

Abstract
The utility and reliability of assessing molecular biomarkers for translational applications on pre-operative core biopsy specimens assume consistency of molecular profiles with larger surgical specimens. Whether DNA methylation in ductal carcinoma in situ (DCIS), measured in core biopsy and surgical specimens are similar, remains unclear. Here, we compared genome-scale DNA methylation measured in matched core biopsy and surgical specimens from DCIS, including specific DNA methylation biomarkers of subsequent invasive cancer. DNA was extracted from guided 2mm cores of formalin fixed paraffin embedded (FFPE) specimens, bisulfite-modified, and measured on the Illumina HumanMethylation450 BeadChip. DNA methylation profiles of core biopsies exhibited high concordance with matched surgical specimens. Within-subject variability in DNA methylation was significantly lower than between-subject variability (all P < 2.20E-16). In 641 CpGs whose methylation was related with increased hazard of invasive breast cancer, lower within-subject than between-subject variability was observed in 92.3% of the study participants (P < 0.05). Between patient-matched core biopsy and surgical specimens, < 0.6% of CpGs measured had changes in median DNA methylation > 15%, and a pathway analysis of these CpGs indicated enrichment for genes related with wound healing. Our results indicate that DNA methylation measured in core biopsies are representative of the matched surgical specimens and suggest that DCIS biomarkers measured in core biopsies can inform clinical decision-making.
Keywords: DNA methylation, Illumina 450k, DCIS core biopsies, Epigenetic biomarkers
1. Introduction
Ductal carcinoma in situ (DCIS) is a non-invasive epithelial lesion associated with increased risk of developing invasive breast cancer 1,2. Standard treatment for DCIS is surgery—local resection or mastectomy—often in combination with radiation 2,3. However, since many patients with a DCIS diagnosis do not develop invasive breast cancer, there is increasing support that surgery may be an overtreatment for some forms of DCIS 4,5.
Molecular profiling, including analysis of DNA methylation profiles, presents new opportunities to identify prognostic biomarkers for DCIS, potentially leading to more informed treatment decisions. Methylation of carbon position 5 of DNA cytosine in the cytosine-phosphate-guanine (CpG) dinucleotide context serves as an epigenetic regulator of gene expression. Promoter methylation is related with repression of gene transcription, while gene-body methylation is often associated with increased gene expression 6,7. Perturbation of DNA methylation is common in breast cancer 8–10. Previously, we characterized the landscape of DNA methylation alterations in DCIS lesions and identified DNA methylation biomarkers related with risk of developing invasive breast cancer 9,10.
Prior studies have shown that breast pathologic features, discrete biomarkers, and certain molecular characteristics are consistent across different tissue specimen types 11–15. However, it remains unclear whether DNA methylation profiles are also consistent between patient-matched DCIS specimens. For example, the biopsy procedure may alter methylation profiles in the subsequent surgical specimen. If the discrepancy between the patient-matched specimens is substantial, it would have implications for the utility of methylation biomarkers in core biopsies that were initially discovered and validated in surgical specimens. Conversely, if methylation levels were consistent between the two specimen types, the use of diagnostic core biopsies would be acceptable for measuring DNA methylation biomarkers to inform pre-surgical clinical decision-making in patients with DCIS.
In this study, we addressed whether DNA methylation profiles between matched core biopsy and surgical excision specimens from patients with DCIS are comparable. We examined DNA methylation profiles in matched specimen pairs from 13 subjects enrolled in the New Hampshire Mammography Network (NHMN) and treated at Dartmouth-Hitchcock Medical Center 10,16.
2. Materials and Methods
2.1 Study population and characteristics
Patients diagnosed with ductal carcinoma in situ (DCIS) identified through the New Hampshire Mammography Network (NHMN), a registry aiming for improved management of breast disease, under the approval of the Dartmouth Committee for Protection of Human Subjects 16. Hematoxylin and eosin (H&E) stained slides were centrally reviewed by a breast pathologist (JDM) who confirmed the diagnosis of DCIS, recorded pathologic features, and selected tissue blocks for analysis.
2.2 DNA extraction, bisulfite modification, and methylation measures
The procedure for DNA extraction and methylation analysis has been described previously 9,10. In brief, 2mm cores were obtained from formalin fixed paraffin embedded (FFPE) tissue blocks. DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) following a lysis step with the TissueLyserII (Qiagen, Valencia, CA). Isolated and purified DNA samples were subjected to bisulfite modification using the EZ DNA methylation kit (Zymo Research, Irvine, CA), treated with Illumina Infinium FFPE restoration solution kit according to the manufacturer’s guidelines. FFPE-restored DNA samples were analyzed on the Illumina Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA), which determines the proportion of methylated alleles (beta-values) at each CpG site 10. Methylation intensity data files were processed by the minfi Bioconductor analysis pipeline (version 1.20.0) 17. Probes failing to meet the detection P-value of 0.05 in greater than 20% of samples were excluded. Samples from two subjects, classified as poor-performing outliers, were also excluded. Quality control and filtering left 478,462 of 485,577 array CpGs with high-quality DNA methylation profiles across 13 pairs of matched specimens.
2.3 Statistical methods
All statistical analyses were performed in R version 3.3.2. To visualize DNA methylation in patient-matched core biopsy and surgical specimens, 1,000, 2,500, and 5,000 most variable CpG sites were selected and subjected to unsupervised hierarchical clustering using the Manhattan distance and average linkage metric. Genomic annotations were downloaded from the University of California Santa Cruz (UCSC) hg19 database. Median DNA methylation beta-values (i.e., proportion of methylated alleles) were determined separately for core biopsy and surgical excision specimens. Methylation along genomic regions that are 2,000 base pairs upstream and downstream of transcription start sites (TSS) was visualized using the Genomation R/Bioconductor package (version 1.6.0) 18.
Within-subject variability was represented by the distribution of Δbeta matched, defined as beta core biopsy – beta matched surgical. Between-subject variability was represented by the distribution of Δbeta permuted, defined as beta core biopsy – beta permuted surgical, where beta permuted surgical is computed by 52 iterations of random sampling with replacement from 12 unmatched surgical specimens. A two-sided Kolmogorov-Simirnov (KS) test was performed to test the difference between Δbeta matched and Δbeta permuted distributions. The significance threshold was set at P = 0.05.
To test whether wound healing and inflammation processes were enriched in surgical specimens relative to patient-matched core biopsies, a Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis19 was performed on genes with | Δbeta matched | > 15%. Unique genes to which the CpGs are mapped were identified by querying the Illumina HumanMethylation450 annotation file. For CpG sites with multiple annotated genes, all such genes were included in the subsequent KEGG pathway analysis.
The relation of DNA methylation with gene expression for protocadherin-family genes was determined by leveraging The Cancer Genome Atlas (TCGA) data 21. TCGA Level 1 Illumina HumanMethylation450 data and Level 3 RNASeqV2 normalized counts were downloaded from the Genomic Data Commons (gdc.cancer.gov). The Level 1 methylation data set was pre-processed in the same manner as the DCIS methylation data set described above. The Level 3 RNASeqV2 data set was analyzed without further processing. TCGA breast invasive carcinoma subjects (n=51) were selected based on the following criteria: 1) female, 2) Caucasian, 3) tumor stage I/Ia/Ib, 4) estrogen receptor (ER) positive, 5) no evidence of metastasis. A linear regression was performed on each of the three CpGs, annotated as gene-body associated sites and shared across all 11 identified protocadherin gene transcripts, against the mRNA expression of each protocadherin transcript.
2.4 Data availability
DCIS surgical specimens are available under Gene Expression Omnibus (GEO) accession GSE6631310. Patient-matched core biopsy specimens are deposited under GEO accession GSE100503. Sample mapping information is available in Supplementary Table 2.
3. Results
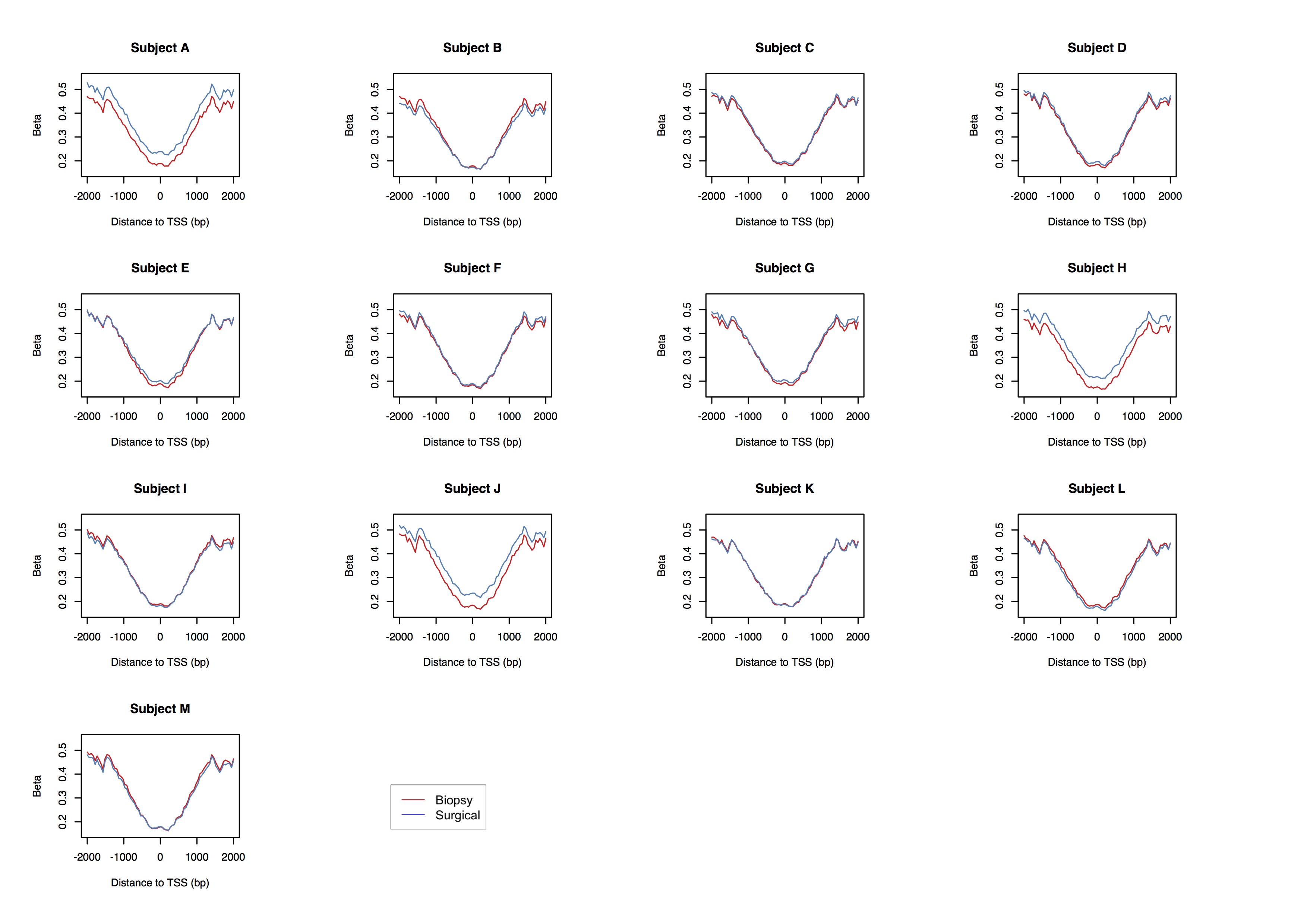
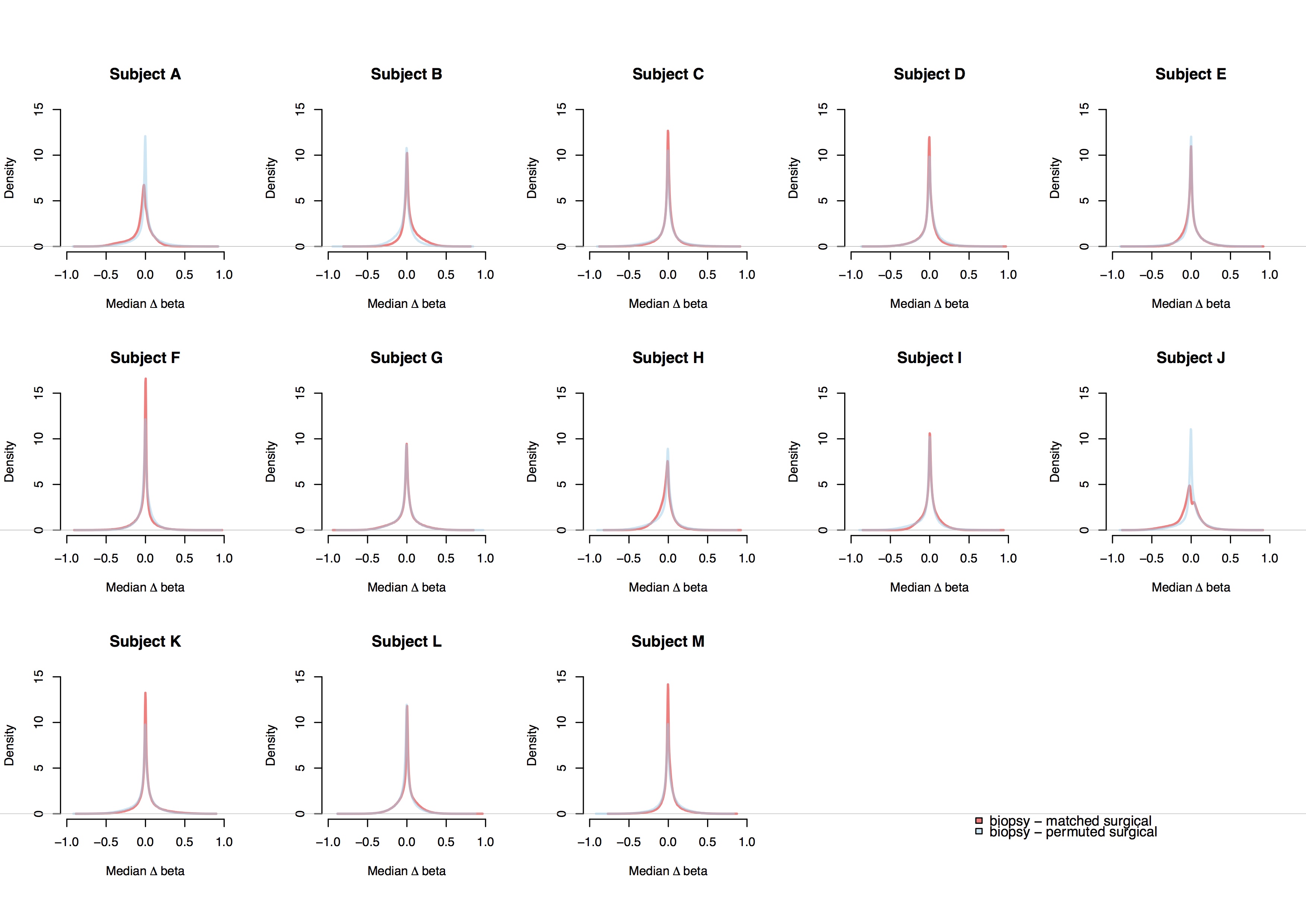
The histopathologic characteristics of the DCIS were similar between core biopsy and surgical specimens (Table 1). Representative images of hematoxylin and eosin (H&E) slides demonstrating DCIS are shown in Figure 1. To determine whether DNA methylation between patient-matched core biopsy and surgical specimens was consistent, we first applied unsupervised hierarchical clustering to 1,000 CpGs with the greatest sample heterogeneity. The resulting heat map in Figure 1C shows that each pair of specimens from the same subject cluster together. As the number of most variable CpGs was increased, cluster structure remained stable (Supplementary Figure 1A–B). Overall distributions of methylation beta-values, which represent the proportion of methylated alleles, were consistent between patient-matched specimens, as indicated by the overlapping density curves (Supplementary Figure 2). DNA methylation profiles between patient-matched specimens over a four-kilobase (kb) window, centered at canonical transcription start sites (TSS), exhibited overlaps (Figure 1D). As expected, methylated cytosines were depleted at, or nearby, TSS (Figure 1D; Supplementary Figure 1C). This pattern was also consistent at the level of individual DCIS subjects (Supplementary Figure 3). To interrogate DNA methylation profiles between patient-matched specimens, we tested the differences between within and between-subject variability in DNA methylation. Within-subject variability was substantially lower than between-subject variability across all 478,462 array-measured CpGs (all KS-test P < 2.20E-16; Supplementary Figure 4).
Table 1.
Study population and clinical characteristics.
| Biopsy n (%) | Surgical n (%) | |
|---|---|---|
| Age, mean ± s.d. | 56.77 ± 10.94 | 56.77 ± 10.94 |
| Family history | ||
| Absent | 8 (61.5) | 8 (61.5) |
| Present | 5 (38.5) | 5 (38.5) |
| Grade | ||
| High | 4 (30.8) | 4 (30.8) |
| Intermediate | 9 (69.2) | 9 (69.2) |
| Architectural pattern | ||
| Comedo | 0 (0.0) | 1 (7.7) |
| Cribriform | 4 (30.8) | 4 (30.8) |
| Solid | 9 (69.2) | 8 (61.5) |
| Necrosis | ||
| Absent | 2 (15.4) | 1 (7.7) |
| Present | 11 (84.6) | 12 (92.3) |
| Periductal sclerosis | ||
| Absent | 6 (46.2) | 8 (61.5) |
| Present | 7 (53.8) | 5 (38.5) |
| Periductal inflammation | ||
| Absent | 7 (53.8) | 10 (76.9) |
| Present | 6 (46.2) | 3 (23.1) |
| Calcifications | ||
| Absent | 2 (15.4) | 4 (30.8) |
| Present | 11 (84.6) | 9 (69.2) |
Figure 1. DCIS histopathology and DNA methylation patterning in patient-matched core biopsy and surgical specimens.

(A) Low-grade DCIS with cribriform and micropapillary patterns. (B) High-grade DCIS with periductal fibrosis and chronic inflammation. (C) Unsupervised hierarchical clustering of 1,000 most variable DNA methylation loci. In the heat map, rows represent methylation beta-values (i.e., proportion of methylated alleles), and columns represent specimens. Horizontal tracking bars denote specimen types and clinical covariates presented in Table 1. (D) Median methylation beta-values plotted relative to canonical transcription start sites (TSS).
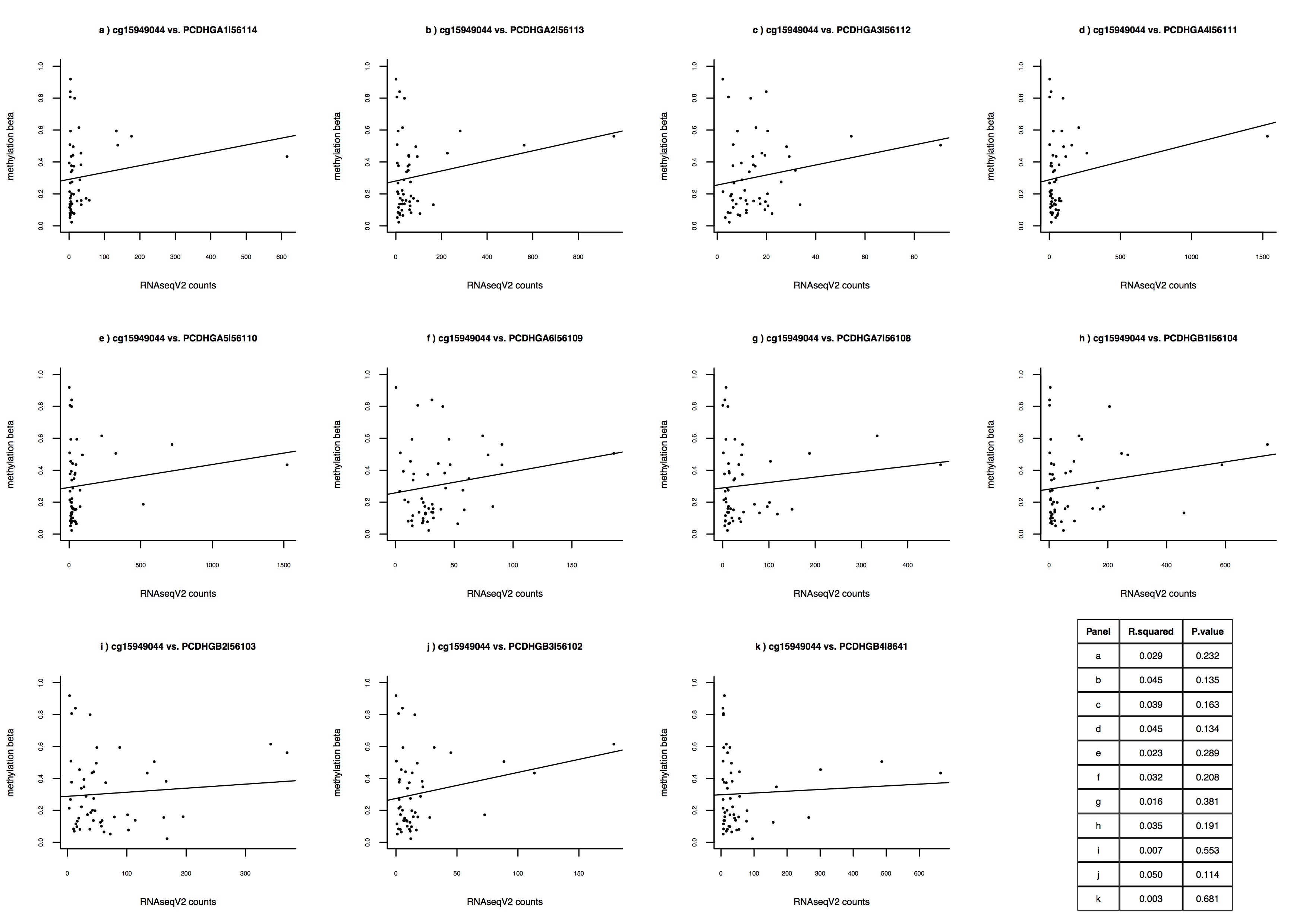
Among the matched pairs, > 99.4% of CpGs had a median methylation difference within 0.15 on the beta-value scale (i.e., proportion of methylated alleles), suggesting consistency between patient-matched specimens (Figure 2A). Despite the broad consistency of methylation in patient-matched specimens, some differential methylation was observed. We defined Δbeta matched as methylation beta-values in surgical specimens subtracted from the matched core biopsies, and focused on CpGs with methylation differences > 15% between matched specimens. We identified 2,170 CpG sites (0.45% of all CpGs measured) associated with 1,184 genes that had higher methylation in surgical specimens, and 372 CpGs (0.07% of all CpGs measured) associated with 270 genes that had higher methylation in core biopsies (Supplementary Table 1). Among the top 25 genes with the highest number of CpGs exhibiting altered methylation in matched specimens, 11 were protocadherin-family genes, which have increased gene expression during wound healing 20. The surgical specimens had increased protocadherin gene-body methylation, consistent with the relation of gene-body methylation with increased gene expression (Supplementary Table 1) 6,7. To further investigate the relation of protocadherin gene-body methylation with gene expression, we analyzed estrogen receptor (ER) positive early-stage breast cancers from The Cancer Genome Atlas (TCGA) 21, and also observed that gene-body methylation of protocadherin genes was related with increased gene expression (Supplementary Figure 5). Further, a Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of genes with increased methylation in surgical specimens identified genes related with cellular adhesion, intracellular signaling, and extracellular matrix (ECM) synthesis pathways (all adjusted P < 0.05; Table 2), whereas genes with increased methylation in core biopsies did not identify significant KEGG pathways.
Figure 2. Ranking of variability in DNA methylation between patient-matched core biopsy and surgical specimens.

Ranking of Δbeta matched (= beta core biopsy – beta matched surgical), the difference in methylation between patient-matched specimens, across (A) all array-measured CpG sites and (B) the 641 biomarker CpGs related with increased risk of invasive breast cancer. Red points above and below the red dotted lines represent CpGs exceeding methylation difference threshold of 15%. Beta-value, proportion of methylated alleles in a sample.
Table 2. Pathways enriched in DCIS surgical excision specimens (FDR-adjusted P < 0.05).
Genes associated with CpG sites with a Δbeta matched (= beta core biopsy – beta matched surgical) less than 0.15 were analyzed by the Kyoto Encyclopedia of Genes and Genomes (KEGG). Top pathways such as no.53, 4514, and 4512 are well known for their roles in wound healing and tissue repair processes. FDR, false discovery rate.
| Pathway ID | Pathway name | Proportion of genes | Raw P-value | FDR-adjusted P-value |
|---|---|---|---|---|
| 53 | Ascorbate and aldarate metabolism | 0.33 | 4.99E-07 | 0.0002 |
| 4940 | Type I diabetes mellitus | 0.23 | 8.29E-06 | 0.0013 |
| 4514 | Cell adhesion molecules (CAMs) | 0.13 | 4.60E-05 | 0.0048 |
| 40 | Pentose and glucuronate interconversions | 0.21 | 2.75E-04 | 0.0095 |
| 860 | Porphyrin and chlorophyll metabolism | 0.19 | 2.44E-04 | 0.0095 |
| 4512 | ECM-receptor interaction | 0.15 | 2.66E-04 | 0.0095 |
| 4724 | Glutamatergic synapse | 0.13 | 2.24E-04 | 0.0095 |
| 5310 | Asthma | 0.23 | 1.37E-04 | 0.0095 |
| 5416 | Viral myocarditis | 0.17 | 1.87E-04 | 0.0095 |
| 983 | Drug metabolism - other enzymes | 0.17 | 4.99E-04 | 0.0155 |
| 4970 | Salivary secretion | 0.13 | 6.67E-04 | 0.0173 |
| 5330 | Allograft rejection | 0.18 | 6.14E-04 | 0.0173 |
| 4080 | Neuroactive ligand-receptor interaction | 0.09 | 7.70E-04 | 0.0184 |
| 4020 | Calcium signaling pathway | 0.10 | 9.45E-04 | 0.0210 |
| 5332 | Graft-versus-host disease | 0.17 | 1.04E-03 | 0.0216 |
| 4713 | Circadian entrainment | 0.12 | 1.23E-03 | 0.0239 |
| 5320 | Autoimmune thyroid disease | 0.15 | 1.44E-03 | 0.0250 |
| 4726 | Serotonergic synapse | 0.12 | 1.90E-03 | 0.0310 |
| 5150 | Staphylococcus aureus infection | 0.14 | 2.15E-03 | 0.0334 |
| 5032 | Morphine addiction | 0.12 | 2.37E-03 | 0.0336 |
| 5200 | Pathways in cancer | 0.08 | 2.37E-03 | 0.0336 |
| 4015 | Rap1 signaling pathway | 0.09 | 2.52E-03 | 0.0340 |
| 140 | Steroid hormone biosynthesis | 0.14 | 2.75E-03 | 0.0343 |
| 4014 | Ras signaling pathway | 0.09 | 2.87E-03 | 0.0343 |
| 4072 | Phospholipase D signaling pathway | 0.19 | 2.79E-03 | 0.0343 |
| 4672 | Intestinal immune network for IgA production | 0.14 | 3.40E-03 | 0.0373 |
| 4730 | Long-term depression | 0.13 | 3.48E-03 | 0.0373 |
| 5033 | Nicotine addiction | 0.15 | 4.09E-03 | 0.0424 |
| 4750 | Inflammatory mediator regulation of TRP channels | 0.11 | 4.75E-03 | 0.0476 |
We recently identified CpGs with altered DNA methylation in DCIS that are related with an increased hazard of invasive breast cancer diagnosis 10. Among the 641 biomarker CpGs that we previously identified as significantly associated with future onset of invasive breast cancer, 631 (> 98%) had consistent methylation between core biopsies and matched surgical specimens (|Δbeta matched| < 0.15; Figure 2B). To evaluate whether DNA methylation measured in core biopsies was representative of that in surgical specimens, we restricted our variability comparisons to the 641 biomarker CpGs. In 12 of 13 (92.3%) subjects, within-subject variability in methylation biomarkers was substantially lower than between-subject variability (KS-test P < 0.05; Figure 3).
Figure 3. Comparison of within and between-subject variability in methylation loci related with invasive breast cancer risk.

Within and between-subject variability in methylation were represented by Δbeta matched (= beta core biopsy – beta matched surgical) and Δbeta permuted (= beta core biopsy – beta permuted surgical), respectively. The difference between Δbeta matched and Δbeta permuted was determined by Kolgomorov-Simirnov (KS) test. The table inset summarizes median and mean KS test P-values for each patient. Beta-value, proportion of methylated alleles in a sample.
4. Discussion
Prior studies have demonstrated that pathologic features, discrete cellular markers, and certain molecular profiles are similar between breast core biopsy and surgical excision specimens 11–15. We extend these observations to include genome-scale DNA methylation profiles as well as specific DNA methylation biomarkers in breast specimens containing ductal carcinoma in situ (DCIS). First, methylation loci that contribute most to sample heterogeneity cluster as patient-matched pairs, suggesting core biopsy and surgical specimens from the same patients are most similar. Second, > 99.4% CpG sites in the genome show consistent methylation in patient-matched specimens. The overlap of DNA methylation distributions between patient-matched specimens was particularly evident along a four-kilobase (kb) window centered at canonical transcription start sites (TSS).
As mentioned above, we previously identified 641 biomarker CpGs associated with DCIS progression to invasive breast cancer 10. Here, we have demonstrated that, in addition to high concordance in the overall methylation profiles, 98% of these biomarker CpG sites are also consistent between patient-matched core biopsy and surgical specimens. This finding lends support to using pre-operative core biopsies for assessing risks of invasive cancer diagnosis through DNA methylation.
A limited number (< 0.6%) of differentially methylated CpG sites between patient-matched specimens. This minor discrepancy could be explained by the biopsy procedure, which introduces a perturbation to the breast tissue microenvironment. We therefore hypothesized that the observed DNA methylation changes occurred in response to the actual biopsy procedure and may upregulate genes associated with wound healing. Our KEGG pathway analysis supported this hypothesis. We observed genes and molecular processes commonly involved in wound healing, including cellular adhesion, extracellular matrix (ECM) remodeling, and inter-cellular communication. Notably, the KEGG pathway with the highest significance—ascorbate metabolism—has long been recognized for its role in wound healing 22. A recent transcriptome-scale study demonstrated that ascorbic acid (Vitamin C) treatment promoted expression of ECM remodeling genes such as integrin alpha 3 (ITGA3). Importantly, ascorbic acid treatment led to increased adhesion properties of fibroblasts, evidence that is in line with our pathway analysis 23.
Prior to the present study, the extent to which a biopsy procedure would affect DCIS DNA methylation measures in a subsequent surgical specimen was unclear. Across both the entire genome and a subset of loci with biomarker potential, we demonstrated that DNA methylation was concordant in patient-matched core biopsy and surgical excision specimens. These findings thus support the potential translational applicability of DNA methylation measures, particularly the known biomarkers, in DCIS core biopsy specimens.
Supplementary Material
Similarity between DCIS core biopsies and surgical specimens in DNA methylation. Unsupervised hierarchical clustering of (A) 2,500 and (B) 5,000 most variable methylation loci. Horizontal tracking bars indicate clinical covariates shown in Table 1. (C) Median methylation beta-values in core biopsies (left) and surgical specimens (right) along a four-kilobase (kb) window centered at canonical TSS. TSS, transcription start site
{kind=link}
Comparison of methylation beta-value distributions in patient-matched core biopsies and surgical specimens. Each sub-plot represents methylation profiles of patient-matched specimen pairs. Curves represent distributions of methylation beta-values. Beta-value, proportion of methylated alleles in a sample.
{kind=link}
DNA methylation by genomic context in individual subjects. Each sub-plot displays distributions of methylation beta-values along a 4-kb window centered at canonical transcription start sites (TSS) in patient-matched specimen pairs. kb, kilobase; beta-value, proportion of methylated alleles in a sample.
{kind=link}
Comparison of within and between-subject variability in all array-measured methylation loci. The Δbeta matched (beta core biopsy – beta matched surgical) and Δbeta permuted (beta core biopsy – beta permuted surgical) distributions represent within and between-subject variability, respectively. For each DCIS patient, Δbeta matched and Δbeta permuted distributions were compared by Kolgomorov-Simirnov (KS) tests with 52 iterations. All KS test P-values < 2.20E-16. Beta-value, proportion of methylated alleles in a sample.
{kind=link}
Relation of methylation of protocadherin gene-body CpGs (A) cg26890354, (B) cg15949044, and (C) cg23445461 with expression of 11 protocadherin mRNAs in Stage I/Ia/Ib, estrogen receptor (ER)-positive patients from The Cancer Genome Atlas (TCGA). Every subpanel (labeled “a” through “k”) represents linear regression of DNA methylation (y-axis) and a given protocadherin mRNA transcript (x-axis). All regression coefficients were positive, indicating a positive association between protocadherin methylation and gene expression. Table insets show the proportion of variance explained (R.squared) and the associated P-value (P.value).
{kind=link}
{kind=link}
{kind=link}
Highlights.
Genome-scale DNA methylation profiles of breast DCIS core biopsies are representative of patient-matched surgical excision specimens.
A small number of loci with differential methylation were related with wound healing.
Our results support translational applications of DNA methylation biomarkers in DCIS core biopsies that can inform clinical decision-making.
Acknowledgments
This work was supported by the U.S. National Institute of Health (P20GM104416, R01DE022772, and 1P01CA154292), the New Hampshire Mammography Registry (U01CA086082), and the Burroughs-Wellcome Fund.
Abbreviations
- CpG
cytosine-phosphate-guanine
- DCIS
ductal carcinoma in situ
- ECM
extracellular matrix
- ER
estrogen receptor
- FFPE
formalin fixed paraffin embedded
- H&E
hematoxylin and eosin
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KS
Kolmogorov-Simirnov (test)
- NHMN
New Hampshire Mammography Network
- TCGA
The Cancer Genome Atlas
- TSS
transcription start site
Footnotes
Disclosures
The authors have no potential conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Cancer Society. Breast Cancer Facts and Figures 2015–16. Atlanta: American Cancer Society, Inc; 2015. [Google Scholar]
- 2.Pinder SE. Ductal carcinoma in situ (DCIS): pathological features, differential diagnosis, prognostic factors and specimen evaluation. Mod Pathol. 2010;23:S8–S13. doi: 10.1038/modpathol.2010.40. [DOI] [PubMed] [Google Scholar]
- 3.Lester SC, Bose S, Chen YY, Connolly JL, de Baca ME, Fitzgibbons PL, Hayes DF, Kleer C, O’Malley FP, Page DL, Smith BL, Weaver DL, Winer E. Protocol for the examination of specimens from patients with ductal carcinoma in situ of the breast. Arch Pathol Lab Med. 2009;133:15–25. doi: 10.5858/133.1.15. [DOI] [PubMed] [Google Scholar]
- 4.Morrow M, Katz SJ. Addressing overtreatment in DCIS: what should physicians do now? J Natl Cancer Inst. 2015;107(12):djv290. doi: 10.1093/jnci/djv290. [DOI] [PubMed] [Google Scholar]
- 5.Park TS, Hwang ES. Current trends in the management of ductal carcinoma in situ. Oncology. 2016;30(9):823–831. [PubMed] [Google Scholar]
- 6.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 7.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pang J, Dobrovic A, Fox SB. DNA methylation in ductal carcinoma in situ of the breast. Breast Cancer Res. 2013;15:206. doi: 10.1186/bcr3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleischer T, Frigessi A, Johnson KC, Edvardsen H, Touleimat N, Klajic J, Riis ML, Haakensen VD, Wärnberg F, Naume B, Helland Å, Børresen-Dale A, Tost J, Christensen BC, Kristensen VN. Genome wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. doi: 10.1186/s13059-014-0435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson KC, Koestler DC, Fleischer T, Chen P, Jensen EG, Marotti JD, Onega T, Kristensen VN, Christensen BC. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. J Clin Epigenet. 2015;7:75. doi: 10.1186/s13148-015-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenner RJ, Bassett LW, Fajardo LL, Dershaw DD, Evans WP, III, Hunt R, Lee C, Tocino I, Fisher P, McCombs M, Jackson VP. Stereotactic Core-Needle Breast Biopsy: A Multi-institutional Prospective Trial 1. Radiol. 2001;218(3):866–872. doi: 10.1148/radiology.218.3.r01mr44866. [DOI] [PubMed] [Google Scholar]
- 12.Ricci MD, Calvano Filho CMC, de Oliveira Filho HR, Filassi JR, Pinotti JA, Baracat EC. Analysis of the concordance rates between core needle biopsy and surgical excision in patients with breast cancer. Rev Assoc Med Bras. 2012;58(5):523–536. [PubMed] [Google Scholar]
- 13.Burge CN, Chang HR, Apple SK. Do the histologic features and results of breast cancer biomarker studies differ between core biopsy and surgical excision specimens? The Breast. 2006;15:167–172. doi: 10.1016/j.breast.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Sikora MJ, Thibert JN, Salter J, Dowsett M, Johnson MD, Rae JM. High-efficiency genotype analysis from formalin-fixed, paraffin-embedded tumor tissues. Pharmacogenomics J. 2011;11(5):348–358. doi: 10.1038/tpj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zanetti-Dällenbach R, Vuaroqueaux V, Wight E, Labuhn M, Singer G, Urban P, Eppenberger U, Holzgreve W, Eppenberger-Castori S. Comparison of gene expression profiles in core biopsies and corresponding surgical breast cancer samples. Breast Cancer Res. 2006;8(4):R51. doi: 10.1186/bcr1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carney PA, Poplack SP, Wells WA, Littenberg B. The New Hampshire Mammography Network: the development and design of a population-based registry. AJR Am J Roentgenol. 1996;167(2):367–372. doi: 10.2214/ajr.167.2.8686606. [DOI] [PubMed] [Google Scholar]
- 17.Aryee M, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akalin A, Franke V, Vlahoviček K, Mason CE, Schübeler D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015;31(7):1127–1129. doi: 10.1093/bioinformatics/btu775. [DOI] [PubMed] [Google Scholar]
- 19.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charles CA, Tomic-Canic M, Vincek V, Nassiri M, Stojadinovic O, Eaglstein WH, Kirsner RS. A gene signature of nonhealing venous ulcers: potential diagnostic markers. J Am Acad Dermatol. 2008;59(5):758–771. doi: 10.1016/j.jaad.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross RL. The effect of ascorbate on wound healing. Int Ophthalmol Clin. 2000;40(4):51–57. doi: 10.1097/00004397-200010000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Paco S, Casserras T, Rodríguez MA, Jou C, Puigdelloses M, Ortez CI, Diaz-Manera J, Gallardo E, Colomer J, Nascimento A, Kalko SG, Jimenez-Mallebrera C. Transcriptome analysis of Ullrich congenital muscular dystrophy fibroblasts reveals a disease extracellular matrix signature and key molecular regulators. PLoS One. 2015;10(12):e0145107. doi: 10.1371/journal.pone.0145107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Similarity between DCIS core biopsies and surgical specimens in DNA methylation. Unsupervised hierarchical clustering of (A) 2,500 and (B) 5,000 most variable methylation loci. Horizontal tracking bars indicate clinical covariates shown in Table 1. (C) Median methylation beta-values in core biopsies (left) and surgical specimens (right) along a four-kilobase (kb) window centered at canonical TSS. TSS, transcription start site
Comparison of methylation beta-value distributions in patient-matched core biopsies and surgical specimens. Each sub-plot represents methylation profiles of patient-matched specimen pairs. Curves represent distributions of methylation beta-values. Beta-value, proportion of methylated alleles in a sample.
DNA methylation by genomic context in individual subjects. Each sub-plot displays distributions of methylation beta-values along a 4-kb window centered at canonical transcription start sites (TSS) in patient-matched specimen pairs. kb, kilobase; beta-value, proportion of methylated alleles in a sample.
Comparison of within and between-subject variability in all array-measured methylation loci. The Δbeta matched (beta core biopsy – beta matched surgical) and Δbeta permuted (beta core biopsy – beta permuted surgical) distributions represent within and between-subject variability, respectively. For each DCIS patient, Δbeta matched and Δbeta permuted distributions were compared by Kolgomorov-Simirnov (KS) tests with 52 iterations. All KS test P-values < 2.20E-16. Beta-value, proportion of methylated alleles in a sample.
Relation of methylation of protocadherin gene-body CpGs (A) cg26890354, (B) cg15949044, and (C) cg23445461 with expression of 11 protocadherin mRNAs in Stage I/Ia/Ib, estrogen receptor (ER)-positive patients from The Cancer Genome Atlas (TCGA). Every subpanel (labeled “a” through “k”) represents linear regression of DNA methylation (y-axis) and a given protocadherin mRNA transcript (x-axis). All regression coefficients were positive, indicating a positive association between protocadherin methylation and gene expression. Table insets show the proportion of variance explained (R.squared) and the associated P-value (P.value).