

Abstract
Objectives
This study aimed to model the population pharmacokinetics of intravenous paracetamol and its major metabolites in neonates and to identify influential patient characteristics, especially those affecting the formation clearance (CLformation) of oxidative pathway metabolites.
Methods
Neonates with a clinical indication for intravenous analgesia received five 15-mg/kg doses of paracetamol at 12-h intervals (<28 weeks’ gestation) or seven 15-mg/kg doses at 8-h intervals (≥28 weeks’ gestation). Plasma and urine were sampled throughout the 72-h study period. Concentration-time data for paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the combined oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine) were simultaneously modeled in NONMEM 7.2.
Results
The model incorporated 259 plasma and 350 urine samples from 35 neonates with a mean gestational age of 33.6 weeks (standard deviation 6.6). CLformation for all metabolites increased with weight; CLformation for glucuronidation and oxidation also increased with postnatal age. At the mean weight (2.3 kg) and postnatal age (7.5 days), CLformation estimates (bootstrap 95% confidence interval; between-subject variability) were 0.049 L/h (0.038–0.062; 62 %) for glucuronidation, 0.21 L/h (0.17–0.24; 33 %) for sulfation, and 0.058 L/h (0.044–0.078; 72 %) for oxidation. Expression of individual oxidation CLformation as a fraction of total individual paracetamol clearance showed that, on average, fractional oxidation CLformation increased <15 % when plotted against weight or postnatal age.
Conclusions
The parent-metabolite model successfully characterized the pharmacokinetics of intravenous paracetamol and its metabolites in neonates. Maturational changes in the fraction of paracetamol undergoing oxidation were small relative to between-subject variability.
1 Introduction
Paracetamol (N-acetyl-p-aminophenol, acetaminophen) is used to manage mild-to-moderate pain in neonates [1, 2]. Intravenous formulations of the drug have recently become available but have rapidly been adopted into clinical practice for applications in which enteral delivery is unsuitable, such as postoperative analgesia [3]. Several neonatal studies have characterized the pharmacokinetics of intravenous paracetamol [4–7].
Paracetamol primarily undergoes hepatic elimination, so markers of hepatic maturation and function are critical for explaining between-subject variability (BSV) in paracetamol pharmacokinetics [4–6, 8, 9]. Metabolism also plays a key role in paracetamol-induced hepatotoxicity, which is a principal safety concern [10, 11]. The non-toxic products of glucuronidation and sulfation are efficiently excreted in urine, but paracetamol also undergoes oxidation by cytochrome P450 (CYP) enzymes, predominantly CYP2E1, to form the reactive intermediate N-acetyl-p-benzoquinone imine (NAPQI). This electrophile can be detoxified by conjugation with glutathione, and rapid, subsequent metabolism of paracetamol-glutathione produces paracetamol-cysteine and paracetamol-N-acetylcysteine. However, sufficiently high doses of paracetamol will saturate the glutathione detoxification pathway. Excess NAPQI binds covalently to hepatic proteins, and toxicity is thought to result from a combination of the inactivation of critical hepatic proteins and oxidative stress [12, 13].
Previous studies have shown that body weight is the principal predictor of intravenous paracetamol pharmacokinetics in neonates [4, 7]. These findings support implementation of a parsimonious neonatal dosing regimen based solely on equivalent per-kilogram dosing, without a requirement for different doses or dosing intervals dependent upon gestational or postmenstrual age [4]; however, these studies only used pharmacokinetic data for the parent drug, which may not reflect maturational differences in the pharmacokinetics of hepatotoxicity-associated metabolites. Unfortunately, neonatal pharmacokinetic data for paracetamol metabolites remain scarce across all routes of administration, and previous studies that have incorporated metabolite data focused only on glucuronide and sulfate conjugates [6, 14–18].
The aim of this study was to develop a parent-metabolite population pharmacokinetic model for intravenous paracetamol in neonates to (1) estimate pharmacokinetic parameters for all major metabolic pathways of paracetamol, (2) quantify BSV in metabolite pharmacokinetics, and (3) identify patient characteristics (covariates) that influence metabolite pharmacokinetic parameters, with a particular focus on formation clearance of the oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine).
2 Methods
2.1 Ethics Approval and Study Registration
This was a prospective, single-center, open-label pharmacokinetic study, which was approved by the Institutional Review Board at the Children’s National Health System (Washington, DC, USA) and was conducted in accordance with good clinical practice. Written informed consent was obtained from a parent or legal guardian prior to study inclusion. The study was registered at ClinicalTrials.gov (NCT01328808).
2.2 Study Population
Patients <28 days’ postnatal age with an indwelling arterial line and a clinical indication for intravenous analgesia who were admitted to intensive care units at the Children’s National Health System were considered for inclusion. Exclusion criteria were severe asphyxia, grade III or IV intraventricular hemorrhage, major congenital malformations or facial malformations (e.g., cleft lip and palate), neurological disorders, receipt of neuromuscular blockers, and hepatic or renal failure, including systemic hypoperfusion. Hepatic and renal failure were defined, respectively, by the presence of abnormal serum liver enzyme levels or abnormal serum creatinine levels for a given gestational and postnatal age.
2.3 Dosing and Sampling Schedule
Intravenous paracetamol (Ofirmev, 10 mg/mL; Mallinckrodt Pharmaceuticals, Dublin, Ireland) was administered by 30-min infusions at 15-mg/kg per dose. Detailed dosing and pharmacokinetic sampling schemes are provided in Fig. S1, Electronic Supplementary Material. Neonates <28 weeks’ gestation received five doses at 12-h intervals; neonates ≥ 28 weeks’ gestation received seven doses at 8-h intervals. Blood samples (0.2 mL) were obtained from arterial lines over the 24 h following the first and final paracetamol doses. Patients were randomly assigned to one of two blood sampling schedules, each consisting of nine to ten collection times. Blood was collected in sodium heparin Vacutainer tubes (BD, Franklin Lakes, NJ, USA) and centrifuged for 10–15 min at 1500×g at 4 °C. Plasma was transferred to cryovials and stored at −70 °C. Urine samples were collected via indwelling catheter (postoperative patients) or from gel-free study diapers (procedural patients; Cuddle Buns Preemie diapers, Small Beginnings Inc., Hesperia, CA, USA) at 3- to 4-h intervals over the 24 h following the first and final paracetamol doses. For samples collected via diaper, urine volume was estimated from the pre- and post-collection difference in diaper weight. Urine samples were not collected from diapers with stool contamination. After urine sample volumes were recorded, 3- to 5-mL aliquots were stored at −70 °C. Study samples were shipped on dry ice to the Center for Human Toxicology at the University of Utah and stored at −80 °C prior to analysis.
2.4 Analytical Methods
Plasma and urinary concentrations of paracetamol, paracetamol-glucuronide, paracetamol-sulfate, paracetamol-cysteine, and paracetamol-N-acetylcysteine were determined by high-performance liquid chromatography-tandem mass spectrometry according to previously reported methods [19]. Mean intra- and inter-assay accuracy ranged from 85 to 111 %, and intra- and inter-assay imprecision did not exceed 15 % coefficient of variation (CV). In plasma, the lower limit of quantification (LLOQ) was 0.05 mg/L for paracetamol, paracetamol-glucuronide, and paracetamol-sulfate, and 0.01 mg/L for paracetamol-cysteine and paracetamol-N-acetylcysteine. In urine, the LLOQ was 0.2 mg/L for paracetamol, 1 mg/L for paracetamol-glucuronide and paracetamol-sulfate, and 0.1 mg/L for paracetamol-cysteine and paracetamol-N-acetylcysteine. One plasma sample (<1 %) and two urine samples (<1 %) were excluded because all analytes were <LLOQ. Three plasma samples (1 %) had paracetamol-N-acetylcysteine concentrations < LLOQ, and two urine samples (<1 %) had one analyte <LLOQ (paracetamol or paracetamol-glucuronide); values of LLOQ ÷ 2 were used in these instances [20].
2.5 Base Model Development
All concentrations were expressed in paracetamol equivalents (mg/L) via conversion based on molecular weights. Following conversion to paracetamol equivalents, paracetamol-cysteine and paracetamol-N-acetylcysteine concentrations for each sample were summed to approximate the total concentration of metabolites derived from CYP-mediated oxidation. The parent-metabolite pharmacokinetic model was developed using NONMEM 7.2 (ICON Development Solutions, Hanover, MD, USA) interfaced with PsN 4.4.0 (psn.sourceforge.net) and Pirana 2.9.0 (pirana-software.com). Urinary concentrations and urine sample volumes were included as NONMEM data items so that the software program could scale appropriately to urinary amounts [21]. Population parameters were estimated using the first-order conditional estimation with interaction method and the ADVAN6 subroutine. The number of significant digits required for convergence (NSIG), predicted values (TOL), and the objective function (SIGL) was set, respectively, to 2, 6, and 6 [22]. Processing and visualization of NONMEM output were performed in R 3.2.1 (CRAN.R-project.org). During covariate analysis, nested models were compared using the objective function value (OFV). At all other stages of development, model discrimination was based on the Akaike information criterion (AIC) [23].
A schematic of the base structure for the parent-metabolite pharmacokinetic model is shown in Fig. 1. The structural model incorporated the rate and duration of the intravenous paracetamol infusion. Paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the combined oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine) were each modeled with a single plasma compartment and subsequent urinary compartment. Similar structural models have been employed to describe the pharmacokinetics of intravenous paracetamol and its metabolites in adult surgical patients [24] and in women during the peripartum period [25]. One-compartment distribution of paracetamol was considered appropriate based on previous work with parent drug data from the same dataset [7]. All formation (hepatic) and renal clearances were modeled as first-order processes. The model structure required the assumption that the pathways illustrated in Fig. 1 account for all elimination of paracetamol and its metabolites. In total, the model was defined by eight differential equations and eleven pharmacokinetic parameters.
Fig. 1.
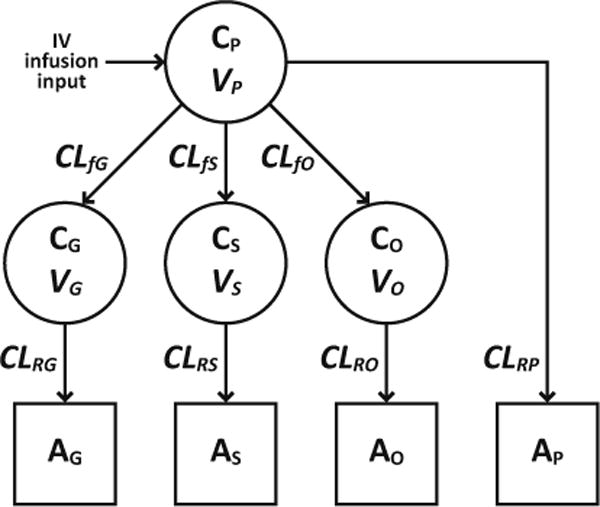
Schematic of the structural pharmacokinetic model for paracetamol and its metabolites in plasma (circles) and urine (squares). All formation and renal clearances were modeled as first-order processes. CP, CG, CS, and CO represent, respectively, plasma concentrations of paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the combined oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine); AP, AG, AS, and AO represent, respectively, urinary amounts of unchanged paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; VP, VG, VS, and VO represent, respectively, volumes of distribution for paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; CLfG, CLfS, and CLfO represent, respectively, formation (hepatic) clearances for paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; CLRP, CLRG, CLRS, and CLRO represent, respectively, renal clearances for unchanged paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites
Random effects were classified as BSV or residual unexplained variability (RUV). Individual pharmacokinetic parameters were assumed to be log-normally distributed, and BSV was modeled exponentially (Eq. 1):
| (1) |
where Pi is the individual pharmacokinetic parameter, θpop is the population value for P, ηi is the between-subject random effect on P for individual i, and ηi is normally distributed with a mean of zero and a variance of ω2. Additive, proportional, and combined additive and proportional functions were tested for incorporation of RUV [26].
2.6 Covariate Analysis
Potential covariates included current body weight, postnatal age, postmenstrual age, indication (postoperative or procedural), sex, race (Caucasian or African American), ethnicity, occasion (first or second), urine flow rate, and estimated glomerular filtration rate (GFR). The first and second occasions were defined, respectively, as < and [42 h after the first paracetamol dose. The urine flow rate covariate was allowed to change over time within each individual, and average flow rate for each 3- to 4-h urine sample was calculated by dividing the sample volume (mL) by the time elapsed during sample collection (h). Estimated GFR was calculated from body length and serum creatinine (modified kinetic Jaffe method) using the updated Schwartz formula [27]. Laboratory samples were obtained within 24 h prior to the first paracetamol dose or during the pharmacokinetic sample collection period. Serum creatinine concentrations obtained at ≤3 days’ postnatal age reflect maternal renal function and were excluded from analysis. During covariate analysis, subjects with missing information for a covariate undergoing evaluation were excluded from both the base and covariate models being tested.
Owing to the large number of potential covariate-pharmacokinetic parameter combinations, only the most physiologically relevant covariate-parameter pairs were considered. Categorical covariates were considered for inclusion using proportional shift models (Eq. 2):
| (2) |
where Pi is the individual pharmacokinetic parameter, θpop is the population value for P when the categorical indicator variable COVi is 0, θcov is the proportional change in θpop when COVi is 1, and ηi is the between-subject random effect on P for individual i.
Current body weight, postnatal age, postmenstrual age, indication, and sex were tested on all pharmacokinetic parameters. For these covariates, continuous variables were normalized to population mean values and tested for inclusion in a power function (Eq. 3):
| (3) |
where Pi is the individual pharmacokinetic parameter for an individual with covariate value COVi, θpop is the population value for P when COVi equals the mean covariate value COVmean, θcov is the covariate effect, and ηi is the between-subject random effect on P for individual i. Given the potential for genetically mediated differences in paracetamol metabolism [28, 29], race and ethnicity were tested on all metabolite formation clearances (Eq. 2). Additionally, occasion was tested on all metabolite formation clearances (Eq. 2) because previous work has suggested that upregulation of paracetamol glucuronidation occurs with repeated administration in adults [30, 31] and in neonates [17, 32]. Finally, urine flow rate and estimated GFR were tested on all renal clearances. Based on a previous pharmacokinetic model of paracetamol, paracetamol-glucuronide, and paracetamol-sulfate in infants, an exponential function was used for incorporation of urine flow rate [33] (Eq. 4):
| (4) |
where Pij is the pharmacokinetic parameter for individual i at time j with urine flow rate UFLOWij, θpop is the population value for P when UFLOWij equals the median urine flow rate UFLOWmed (6.5 mL/h), θcov is the covariate effect, and ηi is the between-subject random effect on P for individual i. Estimated GFR was tested in a mean-centered power function (Eq. 3).
Potential covariates were tested using a modified stepwise forward selection procedure followed by stepwise backward elimination. Changes in OFV were considered significant at p < 0.05 (χ2 distribution, one degree of freedom, ΔOFV >3.84) during forward selection and p < 0.01 (ΔOFV >6.63) during backward elimination [34]. Additionally, covariates were required to provide at least 5 % reduction in BSV or RUV to be added to or retained in the model. The modified forward selection was conducted in a series of rounds. In round 1, weight was tested on all pharmacokinetic parameters and subsequently included on all parameters for which selection criteria were met. In round 2, the remaining covariate-parameter pairs of interest were tested. Round 3 consisted of standard stepwise forward selection using only those covariate-parameter pairs that met selection criteria in round 2. Rounds 2 and 3 were repeated until none of the remaining covariate-parameter pairs met the selection criteria, at which point standard stepwise backward elimination was performed.
2.7 Model Refinement
Following covariate analysis, the model was refined by testing the validity of the default assumption that random effects exhibit no covariance. The extent of covariance in BSV terms was assessed by estimating off-diagonal elements of the Ω matrix. Covariance in RUV terms was evaluated by estimating off-diagonal elements of the Σ matrix, which required use of the NONMEM L2 data item for designation of multivariate observations [21]. Finally, all covariates included in the model were tested by backward elimination to ensure that covariate criteria were still met after model refinement.
2.8 Model Evaluation
A nonparametric bootstrap was performed to assess the stability of the final model and to quantify uncertainty in parameter estimates [35]. Bootstrap datasets (n = 200) were generated in PsN by random sampling with replacement from the original dataset. Visual predictive checks were performed to compare observed plasma concentrations with concentrations obtained from model-based simulation of 1000 datasets [36]. Visual predictive check data were prediction corrected [37] and auto-binned into five or six bins, respectively, over the time following the first and final doses [38]. Additionally, normalized prediction distribution errors (NPDEs) based on 1000 simulations were calculated in NONMEM [39].
3 Results
3.1 Patients and Pharmacokinetic Observations
Demographic characteristics of the 35 study subjects are summarized in Table 1. Most patients (66 %) received the first paracetamol dose within 1 week after birth. All of the postoperative patients had surgery for repair of congenital heart defects, most commonly hypoplastic left heart, coarctation of the aorta, transposition of the great vessels, atrioventricular septal defects, or patent ductus arteriosus. Most of these patients had undergone cardiopulmonary bypass during surgery; however, no patients showed evidence of hepatic or renal injury after surgery.
Table 1.
Demographic characteristics of neonates who received intravenous paracetamol
| Characteristic | N [%] | Mean ± SD | Median [range] |
|---|---|---|---|
| Gestational age (weeks) | 35 [100] | 33.6 ± 6.57 | 37 [23–41] |
| Postnatal agea (days) | 35 [100] | 7.49 ± 5.73 | 6 [1–26] |
| Postmenstrual agea (weeks) | 35 [100] | 34.6 ± 6.28 | 37.6 [23.1–41.6] |
| Current body weighta (kg) | 35 [100] | 2.30 ± 1.22 | 2.80 [0.46–4.20] |
| Current body lengtha (cm) | 34 [97] | 43.4 ± 9.15 | 47.5 [25.0–56.0] |
| Current body weighta (kg) by gestational age subgroup | |||
| Extreme preterm (<28 weeks’ gestation) | 10 [29] | 0.81 ± 0.27 | 0.69 [0.55–1.30] |
| Preterm (<37 weeks’ gestation) | 17 [49] | 1.22 ± 0.76 | 0.96 [0.46–2.80] |
| Full term (37–42 weeks’ gestation) | 18 [51] | 3.32 ± 0.39 | 3.16 [2.70–4.20] |
| Serum creatinineb (mg/dL) | 30 [86] | 0.707 ± 0.242 | 0.65 [0.3–1.1] |
| Estimated GFRc (mL/min per 1.73 m2) | 29 [83] | 30.1 ± 16.6 | 24.1 [12.6–70.9] |
| Primary indication for intravenous paracetamol | |||
| Postoperative analgesia (cardiac surgery) | 19 [54] | ||
| Procedural analgesia | 16 [46] | ||
| Sex | |||
| Male | 20 [57] | ||
| Female | 15 [43] | ||
| Race | |||
| Caucasian | 16 [46] | ||
| African American | 14 [40] | ||
| American Indian/Alaska Native | 1 [3] | ||
| Asian | 1 [3] | ||
| Declined to respond | 3 [9] | ||
| Ethnicity | |||
| Non-Hispanic | 24 [69] | ||
| Hispanic | 8 [23] | ||
| Declined to respond | 3 [9] | ||
GFR glomerular filtration rate, SD standard deviation
On the day of the first paracetamol dose
Serum creatinine levels obtained at ≤3 days’ postnatal age were considered to reflect maternal renal function and were excluded from analysis
Estimated GFR was calculated using the updated Schwartz formula [27]
Eight urine samples were contaminated with stool and were not retained for analysis. Drug and metabolite concentrations were available from 266 plasma samples and 352 urine samples. Six plasma samples (2 %) had implausible drug concentrations (e.g., peak concentrations observed at trough collection times) and were excluded from analysis. One additional plasma sample (< 1 %) and two urine samples (<1 %) were excluded because all analytes were <LLOQ. Thus, 259 plasma samples (median: 8; range: 3–11 samples/patient) and 350 urine samples (median: 11; range: 2–16 samples/patient) were used to develop the population pharmacokinetic model. Figure 2 shows observed plasma concentrations over time following the first and final paracetamol doses.
Fig. 2.
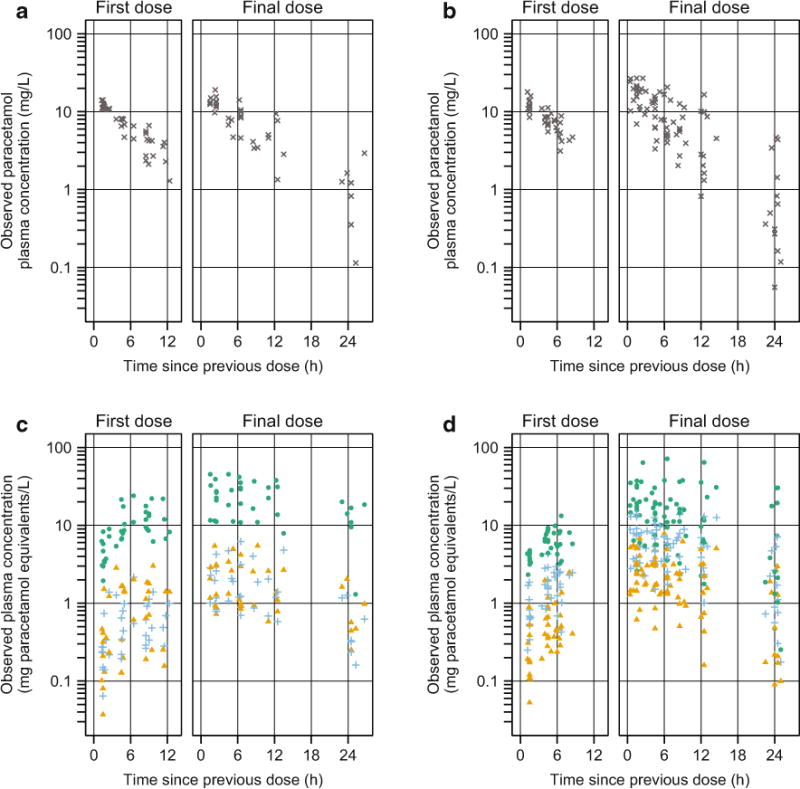
Observed plasma concentrations vs. time for neonates who received five doses at 12-h intervals (a, c 15-mg/kg per dose) and for neonates who received seven doses at 8-h intervals (b, d 15-mg/kg per dose). a, b Show paracetamol concentrations (gray x marks); c, d show concentrations of paracetamol-glucuronide (blue plus signs), paracetamol-sulfate (green circles), and the combined oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine, orange triangles)
3.2 Population Pharmacokinetic Model Development
The combined RUV function provided the lowest AIC and was selected for inclusion in the model. Based on covariate selection criteria, weight was incorporated into the final model on all pharmacokinetic parameters, postnatal age was included on formation clearances of paracetamol-glucuronide and oxidative pathway metabolites, urine flow rate was included on all renal clearances, and indication was included on renal clearance of unchanged paracetamol. No subjects had missing information for any of the covariates that met selection criteria, so the final covariate model incorporated data from all study subjects. Final estimates for covariate effects are provided in Table 2, along with estimates for pharmacokinetic parameters, BSV, RUV, and eta and epsilon shrinkage. Current body weight had a strong influence on all pharmacokinetic parameters. When weight was included on each parameter during round 1 of the modified forward selection process, decreases in OFV ranged from 11.5 to 86.4 and reductions in BSV ranged from 13 to 74 % CV. Urine flow rate was also a highly significant covariate for all renal clearances. In the final model, removal of urine flow rate from each renal clearance produced increases in OFV ranging from 34.0 to 200.3. Effects of postnatal age and indication were more modest than those of weight and urine flow rate. When postnatal age and indication were excluded from the final model, increases in OFV ranged from 10.0 to 20.5 and increases in BSV ranged from 5 to 15 % CV.
Table 2.
Parameter estimates for the final model of paracetamol and its metabolites in neonatesa
| Parameter | Model fit
|
Bootstrap median [95 % CI]b
|
|||
|---|---|---|---|---|---|
| Estimatea | BSV, as %CV | Shrinkage (%) | Estimatea | BSV, as %CV | |
| CLfG (L/h) | 0.049 | 62 | 0 | 0.049 [0.038 to 0.062] | 60 [45–77] |
| Effect of weightc | 2.0 | 2.0 [1.7 to 2.5] | |||
| Effect of PNAc | 0.37 | 0.40 [0.097 to 0.79] | |||
| CLfS (L/h) | 0.21 | 33 | 1 | 0.21 [0.17 to 0.24] | 32 [24–39] |
| Effect of weightc | 0.90 | 0.89 [0.71 to 1.1] | |||
| CLfO (L/h) | 0.058 | 72 | 0 | 0.056 [0.044 to 0.078] | 69 [55–85] |
| Effect of weightc | 1.7 | 1.8 [1.4 to 2.1] | |||
| Effect of PNAc | 0.69 | 0.67 [0.25 to 1.0] | |||
| VP(l) | 2.7 | 11 | 4 | 2.7 [2.6 to 3.2] | 12 [7.7–23] |
| Effect of weightc | 0.90 | 0.90 [0.83 to 1.0] | |||
| VG (L) | 1.2 | 48 | 4 | 1.2 [0.91 to 1.4] | 47 [30–65] |
| Effect of weightc | 1.1 | 1.1 [0.73 to 1.3] | |||
| VS (L) | 0.98 | 36 | 8 | 0.97 [0.77 to 1.2] | 36 [23–50] |
| Effect of weightc | 1.2 | 1.2 [0.88 to 1.4] | |||
| VO (L) | 3.0 | 67 | 3 | 2.9 [2.1 to 4.6] | 62 [23–110] |
| Effect of weightc | 1.5 | 1.5 [1.3 to 1.9] | |||
| CLRP (L/h) | 0.016 | 30 | 6 | 0.016 [0.012 to 0.024] | 30 [18–43] |
| Effect of weightc | 0.64 | 0.62 [0.37 to 1.0] | |||
| Effect of urine flow rated | 0.060 | 0.059 [0.044 to 0.080] | |||
| Effect of postoperative indicatione | −0.57 | −0.57 [−0.76 to −0.34] | |||
| CLRG (L/h) | 0.10 | 37 | 1 | 0.10 [0.079 to 0.12] | 37 [21–47] |
| Effect of weightc | 1.1 | 1.1 [0.76 to 1.3] | |||
| Effect of urine flow rated | 0.015 | 0.016 [0.0020 to 0.029] | |||
| CLRS (L/h) | 0.12 | 54 | 1 | 0.12 [0.085 to 0.14] | 54 [38–70] |
| Effect of weightc | 1.4 | 1.4 [0.99 to 1.7] | |||
| Effect of urine flow rated | 0.015 | 0.016 [0.0027 to 0.031] | |||
| CLRO (L/h) | 0.17 | 43 | 1 | 0.18 [0.14 to 0.21] | 44 [26–62] |
| Effect of weightc | 1.3 | 1.3 [1.1 to 1.6] | |||
| Effect of urine flow rated | 0.032 | 0.032 [0.015 to 0.042] | |||
| Parameter | Model fit
|
Bootstrap median [95 % CI]b | |
|---|---|---|---|
| RUV, as %CV | Shrinkage (%) | RUV, as %CV | |
| Proportional RUV | |||
| Plasma paracetamol | 27 | 5 | 26 [21–32] |
| Plasma paracetamol-glucuronide | 27 | 5 | 27 [23–33] |
| Plasma paracetamol-sulfate | 23 | 5 | 24 [17–30] |
| Plasma oxidative pathway metabolites | 35 | 5 | 34 [27–43] |
| Urinary paracetamol | 49 | 3 | 49 [42–55] |
| Urinary paracetamol-glucuronide | 68 | 3 | 68 [59–76] |
| Urinary paracetamol-sulfate | 70 | 3 | 69 [60–77] |
| Urinary oxidative pathway metabolites | 61 | 3 | 62 [53–70] |
| Parameter | Model fit
|
Bootstrap median [95 % CI]b | |
|---|---|---|---|
| RUV, as SD (mg/L) | Shrinkage (%) | RUV, as SD (mg/L) | |
| Additive RUV | |||
| Plasma paracetamol-sulfate | 1.1 | 5 | 1.1 [0.028–2.0] |
| Urinary paracetamolf | 1.3 | 3 | 1.2 [0.029–1.8] |
| Urinary paracetamol-sulfateg | 14 | 3 | 14 [0.16–35] |
BSV between-subject variability, CI confidence interval, CV coefficient of variation, PNA postnatal age, RUV residual unexplained variability, SD standard deviation. CLfG, CLfS, and CLfO represent, respectively, formation (hepatic) clearances for paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; VP, VG, VS, and VO represent, respectively, volumes of distribution for paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; CLRP, CLRG, CLRS, and CLRO represent, respectively, renal clearances for unchanged paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites
Pharmacokinetic parameter estimates are typical values for patients with a procedural indication at the mean current body weight (2.3 kg), mean PNA (7.5 days), and median urine flow rate (6.5 mL/h)
Bootstrap success rate was 77 % (n = 153 out of 200)
Exponent on mean-centered weight or mean-centered PNA (i.e., the covariate effect, θcov, in Eq. 3)
Coefficient on median-centered urine flow rate in the exponential covariate function (i.e., the covariate effect, θcov, in Eq. 4)
Proportional change in CLRP for patients with a postoperative indication (i.e., the covariate effect, θcov, in Eq. 2)
The mean urinary paracetamol concentration was 14.2 mg/L (SD 11.4)
The mean urinary paracetamol-sulfate concentration was 184 mg paracetamol equivalents/L (SD 174)
Incorporation of covariance estimates for all BSV terms (i.e., a full Ω matrix) improved the model fit compared with the default condition of no covariance (i.e., a diagonal Ω matrix). Use of a full-covariance structure ensured that critical covariance terms would be included, and any ill effects from unnecessary covariance terms were expected to be minimal [40].
Given the physiological basis for the parent-metabolite structural model, it was anticipated that covariance of some parameters would be strong. Correlations in BSV for renal metabolite clearances were particularly high, with correlation coefficients ranging from 0.73 to 0.97. Attempting to estimate a correlation near one can cause numerical instabilities and hinder model convergence. To avoid such problems, correlations in BSV for renal metabolite clearances were fixed to one, as previously reported [41]. Renal clearance of paracetamol-glucuronide was described in an exponential form (Eq. 1). To fix the correlation between random effects for renal clearance of paracetamol-glucuronide and paracetamol-sulfate to one, renal clearance of paracetamol-sulfate was described as follows (Eq. 5):
| (5) |
where CLRS,i is the individual renal clearance of paracetamol-sulfate, is the population value for renal clearance of paracetamol-sulfate, is a scale parameter between the variance of CLRG and the variance of CLRS (Eq. 6), and is the between-subject random effect on renal clearance of paracetamol-glucuronide for individual i. Variance for renal clearance of paracetamol-sulfate could then be determined as follows (Eq. 6):
| (6) |
The same approach was used to fix the correlation between random effects for renal clearance of paracetamol-glucuronide and the oxidative pathway metabolites to one. Model fit suffered slightly when these correlations were fixed (ΔAIC 13.7); however, this worsening of model fit was considered acceptable in exchange for enhanced model stability and a considerable reduction in the number of model parameters (19 fewer parameters). Final estimates for correlation in BSV are provided in Table 3.
Table 3.
Correlation in between-subject variability for the final model of paracetamol and its metabolites in neonatesa
| Parameter | CLfG | CLfS | CLfO | VP | VG | VS | VO | CLRP |
|---|---|---|---|---|---|---|---|---|
| CLfS | 0.37 [0.036 to 0.69] | |||||||
| CLfO | 0.49 [0.16 to 0.80] | 0.054 [−0.33 to 0.54] | ||||||
| VP | 0.35 [− to 0.65] | −0.20 [−0.59 to 0.36] | 0.18 [−0.38 to 0.67] | |||||
| VG | 0.60 [0.14 to 0.83] | 0.032 [−0.30 to 0.50] | 0.42 [−0.0083 to 0.75] | 0.43 [−0.50 to 0.75] | ||||
| VS | 0.19 [−0.35 to 0.77] | 0.64 [0.10 to 0.83] | −0.10 [−0.50 to 0.45] | 0.28 [−0.51 to 0.57] | 0.13 [−0.31 to 0.63] | |||
| VO | −0.10 [−0.42 to 0.59] | −0.40 [−0.72 to 0.37] | 0.44 [−0.24 to 0.75] | 0.53 [−0.37 to 0.85] | 0.15 [−0.14 to 0.57] | −0.14 [−0.47 to 0.44] | ||
| CLRP | 0.041 [−0.46 to 0.51] | −0.033 [−0.41 to 0.49] | −0.14 [−0.51 to 0.43] | 0.20 [−0.44 to 0.72] | −0.036 [−0.59 to 0.42] | −0.0082 [−0.55 to 0.60] | −0.11 [−0.65 to 0.59] | |
| CLRG,CLRS, CLROb | 0.52 [0.29 to 0.73] | 0.73 [0.35 to 0.91] | 0.28 [−0.37 to 0.68] | 0.22 [−0.25 to 0.57] | 0.23 [−0.16 to 0.57] | 0.46 [−0.065 to 0.71] | −0.30 [−0.72 to 0.55] | 0.33 [−0.068 to 0.74] |
CLfG, CLfS, and CLfO represent, respectively, formation (hepatic) clearances for paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; VP, VG, VS, and VO represent, respectively, volumes of distribution for paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites; CLRP, CLRG, CLRS, and CLRO represent, respectively, renal clearances for unchanged paracetamol, paracetamol-glucuronide, paracetamol-sulfate, and the oxidative pathway metabolites
Data are presented as: correlation point estimate from the final model fit [bootstrap 95 % confidence interval]. Bootstrap success rate was 77 % (n = 153 out of 200)
Correlations in between-subject variability for CLRG, clRS, and CLRO were observed to be high and were fixed to one
When covariance estimates on RUV terms from multivariate observations were incorporated into the model, the AIC decreased by 1520.4, indicating a substantial improvement in model fit. Once the covariance terms were included, most additive RUV variance estimates approached zero and could be excluded from the model without compromising model fit. Additive RUV components were retained only for plasma paracetamol-sulfate, urinary paracetamol-sulfate, and urinary paracetamol because the model fit worsened significantly when those terms were removed. The correlation between additive RUV for urinary paracetamol and paracetamol-sulfate was particularly high, with a correlation coefficient estimated at 1.0; therefore, this correlation was fixed to one using the approach shown in Eqs. 5 and 6 to stabilize the model. Final estimates for correlation in proportional RUV are provided in Table 4. NONMEM 7.2 code for the final model, including final equations for each parameter, is provided as an Appendix in the Electronic Supplementary Material.
Table 4.
Correlation in proportional residual unexplained variability for the final model of paracetamol and its metabolites in neonatesa
| Plasma compartments
|
Urine compartments
|
|||||
|---|---|---|---|---|---|---|
| Paracetamol | Paracetamol-glucuronide | Paracetamol-sulfate | Paracetamol | Paracetamol-glucuronide | Paracetamol-sulfate | |
| Paracetamol-glucuronide | 0.38 [0.19–0.54] | 0.58 [0.43–0.72] | ||||
| Paracetamol-sulfate | 0.56 [0.31–0.72] | 0.84 [0.55–0.98] | 0.51 [0.37–0.72] | 0.94 [0.92–0.98] | ||
| Oxidative pathway metabolites | 0.29 [0.10–0.50] | 0.57 [0.30–0.76] | 0.59 [0.28–0.88] | 0.55 [0.39–0.70] | 0.90 [0.87–0.93] | 0.92 [0.89–0.96] |
Data are presented as: correlation point estimate from the final model fit [bootstrap 95 % confidence interval]. Bootstrap success rate was 77 % (n = 153 out of 200)
3.3 Model Evaluation
Standard diagnostic plots of observations vs. predictions are provided in Fig. 3 to illustrate the final model fit. Median bootstrap estimates were very similar to point estimates from the final model fit, and bootstrap 95 % confidence intervals demonstrated reasonably good precision for most parameters (Table 2). Point estimates and bootstrap-derived 95 % confidence intervals are provided in Tables 3 and 4 for correlations in BSV and RUV, respectively. Many BSV correlations were estimated with poor precision (Table 3); however, this was not unexpected given the use of a full Ω matrix. In contrast, correlations in RUV were generally estimated with greater precision than those for BSV, and none of the 95 % confidence intervals crossed zero (Table 4).
Fig. 3.
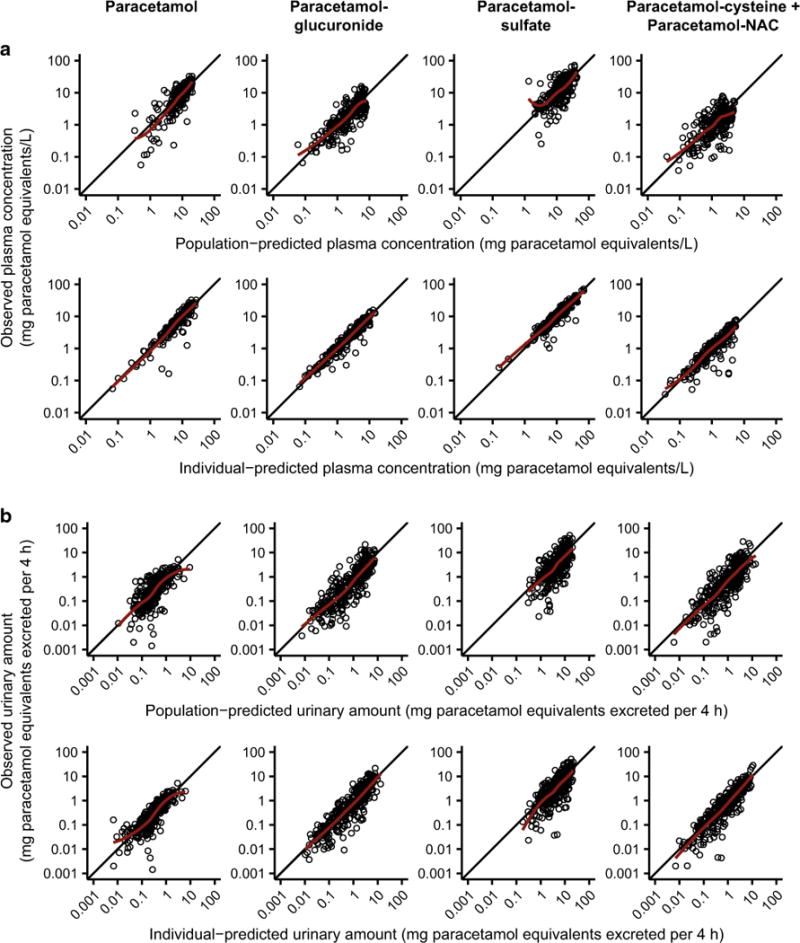
Diagnostic plots for the final model. Observed vs. population-predicted (upper row) and individual-predicted (lower row) plasma concentrations (a), and observed vs. population-predicted (upper row) and individual-predicted (lower row) urinary amounts (b). The solid black lines depict the lines of identity (y = x) and the solid red lines depict the LOESS fits of the data
Out of 200 bootstrap runs, 153 (77 %) minimized successfully, and all others failed owing to rounding errors. Bootstrap summary statistics were derived only from successful runs. However, for most of the parameters listed in Tables 2 and 4, median bootstrap estimates from successful and unsuccessful runs exhibited less than 5 % difference (n = 55 out of 63 parameters, 87 %), and all parameters in Tables 2 and 4 differed by less than 15 %. Discrepancies between successful and unsuccessful runs were more evident for between-subject covariance terms (Table 3): in a comparison of median bootstrap estimates, most of these parameters differed by greater than 15 % (n = 25 out of 36 parameters, 69 %). It is plausible that rounding errors would tend to occur on unnecessary, poorly estimated between-subject covariance terms, thus generating larger discrepancies between successful and unsuccessful runs.
Simulation-based visualizations of model appropriateness were generated with visual predictive checks and NPDEs. Visual predictive checks demonstrated good agreement between observations and model-based simulations for plasma concentrations over time following the first and final paracetamol doses (Fig. 4). NPDE distributions for plasma compartments showed reasonably good agreement with the expected standard normal distribution, but urinary NPDE distributions deviated slightly from expected values (a panels in Figs. S2–S9, Electronic Supplementary Material). Importantly, there were no strong trends in NPDEs when plotted against time since the first dose, population predictions, or influential covariates (panels b–e in Figs. S2–S9, Electronic Supplementary Material).
Fig. 4.
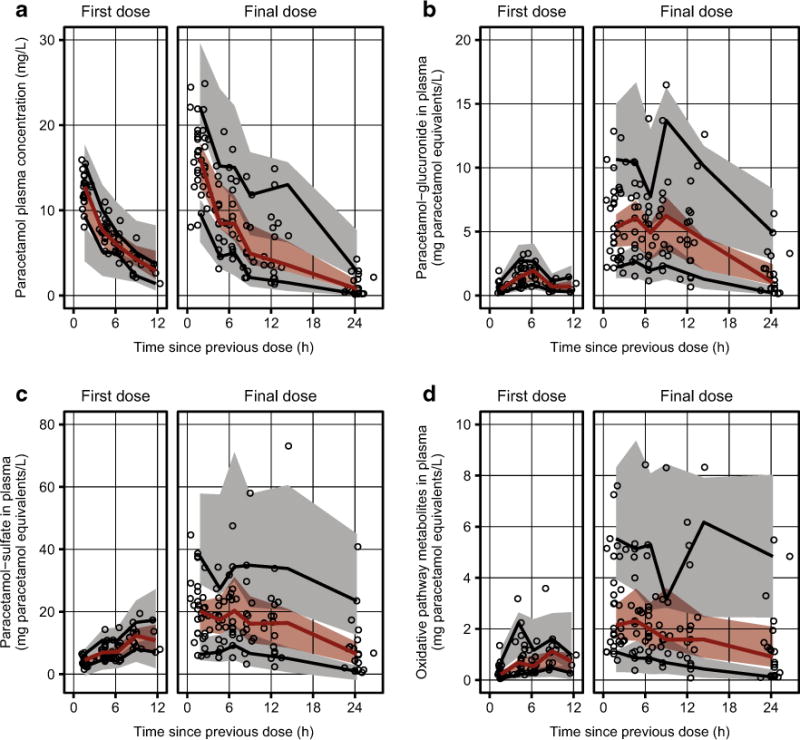
Visual predictive checks of the final model for plasma concentrations of a paracetamol, b paracetamol-glucuronide, c paracetamol-sulfate, and d the combined oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine). Individual observations are depicted as open black circles. The solid red lines depict the observed 50th percentiles, and the solid black lines depict the observed 5th and 95th percentiles. The shaded red regions depict the 95 % confidence intervals surrounding the predicted 50th percentiles, and the shaded gray regions depict the 95 % confidence intervals surrounding the predicted 5th and 95th percentiles. Note that all plotted values reflect prediction-corrected concentrations
Finally, to explore maturational trends in total paracetamol clearance and in the fraction of drug eliminated by the four routes shown in Fig. 1, typical clearance values were plotted over a range of body weights and postnatal ages (Fig. 5), and individual clearance estimates for each pathway were expressed as fractions of total individual paracetamol clearance and plotted against influential covariates (Fig. 6).
Fig. 5.
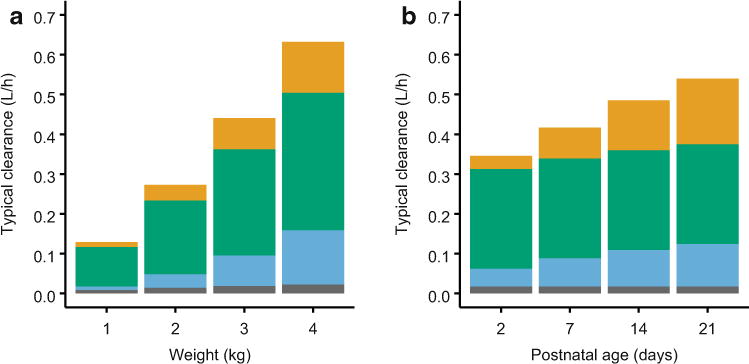
Typical values of total paracetamol clearance from the final model for a patient with a procedural analgesia indication and assuming the median urine flow rate (6.5 mL/h) for a a range of body weights at the median postnatal age of 6 days and b a range of postnatal ages at the median body weight of 2.8 kg. Within each bar, typical clearance values for each pathway are shown (from bottom to top)ingray for renal clearance of unchanged parent drug, blue for glucuronidation, green for sulfation, and orange for oxidation
Fig. 6.
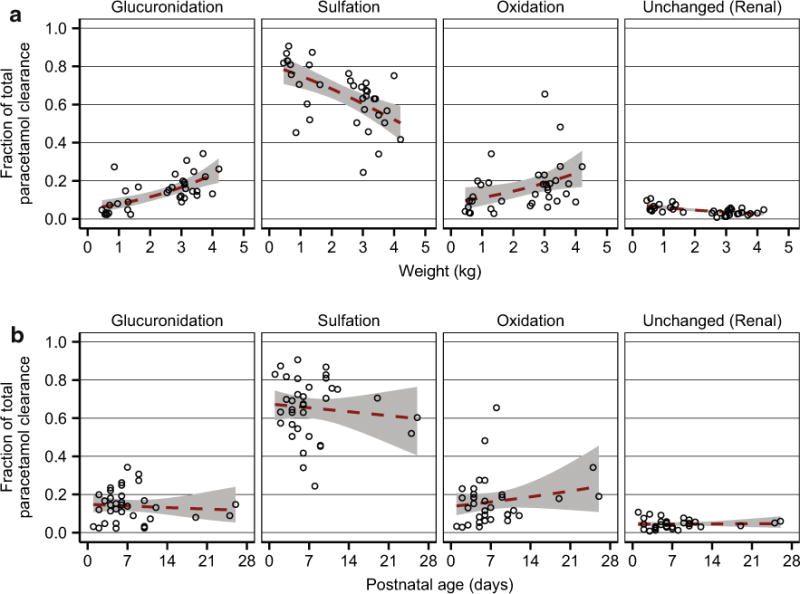
Fraction of total paracetamol clearance accounted for by glucuronidation, sulfation, oxidation, and renal clearance of unchanged parent drug. Fractional clearances for each subject (open black circles) are shown vs. the significant covariates current body weight (a) and postnatal age (b). The dashed red lines depict quasibinomial fits of the data, and the shaded gray regions depict 95 % confidence intervals surrounding the regression curves. Fractional clearances were calculated from individual formation (hepatic) clearance estimates for paracetamol-glucuronide, paracetamol-sulfate, and the combined oxidative pathway metabolites (paracetamol-cysteine and paracetamol-N-acetylcysteine) and from the median individual estimates for renal clearance of unchanged paracetamol
4 Discussion
In neonates, sulfation of paracetamol predominates, and the present observations on the relative elimination of paracetamol by glucuronidation, sulfation, or renal elimination of unchanged parent drug agree with those from prior neonatal pharmacokinetic studies [6, 14–18]. One notable strength of this model was incorporation of both plasma and urinary concentrations of paracetamol and metabolites. Such observations made the model structurally identifiable with respect to metabolite volumes of distribution and renal clearances. Volumes of distribution for paracetamol-glucuronide and paracetamol-sulfate were approximately 40 % of parent drug volume of distribution. This trend is logical given the increased hydrophilicity of these metabolites relative to parent drug, and it is consistent with previous estimates for paracetamol-glucuronide and paracetamol-sulfate volumes of distribution obtained from anephric patients [42]. All three renal metabolite clearances were fairly similar (0.10–0.17 L/h), in agreement with a prior study that found little variation between the same parameters in adult surgical patients [24].
A primary objective of this study was to explore maturational changes in pharmacokinetics of the oxidative pathway metabolites, which serve as markers for the toxic metabolite NAPQI. Weight and postnatal age were identified as covariates that significantly influenced formation clearance of the oxidative pathway metabolites. When weight was incorporated, the OFV decreased by 50.3, and a reduction in BSV of 55 % CV was observed. Inclusion of postnatal age was accompanied by an additional OFV decrease of 20.5 and BSV reduction of 9 % CV. Both weight and postnatal age were also significant covariates on formation clearance of paracetamol-glucuronide. Previous evidence suggests that peripartum or postnatal factors play an important role in initiating hepatic CYP2E1 and UDP-glucuronosyltransferase expression and activity [43–45], which is consistent with the present finding that postnatal age was a significant covariate on formation clearances for these pathways, whereas postmenstrual age was not.
Importantly, the amount of toxic NAPQI formed in a given individual depends not only upon that individual’s capacity for paracetamol oxidation but also on the relative contributions from all paracetamol elimination pathways. Thus, maturational trends in fractional clearance were explored for each elimination route. On average, the fraction of drug undergoing oxidation increased slightly (< 15 %) with increasing weight or postnatal age, but these trends were small relative to BSV (Fig. 6). Unfortunately, there are no established threshold plasma concentrations of paracetamol-cysteine and/or paracetamol-N-acetylcysteine that are known to be associated with the development of paracetamol-induced hepatotoxicity in humans. In cases of overdose, symptoms of toxicity are generally delayed relative to the time of drug ingestion, and patients usually present for hospital admission long after the time of overdose. Characterization of drug and metabolite pharmacokinetics during the time period immediately following toxic doses is, therefore, immensely challenging. A recent study reported plasma concentrations of combined oxidative pathway metabolites (paracetamol-cysteine plus paracetamol-N-acetylcysteine) of approximately 3–4 mg paracetamol equivalents/L in adult overdose patients on the day of hospital admission; however, most of these patients had ingested the drug days earlier, and the metabolite concentrations declined rapidly over the first day of hospitalization [46]. Thus, during the earlier time period following overdose, plasma concentrations of oxidative pathway metabolites were likely substantially higher than those measured upon admission and also substantially higher than those observed in the present study (Fig. 2).
In neonates, total clearance of intravenous paracetamol is primarily influenced by body weight [4, 7, 47, 48], and the present findings expand upon prior knowledge by showing that clearances for all four paracetamol elimination routes were significantly affected by weight. Changes in total paracetamol clearance with increasing postnatal age were also evident but were less pronounced than those for weight (Fig. 5). Intravenous paracetamol has often been administered more conservatively to less mature neonates (e.g., at wider dosing intervals or in lower per-kilogram doses), but a recent analysis of parent drug pharmacokinetics from the same dataset described herein supports the use of a parsimonious regimen based solely on equivalent per-kilogram dosing [7]. The present findings indicate that such a dosing regimen also appears suitable with respect to the pharmacokinetics of hepatotoxicity-associated metabolites.
A previous infant model reported that renal clearances of paracetamol, paracetamol-glucuronide, and paracetamol-sulfate increased with urine flow rate [33]. In the current study, a similar effect was observed for the oxidative pathway metabolites. Additionally, renal clearance of unchanged paracetamol differed significantly between patients with postoperative and procedural indications. However, this finding should be interpreted cautiously. Key patient characteristics were distributed unevenly across the two groups: compared with postoperative patients, procedural patients tended to weigh less and have lower gestational ages and higher postnatal ages.
Urine collection procedures also differed between postoperative and procedural patients (catheter and diaper, respectively). Furthermore, even if this covariate reflects a true physiological effect, it is unlikely to be clinically significant given the low fractional clearance of unchanged drug.
The large BSV observed for most pharmacokinetic parameters highlights the importance of continued caution in administering paracetamol to neonates. In the previous pharmacokinetic analysis of only the parent drug, BSV in total paracetamol clearance was 31 % CV [7], which agrees well with the present estimate of 33 % CV for BSV in sulfation, the predominant elimination pathway. BSV was particularly high for glucuronidation and oxidation (62–72 % CV), as might be expected based on the rapid development of these processes during early life [43, 45]. However, it appeared that BSV in certain pathways might compensate to some extent for BSV in other pathways because the previous estimate of BSV in total clearance (31 % CV) [7] was considerably lower than BSV estimates for glucuronidation and oxidation, even though these pathways contributed substantially to total drug clearance for many of the study subjects. Indeed, the data appear to support this notion of some counterbalancing between extreme BSV values. For instance, across the study population, BSV in formation clearance of paracetamol-sulfate was not significantly correlated with BSV in formation clearance of the oxidative pathway metabolites (Table 3), but two subjects did have very low η values for sulfation (ranked in the bottom three) and very high η values for oxidation (ranked in the top four). Future studies should incorporate additional patient information, such as genetic data, that could further reduce BSV. Urinary RUV also remained fairly high in the final model (Table 2), which was not surprising given that urine sampling introduces more opportunities for error than plasma sampling (e.g., longer collection period and requirement for records of sample start time, end time, and volume). These urine-specific aspects of sample collection might also have contributed to the relatively high correlations in urinary RUV (Table 4).
Interpretation of these findings is subject to several limitations. First, despite good representation of extremely preterm, preterm, and full-term neonates, the sample size was still relatively small, and conclusions drawn from these results must be tempered by potential limitations based on the distributions of demographic characteristics. For instance, a covariate effect of estimated GFR on renal drug and metabolite clearances seems physiologically plausible, but the power to detect such an effect was likely limited because patients with renal dysfunction were not eligible for enrollment. Additionally, body weight and estimated GFR were strongly correlated (r = 0.71), so the ability to detect a distinct covariate effect from estimated GFR would have been further limited after weight was incorporated into the model. Another limitation is that the model relies on the assumption that all elimination of paracetamol and its metabolites occurs via the pathways illustrated in Fig. 1, but small fractions of paracetamol and its metabolites are known to undergo biliary excretion in humans [49, 50]. Unfortunately, the present study design did not allow for direct calculation of the fraction of administered dose recovered in urine. Nevertheless, this assumption does not seem unreasonable based on typical estimates for total paracetamol clearance: in the previous pharmacokinetic analysis of the parent drug alone, total clearance was estimated as 0.35 L/h at the mean subject weight of 2.3 kg [7], and in the present analysis, total clearance estimates obtained by summing the four paracetamol clearance routes were approximately 0.32–0.33 L/h, depending upon indication, at the mean weight of 2.3 kg, mean postnatal age of 7.5 days, and median urine flow rate of 6.5 mL/h (Table 2). The model structure also fails to account for NAPQI that covalently binds proteins to form paracetamol-protein adducts. This fraction of NAPQI is expected to be small relative to the amount conjugated by glutathione; nevertheless, future studies could explore this point more thoroughly by testing for covariate effects on the pharmacokinetics of circulating paracetamol-protein adducts. Finally, although this study contributes critical information regarding the pharmacokinetics of paracetamol metabolites in neonates, pharmacodynamic data for paracetamol in this patient population are still lacking [51].
5 Conclusions
The reported model successfully characterized the pharmacokinetics of intravenous paracetamol and its metabolites in preterm and term neonates. Formation clearance of oxidative pathway metabolites increased with body weight and postnatal age; however, maturational increases in the fraction of drug undergoing oxidation were small relative to BSV.
Supplementary Material
Key Points.
In extremely preterm to full-term neonates, fractional formation clearance of hepatotoxicity-associated metabolites of paracetamol increased slightly with weight and postnatal age, but these maturational changes were small relative to between-subject variability.
Pharmacokinetics of hepatotoxicity-associated metabolites support the use of a parsimonious neonatal dosing regimen based solely on equivalent per-kilogram dosing.
Large between-subject variability in metabolite pharmacokinetics underscores the importance of continued caution in administration of paracetamol to neonates.
Acknowledgments
The authors thank Dr. Syamala Mankala of the Division of Clinical Pharmacology at Children’s National Health System (Washington, DC, USA) for administrative support, Dr. David M. Reith of the Dunedin School of Medicine at the University of Otago (Dunedin, New Zealand) and Dr. Katie H. Owens of the Department of Pharmaceutics at the University of Washington (Seattle, WA, USA) for helpful advice during model development, and Dr. Jessica K. Roberts of the Department of Pharmaceutical Sciences at St. Jude Children’s Research Hospital (Memphis, TN, USA) for constructive feedback during manuscript preparation.
Funding This work was supported by the National Institutes of Health grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD060543; to John van den Anker) and the National Center for Advancing Translational Sciences (UL1TR000075; to Children’s National Health System) and by a contract for analytical laboratory services from McNeil Consumer Healthcare (Division of McNEIL-PPC, Inc., Fort Washington, PA, USA; to Diana Wilkins). Sarah Cook received stipend support from the Howard Hughes Medical Institute (Med into Grad Initiative). Sarah Cook and Chris Stockmann were supported by pre-doctoral fellowships from the American Foundation for Pharmaceutical Education.
Footnotes
Electronic supplementary material The online version of this article (doi: 10.1007/s40262-016-0408-1) contains supplementary material, which is available to authorized users.
Author contributions JNA designed the clinical study; JNA, SSZ, ND, and EFW performed the trial and acquired the clinical data; SFC, ADK, and DGW developed and validated the analytical methods and analyzed the study samples; SFC and CS developed the pharmacokinetic model; CMTS supervised the pharmacokinetic model development; SFC drafted the manuscript. All authors critically revised the manuscript and approved the final version.
Compliance with Ethical Standards
Conflicts of interest Sarah Cook, Chris Stockmann, Samira Samiee-Zafarghandy, Amber King, Nina Deutsch, Elaine Williams, Diana Wilkins, Catherine Sherwin, and John van den Anker have no potential conflicts of interest to declare.
Ethical approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent Informed consent was obtained from a parent or legal guardian of all individual participants included in the study.
References
- 1.Allegaert K, Tibboel D, van den Anker J. Pharmacological treatment of neonatal pain: in search of a new equipoise. Semin Fetal Neonatal Med. 2013;18(1):42–7. doi: 10.1016/j.siny.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacifici GM, Allegaert K. Clinical pharmacology of paracetamol in neonates: a review. Curr Ther Res Clin Exp. 2015;77:24–30. doi: 10.1016/j.curtheres.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duggan ST, Scott LJ. Intravenous paracetamol (acetaminophen) Drugs. 2009;69(1):101–13. doi: 10.2165/00003495-200969010-00007. [DOI] [PubMed] [Google Scholar]
- 4.Allegaert K, Palmer GM, Anderson BJ. The pharmacokinetics of intravenous paracetamol in neonates: size matters most. Arch Dis Child. 2011;96(6):575–80. doi: 10.1136/adc.2010.204552. [DOI] [PubMed] [Google Scholar]
- 5.Zuppa AF, Hammer GB, Barrett JS, Kenney BF, Kassir N, Mouksassi S, et al. Safety and population pharmacokinetic analysis of intravenous acetaminophen in neonates, infants, children, and adolescents with pain or fever. J Pediatr Pharmacol Ther. 2011;16(4):246–61. doi: 10.5863/1551-6776-16.4.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Ganzewinkel C, Derijks L, Anand KJ, van Lingen RA, Neef C, Kramer BW, et al. Multiple intravenous doses of paracetamol result in a predictable pharmacokinetic profile in very preterm infants. Acta Paediatr. 2014;103(6):612–7. doi: 10.1111/apa.12638. [DOI] [PubMed] [Google Scholar]
- 7.Cook SF, Roberts JK, Samiee-Zafarghandy S, Stockmann C, King AD, Deutsch N, et al. Population pharmacokinetics of intravenous paracetamol (acetaminophen) in preterm and term neonates: model development and external evaluation. Clin Pharmacokinet. 2016;55(1):107–19. doi: 10.1007/s40262-015-0301-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson BJ, van Lingen RA, Hansen TG, Lin YC, Holford NH. Acetaminophen developmental pharmacokinetics in premature neonates and infants: a pooled population analysis. Anesthesiology. 2002;96(6):1336–45. doi: 10.1097/00000542-200206000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Allegaert K, Vanhaesebrouck S, Verbesselt R, van den Anker JN. In vivo glucuronidation activity of drugs in neonates: extensive interindividual variability despite their young age. Ther Drug Monit. 2009;31(4):411–5. doi: 10.1097/FTD.0b013e3181a8cc0a. [DOI] [PubMed] [Google Scholar]
- 10.McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30(9):2174–87. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31(12):1499–506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 12.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44(1):88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;196:369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy G, Khanna NN, Soda DM, Tsuzuki O, Stern L. Pharmacokinetics of acetaminophen in the human neonate: formation of acetaminophen glucuronide and sulfate in relation to plasma bilirubin concentration and D-glucaric acid excretion. Pediatrics. 1975;55(6):818–25. [PubMed] [Google Scholar]
- 15.Miller RP, Roberts RJ, Fischer LJ. Acetaminophen elimination kinetics in neonates, children, and adults. Clin Pharmacol Ther. 1976;19(3):284–94. doi: 10.1002/cpt1976193284. [DOI] [PubMed] [Google Scholar]
- 16.van Lingen RA, Deinum JT, Quak JM, Kuizenga AJ, van Dam JG, Anand KJ, et al. Pharmacokinetics and metabolism of rectally administered paracetamol in preterm neonates. Arch Dis Child Fetal Neonatal Ed. 1999;80(1):F59–63. doi: 10.1136/fn.80.1.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allegaert K, de Hoon J, Verbesselt R, Vanhole C, Devlieger H, Tibboel D. Intra- and interindividual variability of glucuronidation of paracetamol during repeated administration of propacetamol in neonates. Acta Paediatr. 2005;94(9):1273–9. doi: 10.1111/j.1651-2227.2005.tb02088.x. [DOI] [PubMed] [Google Scholar]
- 18.Krekels EH, van Ham S, Allegaert K, de Hoon J, Tibboel D, Danhof M, et al. Developmental changes rather than repeated administration drive paracetamol glucuronidation in neonates and infants. Eur J Clin Pharmacol. 2015;71(9):1075–82. doi: 10.1007/s00228-015-1887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook SF, King AD, van den Anker JN, Wilkins DG. Simultaneous quantification of acetaminophen and five acetaminophen metabolites in human plasma and urine by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry: method validation and application to a neonatal pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1007:30–42. doi: 10.1016/j.jchromb.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504. doi: 10.1023/a:1012299115260. [DOI] [PubMed] [Google Scholar]
- 21.Boeckmann AJ, Sheiner LB, Beal SL. NONMEM users guide, part v: introductory guide. San Francisco: NONMEM Project Group, University of California at San Francisco; 2011. [Google Scholar]
- 22.Bauer RJ. NONMEM users guide: introduction to NONMEM 7.2.0. Ellicott City: ICON Development Solutions; 2011. [Google Scholar]
- 23.Ludden TM, Beal SL, Sheiner LB. Comparison of the Akaike information criterion, the Schwarz criterion and the F test as guides to model selection. J Pharmacokinet Biopharm. 1994;22(5):431–45. doi: 10.1007/BF02353864. [DOI] [PubMed] [Google Scholar]
- 24.Owens KH, Murphy PG, Medlicott NJ, Kennedy J, Zacharias M, Curran N, et al. Population pharmacokinetics of intravenous acetaminophen and its metabolites in major surgical patients. J Pharmacokinet Pharmacodyn. 2014;41(3):211–21. doi: 10.1007/s10928-014-9358-0. [DOI] [PubMed] [Google Scholar]
- 25.Kulo A, Peeters MY, Allegaert K, Smits A, de Hoon J, Verbesselt R, et al. Pharmacokinetics of paracetamol and its metabolites in women at delivery and post-partum. Br J Clin Pharmacol. 2013;75(3):850–60. doi: 10.1111/j.1365-2125.2012.04402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development, part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2(4):e38. doi: 10.1038/psp.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20(3):629–37. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao L, Pickering G. Paracetamol metabolism and related genetic differences. Drug Metab Rev. 2011;43(1):41–52. doi: 10.3109/03602532.2010.527984. [DOI] [PubMed] [Google Scholar]
- 29.Krasniak AE, Knipp GT, Svensson CK, Liu W. Pharmacogenomics of acetaminophen in pediatric populations: a moving target. Front Genet. 2014;5:314. doi: 10.3389/fgene.2014.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gelotte CK, Auiler JF, Lynch JM, Temple AR, Slattery JT. Disposition of acetaminophen at 4, 6, and 8 g/day for 3 days in healthy young adults. Clin Pharmacol Ther. 2007;81(6):840–8. doi: 10.1038/sj.clpt.6100121. [DOI] [PubMed] [Google Scholar]
- 31.Owens KH, Medlicott NJ, Zacharias M, Curran N, Chary S, Thompson-Fawcett M, et al. The pharmacokinetic profile of intravenous paracetamol in adult patients undergoing major abdominal surgery. Ther Drug Monit. 2012;34(6):713–21. doi: 10.1097/FTD.0b013e31826a70ea. [DOI] [PubMed] [Google Scholar]
- 32.Allegaert K, Verbesselt R, Rayyan M, Debeer A, de Hoon J. Urinary metabolites to assess in vivo ontogeny of hepatic drug metabolism in early neonatal life. Methods Find Exp Clin Pharmacol. 2007;29(4):251–6. doi: 10.1358/mf.2007.29.4.1106408. [DOI] [PubMed] [Google Scholar]
- 33.van der Marel CD, Anderson BJ, van Lingen RA, Holford NH, Pluim MA, Jansman FG, et al. Paracetamol and metabolite pharmacokinetics in infants. Eur J Clin Pharmacol. 2003;59(3):243–51. doi: 10.1007/s00228-003-0608-0. [DOI] [PubMed] [Google Scholar]
- 34.Owen JS, Fiedler-Kelly J. Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models. London: Wiley; 2014. [Google Scholar]
- 35.Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37(6):486–95. doi: 10.1002/j.1552-4604.1997.tb04326.x. [DOI] [PubMed] [Google Scholar]
- 36.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 37.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brendel K, Comets E, Laffont C, Mentre F. Evaluation of different tests based on observations for external model evaluation of population analyses. J Pharmacokinet Pharmacodyn. 2010;37(1):49–65. doi: 10.1007/s10928-009-9143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holford NHG, Gobburu JVS, Mould DR. Implications of including and excluding correlation of random effects in hierarchical mixed effects pharmacokinetic models. Verona: Population Approach Group in Europe; 2003. [Google Scholar]
- 41.Salinger DH, Blough DK, Vicini P, Anasetti C, O’Donnell PV, Sandmaier BM, et al. A limited sampling schedule to estimate individual pharmacokinetic parameters of fludarabine in hematopoietic cell transplant patients. Clin Cancer Res. 2009;15(16):5280–7. doi: 10.1158/1078-0432.CCR-09-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowenthal DT, Oie S, Van Stone JC, Briggs WA, Levy G. Pharmacokinetics of acetaminophen elimination by anephric patients. J Pharmacol Exp Ther. 1976;196(3):570–8. [PubMed] [Google Scholar]
- 43.Johnsrud EK, Koukouritaki SB, Divakaran K, Brunengraber LL, Hines RN, McCarver DG. Human hepatic CYP2E1 expression during development. J Pharmacol Exp Ther. 2003;307(1):402–7. doi: 10.1124/jpet.102.053124. [DOI] [PubMed] [Google Scholar]
- 44.Vieira I, Sonnier M, Cresteil T. Developmental expression of CYP2E1 in the human liver: hypermethylation control of gene expression during the neonatal period. Eur J Biochem. 1996;238(2):476–83. doi: 10.1111/j.1432-1033.1996.0476z.x. [DOI] [PubMed] [Google Scholar]
- 45.Krekels EH, Danhof M, Tibboel D, Knibbe CA. Ontogeny of hepatic glucuronidation; methods and results. Curr Drug Metab. 2012;13(6):728–43. doi: 10.2174/138920012800840455. [DOI] [PubMed] [Google Scholar]
- 46.Xie Y, McGill MR, Cook SF, Sharpe MR, Winefield RD, Wilkins DG, et al. Time course of acetaminophen-protein adducts and acetaminophen metabolites in circulation of overdose patients and in HepaRG cells. Xenobiotica. 2015;45(10):921–9. doi: 10.3109/00498254.2015.1026426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allegaert K, Anderson BJ, Naulaers G, de Hoon J, Verbesselt R, Debeer A, et al. Intravenous paracetamol (propacetamol) pharmacokinetics in term and preterm neonates. Eur J Clin Pharmacol. 2004;60(3):191–7. doi: 10.1007/s00228-004-0756-x. [DOI] [PubMed] [Google Scholar]
- 48.Palmer GM, Atkins M, Anderson BJ, Smith KR, Culnane TJ, McNally CM, et al. I.V. acetaminophen pharmacokinetics in neonates after multiple doses. Br J Anaesth. 2008;101(4):523–30. doi: 10.1093/bja/aen208. [DOI] [PubMed] [Google Scholar]
- 49.Siegers CP, Loeser W, Gieselmann J, Oltmanns D. Biliary and renal excretion of paracetamol in man. Pharmacology. 1984;29(5):301–3. doi: 10.1159/000138026. [DOI] [PubMed] [Google Scholar]
- 50.Jayasinghe KS, Roberts CJ, Read AE. Is biliary excretion of paracetamol significant in man? Br J Clin Pharmacol. 1986;22(3):363–6. doi: 10.1111/j.1365-2125.1986.tb02902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allegaert K, Naulaers G, Vanhaesebrouck S, Anderson BJ. The paracetamol concentration-effect relation in neonates. Paediatr Anaesth. 2013;23(1):45–50. doi: 10.1111/pan.12076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.