

Abstract
Renal ischemia/reperfusion is a major cause of acute kidney injury. However, the pathogenic mechanisms underlying renal ischemia/reperfusion injury (IRI) are not fully defined. Here, we investigated the role of PTEN, a dual protein/lipid phosphatase, in the development of ischemic AKI in mice. Pharmacological inhibition of PTEN with bpV(HOpic) exacerbated renal dysfunction and promoted tubular damage in mice with IRI compared with vehicle-treated mice with IRI. PTEN inhibition enhanced tubular cell apoptosis in kidneys with IRI, which was associated with excessive caspase-3 activation. Furthermore, PTEN inhibition expanded the infiltration of neutrophils and macrophages into kidneys with IRI, which was accompanied by increased expression of the proinflammatory molecules. These results have demonstrated that PTEN plays a crucial role in the pathogenesis of ischemic acute kidney injury through regulating tubular cell apoptosis and inflammation suggesting PTEN could be a potential therapeutic target for acute kidney injury.
Introduction
Acute kidney injury (AKI) is a common clinical condition associated with increased mortality and morbidity, prolonged hospital stay, and accelerated chronic kidney disease (CKD)1, 2. Ischemia-reperfusion injury (IRI) is a major cause of AKI1, 3. However, the pathogenesis of ischemic AKI is complex and incompletely understood. The pathophysiology of ischemic AKI includes hemodynamic alteration, inflammation, endothelial dysfunction, and epithelial cell injury which eventually result in acute tubular damage and renal dysfunction1, 4, 5. Currently, there is no effective therapy for ischemic AKI. Therefore, a better understanding of the pathogenic mechanisms underlying IRI is essential to develop effective therapy for ischemic AKI.
Phosphatase and tensin homologue deleted from chromosome 10 (PTEN) is a dual protein and phospholipid phosphatase6. The functional role of PTEN includes the regulation of cell growth, adhesion, and migration7, 8. The primary function of PTEN acts as a lipid phosphatase that converts phosphatidylinositol (3,4,5)-triphosphate (PIP3) to phosphatidylinositol (4,5)-triphosphate (PIP2) thereby downregulating phosphatidylinositol 3 kinase (PI3K)/Akt signaling6, 9, 10. Recent studies have shown that PTEN regulates kidney morphology, podocyte injury and renal fibrosis. PTEN loss is associated with increased transforming growth factor (TGF)-β signaling and renal fibrosis11. Mice with proximal tubule deletion of PTEN develop larger kidneys characterized by proximal tubule cell hypertrophy12. We have recently demonstrated that loss of PTEN in podocytes aggravates diabetic nephropathy through regulation of cytoskeletal rearrangement13. However, the functional role of PTEN in ischemic AKI is not known.
In this study, we examined the effect of pharmacological inhibition of PTEN on the pathogenesis of AKI in a mouse model of ischemia-reperfusion injury. Our results show that pharmacological inhibition of PTEN with bpV(HOpic), a selective PTEN inhibitor13, exacerbates ischemic AKI by promoting apoptosis and inflammation.
Results
PTEN is activated in the kidney in response to ischemic AKI
We first determined whether PTEN is induced in the kidney in a mouse model of AKI induced by 30 min of ischemia followed by 24 hr of reperfusion. Sham-operated mice were served as controls. Kidney sections were stained for PTEN. Immunohistochemical staining revealed that PTEN is expressed at a low level in the kidney of sham-operated mice, which was markedly induced in the interstitial cells of the kidney following ischemic AKI (Fig. 1A). We next examined if IRI affects PTEN activity in the kidney using the malachite green phosphatase assay14. The results showed that PTEN activity increased in the kidney following ischemic AKI, which was significantly inhibited by bpV(HOpic) (Fig. 1B). These data indicate that PTEN may regulate the pathogenesis of ischemic AKI.
Figure 1.
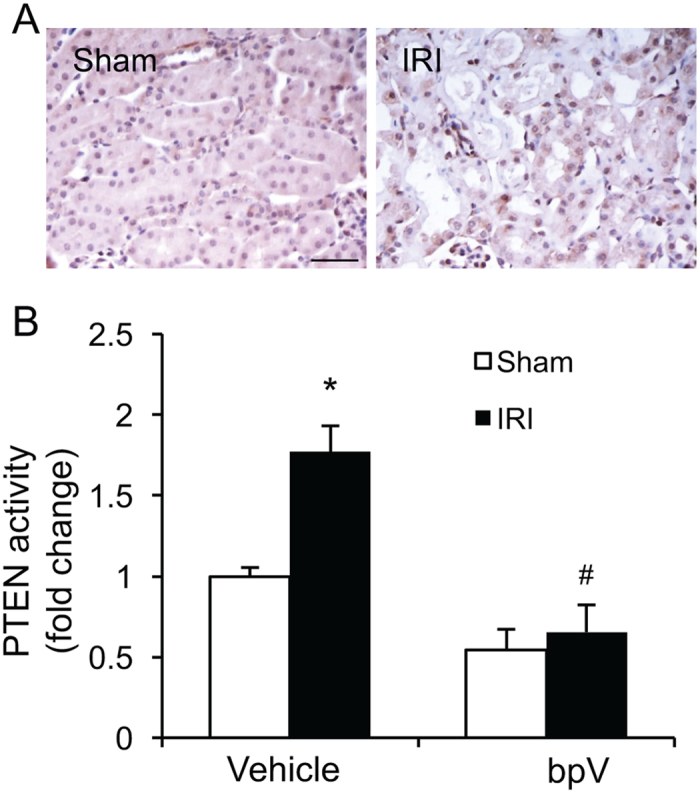
PTEN is activated in the kidney with IRI. (A) Representative photomicrographs of kidney sections stained with PTEN (brown) and counterstained with hematoxylin (blue). Scale bar, 50 μm. (B) PTEN activity was measured in kidney homogenates and expressed as fold changes. *P < 0.05 vs. Vehicle Sham, # P < 0.05 vs. vehicle IRI. n = 6 per group.
PTEN inhibition exacerbates kidney dysfunction following IRI
To determine the role of PTEN in the pathogenesis of ischemic AKI, mice were subjected to 30 min of ischemia, followed by 24 hr of reperfusion. Sham-operated mice were served as controls. PTEN inhibitor, bpV(HOpic) (200 μg/kg), or vehicle was intraperitoneally administered 1 h before ischemia and then administered every 6 h after ischemia for 24 hr. Renal IRI led to significant kidney dysfunction with an increase in serum creatinine and BUN (Fig. 1A and B). In contrast, kidney dysfunction was even worsened in bpV(HOpic) treated mice with much higher elevation of serum creatinine and BUN (Fig. 2A and B). Consistent with the worsening of kidney dysfunction in bpV(HOpic) treated mice with IRI, there was a much greater increase in histological injury of the kidney as reflected by more tubular injury, tubular dilation, and intratubular cast formation in bpV(HOpic) treated mice with IRI compared with vehicle-treated mice with IRI (Fig. 2C and D). These data indicate that PTEN inhibition exacerbates ischemic AKI.
Figure 2.

PTEN inhibition exacerbates kidney injury after IRI. (A) Serum creatinine increased markedly in PTEN inhibitor bpV treated mice with IRI. **P < 0.01 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, ## P < 0.01 vs. Vehicle IRI. n = 6 per group. (B) Blood serum urea nitrogen (BUN) increased significantly in bpV treated mice with IRI. **P < 0.01 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, ## P < 0.01 vs. Vehicle IRI. n = 6 per group. (C) Representative photomicrographs of kidney sections stained with hematoxylin and eosin. Scale bar, 50 μm. (D) Quantitative analysis of tubular damage after IRI. *P < 0.05 vs. Vehicle IRI. n = 6 per group.
PTEN inhibition enhances apoptotic cell death in the kidney with IRI
Tubular epithelial cell apoptosis plays an important role in the pathogenesis of ischemic kidney injury1, 15, 16. Therefore, we determine the effect of PTEN inhibition on tubular epithelial cell apoptosis following IRI. Kidney IRI significantly increased the number of tubular apoptotic cells in the kidneys as assessed by TUNEL staining; whereas bpV(HOpic) treatment further increased the number of tubular apoptotic cells in IRI kidneys (Fig. 3A and B). These data indicate that inhibition of PTEN promotes tubular epithelial cell apoptosis.
Figure 3.

PTEN inhibition promotes tubular epithelial cell apoptosis in IRI kidney. (A) Representative photomicrographs of kidney sections stained for apoptotic cells (brown) and counterstained with methyl green (green) in kidneys. Scale bar, 50 μm. (B) Quantitative analysis of TUNEL-positive cells in the kidneys. **P < 0.01 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, # P < 0.05 vs. Vehicle IRI. n = 6 per group. HPF, high power field; TUNEL, terminal transferase dUTP nick-end labeling.
Caspase 3 is the final effector in apoptotic cell death17. We then determined the effect of PTEN inhibition on caspase 3 activation in the kidney following IRI. Western blot analysis showed that the levels of cleaved caspase 3 in the kidneys were significantly higher in bpV(HOpic)-treated mice after IRI than vehicle-treated mice with IRI (Fig. 4A). Furthermore, caspase 3 activity assay revealed that caspase 3 activity was significantly increased in bpV(HOpic)-treated kidney with IRI compared with vehicle-treated kidney with IRI (Fig. 4B). These data indicate that PTEN inhibition enhances caspase 3 activation in the kidney after IRI.
Figure 4.

PTEN inhibition increases caspase 3 activation in IRI kidney. (A) Representative western blots show cleaved caspase 3 protein levels in the kidneys after sham or IRI. (B) Quantitative analysis of cleaved caspase 3 protein levels in the kidneys. *P < 0.05 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, # P < 0.05 vs. Vehicle IRI. n = 6 per group. (C) Caspase 3 activity is increased in bpV treated IRI kidney than vehicle treated IRI kidney. **P < 0.01 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, # P < 0.05 vs. vehicle IRI. n = 6 per group.
PTEN inhibition augments inflammatory cell infiltration in IRI kidney
Inflammatory cells have been shown to infiltrate the kidney during the injury phase and participate in the pathogenesis of renal IRI18. We next investigated whether PTEN inhibition affects inflammatory cell infiltration into the kidney in response to IRI. Kidney sections were stained with antibodies against MPO, a neutrophil marker, and F4/80, a macrophage marker. The number of neutrophils were increased significantly in the IRI kidneys of mice treated with vehicle, which were further increased in the IRI kidneys of mice treated with bpV(HOpic) (Fig. 5). Similarly, bpV(HOpic) treatment resulted in a significant increase in macrophages in the kidneys of mice with IRI compared with vehicle-treated mice with IRI (Fig. 6). These data suggest that PTEN inhibition promotes inflammatory cell infiltration into the kidney during IRI.
Figure 5.
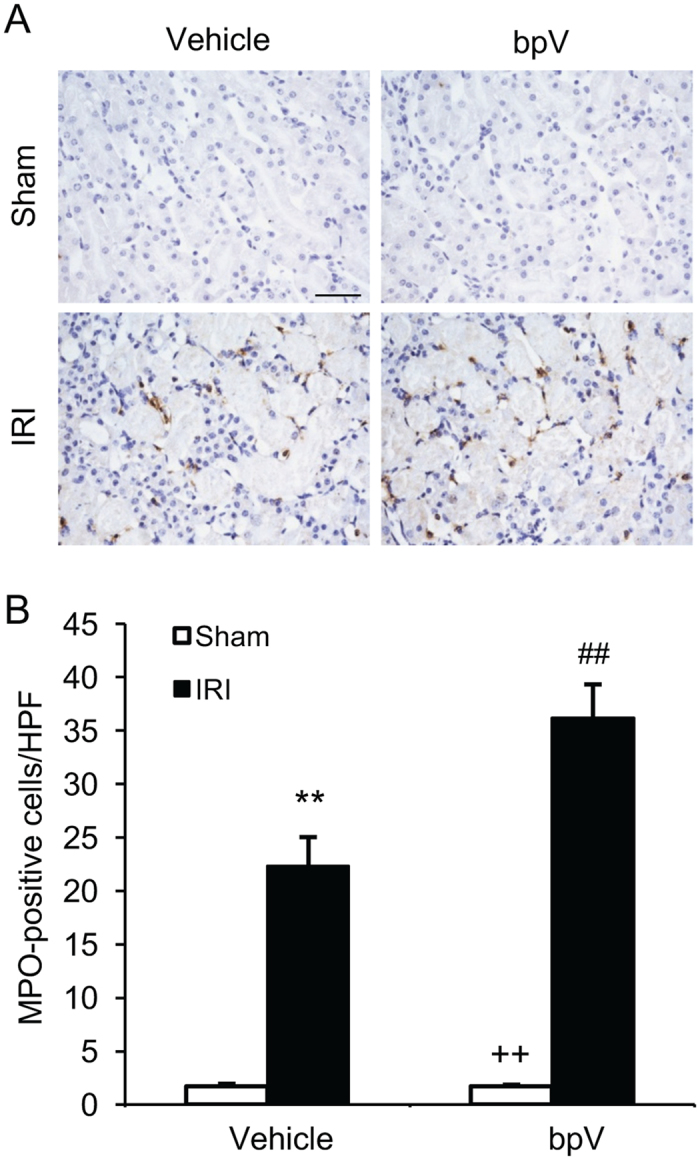
PTEN inhibition promotes neutrophils infiltration into IRI kidney. (A) Representative photomicrographs of kidney sections stained for MPO (a neutrophils marker) (brown) and counterstained with hematoxylin (blue). Scale bar, 50 μm. (B) Quantitative analysis of neutrophils in the kidneys. **P < 0.01 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, ## P < 0.01 vs. Vehicle IRI. n = 6 per group.
Figure 6.
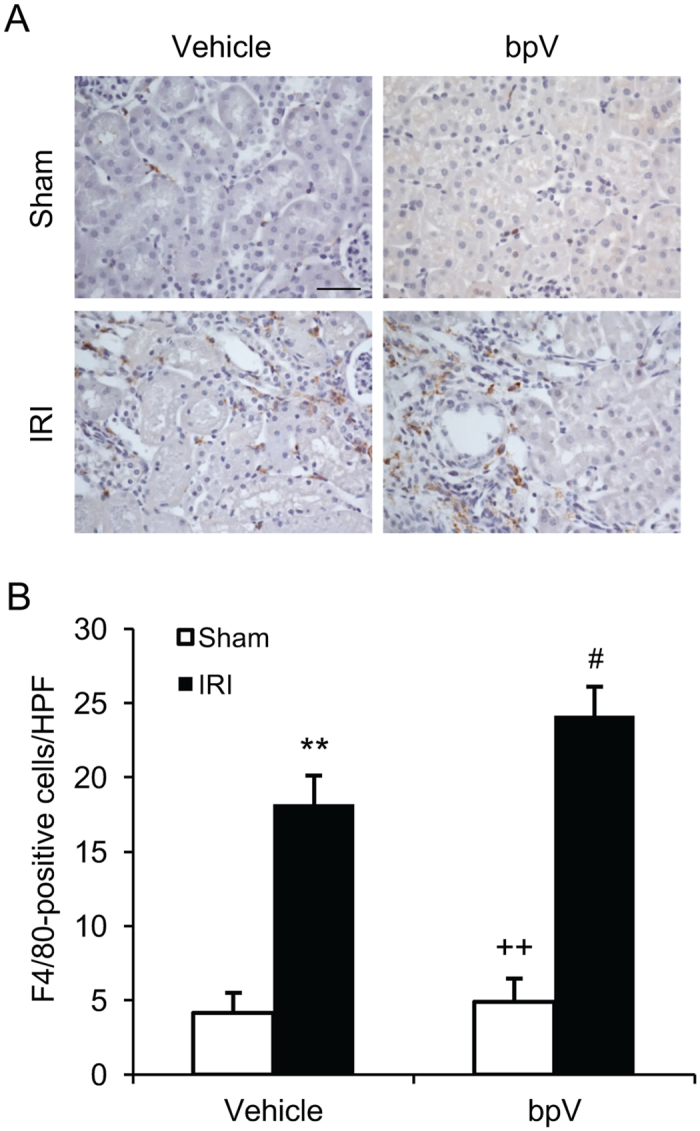
PTEN inhibition enhances macrophage infiltration into IRI kidney. (A) Representative photomicrographs of kidney sections stained for F4/80 (a macrophage marker) (brown) and counterstained with hematoxylin (blue). Scale bar, 50 μm. (B) Quantitative analysis of macrophages in the kidneys. **P < 0.01 vs. Vehicle Sham, ++ P < 0.01 vs. bpV IRI, # P < 0.05 vs. Vehicle IRI. n = 6 per group.
PTEN inhibition upregulates inflammatory mediators in IRI kidney
Inflammatory cytokines and chemokines are crucial mediators in the pathogenesis of ischemic AKI16, 18. We then examined the effect of PTEN inhibition on the expression of inflammatory mediators in the kidney by real-time RT-PCR. The mRNA levels of interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), MCP-1 and MIP-2 were increased significantly in IRI kidneys of mice treated with vehicle compared with vehicle-treated sham–operated controls (Fig. 7). Inhibition of PTEN with bpV(HOpic) further increased the expression levels of these proinflammatory molecules in the IRI kidneys (Fig. 7). These results suggest that PTEN inhibition augments inflammatory chemokine and cytokine expression in the kidney during IRI injury.
Figure 7.

PTEN inhibition increases gene expression of inflammatory molecules in the kidneys with IRI. (A) IL-6 mRNA expression is increased in bpV-treated IRI kidneys compared with vehicle-treated IRI kidneys. **P < 0.01 vs. Vehicle Sham; ++ P < 0.01 vs. bpV IRI; ## P < 0.01 vs. Vehicle IRI. n = 4 per group. (B) TNF-α mRNA expression is enhanced in bpV-treated IRI kidneys compared with vehicle-treated IRI kidneys. **P < 0.01 vs. Vehicle Sham; ++ P < 0.01 vs. bpV IRI; #P < 0.05 vs. Vehicle IRI. n = 4 per group. (C) MCP-1 mRNA expression is increased in bpV-treated IRI kidneys compared with vehicle-treated IRI kidneys. **P < 0.01 vs. Vehicle Sham; ++ P < 0.01 vs. bpV IRI; # P < 0.05 vs. Vehicle IRI. n = 4 per group. (D) MIP-2 mRNA expression is augmented in bpV-treated IRI kidneys compared with vehicle-treated IRI kidneys. **P < 0.001 vs. Vehicle Sham; ++ P < 0.001 vs. bpV IRI; # P < 0.05 vs. Vehicle IRI. n = 4 per group.
Discussion
PTEN has shown to regulate cell growth, adhesion, and migration7, 8. However, its role in ischemic AKI is not known. In the present study, PTEN inhibition with bpV(HOpic) exacerbates kidney dysfunction through augmenting inflammatory cell infiltration and proinflammatory molecule production. Our results identify PTEN as a critical regulator of the pathogenesis of renal IRI. These results suggest that PTEN plays an important role in the pathogenesis of renal IRI through the regulation of inflammation and apoptosis.
PTEN is first identified as a tumor-suppressor gene located on chromosome 10, which is mutated in a large number of tumors19. The crystal structure of PTEN reveals that PTEN contains a phosphatase domain that is similar to protein phosphatases but also has an enlarged active site important for the accommodation of the phosphoinositide substrate20. The principal substrate of PTEN is phosphatidylinositol (3,4,5)-trisphosphate (PIP3) in vitro 10, 20. PIP3 is a lipid second messenger generated by the activation of phosphoinositide 3-kinase (PI3K)20. Thus, PTEN negatively regulated PI3K signaling by dephosphorylating PIP3 and downregulating its downstream effector including AKt/PKB kinase which has potent pro-migration and growth stimulatory effect20. A function role of PTEN phosphatase activity in tumor suppressor has been extensively studied21, 22. Recent evidence indicates that PTEN is involved in kidney development and pathogenesis of kidney disease. For example, one study shows that PTEN loss is associated with increased TGF-β signaling and renal fibrosis11. Another study reports that mice with proximal tubule deletion of PTEN develop larger kidneys characterized by proximal tubule cell hypertrophy12. We have recently demonstrated that loss of PTEN in podocytes aggravates diabetic nephropathy through regulation of cytoskeletal rearrangement13. However, the functional role of PTEN in ischemic AKI is unknown. In the present study, our results have showed that inhibition of PTEN with bpV(HOpic) augments renal dysfunction and promotes tubular damage in mice with IRI compared with vehicle-treated mice with IRI. These results indicate that PTEN inhibition promotes kidney dysfunction in response to IRI.
Apoptosis of tubular epithelial cells contributes to the development of ischemic AKI15, 16. In the present study, IRI resulted in TUNEL-positive apoptotic cell death in the kidney, which is further increased in mice treated with bpV(HOpic). The Caspase family of cysteine proteases mediate the initiation and execution of the mammalian apoptotic cell death program. Caspase-3 is activated by death ligand and mitochondrial pathways. Caspase-3 activation is a key mechanism underlying the pathogenesis of IRI-induced apoptotic cell death in tubular epithelial cells23. In the present study, we have examined the effect of PTEN inhibition with bpV(HOpic) on IRI-induced caspase-3 activation in the kidney. Our results have demonstrated that caspase-3 is significantly activated in the kidney following IRI, whereas bpV(HOpic) treatment leads to greater caspase-3 activation in the kidney with IRI. These data suggest that PTEN inhibits caspase-3 activation and tubular epithelial cell apoptosis in response to IRI.
In response to kidney ischemia-reperfusion injury, innate and adaptive immune cells are recruited into the kidney participating in the pathogenesis of ischemic AKI1, 16, 18. In the present study, we have shown that neutrophils and macrophages infiltrate into the kidney following IRI. PTEN inhibition increases infiltration of neutrophils and macrophages into the kidney with IRI. These data indicate that PTEN inhibition promotes renal IRI through increasing the infiltration of neutrophils and macrophages into the kidney.
Inflammatory cytokines and chemokines are crucial mediators in the pathogenesis of ischemic AKI16, 18. Recent studies have shown that proinflammatory cytokines and chemokines such as TNF-α, IL-6, MCP-1, and MIP-2 contribute to the development of renal IRI. In the present study, we have shown that inflammation molecules such as IL-6, TNF-α, MCP-1 and MIP-2 are elevated in IRI kidney. Inhibition of PTEN with bpV(HOpic) considerably enhances the production of these inflammatory molecules. These results indicate PTEN affects IRI through regulating the inflammatory cells infiltration and inflammatory cytokines production.
In summary, our study shows that PTEN is involved in regulating the pathogenesis of renal IRI. Pharmacological inhibition of PTEN promotes accumulation of circulating inflammatory cells into the kidney and production of inflammatory molecules, which exacerbates the development of ischemic AKI. These data suggest that PTEN activation could be a potential therapeutic approach for ischemic AKI.
Methods
Animals
The animal experiments were conducted according to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. Wild type (WT) C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Male WT mice, 8 to 12 weeks old, weighing 20 to 30 grams, were anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg). Kidneys were exposed through flank incision and were subjected to ischemia by clamping renal pedicles with non-traumatic microaneurism clamps. After 30 minutes, the clamps were removed and blood reflow was confirmed. Body temperature was maintained at 36.5–37.5 °C throughout the procedure. Sham control mice underwent identical surgical procedure but without pedicle clamping. Animals were sacrificed at 24 hours after reperfusion. Kidneys were perfused and harvested16.
PTEN Phosphatase Activity Measurement
Kidney tissues were homogenized in ice-cold lysis buffer (25 mM Tris PH 8.0, 150 mM NaCl, 1%NP-40, 1 mM EDTA, 5% Glycerol and protease inhibitor cocktail). Tissue homogenates were centrifuged at 14000 × g for 10 minutes at 4 °C. The pellets were incubated with PTEN antibody and protein A agarose beads overnight at 4 °C. The beads were washed with PTEN reaction buffer (25 mM Tris-Cl, PH 7.4, 140 mM NaCl, 2.7 mM KCl with 10 mM DTT). After washing, the beads were resuspended in PTEN reaction buffer and PTEN enzyme reaction was initiated by adding PI(3,4,5)P3 substrate for 1 hour at 37 °C. The enzyme reaction was terminated by adding Malachite Green solution for 20 minutes at room temperature. PTEN phosphatase activity was measured at 620 nm using a plate reader.
Renal Function Measurement
Serum creatinine was measured using a creatinine assay kit (BioAssay Systems, Hayward, CA) and blood urea nitrogen was determined fluorometrically as described16, 24, 25.
Renal Morphology
Kidney tissues were fixed in 10% buffered formalin, embedded in paraffin, and cut at 4 µm thickness. After deparaffinization and rehydration, kidney sections were stained with hematoxylin and eosin. Tissue damage was examined in a blinded manner and scored according to the percentage of damaged tubules: 0, no damage; 1, less than 25% damage; 2, 25–50% damage; 3, 50–75% damage; and 4, more than 75% damage as reported16, 26.
Immunohistochemistry
Immunohistochemical staining was performed on paraffin sections. Antigen retrieval was performed with antigen unmasking solution (Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched with 3% H2O2. After blocked with 5% normal serum, slides were incubated with primary antibodies in a humidified chamber overnight. After washing, slides were incubated with appropriate secondary antibodies and ABC solution sequentially per ABC Elite kit (Vector Laboratories, Burlingame, CA). Immunoreactivity was revealed by incubation in DAB solution. Nuclear staining was performed with hematoxylin. The slides were dehydrated, cleared, and mounted. The images were acquired and analyzed by NIS Element software with Nikon microscope image system16.
Caspase 3 Activity Assay
Caspase 3 Activity was measured using Caspase-3 Colorimetric Activity Assay Kit (Millipore, Billerica, MA). Briefly, kidney tissue was homogenized in chilled cell lysis buffer and incubated on ice for 10 minutes. After centrifugation for 1 min at 10,000 × g, reaction buffer containing 10 mM DTT and DEVD-pNA substrate were added to the supernatant and incubated for 1 hour at 37 °C. Samples were read at 405 nm in a microtiter plate reader. Caspase 3 activity was determined by comparing with the level of the control.
Apoptosis Detection
TUNEL assay was performed to evaluate apoptosis using ApopTagR plus Peroxidase in Situ Apoptosis Detection Kit (Millipore, Billerica, MA) according to manufacturer’s instruction. The number of TUNEL-positive cells per high-power field were counted and analyzed in a blinded fashion16, 27.
Quantitative Real-Time RT-PCR
Total RNA was extracted from frozen kidney tissues with TRIzol reagent (Invitrogen, Carlsbad, CA) followed by RNase-free DNase I (Roche, Madison, WI). Aliquots (1 μg) of total RNA were reverse transcribed using SuperScript II reverse transcriptase. Real-time PCR was performed using IQ SYBR green supermix reagent (Bio-Rad, Hercules, CA) with a Bio-Rad real-time PCR machine28–30. The expression levels of the target genes were normalized to GAPDH level in each sample. The primer sequences were: IL-6 - forward, 5′-AGGATACCACTCCCAACAGACCTG-3′, reverse, 5′-CTGCAAGTGCATCATCGTTGTTCA-3′; TNF-α - forward, 5′-CATGAGCACAGAAAGCATGATCCG-3′ reverse, 5′-AAGCAGGAATGAGAAGAGGCTGAG-3′; MCP-1 - forward, 5′-TCACCTGCTGCTACTCATTCACCA-3′, reverse, 5′-TACAGCTTCTTTGGGACACCTGCT-3′; MIP-2 - forward, 5′-AAAGTTTGCCTTGACCCTGAAGCC-3′, reverse, 5′-TCCAGGTCAGTTAGCCTTGCCTTT-3′; GAPDH - forward, 5′-CCAATGTGTCCGTCGCGTGGATCT-3′, reverse, 5′-GTTGAAGTCGCAGGAGACAACC-3′.
Western Blot Analysis
Proteins were extracted using RIPA buffer containing cocktail proteinase inhibitors and quantified with a Bio-Rad protein assay. Equal amounts of protein (50 μg) were separated on SDS-polycrylamide gels and then transferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies overnight followed by incubation with appropriate fluorescence-conjugated secondary antibodies. The proteins of interest were analyzed using an Odyssey IR scanner, and signal intensities were quantified using NIH Image/J software28–35.
Statistical Analysis
All data were expressed as mean ± SEM. Multiple group comparisons were performed by ANOVA followed by the Bonferroni procedure for comparison of means. Comparisons between two groups were analyzed by the two-tailed t test. P < 0.05 was considered statistically significant.
Acknowledgements
We thank Dr. William E. Mitch for helpful discussion. This work was supported by the National Institutes of Health grant R01DK95835 and the U.S. Department of Veterans Affairs Merit Review grant I01BX02650 (to YW). The contents of the article do not represent the views of the U.S. Department of Veterans Affairs or the United States government.
Author Contributions
J.Z. and Y.W. conceived and designed the experiments. J.Z. performed the experiments. J.Z., L.J., and Z.H. analyzed the data. J.Z., L.J., and Y.W. wrote and edited the paper. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siew ED, Davenport A. The growth of acute kidney injury: a rising tide or just closer attention to detail? Kidney Int. 2015;87:46–61. doi: 10.1038/ki.2014.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Y, et al. Renal Ischemia/Reperfusion Injury in Soluble Epoxide Hydrolase-Deficient Mice. PLoS One. 2016;11 doi: 10.1371/journal.pone.0145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 5.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 6.Brenner W, et al. Loss of tumor suppressor protein PTEN during renal carcinogenesis. Int J Cancer. 2002;99:53–57. doi: 10.1002/ijc.10303. [DOI] [PubMed] [Google Scholar]
- 7.Tamura M, et al. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 8.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114:2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 9.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 10.Leslie NR, Downes CP. PTEN: The down side of PI 3-kinase signalling. Cell Signal. 2002;14:285–295. doi: 10.1016/S0898-6568(01)00234-0. [DOI] [PubMed] [Google Scholar]
- 11.Lan R, et al. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol. 2012;302:F1210–1223. doi: 10.1152/ajprenal.00660.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen JK, et al. Phosphatidylinositol 3-kinase signaling determines kidney size. J Clin Invest. 2015;125:2429–2444. doi: 10.1172/JCI78945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin JS, et al. Loss of PTEN promotes podocyte cytoskeletal rearrangement, aggravating diabetic nephropathy. J Pathol. 2015;236:30–40. doi: 10.1002/path.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keyes KT, et al. Pharmacological inhibition of PTEN limits myocardial infarct size and improves left ventricular function postinfarction. Am J Physiol Heart Circ Physiol. 2010;298:H1198–1208. doi: 10.1152/ajpheart.00915.2009. [DOI] [PubMed] [Google Scholar]
- 15.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin X, Chen J, Hu Z, Chan L, Wang Y. Genetic deficiency of adiponectin protects against acute kidney injury. Kidney Int. 2013;83:604–614. doi: 10.1038/ki.2012.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2010;17:1104–1114. doi: 10.1038/cdd.2009.180. [DOI] [PubMed] [Google Scholar]
- 18.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. 2015;11:88–101. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 19.Myers MP, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JO, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/S0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki A, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/S0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 22.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 23.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. The Journal of clinical investigation. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May RC, Kelly RA, Mitch WE. Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J Clin Invest. 1987;79:1099–1103. doi: 10.1172/JCI112924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang H, et al. CXCL16 Deficiency Attenuates Renal Injury and Fibrosis in Salt-Sensitive Hypertension. Sci Rep. 2016;6 doi: 10.1038/srep28715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang, H., Zhang, Z., He, L. & Wang, Y. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget, doi:10.18632/oncotarget.9386 (2016). [DOI] [PMC free article] [PubMed]
- 28.Xia Y, Entman ML, Wang Y. Critical Role of CXCL16 in Hypertensive Kidney Injury and Fibrosis. Hypertension. 2013;62:1129–1137. doi: 10.1161/HYPERTENSIONAHA.113.01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia Y, Jin X, Yan J, Entman ML, Wang Y. CXCR6 Plays a Critical Role in Angiotensin II-Induced Renal Injury and Fibrosis. Arterioscler Thromb Vasc Biol. 2014;34:1422–1428. doi: 10.1161/ATVBAHA.113.303172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, et al. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PLoS One. 2012;7 doi: 10.1371/journal.pone.0052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G, et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol. 2011;22:1876–1886. doi: 10.1681/ASN.2010080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, et al. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol. 2013;24:1644–1659. doi: 10.1681/ASN.2013030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan J, Zhang Z, Yang J, Mitch WE, Wang Y. JAK3/STAT6 Stimulates Bone Marrow-Derived Fibroblast Activation in Renal Fibrosis. J Am Soc Nephrol. 2015;26:3060–3071. doi: 10.1681/ASN.2014070717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia Y, Yan J, Jin X, Entman ML, Wang Y. The chemokine receptor CXCR6 contributes to recruitment of bone marrow-derived fibroblast precursors in renal fibrosis. Kidney Int. 2014;86:327–337. doi: 10.1038/ki.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Z, Jin X, He L, Wang Y. CXCL16 regulates renal injury and fibrosis in experimental renal artery stenosis. Am J Physiol Heart Circ Physiol. 2016;311:H815–821. doi: 10.1152/ajpheart.00948.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]