

Abstract
Purpose of review
Magnesium (Mg2+) imbalances are frequently overlooked. Hypermagnesemia usually occurs in preeclamptic women after Mg2+ therapy or in end-stage renal disease patients, while hypomagnesemia is more common with a prevalence of up to 15% in the general population. Increasing evidence points towards a role for mild to moderate, chronic hypomagnesemia in the pathogenesis of hypertension, type 2 diabetes mellitus, and metabolic syndrome.
Recent findings
The kidneys are the major regulator of total body Mg2+ homeostasis. Over the last decade the identification of the responsible genes in rare genetic disorders has enhanced our understanding of how the kidney handles Mg2+. The different genetic disorders and medications contributing to abnormal Mg2+ homeostasis are reviewed.
Summary
As dysfunctional Mg2+ homeostasis contributes to the development of many common human disorders, serum Mg2+ deserves closer monitoring. Hypomagnesemic patients may be asymptomatic or may have mild symptoms. In severe hypomagnesemia patients may present with neurological symptoms such as seizures, spasms or cramps. Renal symptoms include nephrocalcinosis and impaired renal function. Most conditions affect tubular Mg2+ reabsorption by disturbing the lumen-positive potential in the thick ascending limb or the negative membrane potential in the distal convoluted tubule.
Keywords: Magnesium, kidney, physiology, tubule, nephron
Introduction
Magnesium (Mg2+) is a frequently neglected electrolyte despite being the second most abundant intracellular cation(1). Mg2+ plays an important role in human physiology: it functions in over 600 enzymes as a cofactor, is crucial for nerve conduction and cardiac contractility, enhances resistance of DNA and RNA against oxidative stress by stabilizing their tertiary structure, cell cycle control and cell proliferation depend on Mg2+, and ATP has to bind Mg2+ to be biologically active(2). Nevertheless, general practitioners frequently do not evaluate serum Mg2+(3, 4). Hypermagnesemia occurs usually in preeclamptic women after Mg2+ therapy and in patients with end-stage renal disease (ESRD). Elevated Mg2+ concentrations cause muscle weakness, fatigue and somnolence. Hypomagnesemia is more common and occurs in up to 15% of the general population(5). Mild to moderate hypomagnesemia frequently does not cause symptoms, if more significant muscle spasms, arrhythmia, and seizures occur. Increasing evidence points to chronic hypomagnesemia as a risk factor for developing hypertension, type 2 diabetes mellitus, metabolic syndrome, chronic kidney disease, and cancer(6–16).
Blood Mg2+ concentration reflects the equilibrium between intestinal Mg2+ reabsorption and renal Mg2+ excretion. In the small intestine Mg2+ undergoes reabsorption in a paracellular fashion, in between epithelial cells (17, 18) (Fig. 1). This allows for reabsorption of large amounts of Mg2+ as this mode of reabsorption is non-saturable(18) (Fig. 1A). Mg2+ reabsorption in the cecum and colon occurs in a transcellular fashion via apical Mg2+ channels TRPM6 and TRPM7 which in contrast to paracellular transport is saturable (Fig. 1A, B). Intestinal Mg2+ reabsorption is poorly regulated and depends on Mg2+ intake.
Fig. 1. Intestinal and renal mechanisms of Mg2+ reabsorption.
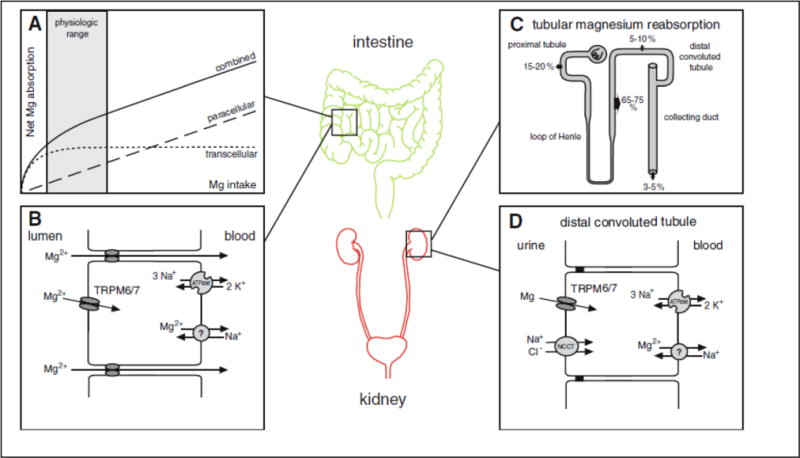
Intestinal Mg2+ reabsorption occurs predominantly in the small intestine, cecum and colon(17, 18) (Fig. 1). Intestinal Mg2+ reabsorption varies between 25–80% with higher Mg2+ reabsorption in low Mg2+ states(17, 18). A) Intestinal Mg2+ reabsorption occurs in a paracellular, non-saturable fashion, while Mg2+ reabsorption in the cecum and colon occurs in a transcellular, saturable fashion. B) Transcellular Mg2+ transport in the intestinal tract occurs via apical Mg2+ channels TRPM6 and TRPM7. It is thought that a Mg2+ ATPase and/or a Mg2+-Na+ exchanger facilitates basolateral Mg2+ extrusion(18). C) About 2.4 g of Mg2+ is filtered by the glomerulus daily. As 95 to 99% of the filtered Mg2+ is reabsorbed along the nephron only approximately 100 mg of Mg2+ is excreted per day. In contrast to many other electrolytes the proximal tubule reabsorbs only 10–25% of Mg2+ and the majority of Mg2+ is reabsorbed in the TAL. D) Similar to the intestinal tract, TRPM6 and TRPM7 channels in the apical membrane mediate transcellular Mg2+ reabsorption in the DCT. Reprinted with permission from(19).
The kidneys are the primary regulators of Mg2+ homeostasis. About 80% of the serum Mg2+ is filtered in the glomerulus and about 95–99% of the filtered Mg2+ is recovered along the nephron(2) (Fig. 1C). The main location for renal Mg2+ reabsorption with 65–75% is the thick ascending limb of Henle (TAL). Here Mg2+ transport occurs in a paracellular mode(20) (Fig. 1C). In the TAL the major driving force for Mg2+ reabsorption is the lumen-positive transepithelial potential difference. The fine-tuning of renal Mg2+ reabsorption occurs in the distal convoluted tubule (DCT) via apical TRPM6 and TRPM7 channels(21) (Fig. 1D). Here, the negative membrane potential is a prerequisite for Mg2+ reabsorption(22, 23). As in the gut it is unclear how Mg2+ is exported on the basolateral side. Hypomagnesemias are due to medication side effects (Table 1), insufficient dietary Mg2+ intake, or rare inherited renal defects, which are divided in hypercalciuric, Gitelman-like, and other hypomagnesemias (Table 2).
Table 1.
Medications contributing to hypomagnesemia.
| Medications contributing to hypomagnesemia | Mechanism |
|---|---|
| Proton pump inhibitors (e.g. omeprazole) | Decreased intestinal Mg2+
reabsorption via TRPM6(18, 24, 25) Tubulo-interstitial nephritis(26) |
| Diuretics (e.g. furosemide, thiazide) | ↓ lumen positive potential difference in TAL blocking Na+ reabsorption in DCT affects membrane potential(27, 28) |
| Platinum derivatives (e.g. cisplatin, carboplatin) | Necrotic nephropathy(29) PT and DCT injury(30–33) |
| Calcineurin inhibitors (e.g. cyclosporine A, tacrolimus) | Downregulation of Claudin-16 downregulation of TRPM6(34–37) |
| Epidermal growth factor receptor inhibitor (e.g. cetuximab) | Blockade of EGF receptor and lack of TRPM6 stimulation(38, 39) |
| Antimicrobials (e.g. aminoglycosides, amphotericin B, pentamidine,…) | PT damage, Fanconi syndrome(40, 41) |
Table 2.
List of inherited forms of hypomagnesemia.
| Disorder | Inheritance | Gene locus | Gene (Protein) |
Function |
|---|---|---|---|---|
| Hypercalciuric hypomagnesemias | ||||
|
| ||||
| Familial hypomagnesemia with hypercalciuria and nephrocalcinosis | AR | 3q28 |
CLDN16 (Claudin-16) |
tight junction protein |
|
| ||||
| Familial hypomagnesemia with hypercalciuria and nephrocalcinosis plus ocular involvement | AR | 1p34 |
CLDN19 (Claudin-19) |
tight junction protein |
|
| ||||
| Classical Bartter syndrome (type 3) | AR | 1p36 |
ClC-Kb (ClC subunit B) |
basolateral Chloride channel |
|
| ||||
| Autosomal dominant hypocalcemia/Bartter syndrome (type 5) | AD | 3q13 |
CASR (CaSR) |
Calcium sensing receptor |
|
| ||||
| Gitelman-like hypomagnesemias | ||||
|
| ||||
| Gitelman syndrome | AR | 16q13 |
SLC12A3 (NCC) |
Na+-Cl− cotransporter |
|
| ||||
| Antenatal Bartter syndrome with sensorineural deafness (type 4) | AR | 1p31 |
BSND (Barttin) |
Subunit of ClC-Ka/b |
|
| ||||
| EAST/SeSAME syndrome | AR | 1q23 |
KCNJ10 (Kir4.1) |
apical potassium channel |
|
| ||||
| Isolated dominant hypomagnesemia | AD | 11q23 |
FXYD2 (FXYD2) |
Na+/K+-ATPase (ɣ subunit) |
| AD | 12p13 |
KCNA1 (Kv1.1) |
apical potassium channel | |
|
| ||||
| HNF1B nephropathy | AD | 17q12 |
HNF1B (HNF1beta) |
transcription factor |
|
| ||||
| Hypomagnesemia after transient neonatal hyperphenylalaninemia | AR | 10q22 |
PCBD1 (PCBD1) |
tetrahydrobiopterin metabolism |
|
| ||||
| Other hypomagnesemias | ||||
|
| ||||
| Isolated recessive hypomagnesemia | AR | 4q25 |
EGF (Pro-EGF) |
epidermal growth factor |
|
| ||||
| Hypomagnesemia with secondary hypocalcemia | AR | 9q22 |
TRPM6 (TRPM6) |
apical Mg2+ channel |
|
| ||||
| Hypomagnesemia with impaired brain development | AD/AR | 10q24 |
CNNM2 (CNNM2) |
Cyclin M2 |
|
| ||||
| Hypomagnesemia with metabolic syndrome | maternal | mtDNA |
MTTI (MTTI) |
mitochondrial tRNA for isoleucine |
| Hyperuricemia, pulmonary hypertension and progressive renal failure (HUPRA) | AR | 19q13 |
SARS2 (SARS2) |
seryl-tRNA synthetase |
Hypercalciuric hypomagnesemias
These disorders share that the gene defect impairs paracellular Mg2+ and Ca2+ reabsorption in the TAL by disrupting the lumen-positive transepithelial potential difference and thereby cause hypercalciuria and hypomagnesemia.
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC)
Symptoms in FHHNC include hypomagnesemia, and hypocalcemia due to urinary Ca2+ and Mg2+ losses, nephrocalcinosis and in some patients eye involvement(42–44). Infrequent symptoms are dRTA, hypocitraturia, nephrolithiasis, hyperuricemia, recurrent UTIs, polyuria and failure to thrive(45, 46). Neurological symptoms due to hypomagnesemia are rare. While patients grow relatively well tooth enamel defects can occur(47, 48). In the teens to twenties patients can develop chronic kidney disease and may require renal replacement therapy(45). Recessive mutations in Claudin-16 (CLDN16) were published(49). A genotype-phenotype correlation for CLDN16 mutations was published with earlier onset of ESRD with two loss of function mutations(45). Patients with FHHNC and ocular involvement were found to have mutations in a related gene called Claudin-19 (CLDN19)(50). Both proteins are strongly expressed in renal tissue, specifically in the TAL(49–51). Claudin-19 is also found in ocular tissue(50). Claudin-16 and 19 both co-localize to tight junctions (TJ) in the TAL and the DCT(49, 50). TJ are protein complexes in between epithelial cells which determine the permeability of the epithelial barrier(51). Both proteins interact with each other and form heterodimers(52, 53). Initially, it was thought that CLDN16 mutations would impair tubular Ca2+ and Mg2+ reabsorption by disturbing a Ca2+ and Mg2+ selective channel(49). However, the mechanism contributing to hypomagnesemia and hypercalciuria turned out to be more complex (Fig. 2A). The lumen-positive potential difference, which is the major driving force for paracellular Ca2+ and Mg2+ reabsorption, is created by two different mechanisms: Secretion of K+ via ROMK, which is driven by the reabsorption of Na+, K+ and Cl− via NKCC2 in the TAL, contributes to the lumen-positive potential. The second component of the lumen-positive potential is called the dilution potential and is explained in detail in Fig. 2A. Mutations in CLDN16 and 19 affect the creation of the dilution potential and result in a less lumen-positive potential(52).
Fig. 2. Mechanisms of hypercalciuric hypomagnesemias.
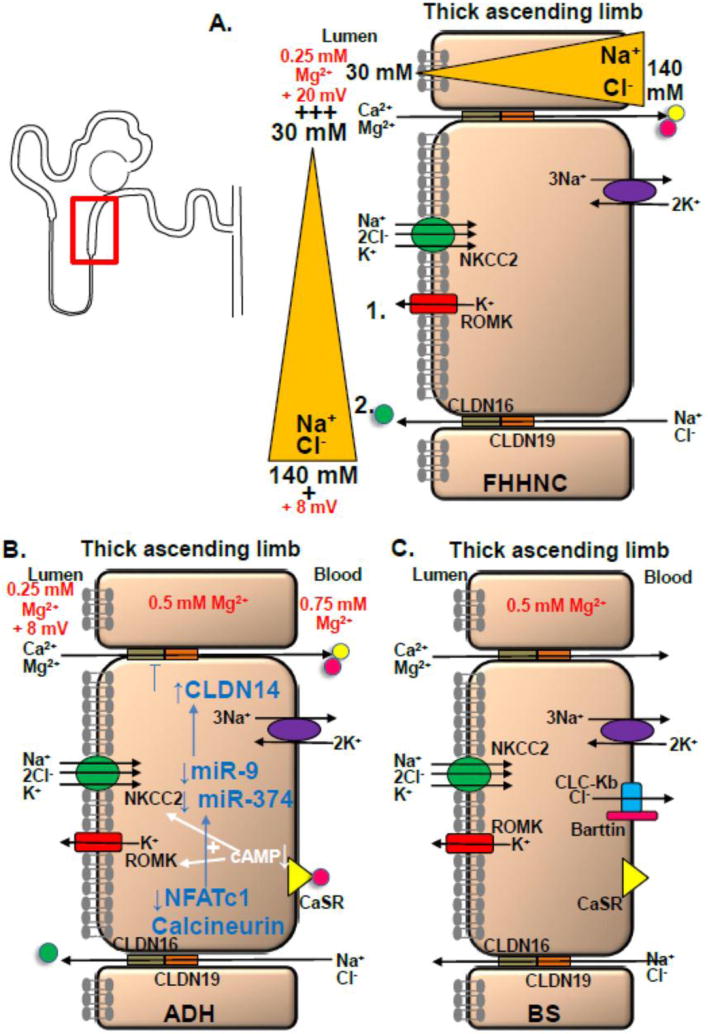
Hypercalcuric hypomagnesemias such as FHHNC, ADH, and BS affect the TAL by disturbing the lumen-positive potential. A) Potassium secretion via ROMK constitutes one component of the lumen-positive potential (1.). The second (2.) component is due to the dilution potential: At the beginning of the TAL luminal Na+ has a concentration of 140 mM which decreases to 30 mM along the downstream TAL. As more Na+ and Cl− is absorbed along the TAL a major Na+ and Cl− gradient between lumen and interstitium is created. A higher Na+ and Cl− concentration in the interstitium result in a driving force for both ions to leak back into the tubular lumen. This is where Claudin-16 and 19 come into play. While Claudin-16 increases the Na+ permeability, Claudin-19 decreases the Cl− permeability, thereby contributing to a high permeability ratio of Na+ to Cl− and providing a strong cation selectivity for Na+ causing the lumen-positive potential to rise from 8 to 20 mV or even higher. B) Different mechanisms contribute to hypomagnesemia in case of CaSR activation including an inhibitable adenylcyclase and activation of inhibitory G proteins resulting in decreased intracellular cAMP levels (cAMP usually enhances NKCC2 and ROMK activities). A novel pathway involves calcineurin signaling and downregulation of NFAT which reduces transcription of two miRNAs called miR9 and miR374 (blue arrows). With CaSR activation there is downregulation of miR9 and 374 and upregulation of Claudin-14 which suppresses the Claudin-16 and 19 complex, thereby interfering with the lumen-positive lumen potential. C) Mutations in CLCNKB and BSND disturb the intracellular Cl− regulation which is thought to affect apical NCC and NKCC2 function. Impaired NCC and NKCC2 function interferes with the generation of the lumen-positive potential and may thereby disturb tubular Mg2+ absorption.
It remains unclear why FHHNC patients develop ESRD. Japanese cattle lacking CLDN16 developed tubulointerstitial nephritis with hypocalcemia but no hypomagnesemia(54, 55). Immature tubular epithelial cells with loss of polarization and renal tubular dysplasia raise concern for a developmental defect. Cldn16 knockout mice displayed hypercalciuria and hypomagnesemia but no renal impairment(56). Cldn19 knockout mice show disorganized tight junctions in Schwann cells, abnormal behavior and neuropathy but no ocular or renal phenotype(57). At this point it is thought that hypercalciuria and nephrocalcinosis contribute to the development of ESRD. Hydrochlorothiazide treatment reduces hypercalciuria but had no effect on slowing down ESRD progression(58).
Autosomal dominant hypocalcemia (ADH)
ADH is characterized by hypocalcemia, hypercalciuria, kidney stones, normal to inappropriately low PTH levels, and about 50% of all ADH patients have hypomagnesemia resulting in seizures and carpopedal spasms(59, 60). ADH is caused by gain-of-function mutations in the calcium sensing receptor (CaSR)(61). In the TAL the CaSR is expressed at the basolateral membrane(62) (Fig. 2B). CASR mutations result in a higher sensitivity of the CaSR contributing to a higher receptor response despite physiological extracellular Ca2+ concentrations, mimicking hypercalcemia(59). Subsequently, PTH is suppressed and renal Ca2+ and Mg2+ absorption is impaired(62, 63). Patients with severe gain-of-function mutations in CaSR can develop Bartter syndrome (BS) (BS type V)(64). Here, gain-of function CASR mutations result in impaired NKCC2 and ROMK function. Different mechanisms of how CaSR activation contributes to urinary Na+, K+ and Cl− losses are discussed (Fig. 2B)(62, 65, 66). All these possibilities decrease the lumen-positive potential.
Recent studies outlined a novel, intricate mechanism how CaSR activation results in renal Ca2+ and Mg2+ wasting involving the calcineurin signaling pathway, NFAT, specific miRNAs, and Claudin-14 (Fig. 2B)(67, 68). CASR stimulation enhances Claudin 14 expression, a negative regulator of Claudins-16 and 19. This diminishes the lumen-positive potential and contributes to urinary Ca2+ and Mg2+ losses. Downregulation of Claudin-14 and upregulation of Claudin-16 was confirmed in a Casr−/− mouse model(69).
Classical Bartter syndrome (cBS)
The features of cBS (or BS type III) are polyuria, renal salt wasting, concentration defect, hypokalemic metabolic alkalosis, and hypercalciuria(70). Later in life patients can develop a Gitelman-like phenotype with hypocalciuria and hypomagnesemia(71, 72). Classical BS is caused by recessive mutations in the CLCNKB gene which encodes the basolateral chloride channel CLC-Kb (Fig. 2C)(73, 74). Another form of antenatal BS (BS with deafness, or BS type IV) is due to recessive mutations in the BSND gene. BSND encodes the protein Barttin which is a subunit of the CLC-Kb channel (Fig. 2C)(75). This form of BS can also result in hypomagnesemia but may initially not display hypercalciuria and is therefore typically included in the Gitelman-like forms of hypomagnesemia (Table 2)(76). Mutations in CLCNKB and BSND disturb the intracellular Cl− regulation. This affects apical NCC and NKCC2 function which subsequently interferes with the generation of the lumen-positive potential and may so disturb tubular Mg2+ absorption(77, 78).
Gitelman-like hypomagnesemias
Mg2+ reabsorption in the DCT depends critically on the negative membrane potential. In this group mutated proteins are involved in Na+, K+, or Cl− transport in the DCT, thereby disturbing the negative membrane potential. All disorders in this group result in hypocalciuria, volume contraction, hypotension, and activation of the renin-angiotensin system (RAS), which then drives K+ and H+ secretion, thus contributing to hypokalemia and metabolic alkalosis (Table 2).
Gitelman syndrome (GS)
GS is one of the most common tubulopathies with a prevalence of 1:40,000(79). GS is characterized by hypokalemic alkalosis, hypocalciuria and hypomagnesemia(80). Symptoms include muscle weakness, fatigue, salt craving, thirst, nocturia, and carpopedal spasms. Clinical presentation is heterogenous ranging from asymptomatic to severe impairment of quality of life(81). Recessive mutations were published in the SLC12A3 gene which encodes the Na-Cl cotransporter NCC(82). In up to 40% of patients the second mutation cannot be detected possibly due to deep intronic location or large genomic rearrangements(83–85). A Ncc knockout mouse model confirmed the human GS phenotype with hypomagnesemia and hypocalciuria but lacked volume contraction and metabolic alkalosis(86, 87). Ncc mutant mice displayed hypotension, volume contraction, metabolic alkalosis, and urinary Mg2+ wasting(88). Hypocalciuria and hypomagnesemia in GS are thought to be the result of compensatory paracellular volume, Na+ and Ca2+ reabsorption in the PT due to volume contraction(28, 87). Regarding hypomagnesemia different hypotheses were proposed including K+ depletion, dysfunctional Mg2+ absorption, atrophy of the DCT and renal Mg2+ wasting. Ncc−/− mice and thiazide treatment in wild-type (WT) mice displayed urinary Mg2+ wasting due to less apical Trpm6 expression(28). GS patients require lifelong Mg2+ and possibly K+ supplementation, as well as spironolactone, amiloride or eplerenone.
EAST syndrome
EAST is an abbreviation for epilepsy, ataxia, sensorineural deafness, and salt loosing tubulopathy(89). These patients present in early infancy with seizures, speech and motor delay, hearing impairments, and cerebellar symptoms such as ataxia, tremor, and dysdiadochokinesia(90). Hypokalemic metabolic alkalosis, hypocalciuria and hypomagnesemia develop during the disease course(91). Recessive mutations in the basolateral, inwardly rectifying K+ channel Kir4.1 were identified(89, 92). Consistent with the disease phenotype Kir4.1 expression was found in the brain, stria vascularis of the inner ear and DCT. Kir4.1−/− mice displayed a phenotype similar to humans(89). Kir4.1 forms heteromeric complexes with another K+ channel called Kir5.1(93). The basolateral Kir4.1/5.1 complex works together with the basolateral Na+/K+-ATPase by recycling K+ ions that entered the cell in exchange for extruding Na+ ions (Fig. 3A)(92, 94). The distribution of extracellular and intracellular K+ is crucial for the generation of the negative membrane potential. The neurological phenotype is due to depolarization of neurons thus lowering the threshold for seizures(92).
Fig. 3. Mechanisms of Gitelman-like hypomagnesemias.
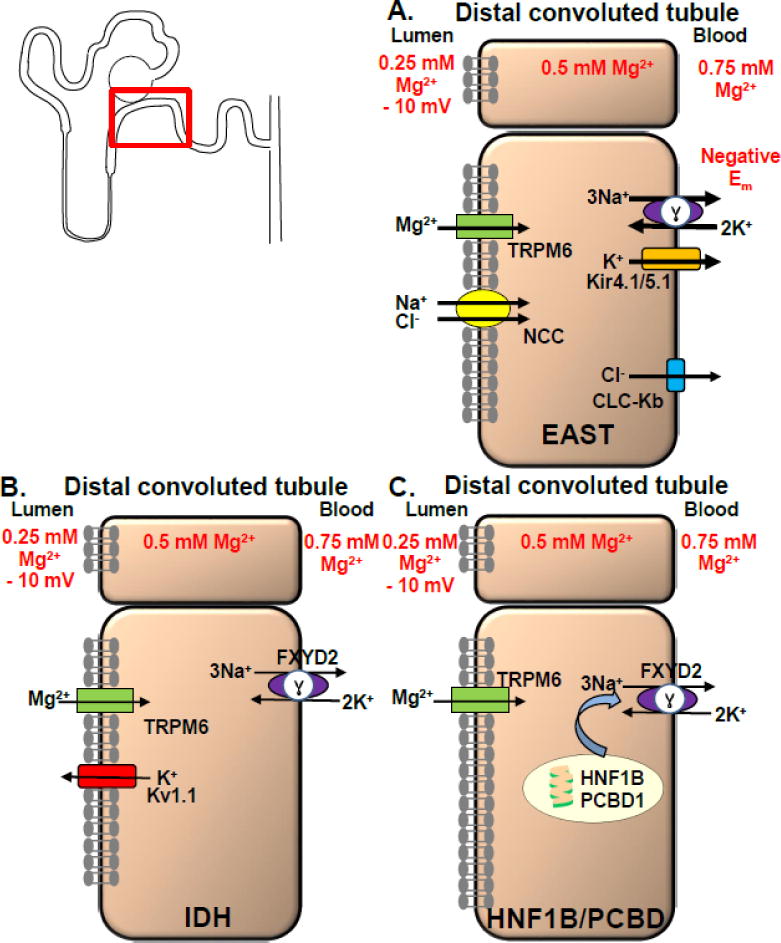
Gitelman-like hypomagnesemias such as EAST, IDH, and HNF1B/PCBD affect the DCT by disturbing the negative membrane potential. A) EAST syndrome: The negative membrane potential promotes basolateral chloride exit via CLC-Kb and apical Mg2+ entrance via TRPM6 due to a favorable electrical gradient. In case of Kir4.1 mutations Na+/K+-ATPase function is decreased and due to loss of K+ recycling via Kir4.1/5.1 a less negative membrane potential is the result. This reduces the basolateral chloride export and inhibits apical reabsorption of Na+ and Cl− via NCC and Mg2+ via TRPM6(66, 92). Urinary salt wasting activates RAS, enhances Na+ reabsorption via ENaC and K+ and H+ secretion in the collecting duct which explains the hypokalemic metabolic alkalosis. B) The mechanism how FXYD2 mutations in IDH cause Mg2+ dysregulation remains unclear. Different hypotheses consider that a dysfunctional γ subunit of the Na+/K+-ATPase may result in reduced intracellular K+ concentration (which may depolarize the apical membrane and so impair Mg2+ reabsorption), altered intracellular Na+ concentration (which could affect a putative basolateral Mg2+-Na+ exchanger), the γ subunit could also be important for the Mg2+-Na+ exchanger, and a defective Na+/K+-ATPase disturbs potentially cellular energy metabolism and may result in a higher risk of apoptosis. For IDH due to KCNA1 mutations: The negative membrane voltage, which is maintained by an apical K+ efflux via Kv1.1, is crucial as Mg2+ uptake from the ultrafiltrate via the TRPM6 channel is driven by a favorable negative membrane voltage. C) HNF1B and PCBD1 form a heteromeric complex and stimulate FXYD2 transcription, which encodes a subunit of the Na+/K+-ATPase.
Isolated dominant hypomagnesemia (IDH)
IDH is characterized by hypocalciuria and hypomagnesemia but lacks hypokalemic metabolic alkalosis, salt wasting or RAS stimulation. Patients present with seizures in childhood, severe hypomagnesemia, developmental delay due to repeated seizures, urinary Mg2+ wasting but normal or even upregulated intestinal Mg2+ absorption. Dominant mutations in two different genes, FXYD2 and KCNA1, cause IDH(95, 96). FXYD2 encodes the γ subunit of the Na+/K+-ATPase (Fig. 3B). FXYD2 enhances ATP and decreases Na+ affinity in a tissue-specific manner(97, 98). Only one mutation (glycine to arginine at position 41) has been identified which has a dominant-negative effect resulting in incorrect trafficking and perinuclear accumulation of the mutant protein(95, 99). Lack of FXYD2 in humans or mice does not cause Mg2+ abnormalities. The mechanism how FXYD2 mutations cause Mg2+ dysregulation remains unclear.
A second gene contributing to IDH was identified with the KCNA1 gene which encodes the apical voltage-gated channel Kv1.1(96). Patients display muscle cramps, tetany, tremors and muscle weakness. Urinary Mg2+ wasting but no abnormalities regarding Ca2+ were found. Ataxia and myokymia, an involuntary form of localized muscle trembling, were found in all affected patients. Interestingly, KNCA1 mutations were previously described in other patients with ataxia and myokymia(100). However, only the N255D (asparagine to aspartic acid at position 255) mutation caused hypomagnesemia(96). Kv1.1 co-localizes with TRPM6 in the DCT but does not regulate TRPM6. The Kv1.1 channel forms tetramers and co-expression of mutant and WT KV1.1 results in a dominant-negative effect of the mutant Kv1.1. As channel trafficking is preserved probably gating of the channel pore is affected resulting in diminished negative membrane potential (Fig. 3B).
HNF1B nephropathy
Mutations in HNF1B result in a spectrum of symptoms including maturity-onset diabetes of the young type 5 (MODY5), hyperechogenic kidneys, multicystic and glomerulocystic kidney disease, renal hypoplasia, renal agenesis, hyperuricemic nephropathy, renal cysts and diabetes syndrome(101–105). About 50% of HNF1B patients have hypomagnesemia due to renal Mg2+ losses and display hypocalciuria(106). Mutations in HNF1B can be inherited in an autosomal dominant fashion or occur de novo. HNF1B encodes the transcription factor hepatocyte nuclear factor 1β, a member of the homeodomain-containing superfamily, which is crucial for renal and pancreatic development. HNF1B regulates other proteins which are involved in renal Mg2+ absorption such as FXYD2(106). Several HNF1B binding sites were identified in the FXYD2 promoter and HNF1B mutations impair FXYD2 transcription(107).
Transient neonatal hyperphenylalaninemia and primapterinuria
Transient neonatal hyperphenylalaninemia and primapterinuria is a benign neonatal syndrome without long-term sequela(108). However, three adults with hypomagnesemia, renal Mg2+ losses, and MODY were found to have recessive mutations in the gene PCBD1 which usually causes the transient metabolic disease(109). The encoded protein Pterin-4α carbinolamine dehydratase forms a heterotetrameric complex by physically interacting with HNF1B and is a crucial dimerization factor(109). Defective dimerization due to mutant PCBD1 results in proteolytic instability of the PCBD1-HNF1B complex and subsequently impaired HNF1B-mediated stimulation of FXYD2 promoter activity (Fig. 3C).
Other hypomagnesemia
This group includes different inherited diseases which may not fit in one of the above listed groups (Table 2).
Isolated recessive hypomagnesemia (IRH)
IRH is caused by recessive mutations in the EGF gene(38). The affected individuals developed seizures in infancy, developmental delay, hypomagnesemia due to renal Mg2+ wasting, but no other electrolyte abnormality(110). A homozygous mutation in the pro-EGF gene, encoding for the epidermal growth factor, was identified(38). Pro-EGF is a type 1 transmembrane protein with 1207 amino acids (aa) which is cleaved to the final EGF (53 aa length). The identified mutation is located in the cytosolic C-terminus within a sorting motif and results in impaired EGF secretion(38). WT EGF is secreted and binds to EGF receptor at the basolateral membrane of the DCT, thereby activating a tyrosine kinase which stimulates TRPM6(38) (Fig. 4). The significance of EGF as an autocrine magnesiotropic hormone was confirmed by the chimeric human/mouse anti-EGF antibody cetuximab in chemotherapy as patients treated with cetuximab develop hypomagnesemia(111).
Fig. 4. Mechanisms of other hypomagnesemias.
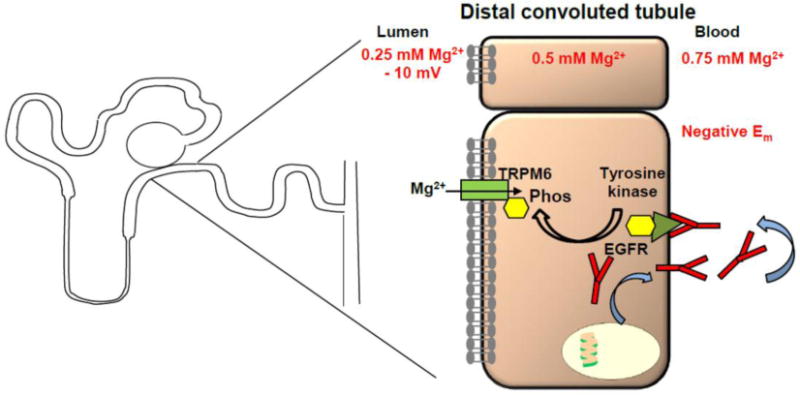
Other hypomagnesemias such as IRH and HSH affect the DCT by impairing EGF secretion or dysfunctional TRPM6 channels. In IRH EGF is less secreted at the basolateral side and therefore there is less autocrine stimulation of the basolateral EGF receptor (EGFR). EGFR stimulates apical TRPM6 channel via phosphorylation by a tyrosine kinase.
Hypomagnesemia with secondary hypocalcemia (HSH)
Patients with this rare condition present with generalized seizures in early infancy, severely low serum Mg2+ (approximately 0.2 mmol/L), and hypocalcemia(112). Perfusion studies in humans indicated an intestinal and renal Mg2+ leak(113). The cause of hypocalcemia is poorly understood but it seems that low Mg2+ impairs PTH secretion(114). Consistent with this finding HSH patients have inappropriately low PTH concentrations. The hypocalcemia does not respond to Ca2+ or vitamin D supplements but only to Mg2+ supplementation(115). Recessive mutations in the gene encoding the transient receptor potential melastatin 6 channel (TRPM6), a member of the TRP family with a C-terminal kinase domain, were published(116, 117). TRPM6 is expressed in the gut (e.g. duodenum, jejunum, colon) and the kidney (e.g. DCT)(118). TRPM6 is homologous to TRPM7, another channel permeable to Ca2+ and Mg2+. Both, TRPM6 and 7 form heteromers, interact and modify each other(119). TRPM6 is regulated my multiple factors including hypomagnesemia, estradiol, and calcineurin inhibitors(35, 120, 121).
Hypomagnesemia with impaired brain development
Patients present with seizures, hypomagnesemia (~0.4–0.5 mmol/L), muscle weakness, vertigo and headaches. Heterozygous and homozygous CNNM2 mutations were found in affected individuals but some mutation carriers remained asymptomatic(122). CNNM2 encodes the transmembrane protein Cyclin M2, which is expressed at the basolateral side of the TAL and DCT. Truncating and missense mutations were published, the latter impaired Mg2+ sensitive current(122, 123). The physiological function of CNNM2 remains unknown, but involves possibly a role as Mg2+ transporter or Mg2+ sensor(122, 124).
Mitochondrial Hypomagnesemia
Metabolic disease with hypercholesterolemia, and hypertension was found linked with hypomagnesemia in a large kindred(125). Interestingly, only females were affected pointing to mitochondrial inheritance. Hypomagnesemia was associated with hypocalciuria indicating involvement of the DCT. A mutation in the mitochondrially encoded isoleucine tRNA gene was found. The mutation affects a thymidine residue adjacent to the anticodon triplet, which is extremely conserved and critical for codon-anticodon recognition. Interestingly, additional mitochondrial disorders have been identified causing hypomagnesemia also involving the TAL(126–128). Both TAL and DCT have high energy requirements but the exact mechanism how mitochondrial disorders contribute to hypomagnesemia remains unclear.
Conclusion
Despite the identification of several genes with rare Mendelian disorders our understanding of renal Mg2+ homeostasis remains very incomplete. Most of the inherited conditions alter the lumen-positive potential in the TAL or the negative membrane potential in the DCT and only few conditions affect directly a Mg2+ channel. As most of the patients present with neurological symptoms practitioners need to be vigilant and neurologists should be educated about the nature of these conditions.
Key Bullet Points.
Mild to moderate chronic hypomagnesemia is a common problem in up to 15% of the general population but symptoms are usually absent or mild.
Because hypomagnesemia is associated with multiple common human disorders (e.g. hypertension, type 2 diabetes mellitus, metabolic syndrome, chronic kidney disease, and cancer) this electrolyte deserves better monitoring.
Hypomagnesemia can be caused by insufficient dietary Mg2+ intake, medication side effects, and rare inherited disorders resulting in urinary Mg2+ wasting.
Inherited forms of hypomagnesemia are divided into hypercalciuric (mostly affecting the TAL), Gitelman-like (mostly affecting the DCT), and other forms of hypomagnesemia (mostly affecting the DCT).
Most forms of inherited hypomagnesemia do not affect directly a Mg2+ channel but rather affect Mg2+ reabsorption by altering the lumen-positive potential difference in the TAL or the negative membrane potential in the DCT.
Acknowledgments
I would like to thank Dr. Michel Baum and Dr. Jyothsna Gattineni for their support with this review.
Financial support and sponsorship
The author is supported by NIH funding (K08-DK095994-05; R03DK111776-01), and the Children’s Clinical Research Advisory Committee (CCRAC), Children’s Medical Center, Dallas.
List of abbreviations
- ADH
autosomal dominant hypocalcemia
- BS
Bartter syndrome
- BSND
Barttin
- Ca2+
calcium
- CaSR
calcium sensing receptor
- Cl−
chloride
- CLDN
claudin
- DCT
distal convoluted tubule
- EAST
epilepsy, ataxia, sensorineural deafness and tubulopathy
- EGF
epidermal growth factor
- ESRD
end-stage renal disease
- FHHNC
familial hypomagnesemia with hypercalciuria and nephrocalciosis
- GS
Gitelman syndrome
- HSH
hypomagnesemia with secondary hypocalcemia
- IDH
isolated dominant hypomagnesemia
- IRH
isolated recessive hypomagnesemia
- K+
potassium
- Kir4.1
inwardly rectifying potassium channel 4.1
- Kv1.1
voltage-gated potassium channel 1.1
- Mg2+
magnesium
- NCC
Na-Cl cotransporter
- NKCC2
Na-K-Cl cotransporter
- NFAT
nuclear factor of activated T-cells
- PT
proximal tubule
- RAS
renin-angiotensin system
- ROMK
renal outer medullary potassium channel
- TAL
thick ascending limb of Henle
- TRPM6
transient receptor potential melastatin 6 channel
Footnotes
Conflicts of interest
No conflict of interest.
References and recommended reading
Papers of particular interest, published within the last 18 months, have been highlighted as:
■ of special interst
■■ of outstanding interest
- 1.Ebel H, Gunther T. Magnesium metabolism: a review. J Clin Chem Clin Biochem. 1980;18(5):257–270. doi: 10.1515/cclm.1980.18.5.257. [DOI] [PubMed] [Google Scholar]
- ■■2.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95(1):1–46. doi: 10.1152/physrev.00012.2014. This is an outstanding review on the role of magnesium in human physiology. [DOI] [PubMed] [Google Scholar]
- 3.Whang R, Ryder KW. Frequency of hypomagnesemia and hypermagnesemia. Requested vs routine. Jama. 1990;263(22):3063–3064. [PubMed] [Google Scholar]
- 4.Pham PC, Pham PA, Pham SV, et al. Hypomagnesemia: a clinical perspective. Int J Nephrol Renovasc Dis. 2014;7:219–230. doi: 10.2147/IJNRD.S42054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schimatschek HF, Rempis R. Prevalence of hypomagnesemia in an unselected German population of 16,000 individuals. Magnes Res. 2001;14(4):283–290. [PubMed] [Google Scholar]
- 6.He K, Liu K, Daviglus ML, et al. Magnesium intake and incidence of metabolic syndrome among young adults. Circulation. 2006;113(13):1675–1682. doi: 10.1161/CIRCULATIONAHA.105.588327. [DOI] [PubMed] [Google Scholar]
- 7.Hopping BN, Erber E, Grandinetti A, et al. Dietary fiber, magnesium, and glycemic load alter risk of type 2 diabetes in a multiethnic cohort in Hawaii. J Nutr. 2010;140(1):68–74. doi: 10.3945/jn.109.112441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez-Ridaura R, Willett WC, Rimm EB, et al. Magnesium intake and risk of type 2 diabetes in men and women. Diabetes Care. 2004;27(1):134–140. doi: 10.2337/diacare.27.1.134. [DOI] [PubMed] [Google Scholar]
- 9.Pham PC, Pham PM, Pham SV, et al. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2007;2(2):366–373. doi: 10.2215/CJN.02960906. [DOI] [PubMed] [Google Scholar]
- ■10.Guerrero-Romero F, Rodriguez-Moran M, Hernandez-Ronquillo G, et al. Low Serum Magnesium Levels and Its Association with High Blood Pressure in Children. J Pediatr. 2016;168:93–98e1. doi: 10.1016/j.jpeds.2015.09.050. The authors describe for the first time the association between hypomagnesemia and hypertension in otherwise healthy children. [DOI] [PubMed] [Google Scholar]
- 11.Del Gobbo LC, Imamura F, Wu JH, et al. Circulating and dietary magnesium and risk of cardiovascular disease: a systematic review and meta-analysis of prospective studies. Am J Clin Nutr. 2013;98(1):160–173. doi: 10.3945/ajcn.112.053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■12.Tin A, Grams ME, Maruthur NM, et al. Results from the Atherosclerosis Risk in Communities study suggest that low serum magnesium is associated with incident kidney disease. Kidney Int. 2015;87(4):820–827. doi: 10.1038/ki.2014.331. This is the first study demonstrating that hypomagnesemia is associated with an increased risk of chronic kidney disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■13.Ulm MA, Watson CH, Vaddadi P, et al. Hypomagnesemia Is Prevalent in Patients Undergoing Gynecologic Surgery by a Gynecologic Oncologist. Int J Gynecol Cancer. 2016;26(7):1320–1326. doi: 10.1097/IGC.0000000000000766. The risk of gynecologic forms of cancer in hypomagnesemic women is discussed. [DOI] [PubMed] [Google Scholar]
- 14.Liao F, Folsom AR, Brancati FL. Is low magnesium concentration a risk factor for coronary heart disease? The Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 1998;136(3):480–490. doi: 10.1016/s0002-8703(98)70224-8. [DOI] [PubMed] [Google Scholar]
- 15.Dai Q, Motley SS, Smith JA, Jr, et al. Blood magnesium, and the interaction with calcium, on the risk of high-grade prostate cancer. PLoS One. 2011;6(4):e18237. doi: 10.1371/journal.pone.0018237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y, Selvaraj S, Varma A, et al. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. J Biol Chem. 2013;288(1):255–263. doi: 10.1074/jbc.M112.393918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Intestinal absorption of magnesium from food and supplements. J Clin Invest. 1991;88(2):396–402. doi: 10.1172/JCI115317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quamme GA. Recent developments in intestinal magnesium absorption. Curr Opin Gastroenterol. 2008;24(2):230–235. doi: 10.1097/MOG.0b013e3282f37b59. [DOI] [PubMed] [Google Scholar]
- 19.Chubanov V, Gudermann T, Schlingmann KP. Essential role for TRPM6 in epithelial magnesium transport and body magnesium homeostasis. Pflugers Arch. 2005;451(1):228–234. doi: 10.1007/s00424-005-1470-y. [DOI] [PubMed] [Google Scholar]
- 20.Shareghi GR, Agus ZS. Magnesium transport in the cortical thick ascending limb of Henle’s loop of the rabbit. J Clin Invest. 1982;69(4):759–769. doi: 10.1172/JCI110514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunette MG, Vigneault N, Carriere S. Micropuncture study of magnesium transport along the nephron in the young rat. Am J Physiol. 1974;227(4):891–896. doi: 10.1152/ajplegacy.1974.227.4.891. [DOI] [PubMed] [Google Scholar]
- 22.Hoenderop JG, Bindels RJ. Calciotropic and magnesiotropic TRP channels. Physiology (Bethesda) 2008;23:32–40. doi: 10.1152/physiol.00039.2007. [DOI] [PubMed] [Google Scholar]
- 23.Hoenderop JG, Bindels RJ. Epithelial Ca2+ and Mg2+ channels in health and disease. J Am Soc Nephrol. 2005;16(1):15–26. doi: 10.1681/ASN.2004070523. [DOI] [PubMed] [Google Scholar]
- 24.Hess MW, Hoenderop JG, Bindels RJ, Drenth JP. Systematic review: hypomagnesaemia induced by proton pump inhibition. Aliment Pharmacol Ther. 2012;36(5):405–413. doi: 10.1111/j.1365-2036.2012.05201.x. [DOI] [PubMed] [Google Scholar]
- 25.Cundy T, Dissanayake A. Severe hypomagnesaemia in long-term users of proton-pump inhibitors. Clin Endocrinol (Oxf) 2008;69(2):338–341. doi: 10.1111/j.1365-2265.2008.03194.x. [DOI] [PubMed] [Google Scholar]
- 26.Ray S, Delaney M, Muller AF. Proton pump inhibitors and acute interstitial nephritis. Bmj. 2010;341:c4412. doi: 10.1136/bmj.c4412. [DOI] [PubMed] [Google Scholar]
- 27.Quamme GA. Effect of furosemide on calcium and magnesium transport in the rat nephron. Am J Physiol. 1981;241(4):F340–347. doi: 10.1152/ajprenal.1981.241.4.F340. [DOI] [PubMed] [Google Scholar]
- 28.Nijenhuis T, Vallon V, van der Kemp AW, et al. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115(6):1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lajer H, Daugaard G. Cisplatin and hypomagnesemia. Cancer Treat Rev. 1999;25(1):47–58. doi: 10.1053/ctrv.1999.0097. [DOI] [PubMed] [Google Scholar]
- 30.Gonzales-Vitale JC, Hayes DM, Cvitkovic E, Sternberg SS. The renal pathology in clinical trials of cis-platinum (II) diamminedichloride. Cancer. 1977;39(4):1362–1371. doi: 10.1002/1097-0142(197704)39:4<1362::aid-cncr2820390403>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 31.Karasawa T, Steyger PS. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol Lett. 2015;237(3):219–227. doi: 10.1016/j.toxlet.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■32.Torres R, Velazquez H, Chang JJ, et al. Three-Dimensional Morphology by Multiphoton Microscopy with Clearing in a Model of Cisplatin-Induced CKD. J Am Soc Nephrol. 2016;27(4):1102–1112. doi: 10.1681/ASN.2015010079. The authors describe a novel application for multiphton microscopy with clearing to provide three dimensional morphology of cisplatin induced chronic kidney disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solanki MH, Chatterjee PK, Gupta M, et al. Magnesium protects against cisplatin-induced acute kidney injury by regulating platinum accumulation. Am J Physiol Renal Physiol. 2014;307(4):F369–384. doi: 10.1152/ajprenal.00127.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazzola BL, Vannini SD, Truttmann AC, et al. Long-term calcineurin inhibition and magnesium balance after renal transplantation. Transpl Int. 2003;16(2):76–81. doi: 10.1007/s00147-002-0479-9. [DOI] [PubMed] [Google Scholar]
- 35.Nijenhuis T, Hoenderop JG, Bindels RJ. Downregulation of Ca(2+) and Mg(2+) transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol. 2004;15(3):549–557. doi: 10.1097/01.asn.0000113318.56023.b6. [DOI] [PubMed] [Google Scholar]
- 36.Ledeganck KJ, Boulet GA, Horvath CA, et al. Expression of renal distal tubule transporters TRPM6 and NCC in a rat model of cyclosporine nephrotoxicity and effect of EGF treatment. Am J Physiol Renal Physiol. 2011;301(3):F486–493. doi: 10.1152/ajprenal.00116.2011. [DOI] [PubMed] [Google Scholar]
- 37.Chang CT, Hung CC, Tian YC, et al. Ciclosporin reduces paracellin-1 expression and magnesium transport in thick ascending limb cells. Nephrol Dial Transplant. 2007;22(4):1033–1040. doi: 10.1093/ndt/gfl817. [DOI] [PubMed] [Google Scholar]
- 38.Groenestege WM, Thebault S, van der Wijst J, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest. 2007;117(8):2260–2267. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thebault S, Alexander RT, Tiel Groenestege WM, et al. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. 2009;20(1):78–85. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexandridis G, Liberopoulos E, Elisaf M. Aminoglycoside-induced reversible tubular dysfunction. Pharmacology. 2003;67(3):118–120. doi: 10.1159/000067797. [DOI] [PubMed] [Google Scholar]
- 41.Nagai J, Tanaka H, Nakanishi N, et al. Role of megalin in renal handling of aminoglycosides. Am J Physiol Renal Physiol. 2001;281(2):F337–344. doi: 10.1152/ajprenal.2001.281.2.F337. [DOI] [PubMed] [Google Scholar]
- 42.Weber S, Schneider L, Peters M, et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2001;12(9):1872–1881. doi: 10.1681/ASN.V1291872. [DOI] [PubMed] [Google Scholar]
- 43.Michelis MF, Drash AL, Linarelli LG, et al. Decreased bicarbonate threshold and renal magnesium wasting in a sibship with distal renal tubular acidosis. (Evaluation of the pathophysiological role of parathyroid hormone) Metabolism. 1972;21(10):905–920. doi: 10.1016/0026-0495(72)90025-x. [DOI] [PubMed] [Google Scholar]
- 44.Claverie-Martin F, Garcia-Nieto V, Loris C, et al. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One. 2013;8(1):e53151. doi: 10.1371/journal.pone.0053151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konrad M, Hou J, Weber S, et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2008;19(1):171–181. doi: 10.1681/ASN.2007060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Godron A, Harambat J, Boccio V, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol. 2012;7(5):801–809. doi: 10.2215/CJN.12841211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■47.Yamaguti PM, Neves FA, Hotton D, et al. Amelogenesis imperfecta in familial hypomagnesaemia and hypercalciuria with nephrocalcinosis caused by CLDN19 gene mutations. J Med Genet. 2016:jmedgenet-2016-103956. doi: 10.1136/jmedgenet-2016-103956. [Epub ahead of print] In this paper this authors expand the phenotype of FFHNC by adding tooth enamel defects. [DOI] [PubMed] [Google Scholar]
- 48.Wolf MT, Dotsch J, Konrad M, et al. Follow-up of five patients with FHHNC due to mutations in the Paracellin-1 gene. Pediatr Nephrol. 2002;17(8):602–608. doi: 10.1007/s00467-002-0884-4. [DOI] [PubMed] [Google Scholar]
- 49.Simon DB, Lu Y, Choate KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285(5424):103–106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 50.Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79(5):949–957. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■51.Yu AS. Claudins and the kidney. J Am Soc Nephrol. 2015;26(1):11–19. doi: 10.1681/ASN.2014030284. This is an excellcent review about the role of claudins in the kidney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hou J, Renigunta A, Konrad M, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest. 2008;118(2):619–628. doi: 10.1172/JCI33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou J, Renigunta A, Gomes AS, et al. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci U S A. 2009;106(36):15350–15355. doi: 10.1073/pnas.0907724106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirano T, Kobayashi N, Itoh T, et al. Null mutation of PCLN-1/Claudin-16 results in bovine chronic interstitial nephritis. Genome Res. 2000;10(5):659–663. doi: 10.1101/gr.10.5.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kobayashi N, Hirano T, Maruyama S, et al. Genetic mapping of a locus associated with bovine chronic interstitial nephritis to chromosome 1. Anim Genet. 2000;31(2):91–95. doi: 10.1046/j.1365-2052.2000.00589.x. [DOI] [PubMed] [Google Scholar]
- 56.Will C, Breiderhoff T, Thumfart J, et al. Targeted deletion of murine Cldn16 identifies extra- and intrarenal compensatory mechanisms of Ca2+ and Mg2+ wasting. Am J Physiol Renal Physiol. 2010;298(5):F1152–1161. doi: 10.1152/ajprenal.00499.2009. [DOI] [PubMed] [Google Scholar]
- 57.Miyamoto T, Morita K, Takemoto D, et al. Tight junctions in Schwann cells of peripheral myelinated axons: a lesson from claudin-19-deficient mice. J Cell Biol. 2005;169(3):527–538. doi: 10.1083/jcb.200501154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zimmermann B, Plank C, Konrad M, et al. Hydrochlorothiazide in CLDN16 mutation. Nephrol Dial Transplant. 2006;21(8):2127–2132. doi: 10.1093/ndt/gfl144. [DOI] [PubMed] [Google Scholar]
- 59.Pearce SH, Williamson C, Kifor O, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335(15):1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- 60.Pollak MR, Brown EM, Chou YH, et al. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75(7):1297–1303. doi: 10.1016/0092-8674(93)90617-y. [DOI] [PubMed] [Google Scholar]
- 61.Pollak MR, Brown EM, Estep HL, et al. Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet. 1994;8(3):303–307. doi: 10.1038/ng1194-303. [DOI] [PubMed] [Google Scholar]
- 62.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81(1):239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 63.Bapty BW, Dai LJ, Ritchie G, et al. Mg2+/Ca2+ sensing inhibits hormone-stimulated Mg2+ uptake in mouse distal convoluted tubule cells. Am J Physiol. 1998;275(3 Pt 2):F353–360. doi: 10.1152/ajprenal.1998.275.3.F353. [DOI] [PubMed] [Google Scholar]
- 64.Watanabe S, Fukumoto S, Chang H, et al. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet. 2002;360(9334):692–694. doi: 10.1016/S0140-6736(02)09842-2. [DOI] [PubMed] [Google Scholar]
- 65.Hebert SC, Brown EM, Harris HW. Role of the Ca(2+)-sensing receptor in divalent mineral ion homeostasis. J Exp Biol. 1997;200(Pt 2):295–302. doi: 10.1242/jeb.200.2.295. [DOI] [PubMed] [Google Scholar]
- 66.Zhang C, Wang L, Zhang J, et al. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1) Proc Natl Acad Sci U S A. 2014;111(32):11864–11869. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gong Y, Renigunta V, Himmerkus N, et al. Claudin-14 regulates renal Ca(+)(+) transport in response to CaSR signalling via a novel microRNA pathway. Embo j. 2012;31(8):1999–2012. doi: 10.1038/emboj.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■68.Gong Y, Himmerkus N, Plain A, et al. Epigenetic regulation of microRNAs controlling CLDN14 expression as a mechanism for renal calcium handling. J Am Soc Nephrol. 2015;26(3):663–676. doi: 10.1681/ASN.2014020129. In this outstanding manuscript a novel mechanism influencing renal calcium reabsorption via claudin-14 is described. The authors delineate the regulation of claudin-14 by two specific miRNAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toka HR, Al-Romaih K, Koshy JM, et al. Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria. J Am Soc Nephrol. 2012;23(11):1879–1890. doi: 10.1681/ASN.2012030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bartter FC, Pronove P, Gill JR, Jr, Maccardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med. 1962;33:811–828. doi: 10.1016/0002-9343(62)90214-0. [DOI] [PubMed] [Google Scholar]
- 71.Zelikovic I, Szargel R, Hawash A, et al. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003;63(1):24–32. doi: 10.1046/j.1523-1755.2003.00730.x. [DOI] [PubMed] [Google Scholar]
- 72.Jeck N, Konrad M, Peters M, et al. Mutations in the chloride channel gene, CLCNKB, leading to a mixed Bartter-Gitelman phenotype. Pediatr Res. 2000;48(6):754–758. doi: 10.1203/00006450-200012000-00009. [DOI] [PubMed] [Google Scholar]
- 73.Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17(2):171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 74.Konrad M, Vollmer M, Lemmink HH, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000;11(8):1449–1459. doi: 10.1681/ASN.V1181449. [DOI] [PubMed] [Google Scholar]
- 75.Birkenhager R, Otto E, Schurmann MJ, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29(3):310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- ■76.Viering DH, de Baaij JH, Walsh SB, et al. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol. 2016 doi: 10.1007/s00467-016-3416-3. A comprehensive review of hereditary hypomagnesemias. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12(5):527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 78.Kleta R, Bockenhauer D. Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol. 2006;104(2):p73–80. doi: 10.1159/000094001. [DOI] [PubMed] [Google Scholar]
- 79.Rudin A. Bartter’s syndrome. A review of 28 patients followed for 10 years. Acta Med Scand. 1988;224(2):165–171. [PubMed] [Google Scholar]
- 80.Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235. [PubMed] [Google Scholar]
- 81.Cruz DN, Shaer AJ, Bia MJ, et al. Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59(2):710–717. doi: 10.1046/j.1523-1755.2001.059002710.x. [DOI] [PubMed] [Google Scholar]
- 82.Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12(1):24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 83.Nozu K, Iijima K, Nozu Y, et al. A deep intronic mutation in the SLC12A3 gene leads to Gitelman syndrome. Pediatr Res. 2009;66(5):590–593. doi: 10.1203/PDR.0b013e3181b9b4d3. [DOI] [PubMed] [Google Scholar]
- 84.Lo YF, Nozu K, Iijima K, et al. Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman’s syndrome. Clin J Am Soc Nephrol. 2011;6(3):630–639. doi: 10.2215/CJN.06730810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22(4):693–703. doi: 10.1681/ASN.2010090907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schultheis PJ, Lorenz JN, Meneton P, et al. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl- cotransporter of the distal convoluted tubule. J Biol Chem. 1998;273(44):29150–29155. doi: 10.1074/jbc.273.44.29150. [DOI] [PubMed] [Google Scholar]
- 87.Loffing J, Vallon V, Loffing-Cueni D, et al. Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman’s syndrome. J Am Soc Nephrol. 2004;15(9):2276–2288. doi: 10.1097/01.ASN.0000138234.18569.63. [DOI] [PubMed] [Google Scholar]
- 88.Yang SS, Lo YF, Yu IS, et al. Generation and analysis of the thiazide-sensitive Na+ -Cl- cotransporter (Ncc/Slc12a3) Ser707X knockin mouse as a model of Gitelman syndrome. Hum Mutat. 2010;31(12):1304–1315. doi: 10.1002/humu.21364. [DOI] [PubMed] [Google Scholar]
- 89.Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360(19):1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cross JH, Arora R, Heckemann RA, et al. Neurological features of epilepsy, ataxia, sensorineural deafness, tubulopathy syndrome. Dev Med Child Neurol. 2013;55(9):846–856. doi: 10.1111/dmcn.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bandulik S, Schmidt K, Bockenhauer D, et al. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch. 2011;461(4):423–435. doi: 10.1007/s00424-010-0915-0. [DOI] [PubMed] [Google Scholar]
- 92.Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parrock S, Hussain S, Issler N, et al. KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol. 2013;123(3–4):7–14. doi: 10.1159/000356353. [DOI] [PubMed] [Google Scholar]
- ■94.Zhang C, Wang L, Su XT, et al. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol. 2015;308(11):F1288–1296. doi: 10.1152/ajprenal.00687.2014. The role of Kir4.1 was studied in wild-type and Kir4.1 knockout mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meij IC, Koenderink JB, van Bokhoven H, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat Genet. 2000;26(3):265–266. doi: 10.1038/81543. [DOI] [PubMed] [Google Scholar]
- 96.Glaudemans B, van der Wijst J, Scola RH, et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119(4):936–942. doi: 10.1172/JCI36948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones DH, Li TY, Arystarkhova E, et al. Na,K-ATPase from mice lacking the gamma subunit (FXYD2) exhibits altered Na+ affinity and decreased thermal stability. J Biol Chem. 2005;280(19):19003–19011. doi: 10.1074/jbc.M500697200. [DOI] [PubMed] [Google Scholar]
- 98.Sweadner KJ, Arystarkhova E, Donnet C, Wetzel RK. FXYD proteins as regulators of the Na,K-ATPase in the kidney. Ann N Y Acad Sci. 2003;986:382–387. doi: 10.1111/j.1749-6632.2003.tb07218.x. [DOI] [PubMed] [Google Scholar]
- ■99.de Baaij JH, Dorresteijn EM, Hennekam EA, et al. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant. 2015;30(6):952–957. doi: 10.1093/ndt/gfv014. This report confirms the significance of the FXYD2 G41R mutation for hypomagnesemia and points out that so far no other FXYD2 mutations has been identified. [DOI] [PubMed] [Google Scholar]
- 100.Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8(2):136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- 101.Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384–385. doi: 10.1038/ng1297-384. [DOI] [PubMed] [Google Scholar]
- ■■102.Clissold RL, Hamilton AJ, Hattersley AT, et al. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol. 2015;11(2):102–112. doi: 10.1038/nrneph.2014.232. An excellent review about the expanding clinical spectrum of HNF1B mutations. [DOI] [PubMed] [Google Scholar]
- ■103.Gondra L, Decramer S, Chalouhi GE, et al. Hyperechogenic kidneys and polyhydramnios associated with HNF1B gene mutation. Pediatr Nephrol. 2016;31(10):1705–1708. doi: 10.1007/s00467-016-3421-6. The authors describe the association of HNF1B mutations with polyhydramnios. [DOI] [PubMed] [Google Scholar]
- 104.Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte Nuclear Factor 1beta-Associated Kidney Disease: More than Renal Cysts and Diabetes. J Am Soc Nephrol. 2016;27(2):345–353. doi: 10.1681/ASN.2015050544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol. 2016;31(5):707–714. doi: 10.1007/s00467-015-3142-2. [DOI] [PubMed] [Google Scholar]
- 106.Adalat S, Woolf AS, Johnstone KA, et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20(5):1123–1131. doi: 10.1681/ASN.2008060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ferre S, Veenstra GJ, Bouwmeester R, et al. HNF-1B specifically regulates the transcription of the gammaa-subunit of the Na+/K+-ATPase. Biochem Biophys Res Commun. 2011;404(1):284–290. doi: 10.1016/j.bbrc.2010.11.108. [DOI] [PubMed] [Google Scholar]
- 108.Thony B, Neuheiser F, Kierat L, et al. Mutations in the pterin-4alpha-carbinolamine dehydratase (PCBD) gene cause a benign form of hyperphenylalaninemia. Hum Genet. 1998;103(2):162–167. doi: 10.1007/s004390050800. [DOI] [PubMed] [Google Scholar]
- 109.Ferre S, de Baaij JH, Ferreira P, et al. Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2014;25(3):574–586. doi: 10.1681/ASN.2013040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Geven WB, Monnens LA, Willems JL, et al. Isolated autosomal recessive renal magnesium loss in two sisters. Clin Genet. 1987;32(6):398–402. doi: 10.1111/j.1399-0004.1987.tb03157.x. [DOI] [PubMed] [Google Scholar]
- 111.Tejpar S, Piessevaux H, Claes K, et al. Magnesium wasting associated with epidermal-growth-factor receptor-targeting antibodies in colorectal cancer: a prospective study. Lancet Oncol. 2007;8(5):387–394. doi: 10.1016/S1470-2045(07)70108-0. [DOI] [PubMed] [Google Scholar]
- 112.Paunier L, Radde IC, Kooh SW, et al. Primary hypomagnesemia with secondary hypocalcemia in an infant. Pediatrics. 1968;41(2):385–402. [PubMed] [Google Scholar]
- 113.Milla PJ, Aggett PJ, Wolff OH, Harries JT. Studies in primary hypomagnesaemia: evidence for defective carrier-mediated small intestinal transport of magnesium. Gut. 1979;20(11):1028–1033. doi: 10.1136/gut.20.11.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Anast CS, Mohs JM, Kaplan SL, Burns TW. Evidence for parathyroid failure in magnesium deficiency. Science. 1972;177(4049):606–608. doi: 10.1126/science.177.4049.606. [DOI] [PubMed] [Google Scholar]
- 115.Shalev H, Phillip M, Galil A, et al. Clinical presentation and outcome in primary familial hypomagnesaemia. Arch Dis Child. 1998;78(2):127–130. doi: 10.1136/adc.78.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schlingmann KP, Weber S, Peters M, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31(2):166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 117.Walder RY, Landau D, Meyer P, et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet. 2002;31(2):171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 118.Voets T, Nilius B, Hoefs S, et al. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279(1):19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 119.Chubanov V, Waldegger S, Mederos y Schnitzler M, et al. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A. 2004;101(9):2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Groenestege WM, Hoenderop JG, van den Heuvel L, et al. The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J Am Soc Nephrol. 2006;17(4):1035–1043. doi: 10.1681/ASN.2005070700. [DOI] [PubMed] [Google Scholar]
- 121.Cao G, van der Wijst J, van der Kemp A, et al. Regulation of the epithelial Mg2+ channel TRPM6 by estrogen and the associated repressor protein of estrogen receptor activity (REA) J Biol Chem. 2009;284(22):14788–14795. doi: 10.1074/jbc.M808752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stuiver M, Lainez S, Will C, et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet. 2011;88(3):333–343. doi: 10.1016/j.ajhg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arjona FJ, de Baaij JH, Schlingmann KP, et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet. 2014;10(4):e1004267. doi: 10.1371/journal.pgen.1004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■124.Sponder G, Mastrototaro L, Kurth K, et al. Human CNNM2 is not a Mg(2+) transporter per se. Pflugers Arch. 2016;468(7):1223–1240. doi: 10.1007/s00424-016-1816-7. The role of CNNM2 in hypomagnesemia is studied in different cell culture systems. [DOI] [PubMed] [Google Scholar]
- 125.Wilson FH, Hariri A, Farhi A, et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306(5699):1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88(2):193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Emma F, Pizzini C, Tessa A, et al. “Bartter-like” phenotype in Kearns-Sayre syndrome. Pediatr Nephrol. 2006;21(3):355–360. doi: 10.1007/s00467-005-2092-5. [DOI] [PubMed] [Google Scholar]
- 128.Goto Y, Itami N, Kajii N, et al. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr. 1990;116(6):904–910. doi: 10.1016/s0022-3476(05)80648-1. [DOI] [PubMed] [Google Scholar]