20.8 | 593.36 | 594.30 | 164 000 ± 21 200 | |
| 26 | 4-GMe-Phac-Val-Arg-4-Amba | 20.1 | 593.36 | 594.20 | 269 300 ± 25 600 | |
| 27 | 5-Ava-Val-Arg-4-Amba | 16.0 | 503.33 | 504.10 | 38 500 ± 2 900 | |
| 28 | 5-Gva-Val-Arg-4-Amba | 17.7 | 545.36 | 546.33 | 2 500 ± 300 | |
| 29 | 5-Ava-Tle-Arg-4-Amba | 17.6 | 517.35 | 518.15 | 26 700 ± 900 | |
| 30 | 5-Gva-Tle-Arg-4-Amba | 19.3 | 559.37 | 560.24 | 1 260±100 | |
(M + 2H)2+/2
Ki values were determined under tight-binding conditions (for inhibitors with Ki values < 170 pM).
Data represent the mean ± SD obtained by at least three independent measurements.
Coefficient of determination
Used abbreviations: Ac, acetyl; AMe-Phac, aminomethyl-phenylacetyl; 5-Ava, 5-aminovaleroyl; N(Ca)Arg, Nα(carbamidoyl)-arginine; GMe-Phac, guanidinomethyl-phenylacetyl; Gva, 5-guanidinovaleroyl; Phac, phenylacetyl; Tle, tert.leucine or tert.butylglycine.
In contrast, compounds 8–15 possess a free amino group at the N-terminus. To prevent potential degradation by aminopeptidases, a DArg residue was coupled as the N-terminal residue in most analogues, a strategy known from the development of icatibant, a bradykinin B2 receptor antagonist approved for the treatment of hereditary angioedema.[19] Inhibitors 8 and 14 containing an N-terminal Arg served as reference compounds.
The Ki values of the compounds above against furin were determined under tight-binding conditions; all represented very effective inhibitors.[20] Compound 5, containing three Arg residues in positions P4-P6, is the most potent derivative of this series (Ki = 13.8 pM). The shorter analogue 4 and derivatives 6 and 7, containing Arg-Ala or Ala-Arg as P5-P6 segment, inhibit furin with slightly reduced Ki values close to 50 pM. However, all of these analogues revealed reduced potency compared to inhibitor 2, which possesses a 4-(guanidinomethyl)phenylacetyl residue in position P5. Fitting accuracy of the measured vs, [I] data pairs to equation (1) for tight-binding inhibitors was lower in the case of compounds 8–15, in particular in the “elbow region” (Figure 2). Yet, curve fitting with tight-binding inhibitors is very sensitive to small errors in the enzyme and/or inhibitor concentrations used. However, a perfect fit was always obtained for inhibitor 3, which was used as a reference on each 96-well plate. These control measurements confirm the correctness of the enzyme concentration used, which was in agreement with a determination made from the point of intersection of the elongated linear part of the tight-binding curve with the abscissa at low inhibitor concentrations.[20] Moreover, to avoid any inaccuracy in the concentrations of the inhibitors their peptide content was determined from the amount of nitrogen obtained by elemental analysis. When considering the appropriate number of TFA counter ions (one TFA molecule per basic group) the determined peptide concentrations were normally in the range between 90 and 95 %. However, even with the corrected inhibitor concentrations, no perfect fitting was possible as indicated by the slightly reduced coefficient of determination (R2) with values between 0.965 and 0.99 (Table 1). In these cases (inhibitors 8–15), the measured data points in the elbow region and at higher inhibitor concentration are below the fitted curve, which indicates that the true inhibitory potency should be even stronger as calculated by the Ki values. We dispensed with the determination of IC50 values, which are often used to describe the potency of inhibitors with unknown binding mechanisms, because it is not recommended for tight-binding inhibitors.[20]
Figure 2.
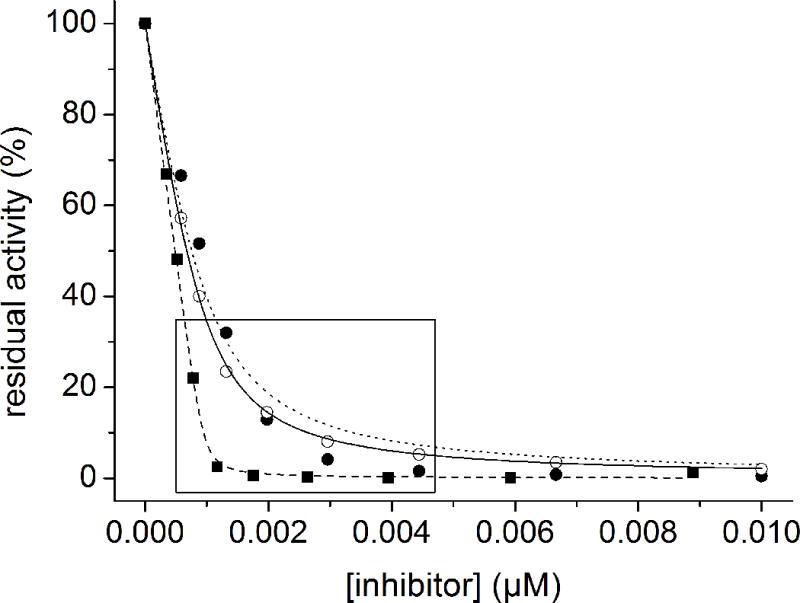
Inhibition of furin (0.95 nM in assay) by compounds 3, 4 and 12 in the presence of the substrate Phac-Arg-Val-Arg-Arg-AMC (12.5 μM in assay). The data were fitted to the equation for reversible tight-binding inhibition (equation 1). The calculated curve of derivative 12 (●, dotted line) deviates significantly from the measured data in the “elbow region” (marked by the box, R2 = 0.974), whereas the calculated curve of inhibitor 4 (○, solid line) and reference compound 3 (■, dashed line; R2 = 0.999) fit well with the experimental data (R2 = 0.999).
The Ki values of derivatives 8 and 9 with an Arg residue in position P5 exhibit a slightly stronger inhibition compared to the elongated compounds 10–12 with DArg at P5. The D-configuration should alter the substrate-analogue backbone conformation and thereby also the side chain orientation, resulting in impaired interactions in the more distal N-terminal binding pockets. In contrast, an identical potency was found for the shorter compounds 13 and 14 comprising only the P5-P1 segment.
To prevent potential degradation by aminopeptidases, the N-terminal α-amino group of compounds 16 and 17 was alternatively converted into a guanidine moiety. The derivative Nα(carbamidoyl)-Arg-Arg-Arg-Val-Arg-4-Amba (compound 16) with a Ki value of 6.2 pM is the most potent inhibitor of this series.
Based on the work of the Day lab inhibitors containing an N-terminal tetra-Leu motif including the previously described inhibitors 18 and 19 were synthesized as further reference compounds.[21,22] Under the used conditions derivative 18 (Ac-(Leu)4-Arg-Val-Lys-4-Amba) and its N-terminal Ac-DLeu analogue 19 inhibit furin with nearly identical Ki values of 491 pM and 449 pM, respectively. Derivative 20, the P3 Tle analogue of compound 18, exhibited a 2.5-fold improved inhibition constant of 193 pM. In inhibitors 21 and 22 the P2 Lys was exchanged with Arg; their ~ 2-fold improved Ki values (164 and 79 pM) confirm previous results obtained with related 4-amidinobenzylamide inhibitors.[11,12]
To complement the size variations, we also attempted to reduce the length of the compounds. For this work, the P5 residue was completely eliminated and the P4 position was modified with various basic residues. In the case of inhibitors 23 and 24, the P3-P1 segment (Val-Arg-4-Amba) was maintained and 3- or 4-aminomethylphenylacetic acid was coupled. Both compounds revealed relatively poor inhibition (Ki values of 48.2 nM and 37.8 nM) compared to the reference compound 1 (which contains a phenylacetyl moiety in P5- and arginine in the P4-position).[11] The conversion of the N-terminal benzylamine into benzylguanidine resulted in a further drop in the potency of compounds 25 and 26. This suggests that the N-terminal guanidinomethylene group on the relatively rigid Phac residue cannot bind to the same position occupied by the P4 arginine side chain in substrate analogue inhibitors.[13,14]
To overcome this problem a more flexible 5-aminovaleroyl residue was incorporated in inhibitors 27 and 29, which was further converted into a 5-guanidinovaleroyl group in compounds 28 and 30. This residue can be considered as a des-aminoArg, and provided significantly stronger furin inhibition with Ki values of 2.54 nM and 1.26 nM for the Val and Tle analogues, respectively.
Antiviral and antibacterial activities in cell culture
In previous cell culture experiments, we have observed that selected 4-amidinobenzylamide-type furin inhibitors exhibit strong antiviral activity against highly pathogenic avian influenza viruses of the strains H5N1 and H7N1 as well as against canine distemper virus.[13,23] Furthermore, these compounds strongly protected cells against anthrax, shiga and diphtheria toxin in cell culture.[16] We wished to extend these studies to alphaviruses.
Inhibition of alphavirus propagation
Alphaviruses are mosquito-transmitted pathogens. Some of them, such as chikungunya virus (CHIKV) and Ross River virus, cause high fever and debilitating joint pain in humans, whereas others like Semliki Forest virus (SFV) are low-pathogenic in humans albeit neuropathogenic in mice. Despite differences in pathogenicity, the alphaviruses are closely related in terms of their molecular biology regarding e.g. replication and protein processing.[24] In the case of SFV and CHIKV, furin cleaves the precursor protein E3-E2 (p62) in a post-Trans Golgi network compartment, generating the mature glycoprotein E2.[25,26] Furin cleavage is a prerequisite for formation of the functional membrane glycoprotein complex E1/E2 that mediates receptor binding and membrane fusion. The inhibition of this processing step could be a suitable approach for the treatment of infections with alphaviruses.
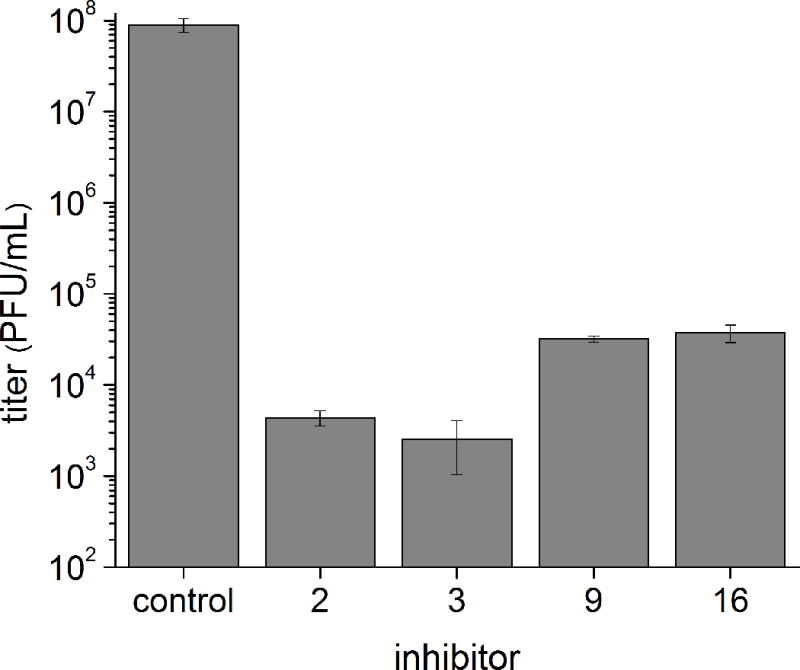
Due to its low pathogenicity, SFV can serve as a convenient model system to study alphaviruses. In a first experiment the reference inhibitors 2 and 3, along with the strongest furin inhibitors of each subgroup (separated by the dashed lines in Table 1, e.g., compound 5 from inhibitors 4–7, compound 9, 16, and 17 from inhibitors 8–17, compounds 21 and 22 from the multi-Leu inhibitors 18–22, and compound 30 from inhibitors 23–30) were screened at a concentration of 25 μM in baby hamster kidney (BHK-21) cells infected with SFV. Virus titers were determined by plaque assay, results are displayed in Figure 3.
Figure 3.

Infection of BHK-21 cells with SFV at a multiplicity of infection (MOI) of 0.01 in presence of the indicated furin inhibitors (25 μM in assay). Viral titers were determined by plaque assay 24 h post-infection. The dashed line illustrates the control titer in the absence of a furin inhibitor. Data represent the mean ± SE for three independent experiments.
No or only negligible reduction in virus titers was found in presence of inhibitors 21, 22 and 30. A relatively weak effect was achieved with inhibitors 5, 9 and 17, which reduced the virus titers by less than one order of magnitude. The strongest antiviral efficacies were determined for the new inhibitor 16 and the reference compounds 2 and 3, where viral titers were severely reduced by more than two orders of magnitude. Under these inhibitor concentrations and conditions, the cells did not exhibit any significant cytopathic effect at 24 h post-infection. The most potent inhibitors 2, 3, 9 and 16 were analyzed further and showed concentration-dependent inhibition of SFV replication (Figure 4).
Figure 4.
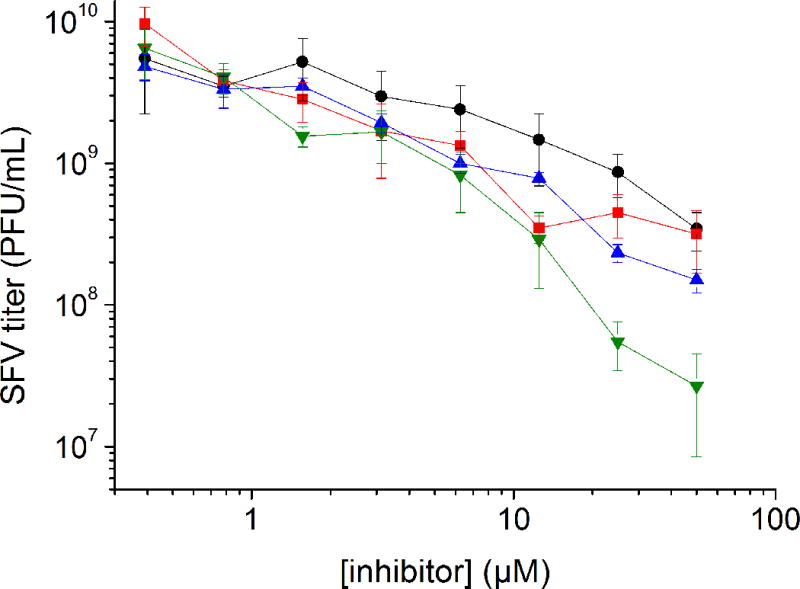
Infection of BHK-21 cells with SFV at an MOI of 0.01 in the presence of the selected furin inhibitors 2 (
 ), 3 (
), 3 (
 ), 9 (
), 9 (
 ) and 16 (●). Titers were determined by plaque assay at 24 h post-infection. Data represent the mean ± SE for three independent experiments.
) and 16 (●). Titers were determined by plaque assay at 24 h post-infection. Data represent the mean ± SE for three independent experiments.
To show inhibition of furin-mediated E3-E2 processing by the selected compounds, BHK cells were infected with SFV at high MOI, subjected to drug treatment and processed for gel electrophoresis and Western blot analysis at 8 h post-infection (Figure 5).
Figure 5.
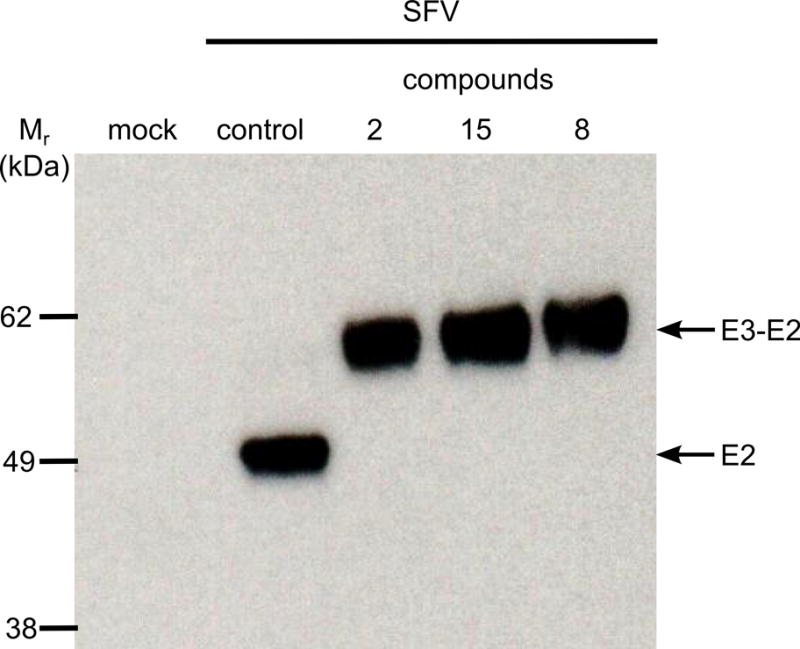
Inhibition of E3-E2 (p62) cleavage by compounds 3, 9 and 16 (25 μM in assay) compared to a control in the absence of inhibitor. BHK-21 cells were infected with SFV at an MOI of 10. Proteins E3-E2 and mature E2 (49 kDa) were immunochemically detected with an E2 antibody after gel electrophoresis followed by Western blot analysis. Relative molecular mass of marker proteins displayed on the left-hand side.
While only mature E2 was detected in SFV-infected cells in the absence of inhibitor, the cleavage of the precursor E3-E2 was very strongly suppressed by all three inhibitors under the conditions used since only the uncleaved precursor could be detected. Subsequently, the most potent inhibitors identified by SFV screening were tested in BHK-21 cells infected with the human-pathogenic CHIKV (Figure 6).
Figure 6.
Infection of BHK-21 cells with CHIKV in the presence of the indicated furin inhibitors (25 μM) at an MOI of 0.1. The titers at 24 h post-infection were determined by plaque assay. Data represent the mean ± SE obtained of at least three independent experiments.
In the presence of these inhibitors, a significant reduction in the virus titer by more than three orders of magnitude was observed. In agreement with the SFV assay data, compounds 9 and 16 were less active compared to the reference inhibitors 2 and 3.
Inhibition of diphtheria toxin action
Diphtheria is a severe infection caused by the exotoxin-producing Corynebacterium diphtheriae. The disease is characterized by pseudomembrane formation in the pharynx and swollen lymph nodes, but can also lead to life-threatening complications like myocarditis or polyneuritis.[27] Although very effective vaccines are available, diphtheria is still a major problem in certain developing countries. The released toxin is cleaved by furin after Arg193 into two subunits, called A and B.[28] Through pore formation, fragment B enables the transport of the catalytically active fragment A into the cytosol of the cell. There, fragment A catalyzes the ADP-ribosylation of elongation factor 2, which leads to the disruption of protein synthesis and the death of the cell.[29]
Selected inhibitors were screened at three concentrations (10, 50 and 200 nM) for their ability to prevent the activation of unnicked diphtheria toxin in Vero cells (Figure 7). The protective factor was calculated as the ratio of toxin concentration inducing 50 % cell death in the presence of inhibitor compared to the control lacking inhibitor.
Figure 7.
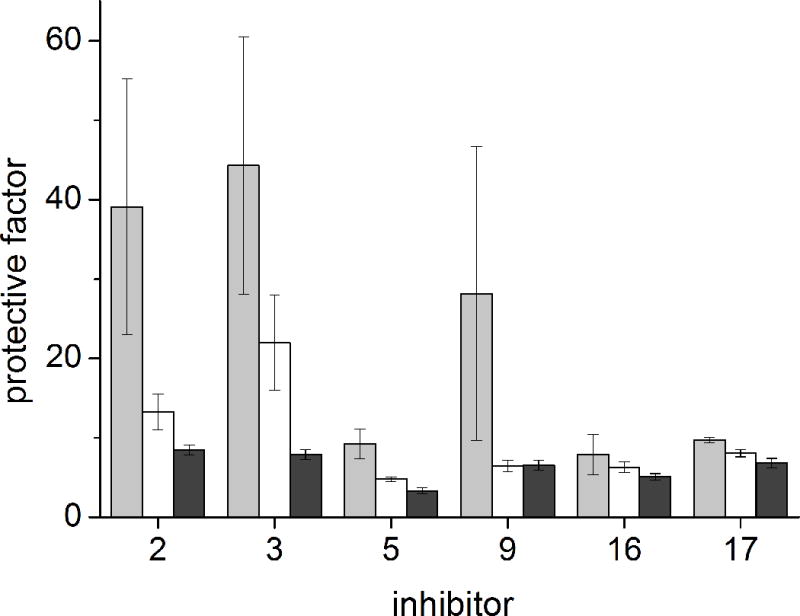
Protection of Vero cells against diphtheria toxin intoxication in the presence of 200 (gray), 50 (white) and 10 nM (black) inhibitor (n = 2).
In the presence of derivatives 5, 16 and 17 protection factors of ~ 10 were observed. At the highest concentration of 200 nM an approximately 3-fold improved effect was observed for inhibitor 9. The best results were achieved with reference compounds 2 and 3 with protection factors around 40 at a concentration of 200 nM.
Discussion
The effective inhibition of furin represents a potential strategy for a short-time treatment of numerous severe infectious diseases.[1,4] So far, the most potent synthetic inhibitors of furin and furin-like PCs contain a C-terminal 4-amidinobenzylamide residue.[13,22] It was known that the incorporation of a basic P5 anchor strongly improves the affinity of this inhibitor type.[13,16] To investigate the influence of a further N-terminal elongation with basic residues, a new inhibitor series was prepared. Compared to compound 1, containing a neutral P5 residue, the coupling of arginine residues in D- and/or L-configuration improved the inhibition of furin providing Ki values ≤ 110 pM. However, fitting of the measured data to the equation for tight binding inhibition (equation 1) yielded lower R2 values in case of compounds 8–15. Surprisingly, this issue was only found for inhibitors containing a free N-terminus. It is hence conceivable that these compounds may bind to furin with different binding modes. Notably, a reverse binding orientation was postulated for related substrate-analogue inhibitors in a previous work.[30] In contrast, a perfect fit was obtained for compounds 4–7 with an N-terminal phenylacetyl residue or for derivatives 16 and 17 exhibiting an N-terminal Nα-guanidino group suggesting a single binding mode as observed in crystal structures with these amidinobenzylamide-derived inhibitors.[14,15] The latter modification contributed to an improved affinity and probably stabilizes the peptides against degradation by aminopeptidases, although this was not tested so far. Besides the elongation with basic residues, compounds with an N-terminal acetylated multi-Leu motif were prepared as references, which were described in a previous work by the Day group.[31] Inhibitors 18 and 19 were resynthesized, their Ki values of ~ 0.5 nM determined in our assay are approximately 10- and 20-fold lower as compared to the inhibition constants described by Kwiatkowska et al..[21,22] These discrepancies are probably caused by different assay conditions. Compound 18 was used as a lead structure for modifications in the P2 and P3 positions. The exchange of the P3-Val to Tle led to a 2.5-fold improved Ki value of 0.19 nM. The incorporation of Arg as the P2-residue provided compounds 21 and 22 with further reduced inhibition constants; both of the latter effects confirming results of previous studies with related inhibitors[11–13] and suggesting that all of these analogues inhibit furin by a similar binding mode.
In contrast to the elongated peptides, the shortened derivatives 23 and 24 containing an N-terminal 4-aminomethyl-Phac residue exhibited reduced potency. The further drop in affinity found for their guanidine analogues 25 and 26 indicates that this more rigid residue cannot adopt a conformation, in which its terminal guanidine occupies the preferred position known from the P4-Arg residue in several crystal structures.[13,14] A significant gain in potency was achieved after incorporation of the more flexible 5-guanidinovaleroyl group as P4 residue, resulting in compounds 28 and 30 with Ki values of 2.5 and 1.3 nM, respectively. Despite the relatively potent furin inhibition (in the low nanomolar range) observed for the shorter analogue 30, we could not detect any significant antiviral activity in the screening against SFV.
In contrast, the novel elongated derivatives 9 and 16 strongly reduced titers of the alphaviruses SFV and CHIKV in cell culture, clearly showing their antiviral activity, although their efficacy was found to be lower than for the reference inhibitors 2 and 3. In contrast to the picomolar binding constants, inhibitor concentrations in the micromolar range were required to significantly reduce the virus titers. The processing of the E3-E2 precursor occurs in a post-TGN compartment, which likely is poorly accessible to highly basic and polar inhibitors. It seems that this drawback can be partially compensated by the very strong affinity of inhibitors 2 and 3, in the low picomolar range. Notably, in a previous study with the irreversible furin inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone a slightly higher concentration of 100 μM was used to demonstrate the inhibition of the CHIKV E3-E2 cleavage.[26]
In comparison, to inhibit the furin-catalyzed cleavage of diphtheria toxin in early endosomes, only nanomolar inhibitor concentrations were needed to obtain significant effects on toxin activation. We speculate that multibasic inhibitors have a tendency to accumulate at the negatively charged cell surface followed by endocytotic uptake, comparable to polybasic cell penetrating peptides.[32] This mechanism could explain the improved potency of these inhibitors when addressing endosomal targets.
Based on present knowledge,[5,10] a multibasic structure is needed to obtain high affinity towards furin. All presently known and more potent synthetic furin inhibitors contain at least three or even more basic residues, which limits their cellular bioavailability and makes them unsuitable for oral administration. Nevertheless, parenteral application should be possible with this inhibitor type, e.g. for the prophylaxis and treatment of eye infections caused by Pseudomonas aeruginosa.[33]
Conclusion
The elongation of the P4-P1 lead segment Arg-Val-Arg-4-amidinobenzylamide with further basic residues provides highly potent furin inhibitors. Some analogues exhibit a significant activity in the used cell culture tests. In contrast, inhibitors lacking a P5 residue, synthesized to decrease molecular weight, still inhibit furin in the low nanomolar range, but do not show antiviral activity. In the present study we could extend the use of our 4-amidinobenzylamide derivatives to the inhibition of alphavirus propagation in a cell culture model. Some of these analogues also protect cells against diphtheria toxin cytotoxicity. Together with our previous data on the inhibition of influenza or canine distemper virus infections, our novel results suggest that effective inhibitors of the 4-amidinobenzylamide type could be suitable candidates as broad-spectrum anti-infectives against furin-dependent pathogens.
Methods
Reagents
Protected amino acids, coupling reagents, resins, further chemicals and solvents were obtained from Orpegen, Bachem, Iris Biotech GmbH, Polypeptides, Fluka, Acros and Aldrich.
Analytical RP-HPLC
Measurements were performed on a Shimadzu LC-10A system with a Nucleodur C18, 5 μm, 100 Å, 4.6 × 250 mm column (Machery-Nagel, Düren, Germany). A linear gradient (increase of 1 % solvent B/min) of acetonitrile (solvent B) in water (solvent A) both containing 0.1 % TFA at a flow rate of 1 ml/min was used (detection at 220 nm).
Preparative RP-HPLC
The final inhibitors were purified (≥ 95% based on the detection at 220 nm) on a preparative HPLC consisting of a Varian PrepStar Model 218 gradient system pump, ProStar Model 320 detector, Varian Model 701 fraction collector and a C18 column (Nucleodur, 5 μm, 100 Å, 32 × 250 mm, Macherey-Nagel, Düren, Germany) using the same solvents as described above. A linear gradient (increase of 0.5 % solvent B/min) at a flow rate of 20 ml/min was used. All peptides were finally obtained as TFA-salts after lyophilization.
Mass spectrometry
The molecular mass of the synthesized compounds was determined on a QTrap 2000 ESI spectrometer (Applied Biosystems, now life technologies, Carlsbad, CA).
Synthesis
The inhibitors were synthesized according to a manual standard Fmoc SPPS protocol in a 2 ml reaction vessel with frit starting with Fmoc-Arg(Pbf)-2-chlorotrityl resin (0.12 g, loading 0.66 mmol/g). The Fmoc group was removed with 20 % piperidine in DMF (5 min for first and 20 min for second cleavage). For the couplings a 4-fold excess of Fmoc-amino acids, HOBt and HBTU, respectively, and 8.0 equivalents of DIPEA was used. The Boc-protected 3- or 4-aminomethyl-phenylacetic acids were only used in a 2-fold excess. The Nα- and side chain-protected intermediates were cleaved from resin under mild acidic conditions (1.5 ml 1 % TFA in DCM, 3 × 30 min) at room temperature, the solutions were immediately neutralized by addition of DIPEA. The solvent was removed in vacuo and the oily residue was dissolved in 2 mL DMF. The solution was treated with 1.0 equivalent 4-amidinobenzylamine × 2 HCl,[11] 3.0 equivalents 6-Cl-HOBt, 1.1 equivalents PyBOP, and 3.0 equivalents DIPEA at 0 °C (pH ~ 8.5). The mixture was stirred for 15 min at 0 °C and at room temperature overnight. The solvent was removed in vacuo and the remaining protecting groups were removed with 2 ml TFA/TIS/H2O (95/2.5/2.5, v/v/v) for 3 h at room temperature. The deprotected intermediate was precipitated in diethyl ether and dried. The precipitate was purified using preparative HPLC and the product lyophilized from water.
Guanylated inhibitors (16, 17, 25, 26, 28, 30) were obtained by treating the corresponding amine intermediates with a large excess of 1H-pyrazole-1-carboxamidine × HCl in 2 mL 1 M Na2CO3.[34] After completion of the conversion, the mixture was acidified with TFA and purified by preparative HPLC, followed by lyophilization of the product from water.
Enzyme kinetic measurements with recombinant soluble human furin
Enzyme kinetic measurements were conducted as described previously.[13] In short, the measurements were performed with a microplate reader (Safire2, Tecan, Switzerland) at λ ex 380 nm and λem 460 nm in black 96-well plates (Nunc, Langenselbold, Germany) at room temperature. A well contained 2 μL inhibitor solution (dissolved in DMSO), 20 μL of the substrate Phac-Arg-Val-Arg-Arg-AMC (dissolved in water, concentrations used in the assay: 5, 20 and 50 μM) and 160 μL buffer (100 mM HEPES, 0.2 % Triton X-100, 2 mM CaCl2, 0.02 % sodium azide und 1 mg/mL BSA, pH 7.0). The measurements were started by addition of 20 μL furin[17] solution (~ 0.95 nM in the assay) and were performed for 30 min. Steady-state rates were calculated from the slopes of the progress curves. Ki values of the inhibitors with inhibition constants > 0.1 nM were determined by fitting of the data to the equation for classical reversible competitive inhibition, as described previously.[11] All data calculations were performed with Origin 8.1.
In the case of a tight-binding behavior, equation 1[35] was used, whereby v0 is the velocity in absence of an inhibitor, It is the total inhibitor concentration, Et is the total enzyme concentration, and Ki* is the apparent inhibition constant at the used substrate concentration of 12.5 μM.
| (1) |
The apparent Ki* was converted into the true Ki value by equation (2).
| (2) |
Analysis of Semliki Forest virus replication in the presence of furin inhibitors
Cells, viruses and infection
Baby hamster kidney (BHK-21) cells (American Type Culture Collection/ATCC CCL-10) were cultured in growth medium (GMEM supplemented with 10 % fetal bovine serum (FBS), 10 % tryptose phosphate broth, 20 mM HEPES pH 7.4, 2 mM L-glutamine, and penicillin-streptomycin [Sigma-Aldrich]) at 37 °C in an atmosphere with 5 % CO2 and 95 % humidity. Semliki Forest virus (SFV) was generated from an SFV4 infectious clone (pSP6-SFV4) on BHK cells as described previously.[36] Subconfluent BHK cells in 12-well plates were infected with SFV in infection medium (DMEM with 0.2 % bovine serum albumin and 20 mM HEPES pH 7.4) at a multiplicity of infection (MOI) of 0.01 for 1 h, subsequently washed with PBS and overlaid with 1 mL of growth medium containing the indicated concentration of furin inhibitor diluted in water. Cell culture supernatants were harvested at 24 h post-infection and kept at −80 °C until titration. Titer determination was performed by plaque assay on BHK cells using standard methodology. Briefly, serial 10-fold dilutions of virus were added to BHK cells (200 μL/12-well plate), which were incubated for 1 h at 37 °C, washed and overlaid with 0.8 % Electran agarose (VWR) in growth medium with 5 % FBS and incubated for 36–48 h at 37 °C, followed by fixation with 10 % formaldehyde in PBS and staining with crystal violet. Titers were calculated from the number of plaques, expressed as plaque-forming units (PFU)/mL; data are the mean of three independent determinations ± standard error (SE).
Western blot
To assess E3–E2 (p62) processing, BHK cells in 6-well plates were infected with SFV at a MOI of 10 for 1 h, followed by washing with PBS and incubation in growth medium with or without furin inhibitors (25 μM) for another 7 h. Cells were lysed with 250 μL lysis buffer (20 mM HEPES pH 7.4, 110 mM potassium acetate, 2 mM magnesium chloride, 0.1 % (v/v) Tween-20 (TBST), 1 % (v/v) Triton X-100, 0.5 % (w/v) sodium deoxycholate and 500 mM sodium chlorid,[37] supplemented with 1× Complete protease inhibitor and PhosSTOP phosphatase inhibitor cocktails (Roche) on ice. Lysates were cleared by centrifugation at 20,000 × g for 10 min at 4 °C, mixed with 4 × reducing NuPAGE lithium dodecyl sulphate (LDS) sample buffer (Life Technologies) and boiled (95 °C, 5 min). Samples were electrophoresed using precast Novex NuPAGE Bis-Tris (4–12% polyacrylamide) gels (Life Technologies) and subsequently transferred onto Hybond P polyvinylidene difluoride membranes (GE Healthcare). Membranes were blocked with 5 % skim milk powder in Tris-buffered saline with 0.1% (v/v) Tween20 (TBST), followed by incubation with mouse-anti-SFV-E2 antisera (obtained from Peter Liljeström, Karolinska Institutet, Stockholm) in blocking buffer for 16 h at 4 °C, washing with TBST, incubation with a horseradish peroxidase-coupled goat-anti-mouse secondary antibody (Sigma, 1 h, room temperature), further washing and finally detection using enhanced chemiluminescence (ECL, Pierce) with Hyperfilm chemiluminescence film (GE Healthcare).
Analysis of chikungunya virus replication in the presence of furin inhibitors
Cells, viruses and infection
BHK-21 cells in 12-well plates were infected with CHIKV-wt, derived from the wild-type infectious clone CHIKV LR2006-OPY1,[38] at a multiplicity of infection (MOI) of 0.1 for 1 h, followed by medium change to growth medium supplemented with the indicated furin inhibitor at a final concentration of 25 μM. Supernatants were harvested at 24 h post-infection and the virus titered in BHK-21 cells.
Inhibition of diphtheria toxin action
Vero cells used were maintained in DMEM[39] and seeded into 24-well plates at a density of 5 × 104 cells/well 1 day before the experiment. The cells were preincubated in the absence or presence of selected inhibitors (200 nM and 100 nM) in leucine and serum free medium for 30 minutes. Then, various concentrations of unnicked diphtheria toxin (Merck KGaA, Darmstadt, Germany; 0.1, 1, 10, and 100 ng/ml in the assay) were added. Three hours later, the cells were incubated in HEPES medium containing 1 μCi/ml [3H]leucine (Hartmann Analytic, Braunschweig, Germany) lacking unlabeled leucine for 20 min. Afterwards the medium was removed. The cells were washed twice in 5% trichloroacetic acid and dissolved in 0.1 M KOH, followed by measurement of the radioactivity.
Supplementary Material
Acknowledgments
The work was supported by a NIH grant DA05084 for Iris Lindberg.
Abbreviations
- Ac
acetyl
- amba
amidinobenzylamide
- AMC
7-amido-4-methylcoumarin
- AMe-Phac
aminomethyl-phenylacetyl
- Ava
5-aminovaleroyl
- Boc
t-butoxycarbonyl
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DMF
N,N-dimethylformamide
- FBS
fetal bovine serum
- Fmoc
N-(9-fluorenyl)methoxycarbonyl
- GMEM
Glasgow’s Modified Eagle’s Medium
- GMe-Phac
guanidinomethyl-phenylacetyl
- Gva
5-guanidinovaleroyl
- N(Ca)Arg
Nα(carbamidoyl)-arginine
- PBS
phosphate-buffered saline
- PC
proprotein convertase
- Phac
phenylacetyl
- PyBOP
benzotriazol-1-yl-N-oxy-tris(pyrrolidino)-phosphonium hexafluorophosphate
- TFA
trifluoroacetic acid
- TIS
triisopropylsilane
- Tle
tert.leucine or tert.butylglycine
Footnotes
Declaration of interest
The authors report no conflicts of interest.
References
- 1.Thomas G. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rockwell NC, Krysan DJ, Komiyama T, Fuller RS. Chem Rev (Chemical reviews) 2002;102:4525–4548. doi: 10.1021/cr010168i. [DOI] [PubMed] [Google Scholar]
- 3.Chrétien M, Seidah NG, Basak A, Mbikay M. Expert Opin Ther Targets. 2008;12:1289–1300. doi: 10.1517/14728222.12.10.1289. [DOI] [PubMed] [Google Scholar]
- 4.Seidah NG, Prat A. Nat Rev Drug Discovery. 2012;11:367–383. doi: 10.1038/nrd3699. [DOI] [PubMed] [Google Scholar]
- 5.Couture F, D’Anjou F, Day R. Biomol Concepts. 2011;2:421–438. doi: 10.1515/bmc.2011.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasquato A, Ramos da Palma Joel, Galan C, Seidah NG, Kunz S. Antiviral Res. 2013;99:49–60. doi: 10.1016/j.antiviral.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Roebroek Anton JM, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, Lauwers A, Van de Ven Wim JM, Hartmann D, Creemers John WM. J Biol Chem. 2004;279:53442–53450. doi: 10.1074/jbc.M407152200. [DOI] [PubMed] [Google Scholar]
- 8.a) Sarac MS, Cameron A, Lindberg I. Infect Immun. 2002;70:7136–7139. doi: 10.1128/IAI.70.12.7136-7139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sarac MS, Peinado JR, Leppla SH, Lindberg I. Infect Immun. 2004;72:602–605. doi: 10.1128/IAI.72.1.602-605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basak A. J Mol Med (Heidelberg, Ger) 2005;83:844–855. doi: 10.1007/s00109-005-0710-0. [DOI] [PubMed] [Google Scholar]
- 10.Chandra De U, Mishra P, Rudra Pal P, Dinda B, Basak A. Non-peptide inhibitors of proprotein convertase subtilisin kexins (PCSKs) An overall review of existing and new data. Morgan & Claypool; San Rafael, Calif: 2012. [Google Scholar]
- 11.Becker GL, Sielaff F, Than ME, Lindberg I, Routhier S, Day R, Lu Y, Garten W, Steinmetzer T. J Med Chem. 2010;53:1067–1075. doi: 10.1021/jm9012455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker GL, Hardes K, Steinmetzer T. Bioorg Med Chem Lett. 2011;21:4695–4697. doi: 10.1016/j.bmcl.2011.06.091. [DOI] [PubMed] [Google Scholar]
- 13.Hardes K, Becker GL, Lu Y, Dahms SO, Kohler S, Beyer W, Sandvig K, Yamamoto H, Lindberg I, Walz L, et al. ChemMedChem. 2015;10:1218–1231. doi: 10.1002/cmdc.201500103. [DOI] [PubMed] [Google Scholar]
- 14.Dahms SO, Hardes K, Becker GL, Steinmetzer T, Brandstetter H, Than ME. ACS Chem Biol. 2014;9:1113–1118. doi: 10.1021/cb500087x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahms SO, Arciniega M, Steinmetzer T, Huber R, Than ME. Proc Natl Acad Sci USA. 2016;113:11196–11201. doi: 10.1073/pnas.1613630113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker GL, Lu Y, Hardes K, Strehlow B, Levesque C, Lindberg I, Sandvig K, Bakowsky U, Day R, Garten W, et al. J Biol Chem. 2012;287:21992–22003. doi: 10.1074/jbc.M111.332643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kacprzak MM, Peinado JR, Than ME, Appel J, Henrich S, Lipkind G, Houghten RA, Bode W, Lindberg I. J Biol Chem. 2004;279:36788–36794. doi: 10.1074/jbc.M400484200. [DOI] [PubMed] [Google Scholar]
- 18.Gagnon H, Beauchemin S, Kwiatkowska A, Couture F, D’Anjou F, Levesque C, Dufour F, Desbiens AR, Vaillancourt R, Bernard S, et al. J Med Chem. 2014;57:29–41. doi: 10.1021/jm400633d. [DOI] [PubMed] [Google Scholar]
- 19.a) Hock FJ, Wirth K, Albus U, Linz W, Gerhards HJ, Wiemer G, Henke S, Breipohl G, Konig W, Knolle J. Br J Pharmacol. 1991;102:769–773. doi: 10.1111/j.1476-5381.1991.tb12248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wirth K, Hock FJ, Albus U, Linz W, Alpermann HG, Anagnostopoulos H, Henk S, Breipohl G, Konig W, Knolle J. Br J Pharmacol. 1991;102:774–777. doi: 10.1111/j.1476-5381.1991.tb12249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery A Primer for Medicinal Chemists and Pharmacologists. John Wiley & Sons; Hoboken: 2005. [PubMed] [Google Scholar]
- 21.Kwiatkowska A, Couture F, Levesque C, Ly K, Desjardins R, Beauchemin S, Prahl A, Lammek B, Neugebauer W, Dory YL, et al. J Med Chem. 2014;57:98–109. doi: 10.1021/jm401457n. [DOI] [PubMed] [Google Scholar]
- 22.Kwiatkowska A, Couture F, Levesque C, Ly K, Beauchemin S, Desjardins R, Neugebauer W, Dory YL, Day R. ChemMedChem. 2016;11:289–301. doi: 10.1002/cmdc.201500532. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, Hardes K, Dahms SO, Bottcher-Friebertshauser E, Steinmetzer T, Than ME, Klenk HD, Garten W. Antiviral Res. 2015;120:89–100. doi: 10.1016/j.antiviral.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Jose J, Snyder JE, Kuhn RJ. Future Microbiol. 2009;4:837–856. doi: 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Fugère M, Day R, Kielian M. J Virol. 2003;77:2981–2989. doi: 10.1128/JVI.77.5.2981-2989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozden S, Lucas-Hourani M, Ceccaldi PE, Basak A, Valentine M, Benjannet S, Hamelin J, Jacob Y, Mamchaoui K, Mouly V, et al. J Biol Chem. 2008;283:21899–21908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- 27.Hadfield TL, McEvoy P, Polotsky Y, Tzinserling VA, Yakovlev AA. J Infect Dis. 2000;181(Suppl 1):S116–20. doi: 10.1086/315551. [DOI] [PubMed] [Google Scholar]
- 28.Tsuneoka M, Nakayama K, Hatsuzawa K, Komada M, Kitamura N, Mekada E. J Biol Chem. 1993;268:26461–26465. [PubMed] [Google Scholar]
- 29.Collier RJ. Toxicon. 2001;39:1793–1803. doi: 10.1016/s0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Vallejo F, Martinez-Mayorga K. Bioorg Med Chem. 2012;20:4462–4471. doi: 10.1016/j.bmc.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 31.Levesque C, Fugère M, Kwiatkowska A, Couture F, Desjardins R, Routhier S, Moussette P, Prahl A, Lammek B, Appel JR, et al. J Med Chem. 2012;55:10501–10511. doi: 10.1021/jm3011178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a) Fuchs SM, Raines RT. Cell Mol Life Sci. 2006;63:1819–1822. doi: 10.1007/s00018-006-6170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Katayama S, Hirose H, Takayama K, Nakase I, Futaki S. J Controlled Release. 2011;149:29–35. doi: 10.1016/j.jconrel.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 33.a) Karicherla P, Hobden JA. Invest Ophthalmol Visual Sci. 2009;50:256–262. doi: 10.1167/iovs.08-2344. [DOI] [PubMed] [Google Scholar]; b) Karicherla P, Hobden JA. Curr Eye Res. 2010;35:220–224. doi: 10.3109/02713680903487609. [DOI] [PubMed] [Google Scholar]
- 34.Bernatowicz MS, Wu Y, Matsueda GR. J Org Chem. 1992;57:2497–2502. [Google Scholar]
- 35.Williams JW, Morrison JF. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 36.Liljeström P, Lusa S, Huylebroeck D, Garoff H. J Virol. 1991;65:4107–4113. doi: 10.1128/jvi.65.8.4107-4113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cristea IM, Carroll JWN, Rout MP, Rice CM, Chait BT, MacDonald MR. J Biol Chem. 2006;281:30269–30278. doi: 10.1074/jbc.M603980200. [DOI] [PubMed] [Google Scholar]
- 38.Pohjala L, Utt A, Varjak M, Lulla A, Merits A, Ahola T, Tammela P. PloS one. 2011;6:e28923. doi: 10.1371/journal.pone.0028923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurmanova A, Llorente A, Polesskaya A, Garred O, Olsnes S, Kozlov J, Sandvig K. Biochem Biophys Res Commun. 2007;357:144–149. doi: 10.1016/j.bbrc.2007.03.110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.