

Abstract
α3β4 nAChRs have been implicated in various pathophysiological conditions. However, the expression profile of α3β4 nAChRs and α6/α3β4 nAChRs overlap in a variety of tissues. To distinguish between these two subtypes, we redesigned peptide 1 (α-conotoxin TxID), which inhibits α3β4 and α6/α3β4 nAChR subtypes. We systematically mutated 1 to evaluate analog selectivity for α3β4 vs. α6/α3β4 nAChRs expressed in Xenopus laevis oocytes. One analog, peptide 7 ([S9A]TxID), had 46-fold greater potency for α3β4 versus α6/α3β4 nAChRs. Peptide 7 had IC50’s>10 μM for other nAChR subtypes. Molecular dynamics simulations suggested that Ser-9 of TxID was involved in a weak hydrogen bond with β4 Lys-81 in the α6β4 binding site but not in the α3β4 binding site. When Ser-9 was substituted by an Ala, this hydrogen bond interaction was disrupted. These results provide further molecular insights into the selectivity of 7 and provide a guide for designing ligands that block α3β4 nAChRs.
TOC image

INTRODUCTION
Nicotinic acetylcholine receptors (nAChRs) are ionotropic channels belonging to the Cys-loop superfamily of ligand-gated ion channels and have a pentameric protein structure.1,2 Generally, nAChRs are classified into muscle-type or neuronal-type receptors based on anatomical locations, though nAChR subunit expression is being increasingly reported in non-muscle and non-neural tissues. Muscle-type receptors consist of four different subunits assembled with an (α1)2β1δε stoichiometry.3 By contrast, neuronal nAChRs are composed of a combination of α (α2–α10) and β (β2–β4) subunits, which can assemble to form homo- or hetero-pentamers. nAChRs are implicated in normal physiology and the pathophysiology of a number of disease states, including pain, Parkinson’s disease, Alzheimer’s disease, schizophrenia, epilepsy, dementia and cancer.3,4 α4β2* (* indicates possible presence of additional subunits) nAChRs are the most abundant heteromeric subtype in the mammalian brain and receptors containing the α4 nAChR subunit are widely distributed.5 In contrast receptors containing the α6 nAChR subunit have a more restricted distribution with concentration in catecholaminergic and visual pathways.6 α4β2* and α6β2* are both strongly implicated in tobacco addiction and compounds with activity at these receptor subtypes are utilized or in development as anti-smoking drugs.4 nAChR subtypes that contain the α3 subunit are present in autonomic ganglia and modulate cardiovascular functions. The α3* subtypes of nAChRs are also implicated in modulating pain sensations.7 In addition, the α3β4 nAChR has been shown to be associated with nicotine addiction and drug abuse.8,9 Furthermore, several studies have shown that hypothalamic α3β4 nAChRs are implicated in nicotinic-induced decreases in food intake, and might therefore provide clues to treating food overconsumption and obesity.10 Thus, strategies to selectively modulate α3β4 nAChR function are of considerable interest since all the available ligands lack good selectivity between α3β4 and the closely related α6β4 nAChR subtypes, limiting our understanding of the physiological roles of these receptors.
Cone snails are venomous marine molluscs that have evolved a diverse array of structured peptide toxins for capturing their prey or defending against predators. Because conotoxins are potent antagonists of a range of voltage- or ligand-gated ion channels, they are widely used as valuable tools in ion channel research, and several have direct diagnostic or therapeutic potential. Their diverse sequences are documented in the ConoServer database.11 Most of the pharmacologically characterized conotoxins are nAChR antagonists, and are therefore classified as α-conotoxins. The α-conotoxins typically comprise 12–20 amino acids and have exquisite nAChR subtype selectivity, thus enabling dissection of the functional roles of nAChR subtypes. Peptide 1 (α-Conotoxin TxID) cloned from Conus textile is one of the most potent α3β4 nAChR antagonists, and is potentially a valuable tool for elucidating the diverse roles of α3β4 nAChRs.12 In this study we used positional-scanning mutagenesis of peptide 1 (Figure 1) to identify critical residues that confer potency for α3β4 nAChRs. We counter-screened analogs against α6/α3β4 nAChRs (α6/α3 designates a chimeric subunit that contains the extracellular ligand binding domain of α6 linked to the membrane embedded and intracellular portions of the α3 subunit).13 An analog with significantly improved selectivity for α3β4 versus α6/α3β4 nAChRs was identified.
Figure 1.

The sequences of peptide 1 and its analogs. The disulfide connectivity of Cys (I–III, II–IV) is schematically illustrated with lines connecting the Cys residues. Substituted amino acids that were mutated to explore structure-activity relationships are marked in red. (The * indicates a C-terminal amide). Also shown for comparison is peptide 17 (α-conotoxin AuIB),14 for which we compare structure and activity data. aCompound number.
RESULTS
Synthesis of wild type peptide 1 and analogs
Peptide 1 is a 15-amino acid peptide, which belongs to the α-4/6 subclass of conotoxins and its nucleic acid encoding sequence was first determined from Conus textile.12 In addition to its presumed natural function in prey capture, it is a potent and selective antagonist of rat α3β4 nAChRs. Peptide 1 also shows activity at rat α6/α3β4 nAChRs, albeit with 10-fold less potency than at the α3β4 subtype. To understand the structure–activity relationships of peptide 1, we designed and synthesized a suite of Ala-substituted peptide 1 analogs (2–16). In the course of these experiments we noticed that the methionine residue of peptide 1 at position 11 (Met-11) was easily oxidized into a methionine sulfoxide residue, leading to a reduction of activity (data not shown). We therefore substituted Met-11 with isoleucine. The side chains of Met and Ile amino acids are both hydrophobic but Ile is not susceptible to oxidation. Sequence comparison (Figure 1) between peptide 1 and 17, which is similar in structure but less potent at the α3β4 subtype, suggested that position 14 could be important for affinity.14 We thus tested the importance of position 14 by substituting Ile-14 with an Asp (as found in peptide 17), which is negatively charged, as well as an Arg residue, which is positively charged, and a Tyr residue, which is a bulky aromatic residue with hydrogen bond donor-acceptor properties often found on the surface of proteins.
For all of these mutants the peptide chain was successfully synthesized using Fmoc chemistry. The Cys residues were orthogonally protected using acid labile S-trityl (Trt) and acid-stable S-acetamidomethyl (Acm) groups. After cleavage of the assembled peptide chain from the resin, the Trt protecting groups on Cys I and Cys III were removed and the cysteines oxidized by treatment with ferricyanide. The second disulfide bridge was then formed between Cys-II and Cys-IV after Acm deprotection using iodine oxidation: the monocyclic conopeptide was trickled into an equal volume of a solution of iodine (10 mM) in H2O/trifluoroacetic acid/acetonitrile (78:2:20 by volume). The reaction was allowed to proceed for 10 min and then terminated by the addition of ascorbic acid. The solution was diluted 20-fold with 0.1% trifluoroacetic acid and the bicyclic peptide was purified by high-performance liquid chromatography (HPLC).15 This two-step oxidative folding strategy resulted in a single disulfide bond isomer (Figure 2, Figure S1). The observed molecular mass (1488.6 Da) of peptide 1 was consistent with the calculated mass (1488.6 Da) (Figure 2B). Similarly, the observed molecular mass of peptide 7 was 1472.6 Da, consistent with the calculated mass (1472.6 Da) (Figure 2D). Other mutants were synthesized by the same method and their HPLC and mass spectrometry (MS) profiles are provided in Supplemental Information Figure S1.
Figure 2.

HPLC chromatograms and mass spectra of peptide 1 and 7. The peptides were analyzed on a reversed phase analytical Vydac C18 HPLC column using a linear gradient of 10% buffer B to 40% buffer B over 20 min, where buffer A = 0.65% trifluoroacetic acid (TFA), remainder H2O, B = 0.5% TFA, 90% acetonitrile, remainder H2O. Absorbance was monitored at 214 nm. (A) HPLC chromatogram of peptide 1; (B) electrospray ionization mass spectrometry (ESI-MS) data for peptide 1 with observed monoisotopic mass of 1488.6 Da; (C) HPLC chromatogram of peptide 7; (D) ESI-MS data for peptide 1 with an observed monoisotopic mass of 1472.6 Da.
Electrophysiological Assay of Mutagenesis of Rat nAChRs Expressed in Xenopus laevis Oocytes
To better understand which positions are responsible for the activity of peptide 1, we performed alanine scanning mutagenesis. Except for the four Cys residues and Ala-10, each of the original residues was substituted by Ala. The peptide 1 analogs were tested on nAChR subtypes at concentrations up to 10 μM. The activities of peptide 4 ([H5A]TxID), 5 ([P6A]TxID), 6 ([V7A]TxID), 8 ([M11A]TxID) and 10 ([P13A]TxID) analogs at the α3β4 nAChR subtype were reduced by greater than 1000-fold compared to native peptide 1. The activities of these five analogs on α6/α3β4 nAChRs were also abolished. In contrast, several analogs retained high potency for α3β4 nAChRs and α6/α3β4 nAChRs. We therefore investigated the concentration-response relationships of peptide 1 and those analogs that showed antagonist activity at the α3β4 and α6/α3β4 nAChR subtypes (Table 1). Replacing the first glycine residue with an alanine substantially decreased potency at the α3β4 subtype. The G1A substitution also caused a decreased inhibition at the α6/α3β4 nAChR by 8-fold compared to the parent peptide. The activities of 3, 9 and position 14 analogs displayed similar potency at the α3β4 subtype compared to the parent peptide. Peptide 12, in which the easily oxidized methionine residue was substituted by an isoleucine amino acid, had 20-fold reduced potency at α3β4 nAChR, with an IC50 of 74.9 nM. By contrast, peptide 3, 9, 12 and the four position 14 analogs globally retained activity at the α6/α3β4 nAChR subtype. None of the substitutions at positions 1, 4, 11, 12, 13 and 14 resulted in increased selectivity for the α3β4 subtype over the α6/α3β4 subtype compared to peptide 1.
Table 1.
Potencies of peptide 1 and its analogs on rat α3β4 and α6/α3β4 nAChR subtypes expressed in Xenopus laevis oocytes
| Peptide | IC50 (nM) (95% CI) α3β4 | nH | α3β4IC50 ratio relative to 1 | IC50 (nM) (95% CI) α6/α3β4 | nH | α6/α3β4 IC50 ratio relative to 1 | Selectivitya |
|---|---|---|---|---|---|---|---|
| 1 | 3.6 (1.8–7.3) | 0.71 (0.35–1.10) | 1.0 | 33.9 (23.6–48.7) | 1.10 (0.78–1.50) | 1.0 | 9.4 |
| 2 | 61.9 (32.4–118.0) | 0.59 (0.36–0.83) | 17 | 278 (154–503) | 1.0 (0.50–1.50)- | 8.2 | 4.5 |
| 3 | 10.8 (8.6–13.4) | 1.16 (0.763–1.56) | 3.0 | 64.1 (41.6–98.7) | 0.87 (0.54–1.21) | 1.9 | 5.9 |
| 4 | >10 μM | – | >10 μM | – | – | – | |
| 5 | >10 μM | – | – | >10 μM | – | – | – |
| 6 | >10 μM | – | – | >10 μM | – | – | – |
| 7 | 3.9 (2.5–5.9) | 0.74 (0.52–0.96) | 1.1 | 178.1 (137.0–231.5) | 0.85 (0.67–1.03) | 5.2 | 46 |
| 8 | >10 μM | – | – | >10 μM | – | – | – |
| 9 | 17.4 (8.6–3.5) | 1.12 (0.26–1.97) | 4.8 | 39.3 (25.6–59.8) | 1.03 (0.66–1.41) | 1.2 | 2.3 |
| 11 | 16.1 (9.1–28.5) | 1.05 (0.39–1.70) | 4.4 | 45.8 (33.7–62.2) | 1.04 (0.77–1.32) | 1.3 | 2.8 |
| 12 | 74.9 (55.0–102.1) | 1.03 (0.75–1.31) | 21 | 50.4 (31.3–81.2) | 0.85 (0.52–1.18) | 1.5 | 0.67 |
| 13 | 30.7 (14.1–66.8) | 0.85 (0.34–1.37) | 8.4 | 101.0 (55.8–183.2) | 0.91 (0.45–1.37) | 3.0 | 3.3 |
| 10 | >10 μM | – | – | >10 μM | – | – | – |
| 14 | 9.8 (6.6–14.4) | 1.33 (0.387–2.26) | 2.7 | 40.8 (32.9–50.5) | 0.84 (0.69–0.98) | 1.2 | 4.2 |
| 15 | 10.0 (6.4–15.6) | 0.75 (0.52–0.98) | 2.7 | 38.7 (23.0–65.0) | 0.68 (0.42–0.93) | 1.1 | 3.9 |
| 16 | 9.2 (5.0–16.9) | 0.75 (0.38–1.12) | 2.5 | 67.4 (42.8–106) | 1.02 (0.53–1.50) | 2.0 | 7.3 |
IC50 values (nM) with 95% CI and Hill slope (nH) obtained from concentration–response curves for peptide 1 and analogues at α3β4 and α6/α3β4 nAChR subtypes. All data represent mean of n = 6–21 oocytes.
Ratio of α6/α3β4:α3β4 IC50 values.
Remarkably, the S9A substitution had little impact on the activity at α3β4 but led to a 5-fold decrease in inhibition of α6/α3β4, creating a 46-fold selective compound for α3β4 compared to α6/α3β4 nAChRs. Peptide 7 is the only compound in this study showing a higher α3β4-selectivity than the parent peptide 1 (Figures 3 and 4). Representative traces of ACh-evoked currents for α3β4 and α6/α3β4 nAChRs inhibited by peptide 1 and 7 are shown in Figure 4.
Figure 3.
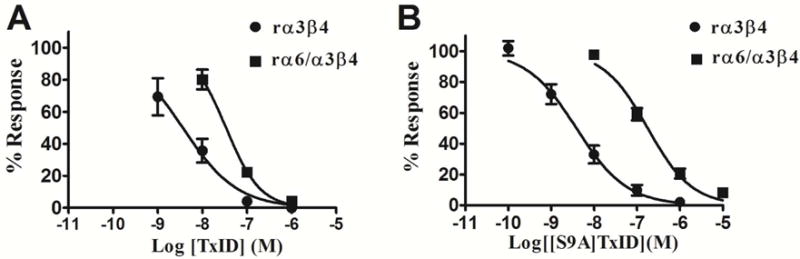
Concentration-response analysis for inhibition of α3β4 and α6/α3β4 nAChRs by pepitde 1 and 7. (A) Pepitde 1 selectively blocks α3β4 nAChR vs α6/α3β4 nAChR by only 10-fold; (B) Peptide 7 selectively blocks α3β4 nAChR vs α6/α3β4 nAChR by 46-fold. The IC50 values are in Table 1. Error bars represent S.E.M. values from 4–5 oocytes for each experimental determination.
Figure 4.

Block by peptide 1 and 7 of rat α3β4 and α6/α3β4 nAChRs. Xenopus laevis oocytes expressing the indicated nAChRs were voltage-clamped at −70 mV and subjected to a 1 s pulse of ACh every minute as described in the Experimental Section. A representative response in a single oocyte is shown. After control responses to ACh, the oocyte was exposed to 10 nM or 100 nM toxin for 5 min (arrow). The toxin was then washed out, and the response to ACh was again measured. “C” indicates control responses to ACh.
As described above, substituting Ser-9 by an Ala amino acid in peptide 1 increased selective inhibitory activity for the α3β4 subtype over α6/α3β4 subtype. We subsequently demonstrated that peptide 7 did not inhibit a range of other subtypes at 10 μM, including α7, α9α10, α1β1δε, α2β2,α2β4, α3β2, α4β2, and α4β4 (Table 2).
Table 2.
IC50 for block of nAChR subtypes by α-conotoxins 1 and peptide 7.
| Peptide | Subtype | IC50 [nM] (95% CI) |
|---|---|---|
| 1 | α3β4 | 3.6 (1.8–7.3) |
| α6/α3β4 | 33.9 (23.6–48.7) | |
| 7 | α3β4 | 3.9 (2.5–5.9) |
| α6/α3β4 | 178.1 (137.0–231.5) | |
| 7 | α7 | >10,000a |
| α9α10 | >10,000a | |
| Mα1β1δε | >10,000a | |
| α2β2 | >10,000a | |
| α2β4 | >10,000a | |
| α3β2 | >10,000a | |
| α4β2 | >10,000a | |
| α4β4 | >10,000a |
Less than 50% block at 10 μM. All receptors are rat except for α1β1δε, which is mouse.
NMR Spectroscopy
NMR spectroscopy analysis revealed that peptide 7 exists as two isomers in aqueous solution at 308 K in a ratio of approximately 70:30. The conformational heterogeneity appears to arise from cis-trans isomerization of the peptide bonds preceding the Pro residues at positions 6 and 13. Isomer 1 was assigned as the trans-trans isomer based upon NOEs observed between the Hαi-1 – HδPi protons of Pro6 and Pro13 and their preceding residues. Complete assignment of the spectral peaks corresponding to this isomer was possible, with the exception of the N-terminal glycine residue. By contrast, isomer 2 could only be partially assigned, particularly towards the N- and C-termini of the peptide. This isomer was determined to be a cis-trans isomer because of the lack of any NOE between Hα5 and Hδ6 protons, and consistency with previous findings for the wild-type peptide.12
Secondary shift analysis (Figure 5), was used to compare the secondary structure elements of peptide 7 with those of peptide 1.12 Deviations of αH shifts greater than 0.1 ppm from random coil values16 are typically indicative of structured peptides, with positive values present for β-type structure and negative values for helical structures. Figure 5 reveals no significant difference in the secondary structure of the trans-trans form (isomer 1) of 7 compared to the trans-trans isomer of 1. Indeed, the mutation of Ser-9 to Ala-9 appears to strengthen the helical nature across the middle of this peptide. By contrast, the cis-trans isomer of both peptide 1 and its [S9A] derivative, both appear to adopt more a random coil structure (data not shown).
Figure 5.

Comparison of NMR spectroscopy of the peptide 7 and wild-type peptide 1. (A) Secondary chemical shift analysis of peptide 7 isomer 1 (black) compared to that of isomer 1 of peptide 1 (grey). (B) Homology models of the two peptide 7 isomers (grey) compared to the NMR solution structures of the wild-type isomers (white).12 The amino acid sequence of peptide 7 is displayed, with the substituted residue for the mutant shown in brackets. The homology models were refined using extensive energy minimization in explicit water. The models suggest that the three-dimensional structure of the isomers is essentially not affected by the S9A substitution.
DISCUSSION
In this paper we report the synthesis, electrophysiological characterization and structural characterization of a series of mutants of the α-conotoxin 1. First, using alanine scanning and site-directed mutagenesis of the non-cysteine residues, we identified five key residues in the peptide 1 sequence, i.e. His-5, Pro-6, Val-7, Met-11 and Pro-13, which when substituted to Ala caused total loss of inhibition at both α3β4 and α6/α3β4 nAChRs. Compared to native peptide 1, the substitution G1A caused a 17-fold decrease in activity at the α3β4 nAChR. Other Ala substitutions, i.e. of residues Ser-4, Ser-9, Ser-12 or Ile-14, resulted in more modest reductions in activity for the α3β4 nAChR (Table 1). Notably, the S9A substitution resulted in little or no loss of potency for the α3β4 nAChR. When these mutants were tested on rat α6/α3β4nAChRs, there was a general decrease in activity. Ser for Ala substitution in the 9th position resulted in a ~46-fold preference for α3β4 versus the very closely related α6/α3β4 nAChR subtype. Recently, the α3β4 subtype has received considerable attention by being implicated in lung cancer and in nicotine addiction.4 Several α-conotoxins have been identified as antagonists of the α3β4 nAChR. However, most of them target a broad range of subtypes, especially for α6 containing subtypes, as shown in Table 3. α-Conotoxin 17 from Conus aulicus was the first conopeptide that selectively blocks α3β4 nAChR, but its low potency has limited its use in physiological studies.14 [N11,12A]RegIIA, a mutant of α-conotoxin RegIIA blocks α3β4 nAChR with an IC50 of 370 nM, and also inhibits the α6/α3β4β3 nAChR with an IC50 of 5100 nM.17 Interestingly, a α-4/4-conotoxin BuIA analog, [T5A, P6O]BuIA, is a potent antagonist of α6/α3β4 nAChRs with IC50 58.1 nM whereas it blocks rat α3β4 nAChRs with an IC50 of 1200 nM (Table 3).18 The current study suggests that 7 has an opposite nAChR subunit selectivity to that of [T5A, P6O]BuIA, which may reveal further insights into how conotoxins discriminate between α3β4 and α6/α3β4 nAChRs. Another BuIA analogue, TP-2212-59, in which two amino acids are substituted by 2-aminobutyric acid and norvaline, is the most potent α3β4 nAChR antagonist with an IC50 of 2.3 nM,19 which is similar to native peptide 1 and 7. But the activity of TP-2212-59 was not reported for α6-containing nAChRs. Thus, Peptide 7 has the greatest selectivity for the α3β4 vs. α6/α3β4 nAChR compared to previously characterized α-conotoxins. We are not aware of any compounds that have greater selectivity than peptide 7 for α3β4 vs. α6/α3β4 nAChRs.
Table 3.
α-Conotoxins that target the α3β4 and α6/α3β4 nAChRs.
| Name | Sequence | Source | IC50 [nM] α3β4 |
IC50 [nM] α6/α3β4 |
Ratioa | nAChR subtype selectivity | Ref. |
|---|---|---|---|---|---|---|---|
| Peptide 1 | GCCSHPVCSAMSP-IC* | C.textile | 3.6 | 33.9 | 9.4 | α3β4>α6/α3β4>α2β4 | 12 |
| Peptide 7 | GCCSHPVCAAMSP-IC* | synthetic | 3.9 | 178 | 46 | α3β4>α6/α3β4 | This work |
| Peptide 17 | GCCSYPPCFATNP-DC* | C.aulicus | 750 | ND | − | ND | 14 |
| RegIIA | GCCSHPACNVNNPHIC* | C.regius | 47.3 | 147 | 3.1 | α3β2>α3β4≈α6β2>α7>α6β4 | 20 |
| [N11,12A]RegIIA | GCCSHPACNVAAPHIC* | synthetic | 370 | 5100 | 14 | α3β4>α6/α3β4 | 17 |
| PeIA | GCCSHPACSVNHPELC* | C.pergrandis | 480 | 1500 | 3.1 | α9α10>α3β2>α6β2>α3β4>α7 | 21 |
| PIA | RDPCCSNPVCTVHNPQIC | C.purpurascens | 518 | 30 | 0.058 | α6/α3β2>>α6/α3β4≈α3β2>α3β4 | 13 |
| BuIA | GCCSTPPCAVLY—C* | C. bullatus | 27.7 | 2.1 | 0.076 | α6/α3β2>α6/α3β4>α3β2>α3β4 | 22 |
| [T5A, P6O]BuIA | GCCSAOPCAVLY—C* | synthetic | 1200 | 58.1 | 0.048 | α6/α3β4>α3β4 | 18 |
| TP-2212-59 | GCCSHPBCFBZY—C* | synthetic | 2.3 | ND | – | ND | 19 |
Indicates a C-terminal amide.
Ratio of α6/α3β4:α3β4 IC50 values. B is 2-aminobutyric acid, Z is norvaline. ND, not determined.
NMR structural studies of peptide 7 revealed the presence of two major conformational isomers, as was found for peptide 1.12 The isomerization derives from cis-trans conformational interchange of the His-5/Pro-6 peptide bond, with Ser-12/Pro-13 apparently fixed in the trans form. Isomer 1 corresponds to a trans-trans arrangement of the two Pro residues whereas isomer 2 has a cis-trans configuration. A similar pattern of isomerisation was observed in the wild type peptide, although interestingly, the mutation of Ser-9 to Ala-9 appears to slightly favor an increased population of the trans-trans isomer versus the cis-trans isomer (70:30 in the mutant vs 40:60 in the wild type peptide, respectively). Secondary shift analysis confirms that the major (i.e. most populated) isomer of peptide 7 adopts a solution structure that is highly similar to isomer 1 of peptide 1. That structure comprises a short 3,10-helix that spans residues 8–10, preceded by a short turn, and is braced by the globular disulfide connectivity. The turn in loop 1 followed by a helical segment is a conserved motif displayed by all α-conotoxins, and the interaction of this motif with nAChRs is one of the best understood interactions between toxins and proteins,23 and for example, this motif is also displayed by snake toxins binding to nAChRs. The binding mode of this motif is similar in crystal structures of α-conotoxins (PDB identifiers 2br8, 2byp, 2c9t, 2uz6, 4ez1) and snake toxins (PDB identifiers 1yi5, 2qc1, 4uy2, 4hqp) binding to the acetylcholine binding protein, which is a structural surrogate of the ligand binding domain of nAChRs. The turn is tightly packed at the bottom of the acetylcholine binding pocket in the “aromatic box”, a conserved feature of the agonist binding site. Thus isomer 1 is the likely binding conformation of peptide 1. In contrast to isomer 1, isomer 2 of peptide 1 does not display this structural motif,12 suggesting that it does not bind the orthosteric binding site. We therefore created molecular models of the complex between peptide 1 isomer 1 and nAChRs to study the functional impact of substitutions.
A comparison of molecular models of 1/α3β4 and 1/α6β4 complexes suggests that the binding modes are slightly different despite nearly all positions in the binding site being conserved between the two receptors (Figure 6A–B). This difference in binding modes originates from a slightly different shape of the binding sites which, in turn, probably arises from sequence differences between the α3 and α6 subunits at positions proximal to the binding site. During the molecular dynamics simulation, the side chain of peptide 1 Ser-9 was involved in a weak hydrogen bond with β4 Lys-81 in the α6β4 binding site but not in the α3β4 binding site (Figure 6C). The substitution of Ser by an Ala at position 9 of peptide 1 would disrupt this hydrogen bond interaction, and the experimentally measured five-fold decreased potency, accounting for a loss of around ~1 kcal/mol at 300 K, is consistent with a loss of a hydrogen bond.24 By contrast, the hydroxyl group of peptide 1 Ser-4 in the α3β4 binding site is not involved in any interaction with the receptor, and therefore the S9A substitution is predicted to have no impact, which is also consistent with experimental measurements.
Figure 6.

Comparison of molecular models of the binding mode of pepitde 1 at the α3β4 (A) and α6β4(B) interface, and distance between side chain hydroxyl of peptide 1 Ser-9 and side chain nitrogen of β4 Lys-81 during 50 ns molecular dynamics (MD) simulations. The molecular models shown are the final frames of the 50 ns MD simulations, and the α3 subunit is drawn in pink, the α6 is in blue and the β4 subunits are in green. A hydrogen bond between Ser-9 and Lys-81 is indicated with a dotted line. Positions of β4 subunits are numbered according to the full-length sequence of rat β4 subunit precursor sequence (UniProt identifier P12392).
Substitution of Gly-1 by an Ala led to a 17-fold or 8-fold reduction of inhibition of α3β4 or α6/α3β4, respectively. The models suggest a possible charge interaction at the interface with the negatively charged Asp-192 located on the β4 subunit, and this interaction could be weakened by a change of backbone conformation of 1 compared to the parent peptide. The substitution of Met-11 by Ile resulted in 20-fold decreased activity at the α3β4 nAChR but did not reduce inhibition of the α6/α3β4 nAChR. According to the model, Met-11 contacts Cys-218 of the C-loop of the α3 subunit and the substitution M11A would probably result in a change of binding mode because of the steric hindrance caused by the larger side chain. By contrast, Met-11 in the α6β4 model could be substituted by an Ile residue without creating any steric clash with the α6 subunit in its bound conformation with wild-type peptide 1, suggesting that this substitution would not have a detrimental effect.
In conclusion, our study has identified critical residues for specific interactions of peptide 1 with the α3β4 nAChR. We discovered and characterized a new mutant peptide 7 that is 46-fold more potent at blocking α3β4 than α6/α3β4 nAChR subtypes. This selectivity provides a template for the further design and development of ligands to discriminate between α3β4 and α6/α3β4 nAChR subtypes.
EXPERIMENTAL SECTION
Materials
Protected Fmoc-amino acid derivatives were purchased from GL Biochem (Shanghai, China). The following side chain protected amino acids were used: Cys(Acm), Cys(Trt), His(Trt), Tyr(tBu), Trp(Boc), Arg(Pbf), Asp(OtBu), Ser(tBu). All other Fmoc amino acids were unprotected. Dimethylformamide (DMF), dichloromethane (DCM), diisopropylethylamine (DIEA), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), ethanedithiol(EDT), thioanisole for peptide synthesis were from Applied Biosystems (Foster City, CA, USA) and GL Biochem (Shanghai, China). Acetonitrile (HPLC grade) was purchased from Fisher Scientific (Pittsburg, PA, USA). Trifluoroacetic acid was from Tedia Company (Fairfield, OH, USA). All other chemicals used were of analytical grade. Acetylcholine chloride, atropine, and bovine serum albumin were obtained from Sigma (St. Louis, MO, USA). Reversed-phase HPLC analytical Vydac C18 (5 μm, 4.6 mm × 250 mm) and preparative C18 Vydac columns (10 μm, 22 mm × 250 mm) were obtained from Grace Vydac (Hesperia, CA, USA). Clones of rat nAChR subunits and mouse muscle subunits (α1β1δε) were kindly provided by S. Heinemann (Salk Institute, San Diego, CA, USA).
Chemical Synthesis of peptide 1 and its mutants
All of the α-conotoxin 1 analogs were synthesized on an automatic ABI 433A peptide synthesizer using the ABI FastMoc protocols Fmoc (N-(9-fluorenyl)methoxycarbonyl) amino acid derivatives, activated in situ using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate/1-hydroxybenzotriazole and N,N9-diisopropylethylamine in N-methylpyrrolidinone, were used in the coupling steps. The peptide was deprotected and cleaved from the resin by treatment with reagent K (trifluoroacetic acid/water/ethanedithiol/phenol/thioanisole; 90:5:2.5:7.5:5, v/v/v/v/v) for 2 h at room temperature. The released peptides were precipitated and washed several times with cold ether. Then crude peptides dissolved in B60 (60% CH3CN, 40% H2O, 0.05% TFA). Then these peptides were purified by reversed phase-high performance liquid chromatography (RP-HPLC) on a Vydac C18 column using a gradient of 10–50% buffer B for 40 min and the eluant was monitored at 214 nm. Electrospray-mass spectroscopy (ESI-MS) confirmed the molecular mass of the fractions collected. A two-step oxidation protocol was used to synthesize α-conotoxin 1 and mutants as described previously.15 In this protocol, linear and folded peptides were purified by HPLC on a reversed-phase C18 Vydac column. HPLC elution conditions included a linear gradient of 10–40% Buffer B over 20 min. Buffer A was 0.1% trifluoroacetic acid in H2O. Buffer B was 0.05% TFA, 90% acetonitrile in H2O. Absorbance was monitored at 214 nm. The purity of the monocyclic and bicyclic peptides was determined by monitoring absorbance at 214 nm during HPLC (⩾95% purity). Mass spectrometry was utilized to confirm the identity of the products.
Electrophysiology
The cRNAs of rat nAChR subunits were obtained by in vitro transcription using the mMessage mMachine SP6 kit (Ambion, Austin, TX, USA). The cRNAs were purified using the MEGAclear™ kit (Ambion). The concentration of each cRNA was determined by Smart SpecTM plus Spectrophotometer (Bio-Rad). Oocytes were harvested and injected with cRNA encoding nAChR subunits as described previously.25 Briefly, oocytes were transferred to the recording chamber (~30 μL in volume) and gravity-perfused at 2 mL/min with ND-96 buffer (96 mM NaCl, 2.0 mM KCl, 1.0 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, pH 7.1–7.5) containing 1 μM atropine and 0.1 mg/mL bovine serum albumin (BSA). ACh-gated currents were obtained with a two-electrode voltage-clamp amplifier (model OC-725B, Warner Instruments, Hamden, CT, USA). The oocytes were clamped at a holding potential of −70 mV. Oocytes were gravity-perfused with standard ND-96 solution and stimulated with 1-s pulses of ACh once every minute. For screening the specific of nAChR subtype, the toxins concentration was 10 μM or lower. Once a stable baseline was achieved, added 3 μL of different concentration toxin (0.1 nM-10 μM) to the chamber and waited for 5 min, then applied perfusion system, during which 1-s pulses of 10 μM ACh were applied every one minute. The concentration-response data were fitted to the equation, % response=100/{1 + ([toxin]/IC50)nH}, where nH is the Hill coefficient, by nonlinear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Each data point of a concentration-response curve represents the average ± S.E.M. of 6 to 21 oocytes.
NMR Spectroscopy
NMR spectra were recorded as described previously for peptide 1.12 Peptide 7(0.9 mg) was dissolved in 500 μL of 10% D2O/90% H2O (~pH 4) and spectra were recorded at 308 K on a Bruker Advance 600 MHz spectrometer. One-dimensional proton spectra and two-dimensional TOCSY (mixing time of 80 ms) and NOESY (mixing time of 200 ms) spectra were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) at 0 ppm.
Molecular modeling
Models of the interactions between peptide 1 and rat α3β4 or α6β4 nAChR ligand binding domains were generated using the same strategy as previously.26 Initial models were built by homology using Modeller9v1327 and two templates: the crystal structure of AChBP bound to conotoxin TxIA variant (PDB identifier 2uz628) and the crystal structure of the isolated human α9 subunit (PDB identifier 4d0129). The two nAChR subtypes were assumed to have a stoichiometry of two α subunits and three β subunits and the toxin was assumed to occupy the orthosteric binding site, which is located at the interface between an α (principal) subunit and a β (complementary) subunit. These models were then refined using molecular dynamics (MD) with the amber ff99SB-ILDN30 force field and the Gromacs 5.1 MD engine.31 The system was solvated in a dodecahedron box with ~28,000 TIP3P water molecules and sodium and chloride ions were added to reach a concentration of 150 mM NaCl. Additional sodium ions were added to neutralize the system. The system was equilibrated over 5 ns while progressively releasing position restraints on the protein atoms. The production run lasted 50 ns and employed a velocity rescaling scheme thermostat32 to maintain the temperature at 300 K and a Parrinello-Rahman barostat33 to maintain atmospheric pressure. The particle mesh Ewald algorithm34 was used to simulate long-range electrostatic interactions. Three MD simulations were carried out for each of the two systems.
Supplementary Material
Acknowledgments
This work was supported, in part, by the Major International Joint Research Project of National Natural Science Foundation of China (81420108028), National Natural Science Foundation of China (81660585, 41366002), and Changjiang Scholars and Innovative Research Team in University Grant (IRT_15R15). Work on conotoxins at UQ is funded by an ARC Australian Laureate Fellowship (FL150100146) and an ARC Discovery grant (DP150103990). This work was also supported by National Institutes of Health Grants GM103801 and GM48677 to JMM. We thank Baldomero Olivera, Cheryl Dowell, Sean Christensen, Layla Azam and Doju Yoshikami for advice and help.
ABBREVIATIONS
- CTx
conotoxin
- nAChR
nicotinic acetylcholine receptor
- TFA
trifluoroacetic acid
- RP-HPLC
reversed-phase high-performance liquid chromatography
- ESI-MS
Electrospray-mass spectroscopy
- BSA
bovine serum albumin
- DMF
Dimethylformamide
- DCM
dichloromethane
- DIEA
diisopropylethylamine
- HBTU
2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- EDT
ethanedithiol
- Fmoc
N-(9-fluorenyl)methoxycarbonyl
- AChBP
acetylcholine-binding protein
Footnotes
Additional figures illustrating chromatographic and mass properties of analogs of α-conotoxin 1. The two coordinate files correspond to the final frames of 50 ns molecular dynamics simulations of the complexes between conotoxin 1 and either α3β4 or α6β4 nAChR subtypes. The A and B chains are the ligand binding domain regions of the α and β subunits respectively. The C chain is the conotoxin 1.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56:237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 4.Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Zoli M, Pistillo F, Gotti C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology. 2015;96:302–311. doi: 10.1016/j.neuropharm.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- 7.Vincler M, Eisenach JC. Plasticity of spinal nicotinic acetylcholine receptors following spinal nerve ligation. Neurosci Res. 2004;48:139–145. doi: 10.1016/j.neures.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Muldoon PP, Jackson KJ, Perez E, Harenza JL, Molas S, Rais B, Anwar H, Zaveri NT, Maldonado R, Maskos U, McIntosh JM, Dierssen M, Miles MF, Chen X, De Biasi M, Damaj MI. The alpha3beta4* nicotinic ACh receptor subtype mediates physical dependence to morphine: mouse human studies. Br J Pharmacol. 2014;171:3845–3857. doi: 10.1111/bph.12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoker AK, Markou A. Unraveling the neurobiology of nicotine dependence using genetically engineered mice. Curr Opin Neurobiol. 2013;23:493–499. doi: 10.1016/j.conb.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mineur YS, Abizaid A, Rao Y, Salas R, DiLeone RJ, Gundisch D, Diano S, De Biasi M, Horvath TL, Gao XB, Picciotto MR. Nicotine decreases food intake through activation of POMC neurons. Science. 2011;332:1330–1332. doi: 10.1126/science.1201889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaas Q, Yu R, Jin AH, Dutertre S, Craik DJ. ConoServer: updated content knowledge and discovery tools in the conopeptide database. Nucleic Acids Res. 2012;40:D325–D330. doi: 10.1093/nar/gkr886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo S, Zhangsun D, Zhu X, Wu Y, Hu Y, Christensen S, Harvey PJ, Akcan M, Craik DJ, McIntosh JM. Characterization of a novel alpha-conotoxin TxID from Conus textile that potently blocks rat alpha3beta4 nicotinic acetylcholine receptors. J Med Chem. 2013;56:9655–9663. doi: 10.1021/jm401254c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A, Yoshikami D, Lindstrom JM, McIntosh JM. Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J Neurosci. 2003;23:8445–8452. doi: 10.1523/JNEUROSCI.23-24-08445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM, McIntosh JM. alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors nicotine-evoked norepinephrine release. J Neurosci. 1998;18:8571–8579. doi: 10.1523/JNEUROSCI.18-21-08571.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo S, Akondi KB, Zhangsun D, Wu Y, Zhu X, Hu Y, Christensen S, Dowell C, Daly NL, Craik DJ, Wang CI, Lewis RJ, Alewood PF, Michael McIntosh J. Atypical alpha-conotoxin LtIA from Conus litteratus targets a novel microsite of the alpha3beta2 nicotinic receptor. J Biol Chem. 2010;285:12355–12366. doi: 10.1074/jbc.M109.079012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. (1)H, (13)C and (15)N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5:332. doi: 10.1007/BF00211764. [DOI] [PubMed] [Google Scholar]
- 17.Kompella SN, Hung A, Clark RJ, Mari F, Adams DJ. Alanine scan of alpha-conotoxin RegIIA reveals a selective alpha3beta4 nicotinic acetylcholine receptor antagonist. J Biol Chem. 2015;290:1039–1048. doi: 10.1074/jbc.M114.605592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azam L, Maskos U, Changeux JP, Dowell CD, Christensen S, De Biasi M, McIntosh JM. alpha-Conotoxin BuIA[T5A;P6O]: a novel ligand that discriminates between alpha6beta4 alpha6beta2 nicotinic acetylcholine receptors blocks nicotine-stimulated norepinephrine release. FASEB J. 2010;24:5113–5123. doi: 10.1096/fj.10-166272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang YP, Banerjee J, Dowell C, Wu J, Gyanda R, Houghten RA, Toll L, McIntosh JM, Armishaw CJ. Discovery of a potent selective alpha3beta4 nicotinic acetylcholine receptor antagonist from an alpha-conotoxin synthetic combinatorial library. J Med Chem. 2014;57:3511–3521. doi: 10.1021/jm500183r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franco A, Kompella SN, Akondi KB, Melaun C, Daly NL, Luetje CW, Alewood PF, Craik DJ, Adams DJ, Mari F. RegIIA: an alpha4/7-conotoxin from the venom of Conus regius that potently blocks alpha3beta4 nAChRs. Biochem Pharmacol. 2012;83:419–426. doi: 10.1016/j.bcp.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 21.McIntosh JM, Plazas PV, Watkins M, Gomez-Casati ME, Olivera BM, Elgoyhen AB. A novel alpha-conotoxin PeIA cloned from Conus pergrandis discriminates between rat alpha9alpha10 alpha7 nicotinic cholinergic receptors. J Biol Chem. 2005;280:30107–30112. doi: 10.1074/jbc.M504102200. [DOI] [PubMed] [Google Scholar]
- 22.Azam L, Dowell C, Watkins M, Stitzel JA, Olivera BM, McIntosh JM. Alpha-conotoxin BuIA a novel peptide from Conus bullatus distinguishes among neuronal nicotinic acetylcholine receptors. J Biol Chem. 2005;280:80–87. doi: 10.1074/jbc.M406281200. [DOI] [PubMed] [Google Scholar]
- 23.Akondi KB, Muttenthaler M, Dutertre S, Kaas Q, Craik DJ, Lewis RJ, Alewood PF. Discovery synthesis and structure-activity relationships of conotoxins. Chem Rev. 2014;114:5815–5847. doi: 10.1021/cr400401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowie JU. Membrane protein folding: how important are hydrogen bonds? Curr Opin Struc Biol. 2011;21:42–49. doi: 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo S, Zhangsun D, Wu Y, Zhu X, Hu Y, McIntyre M, Christensen S, Akcan M, Craik DJ, McIntosh JM. Characterization of a novel alpha-conotoxin from conus textile that selectively targets alpha6/alpha3beta2beta3 nicotinic acetylcholine receptors. J Biol Chem. 2013;288:894–902. doi: 10.1074/jbc.M112.427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu R, Kaas Q, Craik DJ. Delineation of the unbinding pathway of alpha-conotoxin ImI from the alpha7 nicotinic acetylcholine receptor. J Phys Chem B. 2012;116:6097–6105. doi: 10.1021/jp301352d. [DOI] [PubMed] [Google Scholar]
- 27.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 28.Dutertre S, Ulens C, Buttner R, Fish A, van Elk R, Kendel Y, Hopping G, Alewood PF, Schroeder C, Nicke A, Smit AB, Sixma TK, Lewis RJ. AChBP-targeted alpha-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007;26:3858–3867. doi: 10.1038/sj.emboj.7601785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zouridakis M, Giastas P, Zarkadas E, Chroni-Tzartou D, Bregestovski P, Tzartos SJ. Crystal structures of free antagonist-bound states of human alpha9 nicotinic receptor extracellular domain. Nat Struct Mol Biol. 2014;21:976–980. doi: 10.1038/nsmb.2900. [DOI] [PubMed] [Google Scholar]
- 30.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101–014107. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 33.Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 34.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.