

Abstract
Background
To identify the effect of apigenin on cognitive deficits of rats after cerebral ischemia and reperfusion injury, and to investigate the potential molecular mechanisms.
Material/Methods
The rats were given sodium butyrate (NaB) or apigenin (20 or 40 mg/kg) for 28 days. Cognition was investigated by the Morris water maze (MWM) test. On day 28, the rats were euthanized and their hippocampal brain regions were used to identify biochemical and neurochemical alterations. The content of histone deacetylase (HDAC) was measured by enzyme-linked immunosorbent assay (ELISA). Western blot analysis was performed to determine the levels of BDNF, phosphorylated cAMP response element-binding protein (pCREB), acetylated H3, and acetylated H4. The mRNA expressions of brain-derived neurotrophic factor (BDNF) and synapsin-I (Syn-I) were examined by polymerase chain reaction (PCR).
Results
The rats with chronic administration of apigenin (20 and 40 mg/kg) showed better performance in the MWM task than the model rats; there was no significant difference between the apigenin-treated and NaB-treated rats. At the higher apigenin dose of 40 mg/kg, the HDAC content was decreased, the BDNF level was markedly increased, and acetylated H3 and acetylated H4 expressions and Syn-I expressions in the hippocampus was upregulated compared with the model group. Apigenin at 20 mg/kg did not show reversal of the neurochemical alterations.
Conclusions
The improvement effect of apigenin on cognitive impairments after cerebral ischemia and reperfusion injury may involve multiple mechanisms, such as the inhibition of HDAC, induction of BDNF and Syn-I expression, and regulation of histone acetylation.
MeSH Keywords: Apigenin; Brain-Derived Neurotrophic Factor; Histone Deacetylases; Infarction, Middle Cerebral Artery; Memory Disorders
Background
Stroke is a leading cause of morbidity and mortality worldwide. Cognitive deficit is frequent after ischemic stroke. Cognitive impairment can increase disability and indirectly affect functional recovery after stroke, and result in reduced participation in rehabilitation and poor adherence to treatment guidelines [1]. Because of falling mortality rates after stroke, the burden of post-stroke cognitive deficits has increased, and the present therapies for treatment of post-stroke cognitive disorders are not effective. Therefore, further research and development of more efficacious treatments for post-stroke cognitive disorders are imperative.
Cognitive function is one of the most important functions of the brain. However, the mechanisms involved in post-stroke cognitive impairment are complex. Accumulating evidence suggests a critical role for epigenetic regulation of cognition formation. Among these findings, histone acetylation and chromatin homeostasis in the central nervous system has been shown to have a very close relationship with cognitive function [2]. One of the key processes implicated in memory processes is remodeling of chromatin. Histone acetylation is thought to activate transcription by relaxing chromatin. In contrast, histone deacetylation confers on chromatin a role in the expression of gene transcription [3]. Interestingly, histone acetylases and histone deacetylases (HDACs) have functional roles in long-term memory by allowing transcription of genes that support memory. Moreover, treatment with HDAC inhibitors improved long-term memory performances in hippocampus-dependent paradigms, such as the Morris water maze (MWM) task [4]. The class I (HDAC1, 2, 3, 8)-specific HDAC inhibitor sodium butyrate (NaB) was found to rescue memory deficits in rodents [5]. Similarly, treatment with other HDAC inhibitors has been shown to enhance memory reconsolidation [6].
Previous studies have shown that histone acetylation controlled the expression of various proteins associated with cognition [7]. Brain-derived neurotrophic factor (BDNF) is one of the proteins correlated with post-stroke cognitive function recovery. It has been reported that decreased hippocampal BDNF expression aggravates cognitive impairment in both stroke and depression models [8]. The HDAC inhibitor NaB downregulated histone acetylation and improved the behavioral performance of rats in a middle cerebral artery occlusion (MCAO) model, while blockade of BDNF-TrkB pathways abolished this improvement of cognition [9]. It has been shown that the effect of histone acetylation on cognition occurred via the BDNF pathway. Histone acetylation can increase the expression of BDNF, and BDNF can increase histone acetylation, which is regulated by phosphorylated cAMP response element-binding protein (pCREB) [10]. pCREB binds to the promoter regions of many genes associated with memory and synaptic plasticity. Synaptic plasticity is critical to memory formation and storage. synapsin-I (Syn-I) is a marker of synapse formation. Ricobaraza et al. [11] reported that a nonselective HDAC inhibitor reversed spatial learning and memory deficits in an established mouse model of Alzheimer’s disease, and also activated the transcription of synaptic plasticity markers.
In recent years, there has been increasing interest in the identification of novel lead compounds from natural sources, considering their safety profiles. Flavonoids are the largest group of polyphenols in many plants and are known to have several physiological benefits, especially on free radical scavenging, cognitive impairment, learning, and memory [12]. Apigenin belongs to a low-toxicity and non-mutagenic flavone subclass of flavonoids. Apigenin can inhibit the activation of cytokines and NO production, protect Alzheimer’s disease neurons from inflammatory induced stress and neurite retraction, and reduced neuronal hyper-excitability and apoptosis [13]. Han et al. reported that apigenin alleviates KA-induced exictotoxicity by quenching ROS as well as inhibiting GSH depletion in hippocampal neurons [14]. A previous study showed that apigenin had anti-apoptosis and antioxidative properties via affecting the expression of Nrf2 and p53, and their downstream target gene transcription in oxygen and glucose deprivation/reperfusion (OGD/R) model, which mimic the MCAO model [15]. Another research reported that apigenin regulated the expression of adhesion molecules in reactive astrocytes during stroke [16]. The aforementioned studies suggested that apigenin possesses anti-inflammatory, antioxidant and neuroprotective properties. In our previous studies, we verified that apigenin had antioxidant and anti-inflammatory effects after stroke. In addition, there was a study reported that apigenin improved performance in the MWM task and stimulated neurogenesis in the hippocampal region of the rodent brain [17]. Apigenin may ameliorate Alzheimer’s disease-associated learning and memory impairment by relieving the Aβ burden, suppressing the amyloidogenic process, inhibiting oxidative stress, and restoring the ERK/CREB/BDNF pathway [18]. The available reports on apigenin suggest that it could be useful for prevention of cognitive deficits after brain injury. However, the precise mechanisms of apigenin in improving cognition after stroke need further discussed.
In this study, we simulated the real sense of stroke patients by using middle cerebral artery occlusion (MCAO) model, and evaluated the improvement effects of apigenin on cognition after ischemia and reperfusion injury of rats. Furthermore, we investigated the underlying molecular mechanisms of this action. The results showed that apigenin ameliorates rats cognitive deficits after cerebral ischemia and reperfusion injury, and that the mechanisms of this function may include modulation of histone acetylation and promotion of BDNF and Syn-I expressions.
Material and Methods
Animals
Adult male Sprague-Dawley rats, about 3 to 4 months old, weighing 250±20 g, were procured from Beijing HuaFuKang Biological Technology Co. Ltd. (China). The animals were housed in a well-controlled atmosphere with room temperature of 25±1°C and relative humidity of 60%. All experiments involving animals were approved by the guidelines of the Institute for Laboratory Animal Research of the Third Military Medical University, and were executed in accordance with the Guide for the Care and Use of Laboratory Animals. The study was approved by the Animal Research Committee of Whenzhou Medical University (License No. WYDW2013-0039).
Drugs and chemicals
Apigenin (≥99% purity) was purchased from Shangxi Huike Biotechnology Co. Ltd. (China). NaB was purchased from Sigma Chemical Company (USA). HDAC ELISA Kits were purchased from Shanghai Xitang Biological Technology Co. Ltd. (China). BDNF, Syn-I, pCREB, histone H3, and histone H4 were purchased from Abcam Chemical Company (USA). Unless otherwise stated, all chemicals and biochemical reagents used in the study were of the highest analytical grade. Solutions of the drugs and chemicals were freshly prepared before use.
MCAO model
The MCAO model was established as previously reported [19]. In short, rats were anesthetized with intra-peritoneal injection of 10% chloral hydrate (350 mg/kg). The left common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were surgically exposed. A filament was gently inserted into the ICA to occlude the left middle cerebral artery (MCA) at its origin (18–20 mm). Reperfusion was achieved by removing the suture until the tip cleared the lumen of the ECA after 1.5 hours of occlusion. Rectal temperature was monitored throughout the operation and maintained at 37°C with a circulating heating pad. Sham control rats were subjected to cutting of the skin, and sewn up immediately after separating the left carotid artery. After recovery from anesthesia, all rats were returned to their cages with free access to food and water.
Animal grouping and treatment
After seven days of adaptation, the rats were randomly assigned to five groups (n=14 rats/group) as follows: sham group; model group; apigenin-treated groups (20 or 40 mg/kg); NaB-treated group (300 mg/kg). The rats in the apigenin- and NaB-treated groups received an intraperitoneal injection of apigenin or NaB every 24 hours following reperfusion. The rats in the sham group and model group were injected with an equal volume of normal saline at the same times following reperfusion. The rats in each group received gentamicin 8,000 U by intraperitoneal injection to prevent infection in the first three days (Figure 1).
Figure 1.
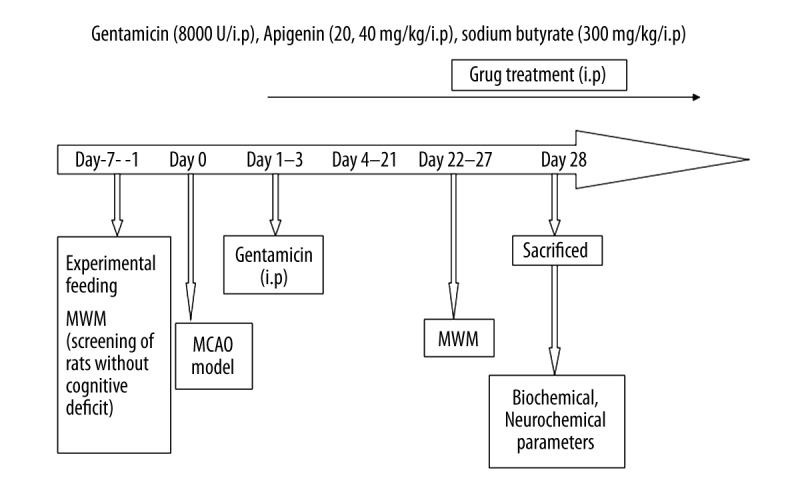
Experimental procedure and treatment schedule. MWM – Morris water maze; MCAO – middle cerebral artery occlusion.
Behavioral assessment
Neurological behavior assessment
The neurological behavior was assessed at one day after MCAO. The Zea Longa score [19] was determined according to the following scoring system: 0, no deficits; 1, unable to extend the right forelimb; 2, decreased grip of the right forelimb; 3, mild circling to the right; 4, spontaneous right circling; and 5, falling to the contralateral side. Rats with scores 1 to 4 formed the efficacy model and were used in the following experiments.
Morris water maze task
The effects of apigenin treatment on spatial learning and memory were assessed by the MWM task [20]. For the first five days, the rats were trained in the MWM with a platform, and the time required to find the hidden platform was recorded as the escape latency. On day 5 in the afternoon, the time spent in the platform quadrant was recorded. A stainless-steel pool with 150 cm in diameter and 50 cm in height containing a submerged escape platform (9 cm in diameter) under 2 cm of the water surface was used. The water temperature was kept at 23°C to 25°C. The tank was theoretically separated into four equal quadrants. The rats were allowed to swim freely for 60 seconds before the formal training. During the following 5 consecutive days, the rats were trained to find the platform four times per day. If the rats could arrive at the platform within the 90 seconds, they were required to remain on the platform for 10 seconds. If the rats could not find the platform, they were guided to the platform for 10 seconds and the time scores were recorded as 90 seconds. A single probe trial was conducted in the last trial on day 5 in the afternoon. The platform was removed and the rats were placed into the pool from the quadrant opposite the training quadrant. The time spent by the rats in the quadrant where the platform had been situated was recorded, in order to test their ability to memorize the location of the platform.
Immunohistochemistry and Nissl staining
Serial paraffin sections (5 μm) containing the hippocampal CA1 area were examined. The brain sections from one rat in each group were subjected to hematoxylin and eosin staining, and observed by light microscopy. Briefly, the hippocampus tissues were extracted and fixed in 4% paraformaldehyde, followed by gradient ethanol dehydration and xylene transparency, and then the tissues were embedded in paraffin and transferred into an embedding device. The paraffin-embedded tissue samples were sliced by a paraffin slicing machine. Next, sections were dewaxed to water, stained with hematoxylin for 10 minutes, flushed with water, rinsed with 1% hydrochloric acid alcohol for 10 minutes, and stained with eosin for 30 seconds for coloration, followed by ethanol dehydration, xylene transparency, and neutral gum sealing. Finally, the slides were mounted and analyzed under a bright-field microscope.
The hippocampus paraffin sections of one rat from each group were subjected to Nissl staining. Briefly, the rat brain sections were dewaxed to water, immersed for 5 minutes in Nissl staining solution, and washed with distilled water two times, followed by ethanol dehydration, xylene transparency, and neutral gum sealing. Finally, the slides were mounted and analyzed under a bright-field microscope.
Enzyme-linked immunosorbent assay (ELISA)
The hippocampal regions from the dissected brains were homogenized in 5 M guanidine/5 mM Tris buffer (pH 8.0), and then diluted with chilled 0.25% casein-blocking buffer containing 0.5 M guanidine and protease inhibitors. The total HDAC content were quantified using ELISA kits (Catalog number: F15657, Shanghai Xitang Biological Technology Co. Ltd., Shanghai, China) according to the manufacturer’s protocols. Data were calculated by the following formula: HDAC content=(ODsample–ODcontrol)×dilution ratio, where ODcontrol and ODsample are the optical density (OD) of the blank control and intervention groups, respectively.
Real-time polymerase chain reaction (RT-PCR) analysis of BDNF and Syn-I
Total RNA was isolated and purified using a TRIzol Plus RNA Purification Kit, according to the manufacturer’s instructions. RNA was reverse-transcribed into cDNA that was utilized as a template in subsequent qPCR amplifications using a SYBR Green I Kit. The qPCR primers and their products included: BDNF (forward: 5′-AGCCTCCTCTGCTCTTTCTGC-3′; reverse: 5′-CTCGCTAATACTGTCACACACGC-3′; product: 131 bp); Syn-I (forward: 5′-GACAACCAACATGACTTCCAGG-3′; reverse: 5′-TTCCAGTTCCCTGACACTGATG-3′; product: 161 bp). Each experiment was repeated at least three times.
Western blot analysis of BDNF, pCREB, acetylated H3, and acetylated H4
Hippocampal proteins were separated on sodium dodecyl sulfate polyacrylamide gels (pCREB: 12%; BDNF: 15%; acetylated H3 and acetylated H4: 15%), and then electrophoretically transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat dry milk for two hours, incubated with anti-BDNF (1: 500), anti-pCREB (1: 500), anti-acetylated H3, and anti-acetylated H4 (1: 500) antibodies at 4°C overnight, and then incubated with secondary antibodies (1: 3,000) for 1.5 hours. The bands were visualized by enhanced chemiluminescence. The relative quantities of the target proteins were analyzed using Quantity One software and normalized to the loading control. Each experiment was repeated at least three times.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM). The results were analyzed by SPSS 20.0 software. The mean values were derived from at least three independent experiments. Statistical comparisons between experimental groups were performed by one-way analysis of variance. The level of statistical significance was set at p<0.05.
Results
Histopathological changes in the hippocampus of MCAO rats
The hippocampal neurons in the sham group were in neat rows, close together, and of normal cell morphology and size (Figure 2A). The hippocampal neurons in the model group were arranged loosely, and exhibited cell body shrinkage, partial nuclear pyknosis, nuclear fragmentation, and nucleolar blurring and even disappearance (Figure 2B). The intervention groups (apigenin and NaB) showed alleviation of the hippocampal neuron damage after ischemia and reperfusion injury compared with the model group (Figure 2C–2E). Nissl staining showed that the Nissl bodies of the nerve cells were blue, and that the neurons were distributed around the nucleus in an orderly manner, had a complete and normal cellular structure, and clear nuclei in the sham group (Figure 3A). The Nissl bodies in the model group were released, the cyton of the neurons was shrunk, there was neuronal degeneration, and the neurons were decreased (Figure 3B). The neuronal changes in the intervention groups (apigenin and NaB butyrate) revealed alleviation of the hippocampal neuron damage after ischemia and reperfusion injury compared with the model group (Figure 3C–3E).
Figure 2.

Microscopic observation of nerve cells in the hippocampus of rats after MCAO (HE staining, 400×). (A) Hippocampal neurons in the sham group. The neurons in the hippocampus were in neat rows, close together, and of normal cell morphology and size. (B) Hippocampal neurons in the model group. The neurons in the hippocampus were arranged loosely, with cell body shrinkage, partial nuclear pyknosis, nuclear fragmentation, and nucleolar blurring and even disappearance. (C) Hippocampal neurons in the NaB group. (D) Hippocampal neurons in the 20 mg/kg apigenin group. (E) Hippocampal neurons in the 40 mg/kg apigenin group. As shown in panels (C–E), the hippocampal neuron damage was alleviated after ischemia and reperfusion injury in the three intervention groups.
Figure 3.

Microscopic observation of nerve cells in the hippocampus of rats after MCAO (Nissl staining, 400×). (A) Hippocampal neurons in the sham group. The Nissl staining showed that the Nissl bodies of nerve cells were blue, and that they were distributed around the nucleus in an orderly manner, with complete and normal cellular structure and clear nuclei. (B) Hippocampal neurons in the model group. The Nissl bodies in the model group were released, the cyton of the neurons was shrunk, there was neuronal degeneration, and the neurons were decreased. (C) Hippocampal neurons in the NaB group. (D) Hippocampal neurons in the 20 mg/kg apigenin group. (E) Hippocampal neurons in the 40 mg/kg apigenin group. As shown in panels (C–E), the hippocampal neuron damage was alleviated after ischemia and reperfusion injury in the three intervention groups.
Effect of apigenin on memory performance in the MWM task in MCAO rats
The results for the MWM task showed that the sham-operated rats could effectively learn how to reach the submerged platform (Figure 4A). The time spent finding the submerged platform by the rats in the sham group decreased significantly from days 23–27 postoperatively. On each observation day of the MWM task, the rats in the model and intervention (NaB and apigenin) groups required more time to find the submerged platform, compared to the rats of sham group, but no significant difference was observed on days 23–25 post-ischemia (p>0.05). On day 26 and 27 post-ischemia, there was a significant difference in the time spent searching for the submerged platform between the model rats and the sham rats (p<0.01). On day 26 and 27 post-ischemia, the model rats showed a trend toward a longer time to find the platform relative to the NaB-treated and 40 mg/kg apigenin-treated rats (p<0.05 and p<0.01, respectively). The 20 mg/kg apigenin-treated rats showed a trend toward a shorter time to find the platform compared with the model rats on day 27 post-ischemia (p<0.05), but not on day 26 post-ischemia (p>0.05). A significant between-group effect on the time spent in the correct quadrant was noted in the testing of spatial memory. In comparison with sham control rats, the model rats spent significantly less time in the target quadrant (p<0.01), thus indicating poorer learning ability after MCAO. The rats with chronic administration of NaB and apigenin spent more time in the target quadrant compared with the model rats (p<0.05), indicating improved consolidation of memory. Apigenin attenuated the memory acquisition deficit, but the attenuation effect was not dose-dependent. In addition, there was no significant difference in the time spent in the correct quadrant between the NaB-treated and apigenin-treated rats (p>0.05) (Figure 4B).
Figure 4.

Spatial learning and memory performance in rats after MCAO. (A) Effect of apigenin on memory performance (mean escape latency) in the MWM task by MCAO rats. Data are expressed as mean ± SEM and were analyzed by one-way ANOVA (n=14). ** p<0.01 vs. sham group, ## p<0.01 vs. model group, # p<0.05 vs. model group. (B) Effect of apigenin on time spent in the target quadrant by MCAO rats. Data are expressed as mean ±SEM and were analyzed by one-way ANOVA (n=14). ** p<0.01 vs. sham group, # p<0.05 vs. model group.
Effect of apigenin on HDAC content in MCAO rats
HDAC content was significantly increased after ischemic and reperfusion injury, compared with those of rats in sham group (p<0.01). Chronic administration of apigenin (40 mg/kg) and NaB decreased the HDAC content (p<0.05). There was no significant difference in the HDAC content between the rats treated with apigenin (40 mg/kg) and NaB. Apigenin at 20 mg/kg had no effect on the content of HDAC (Figure 5).
Figure 5.
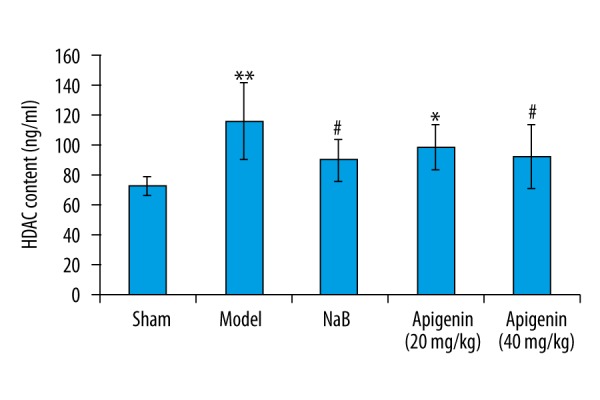
Effect of apigenin on hippocampal HDAC content in MCAO rats. The total HDAC content were quantified using ELISA kits, according to the manufacturer’s protocols. Data are expressed as mean ±SEM and were analyzed by one-way ANOVA (n=6). ** p<0.01 vs. sham group, * p<0.05 vs. sham group, # p<0.05 vs. model group.
Effect of apigenin on acetylated H3 and acetylated H4 expression in MCAO rats
The expressions of acetylated H3 and acetylated H4 were decreased after MCAO, compared with those of rats in sham group (p<0.01). Chronic administration of NaB and apigenin (40 mg/kg) restored the protein levels of acetylated H3 and acetylated H4, compared with those of rats in model group (p<0.01 and p<0.05, respectively). There was no significant difference in acetylated H3 and acetylated H4 expressions between the NaB-treated and apigenin (40 mg/kg)-treated groups (p>0.05). Apigenin at 20 mg/kg had no effect on acetylated H3 and acetylated H4 expressions, compared with those of rats in model group (p > 0.05) (Figure 6A, 6B).
Figure 6.

Effect of apigenin on the expressions of acetylated H3, acetylated H4, BDNF and pCREB in the hippocampus of rats after MCAO. (A) Western blot analysis of protein levels of acetylated H3 and acetylated H4, BDNF and pCREB in the hippocampus. (B) Effect of apigenin on the protein expressions of acetylated H3 and acetylated H4 in the hippocampus. Data are expressed as mean ±SEM and were analyzed by one-way ANOVA (n=3). ** p<0.01 vs. sham group, * p<0.05 vs. sham group, ## p<0.01 vs. model group, # p<0.05 vs. model group, @ p<0.05 vs. NaB group. (C) Effect of apigenin on BDNF mRNA and protein expressions in the hippocampus. Data are expressed as mean ±SEM and were analyzed by one-way ANOVA (n=3). ** p<0.01 vs. sham group, * p<0.05 vs. sham group, ## p<0.01 vs. model group, #p < 0.05 vs. model group, @ p<0.05 vs. NaB group, && p<0.01 vs. apigenin (20 mg/kg) group. (D) Effect of apigenin on pCREB protein expression in the hippocampus. Data are expressed as mean ±SEM and were analyzed by one-way ANOVA (n=3). ** p<0.01 vs. sham group, # p<0.05 vs. model group.
Effect of apigenin on brain BDNF levels in MCAO rats
The mRNA and protein expressions of BDNF were down-regulated after MCAO, compared with those of rats in sham group (p<0.01). Treatment with apigenin (20 and 40 mg/kg) and NaB significantly increased the mRNA level of BDNF (p<0.01 and p<0.05). The BDNF protein level was also up-regulated after treatment with apigenin and NaB (Figure 6A, 6C).
Effect of apigenin on pCREB levels in MCAO rats
The level of pCREB was distinctly declined after MCAO, compared with those of rats in sham group (p<0.01). Treatment with NaB obviously increased the levels of pCREB in the hippocampus, compared with those of rats in model group (p<0.05). However, apigenin showed no effect on pCREB expression in the hippocampus (p>0.05) (Figure 6A, 6D).
Effect of apigenin on Syn-I levels in MCAO rats
The level of Syn-I was obviously down-regulated after MCAO, compared with those of rats in sham group (p<0.01). Intervention with NaB and apigenin (40 mg/kg) obviously increased the level of Syn-I in the hippocampus, compared with those of rats in model group (p<0.05). There was no significant difference in Syn-I expression between the NaB-treated and 40 mg/kg apigenin-treated groups (p>0.05). Apigenin at 20 mg/kg had no effect on the expression of Syn-I (Figure 7).
Figure 7.
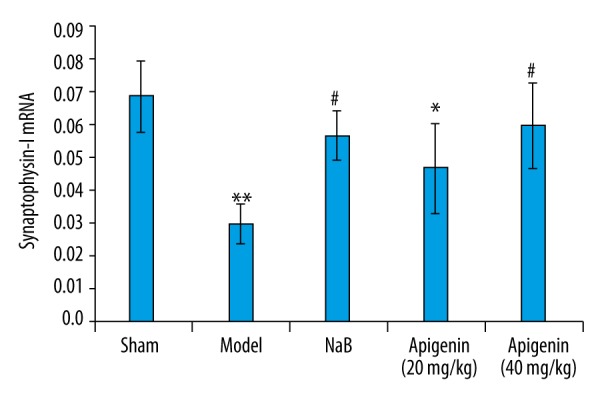
Effect of apigenin on Syn-I mRNA expression in the hippocampus of rats after MCAO. Data are expressed as mean ±SEM and were analyzed by one-way ANOVA (n=3). ** p<0.01 vs. sham group, * p<0.05 vs. sham group, # p<0.05 vs. model group.
Discussion
Cognitive deficits often develop in stroke victims and are closely related to their quality of life. Apigenin is a naturally-occurring medicine that shows a neuroprotective effect. Despite investigations on apigenin for its effect on promoting cognitive rehabilitation by interacting with Aβ peptides, no preexisting studies have reported its effects on cognitive function in the MCAO model. The present study is the first to comprehensively characterize the therapeutic efficacy of apigenin on cognition in ischemic stroke model of rats. Moreover, the work presented here seeks to continue exploration of apigenin as a therapeutic option as well as to identify the potential mechanisms by which it exerts its therapeutic effects on ischemia-induced cognitive deficits. The results of our study suggest that the underlying mechanisms by which apigenin improve cognition may involve modulation of histone acetylation, enhancement of neurotrophic activity, and promotion of synaptic protein expression in the ischemic hippocampus.
Brain ischemia and reperfusion injury caused significant learning and memory impairment as observed in the MWM task. In our study, we found that the apigenin-treated rats showed better performance in the MWM task than the model rats. The dose-response effect of apigenin administration was evaluated after MCAO in rats with the goal of determining the minimum exposure levels that can still provide functional improvements. The results showed that two doses of apigenin (20 and 40 mg/kg) showed improvement in cognition. Notably, no significant difference in the MWM task was observed between the apigenin-treated and NaB-treated rats, suggesting that the effect of apigenin on improving cognition was the same as the effect of NaB.
Recently, histone acetylation, which is regulated by HDACs, has been implicated in memory formation [21,22]. Furthermore, previous findings argued in favor of an important role for histone acetylation in memory consolidation, and more particularly during the reconsolidation of spatial memory in mice [23]. HDAC inhibitors, such as NaB, have been shown to exert strong neuroprotective effects against brain injury, and to significantly improve memory disorders. It was reported that NaB promoted both initial consolidation and reconsolidation of spatial memory in mice. NaB also promoted neurogenesis and behavioral improvement in an experimental ischemia model [24]. In the present study, NaB was used as a controlled intervention agent. Treatment with NaB and apigenin at 40 mg/kg substantially attenuated the ischemia-induced loss of histone acetylation. The up-regulation of histone acetylation could be related to the inhibition of HDAC following ischemia and reperfusion injury. Our results showed that brain ischemic injury caused a significant increase in hippocampal HDAC content and decreased the acetylated H3 and acetylated H4 levels in rats. Apigenin restrained the expression of HDAC and restored the acetylated H3 and acetylated H4 levels, suggesting a role in the modulation of histone acetylation.
Inhibition of HDAC activity can modulate hippocampus-dependent long-term memory in a CREB binding protein dependent manner [25]. The enhancement of hippocampus-dependent memory and hippocampal synaptic plasticity by HDAC inhibitors is mediated by the transcription factor pCREB. HDACs play a role in the regulation of CREB target gene transcription, indicating that CREB-dependent genes such as cFos and Zif268 may also be affected by NaB [26]. In this study, pCREB exhibited a remarkable reduction in MCAO rats and was improved by NaB. However, the present results showed that apigenin did not affect pCREB expression after MCAO. This finding might be related to the small size of the sample, and further studies are required to address this point.
It is well known that neurons require neurotrophic support for their development and survival. BDNF is one of the many effectors of CREB phosphorylation regulation and participates in learning and memory processes. Apigenin was shown to increase BDNF expression in Parkinson’s model of rats, accompanied by cognitive improvement [27]. In our study, we observed that treatment with apigenin and NaB increased BDNF levels in the ischemic rat brain. Interestingly, at the higher apigenin dose of 40 mg/kg, the BDNF protein level was markedly increased compared with the model group, while apigenin at 20 mg/kg had no such effect. These findings suggest that apigenin at the higher dose has neurotrophic potential and may be useful for cognition recovery after ischemic brain injury. However, the mRNA expression of BDNF was up-regulated by both doses of apigenin. Therefore, we speculate that the translation, post-translation processing, and modification of BDNF protein require the higher dose of apigenin.
Gene transcription is required for long-lasting forms of synaptic plasticity and memory storage. A previous report showed that spatial learning increased Syn-I mRNA and protein expressions [28], indicating that Syn-I is associated with learning and memory. Ischemia can cause damage to the expression of Syn-I [29]. A previous study found that hippocampal administration of NaB enhanced the levels of gene expression of synaptic plasticity markers [11]. Our results showed that NaB up-regulated Syn-I expression. Furthermore, the present study showed that apigenin at the higher dose increased Syn-I expression, which may be related to cognition.
Conclusions
In the present study, apigenin at 40 mg/kg was shown to promote histone acetylation, increase BDNF and Syn-I expressions, and obviously improve the cognitive deficits, being the same as the functions of NaB in ischemia and reperfusion brain-injured rats. Overall, regarding biochemical markers, the lower dose of apigenin did not show reversal of the neurochemical alterations. However, apigenin at the two doses showed the same effects on behavioral activity, which could be related to the additive effect on the neuroprotective (antioxidant, antineuroinflammatory) potential.
Our results can partly explain the effects of apigenin as a protective molecule in alleviating cognitive deficits and its underlying mechanisms in MCAO rats. However, further exploration of these potential mechanisms is required.
Footnotes
Conflict of interest
The author confirms that this article content has no conflict of interest.
Source of support: This work was supported by the Medical research project of Zhejiang Province (2016KYB201), Wenzhou Municipal Science and Technology Bureau Project funding (Y20150017), Natural Science Foundation of Zhejiang Province Project (No. Y12H170002), Research fund for public welfare industry of Ministry of Health of the People’s Republic of China (201302002) and Key Construction Disciplines (Children’s Rehabilitation) in Second Affiliated Hospital of Wenzhou Medical University
References
- 1.Cumming TB, Marshall RS, Lazar RM. Stroke, cognitive deficits, and rehabilitation: Still an incomplete picture. Int J Stroke. 2013;8(1):38–45. doi: 10.1111/j.1747-4949.2012.00972.x. [DOI] [PubMed] [Google Scholar]
- 2.Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31(12):605–17. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Peixoto L, Abel T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology. 2013;38(1):62–76. doi: 10.1038/npp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan JS, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55(6):942–57. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maddox SA, Schafe GE. Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn Mem. 2011;18(9):579–93. doi: 10.1101/lm.2243411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma SK. Protein acetylation in synaptic plasticity and memory. Neurosci Biobehav Rev. 2010;34(8):1234–40. doi: 10.1016/j.neubiorev.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZH, Wu LN, Song JG, Li WQ. Correlations between cognitive impairment and brain?derived neurotrophic factor expression in the hippocampus of post-stroke depression rats. Mol Med Rep. 2012;6(4):889–93. doi: 10.3892/mmr.2012.1009. [DOI] [PubMed] [Google Scholar]
- 9.Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci. 2007;27(46):12732–42. doi: 10.1523/JNEUROSCI.2522-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen DY, Bambah-Mukku D, Pollonini G, et al. Glucocorticoid receptors recruit the CaMKIIalpha-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci. 2012;75:1707–14. doi: 10.1038/nn.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricobaraza A, Cuadrado-Tejedor M, Pérez-Mediavilla A, et al. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology. 2009;34(7):1721–32. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 12.Bhullar KS, Rupasinghe HP. Polyphenols: Multipotent therapeutic agents in neurodegenerative diseases. Oxid Med Cell Longev. 2013;2013:891748. doi: 10.1155/2013/891748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balez R, Steiner N, Engel M, et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci Rep. 2016;6:31450. doi: 10.1038/srep31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han JY, Ahn SY, Kim CS, et al. Protection of apigenin against kainate-induced excitotoxicity by anti-oxidative effects. Biol Pharm Bull. 2012;35(9):1440–46. doi: 10.1248/bpb.b110686. [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Kong S, Chen W, et al. Apigenin mediated protection of OGD-evoked neuron-like injury in differentiated PC12 cells. Neurochem Res. 2014;39(11):2197–210. doi: 10.1007/s11064-014-1421-0. [DOI] [PubMed] [Google Scholar]
- 16.Yamagata K, Kitazawa T, Shinoda M, et al. Stroke status evoked adhesion molecule genetic alteration in astrocytes isolated from stroke-prone spontaneously hypertensive rats and the apigenin inhibition of their expression. Stroke Res Treat. 2010;2010:386389. doi: 10.4061/2010/386389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taupin P. Apigenin and related compounds stimulate adult neurogenesis. Mars, Inc., the Salk Institute for Biological Studies: WO2008147483. Expert Opin Ther Pat. 2009;19(4):523–27. doi: 10.1517/13543770902721279. [DOI] [PubMed] [Google Scholar]
- 18.Zhao L, Wang JL, Liu R, et al. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules. 2013;18(8):9949–65. doi: 10.3390/molecules18089949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 20.Foster TC, Kumar A. Susceptibility to induction of long-term depression is associated with impaired memory in aged Fischer 344 rats. Neurobiol Learn Mem. 2007;87(4):522–35. doi: 10.1016/j.nlm.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris MJ, Mahgoub M, Na ES, et al. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J Neurosci. 2013;33(15):6401–11. doi: 10.1523/JNEUROSCI.1001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fass DM, Reis SA, Ghosh B, et al. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology. 2013;64:81–96. doi: 10.1016/j.neuropharm.2012.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villain H, Florian C, Roullet P. HDAC inhibition promotes both initial consolidation and reconsolidation of spatial memory in mice. Sci Rep. 2016;6:27015. doi: 10.1038/srep27015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HJ, Chuang DM. HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am J Transl Res. 2014;6(3):206–23. [PMC free article] [PubMed] [Google Scholar]
- 25.Haettig J, Stefanko DP, Multani ML, et al. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn Mem. 2011;18(2):71–79. doi: 10.1101/lm.1986911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fass DM, Butler JE, Goodman RH. Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem. 2003;278(44):43014–19. doi: 10.1074/jbc.M305905200. [DOI] [PubMed] [Google Scholar]
- 27.Patil SP, Jain PD, Sancheti JS, et al. Neuroprotective and neurotrophic effects of Apigenin and Luteolin in MPTP induced parkinsonism in mice. Neuropharmacology. 2014;86:192–202. doi: 10.1016/j.neuropharm.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Gómez-Pinilla F, So V, Kesslak JP. Spatial learning induces neurotrophin receptor and synapsin I in the hippocampus. Brain Res. 2001;904(1):13–19. doi: 10.1016/s0006-8993(01)02394-0. [DOI] [PubMed] [Google Scholar]
- 29.Pagnussat AS, Simao F, Anastacio JR, et al. Effects of skilled and unskilled training on functional recovery and brain plasticity after focal ischemia in adult rats. Brain Res. 2012;1486:53–61. doi: 10.1016/j.brainres.2012.09.019. [DOI] [PubMed] [Google Scholar]