

Abstract
Low-density lipoprotein (LDL) aggregation is central in triggering atherogenesis. A minor fraction of electronegative plasma LDL, termed LDL(−), plays a special role in atherogenesis. To better understand this role, we analyzed the kinetics of aggregation, fusion and disintegration of human LDL and its fractions, LDL(+) and LDL(−). Thermal denaturation of LDL was monitored by spectroscopy and electron microscopy. Initially, LDL(−) aggregated and fused faster than LDL(+), but later the order reversed. Most LDL(+) disintegrated and precipitated upon prolonged heating. In contrast, LDL(−) partially retained lipoprotein morphology and formed soluble aggregates. Biochemical analysis of all fractions showed no significant degradation of major lipids, mild phospholipid oxidation, and anincrease in non-esterified fatty acid (NEFA) upon thermal denaturation. The main baseline difference between LDL subfractions was higher content of NEFA in LDL(−). Since NEFA promote lipoprotein fusion, increased NEFA content can explain rapid initial aggregation and fusion of LDL(−) but not its resistance to extensive disintegration. Partial hydrolysis of apoB upon heating was similar in LDL subfractions, suggesting that minor proteins importantly modulate LDL disintegration. Unlike LDL(+), LDL(−) contains small amounts of apoA-I and apoJ. Addition of exogenous apoA-I to LDL(+) hampered lipoprotein aggregation, fusion and precipitation, while depletion of endogenous apoJ had an opposite effect. Therefore, the initial rapid aggregation of LDL(−) is apparently counterbalanced by the stabilizing effects of minor proteins such as apoA-I and apoJ. These results help identify key determinants for LDL aggregation, fusion and coalescence into lipid droplets in vivo.
Keywords: Electronegative LDL, Lipoprotein aggregation, fusion and droplet formation, Thermal denaturation, Atherogenesis, Apolipoprotein A-I, Apolipoprotein J
1. Introduction
The response-to-retention hypothesis states that the initiating event in the development of atherosclerotic lesions is the subendothelial retention of LDL, a process mediated by the binding of LDL to arterial proteoglycans (PGs) [1,2]. This binding is triggered by chemical modifications of LDL such as lipolysis, proteolysis or oxidation, which promote LDL aggregation [3,4]. Aggregated LDL particles contain multiple PG binding sites and bind to PGs in the arterial wall matrix with higher affinity than their native non-aggregated counterparts. LDL aggregation can evolve to irreversible LDL fusion (i.e. formation of larger lipoprotein-like particles) followed by lipoprotein disintegration and release of core lipids (or rupture) and coalescence into lipid vesicles and droplets that are too large to exit the subendothelial space of the arterial wall [3,5]. In this environment, macrophages internalize entrapped lipoproteins, leading to the formation of foam cells overloaded with cholesterol, a hallmark of atherosclerotic lesions.
Since LDL aggregation and fusion are important early steps in atherogenesis, extensive studies have been conducted to understand molecular mechanisms of these processes, with a particular focus on the action of lipases and proteases whose expression is increased in atherosclerotic lesions [5–10]. However, other aspects, such as the relative susceptibility of specific LDL subfractions to aggregation and fusion, received relatively little attention [5,11,12]. Our focus here is on electronegative LDL, or LDL(−), which is a modified subfraction of plasma LDL with inflammatory and apoptotic properties [13]. The proportion of LDL(−) in plasma is increased under conditions of high risk of cardiovascular disease [14,15]. LDL(−) has increased propensity to aggregate; in fact, a minor sub-population of LDL(−) is aggregated in plasma [16,17]. Moreover, LDL(−) can promote in vitro aggregation of monomeric, non-electronegative native LDL particles, termed LDL(+) [17–19]. Together, these studies suggest that, although LDL(−) constitutes only 3–5% of total plasma LDL, it may importantly contribute to atherogenesis by promoting LDL aggregation and favoring sub-endothelial retention of LDL. An unique feature of LDL(−) is a relatively high content of non-apoB proteins [20]. The role of these proteins in the aggregation behavior of LDL(−) is unknown, but some of them, such as apoJ and apoA-I, can prevent aggregation, fusion and rupture of total LDL [21,22]. In the current work, we analyzed the role of apoJ and apoA-I in aggregation of LDL subclasses.
Thermal analysis is widely used to determine structural stability and susceptibility to aggregation of proteins and lipoproteins [23]. Our previous thermal denaturation studies revealed that stability of lipoproteins is determined by kinetic barriers, and suggested that similar barriers modulate lipoprotein remodeling and fusion in vivo [11,24– 26]. Importantly, thermal denaturation of lipoproteins such as LDL mimics key aspects of their aggregation, fusion and rupture in vivo, and the products of the heat-induced LDL fusion and rupture are similar to LDL-derived extracellular deposits in atherosclerotic lesions ([5] and references therein). This prompted us to propose that perturbations of a lipoprotein assembly by various means (mechanical, thermal, enzymatic, oxidative etc.) produce a finite number of similar structural responses such as aggregation, fusion and rupture. This concept is illustrated in the Supplemental Fig. S1 showing gel filtration profiles of LDL that have been subjected to lipolysis by sphingomyelinase or phospholipase C or incubation at 37 °C, pH 6 to mimic in vivo conditions in atherosclerotic plaques, as well as to vortexing or heating. Similar responses of LDL assembly to these perturbations suggest that the general trends revealed in thermal denaturation studies are applicable to LDL remodeling at ambient conditions in vivo. Hence, LDL are exposed to high temperatures as a means to accelerate LDL remodeling and quantify its kinetics.
In the current work, we use thermal denaturation as a tool to determine structural stability and susceptibility to aggregation, fusion and disintegration of LDL(−) and LDL(+). The results reveal that, unlike LDL(+) or LDL(total), which form large insoluble particles upon complete thermal denaturation, LDL(−) shows faster initial aggregation but much less extensive rupture and hence, stays longer in solution in the aggregated form. We propose an explanation for these surprising findings based on the distinct biochemical composition of LDL(−), and discuss their implications for the role of LDL(−) in atherogenesis.
2. Material and methods
2.1. Lipoprotein isolation
Plasma of healthy normolipemic volunteers was obtained in the Lipid Laboratory of Hospital de Sant Pau with their written informed consent upon approval by the institutional ethics committee. Total LDL was isolated from pooled plasma by sequential ultracentrifugation (density range 1.019–1.050 g/mL) using KBr gradients at 4 °C. All density solutions contained 1 mM EDTA and 2 μM BHT. LDL were dialyzed against buffer A (10 mM Tris, 1 mM EDTA, pH 7.4) and stored at −80 °C in 10% sucrose solution until use.
Total LDL was filtered using 0.2 μm filter and subfractioned into LDL(+) and LDL(−) by stepwise anion-exchange chromatography using HiPrep Q HP 16/10 column (GE Healthcare) [27]. LDL(total), LDL(+) and LDL(−) were then dialyzed against standard phosphate buffer (20 mM Na phosphate, pH 7.0) and used for further experiments unless otherwise stated. The protein concentration in LDL subfractions was measured by using a modified colorimetric Lowry assay. LDL concentration is expressed as mg protein/mL, unless otherwise indicated. ApoJ-containing LDL (LDL/J+) and apoJ-depleted LDL (LDL/J-) were obtained by affinity chromatography in NHS-HiTrap columns to which an apoJ-specific antibody was attached as described [21]. For biochemical heat-induced changes, LDL samples (0.35 mg protein/mL) in standard phosphate buffer were incubated at 82 °C for 15, 30 or 90 min (end-point). A subset of samples was centrifuged at 14,000 rpm for 10 min at 4 °C to determine the composition of soluble and insoluble particles.
2.2. Chemical characterization of LDL subfractions
Major lipid and protein composition of LDL subfractions was measured by commercial methods adapted to a Cobas 6000/c501 autoanalyzer. Total cholesterol, triglycerides and apolipoprotein (apo) B reagents were from Roche Diagnostics. Phospholipids and non-esterified fatty acids (NEFA) reagents were from Wako Chemicals. Phosphatidylcholine (PC) and sphingomyelin (SM) content was evaluated by normal-phase HPLC in a System Gold chromatograph (Beckman) equipped with a photodiode array detector, as described [28]. Dipalmitoyl-glycero-phosphodymethyl ethanolamine (DGPE, ref. P0399 Sigma) was used as an internal standard. The peak areas corresponding to PC and SM were quantified at 205 nm. The ratio of the absorbance at 205 nm to 234 nm was used as an index of PC oxidation [29].
Non-denaturing gradient (2–16%) gel electrophoresis (NGGE) was used to assess particle size. NGGE were run at 100 V for 4–6 h with 1% Sudan black pre-stained LDL subfractions, as described [30]. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE, 10% acrylamide) was used to assess apoB degradation and the presence of minor apolipoproteins in LDL. SDS gels were run at 100 V for 1.5–2 h, and were stained with Coomassie blue. ApoB, apoA-I and apoJ in LDL subfractions were detected by Western blotting after protein transfer from SDS PAGE to nitrocellulose. Proteins were transferred at 30 V for 1 h, incubated in blocking buffer (50 mM Tris, 500 mM NaCl (TBS), pH 7.4 containing 0.1% casein) for 30 min, washed 3 times with TTBS (TBS containing 0.1% Tween 20) and probed with polyclonal antibody anti-apoB (dilution 1/5000, Acris Ab), anti-apoA-I (dilution 1/1000, Acris Ab) and anti-apoJ (dilution 1/1000, Novus Biologicals) for 2 h, followed by horseradish peroxidase-conjugated anti-goat secondary antibody (dilution 1/5000, Jackson Immuno Research). Nitrocellulose was revealed with Immun-Star developing kit (BioRad).
2.3. Circular dichroism spectroscopy
CD data were recorded by using an AVIV 400 or an AVIV 62DS spectropolarimeter following published protocols [11] with minor modifications. Briefly, to compare the secondary structure in monomeric and in minimally aggregated LDL subfractions, far-UV CD spectra were recorded in the wavelength range 190–250 nm from LDL samples containing 0.1 mg protein/mL in standard buffer (20 mM Na phosphate, pH 7.0). Far-UV CD data were normalized to protein concentration and expressed in units of molar residue ellipticity, [Θ]. Since aggregated LDL are expected to cause spectral distortions in far-UV [31], no quantitative secondary structural analysis based on far-UV CD spectra was performed. ORIGIN software was used for CD data analysis and display.
2.4. Thermal denaturation studies
Structural stability of LDL subfractions was assessed in the heating experiments by simultaneously monitoring near-UV CD and turbidity (dynode voltage) at 320 nm; for methodological details see references [11,23,32]. Previously we showed that heating causes a large increase in turbidity, which reflects LDL aggregation and fusion of a subset of particles followed by lipoprotein disintegration and coalescence into droplets (rupture). The denatured sample represented a heterogeneous mixture of intact-size LDL, fused LDL and ruptured particles as observed by TEM [5,11,30]. Furthermore, lipid re-packing upon lipoprotein disintegration induces a large negative near-UVCDpeak circa 320 nm, which was used to monitor LDL rupture [32]. Such a signal was detected previously in our thermal denaturation studies of LDL, VLDL and HDL [32–35]. In each case, the formation of this negative near-UV CD peak coincided with the observation of lipoprotein rupture by TEM and differential scanning calorimetry, and was followed by sample precipitation. The only lipoprotein moieties that can show induced CD at these long wavelengths are apolar core lipids. Therefore, the induced CD signal circa 320 nm has been attributed to re-packing of apolar core lipids upon lipoprotein rupture and coalescence into droplets. In the current study, CD at 320 nm was used to monitor heat-induced LDL rupture.
Melting and kinetic experiments were performed to assess LDL stability following established protocols [11] with slight modifications. Briefly, in melting experiments, LDL samples (0.35 mg protein/mL in standard buffer) were heated and cooled from 25 °C to 98 °C at a constant rate of 11 °C/h. Turbidity and CD signal at 320 nm were monitored as a function of temperature. In kinetic temperature-jump (T-jump) experiments, the sample temperature was rapidly increased from 25 °C to a higher constant value (ranging from 75 °C to 95 °C) to trigger LDL fusion and rupture, which were monitored by turbidity and CD at 320 nm as a function of time. The denaturation kinetics of LDL subfractions was quantified by using an Arrhenius analysis of the T-jump data (Supplemental Fig. S2).
2.5. Transmission electron microscopy
LDL subfractions that have been subjected to various thermal treatments were visualized at 22 °C by negative staining TEM using a CM12 transmission electron microscope (Phillips Electron Optics) as described previously [36]. PHOTOSHOP and EXCEL software were used for the analysis of particle size distribution, and GraphPad Prism 5.0 software was used to display the results; 50–300 particles were used for such analysis.
2.6. Statistical analysis
Results were expressed as mean ± SEM or as mean±SD,asindicat-ed. Statistically significant differences were assessed by paired non-parametric test (Wilcoxon). P-values of <0.05 were considered significant. GraphPad Prism 5.0 software was used for the statistical analysis and data display, unless otherwise stated.
3. Results
3.1. Isolation and characterization of LDL subfractions
Supplemental Fig. S3A shows a representative anion-exchange chromatogram separating LDL subfractions. The proportion of LDL(−) was 3–5% in all batches explored. Fig. S3B shows a non-denaturing polyacrylamide gradient gel electrophoresis (NGGE) of LDL subfractions. In contrast to LDL(+), which presented a narrow band of homogeneous particles corresponding to the LDL monomer, LDL(−) showed a broader band including both larger and smaller particles. This observation was supported by electron micrographs of LDL(+) particles, which were homogeneous in size, and LDL(−), which contained both larger and smaller particles (Fig. S3A, inserts). Basal lipid composition in LDL subfractions is shown in Supplemental Table S1.
3.2. Differences in the structural stability of LDL(−) and LDL(+)
Fig. 1A shows far-UV circular dichroism (CD) spectra of intact LDL subfractions. CD intensity across all wavelengths decreased in order LDL(+) > LDL(total) > LDL(−), and the ratio of the positive signal at 193 nm to the negative signal at 222 nm decreased from about 1.3 in LDL(+) to 1.0 in LDL(−). Such spectral changes are consistent with the previous studies suggesting reduced secondary structural content in apoB on LDL(−) [47,48]. However, spectral distortions due to UV light scattering from LDL(−) aggregates may also contribute to the observed effects (see Supplemental Fig. S4 for detail).
Fig. 1.
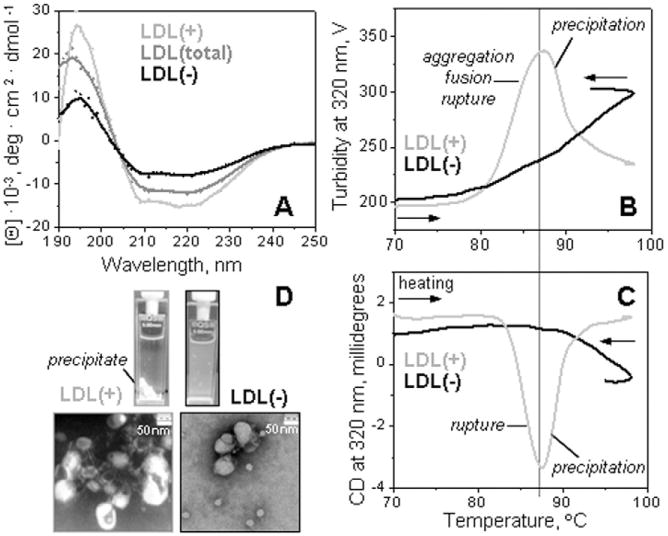
Secondary structure and thermal stability of LDL fractions monitored by circular dichroism spectroscopy. Far-UV CD spectra of intact LDL(total), LDL(+), and LDL(−) (A). The melting data recorded of LDL(+) and LDL(−) at 320 nm by turbidity to monitor changes in the particle size (B) and by near-UV CD to monitor lipid re-packing upon lipoprotein disintegration and release of core lipids (C). The data were recorded using LDL samples under standard conditions (0.35 mg protein/mL, 20 mM Na phosphate buffer, pH 7.0). The melting data in panels B and C were recorded simultaneously during sample heating and cooling at a constant rate of 11 °C/h. Steep decline in CD and turbidity amplitude observed upon heating of LDL(+) above 88 °C is due to sample precipitation (D). The melting data of LDL(total) (not shown) were similar to those of LDL(+) within the error of their experimental determination.
To test for relative structural stability of LDL subclasses, samples of LDL(total), LDL(+) and LDL(−) under otherwise identical conditions of protein and buffer concentration, buffer composition and pH were heated and cooled from 5 °C to 95 °C at a constant rate of 11 °C/h. The turbidity data were recorded at 320 nm to monitor changes in the particle size due to combined effects of aggregation, fusion and rupture. Simultaneously, near-UV CD data were recorded at 320 nm to monitor LDL rupture.
Representative melting data are shown in Fig. 1B, C. As expected, the data of LDL(+), which comprise ∼95% of total LDL, were very similar to those of LDL(total) (not shown to avoid overlap). A steep increase in turbidity was observed in LDL(+) upon heating above 80 °C (Fig. 1B, light-grey line), reflecting an increase in the particle size due to the heat-induced aggregation, fusion and rupture. This was closely followed by a development of a negative near-UV CD signal upon LDL rupture (Fig. 1C, light-grey line). These changes were followed by a sharp drop in turbidity and in near-UV CD amplitude above ∼85 °C, reflecting sample loss upon precipitation that was evident from the visual appearance of the sample (Fig. 1D). These results are in good agreement with our previous studies of LDL(total) [11,32].
Surprisingly, the melting data of LDL(−) were distinctly different from those of LDL(+) or LDL(total) (Fig. 1B, C). Although the onset of the morphological transition was observed at comparable temperatures in various LDL fractions, only LDL(−) showed a relatively broad and shallow transition (i.e. low apparent cooperativity). The signal amplitude in this transition was reduced in LDL(−) compared to LDL(+), and the signal did not reach a peak (i.e. the onset of precipitation) in the temperature range explored up to 98 °C. The latter was supported by the visual observation: in contrast to LDL(+) and LDL(total) that undergo massive irreversible precipitation upon heating above 88 °C, LDL(−) remained in solution even upon heating to 98 °C (Fig. 1D). Visualization of LDL samples by transmission electron microscopy (TEM) after heating to 98 °C followed by cooling confirmed these observations: essentially all lipoproteins in LDL(+) or LDL(total) have coalesced into aggregated lipid droplets, while LDL(−) formed smaller less aggregated droplets and even showed a substantial amount of monomeric particles in normal LDL size range (Fig. 1D).
In summary, our CD and turbidity melting data corroborated by TEM and visual inspection (Fig. 1B–D) revealed that, compared to LDL(+) and LDL(total), LDL(−) shows less extensive and less cooperative thermal remodeling. This result was unexpected, since previous studies consistently showed that LDL(−) is more prone to spontaneous aggregation at near-physiologic conditions [12,29,37,38]. To confirm this surprising result, CD and turbidity melting data were recorded from LDL(+) and LDL(−) that have been isolated from fresh single-donor plasma (Supplemental Fig. S5). The results showed that thermal behavior of such freshly isolated fractions was very similar to that observed in LDL fractions isolated from pooled plasma which have been frozen and thawed prior to CD experiments (Fig. 1B, C). Therefore, the distinct thermal behavior of LDL(−) is not an artifact of the sample preparation. Next, the melting data of LDL(total) were recorded in a broad range of sample concentrations (Supplemental Fig. S6). The results clearly showed that the distinct thermal behavior of LDL(+) and LDL(−) cannot be due to small variations in their protein concentrations and hence, is an intrinsic property of these subclasses.
3.3. LDL(−) and LDL(+) show distinct aggregation kinetics
Since structural stability of LDL and other lipoproteins is modulated by kinetic barriers, kinetic experiments are particularly useful for discerning small differences in stability of lipoprotein subfractions [11]. To compare transition kinetics in LDL(−) and LDL(+), we conducted temperature jump (T-jump) experiments. LDL denaturation was triggered by a rapid increase in temperature to a final constant value, and the time course of the transition was monitored by turbidity and by CD at 320 nm. Fig. 2 shows representative kinetic data recorded in such T-jumps from 25 °C to 82 °C of LDL(+) and LDL(−) under identical conditions (0.35 mg/mL apoB, 20 mM Na phosphate, pH 7.0).
Fig. 2.
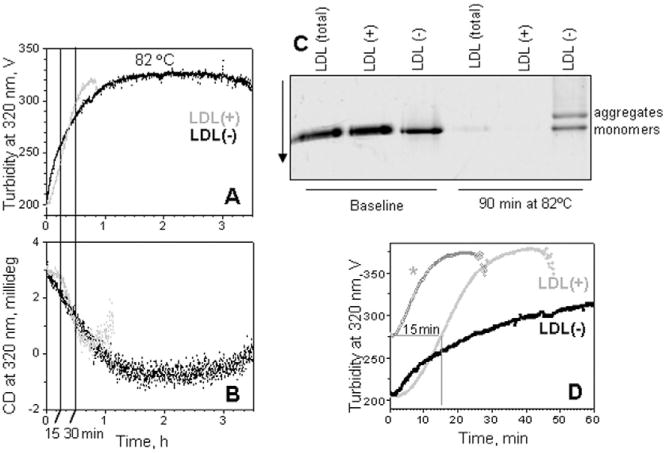
Kinetics of the heat-induced aggregation, fusion and rupture of LDL(+) and LDL(−). All data were recorded from LDL samples under standard conditions in a temperature jump from 25 °C to 82 °C. (A) The time course of the transition was monitored at 320 nm by turbidity for increase in the particle size. (B) Simultaneously, near-UV CD at 320 nm signal was recorded to monitor lipid re-packing upon LDL rupture and coalescence into lipid droplets. For LDL(+), the signal loss due to sample precipitation was observed after ∼60 min. (C) NGGE of LDL subfractions at a baseline and after 90 min of thermal denaturation. (D) Repetitive kinetic data recorded from LDL(+). The sample was subjected to a T-jump from 25 °C to 82 °C (light-grey). An identical sample was subjected to similar T-jump for 15 min, followed by rapid sample cooling on ice, equilibration at 25 °C, and the second consecutive T-jump to 82 °C (grey, *).
Interestingly, LDL(−) showed distinct transition kinetics. In contrast to LDL(total) [11] and LDL(+) (Fig. 2, light-grey line), which showed a sigmoidal transition time course, LDL(−) transition lacked the lag phase and was non-sigmoidal. Instead, at early transition stages (approximately the first 20 min in Fig. 2A, B), LDL(−) showed a much faster increase in turbidity and in the negative near-UV CD as compared to LDL(+) or LDL(total). These rapid initial changes were followed by much slower changes observed in LDL(−) over longer time (hours). Notably, both kinetic and melting data recorded by CD and turbidity (Figs. 1, 2), as well as the visual appearance of the samples consistently showed that LDL(−) remained soluble at high temperatures while LDL(+) and LDL(total) precipitated. Furthermore, NGGE clearly showed precipitation of most LDL(total) and all LDL(+) particles after 90 min incubation; in contrast, some LDL(−) particles were in the size range corresponding to LDL monomers or small aggregates (Fig. 2C). Collectively, these data indicate that LDL(−) is resistant to complete disintegration and precipitation at high temperatures.
3.4. Transmission electron microscopy analysis
To visualize particle morphology at various stages of thermal remodeling, LDL fractions were incubated at 82 °C as described in Fig. 2 legend either for 15 min (where changes in turbidity but not in near-UV CD were observed) or for 30 min (where changes both in turbidity and in near-UV CD were detected). Next, the samples were pooled, briefly placed on ice to stop the structural transition, and deposited on the grids at 22 °C, followed by negative stain TEM analysis. Fig. 3 shows electron micrographs of LDL fractions at a baseline, i.e. prior to heating (left), or incubated at 82 °C for either 15 (middle) or 30 min (right). As previously described, baseline LDL(−) was much more heterogeneous in size as compared to LDL (total),while LDL(+) was highly homogeneous (Fig. 3 and Supplemental Fig. S7).
Fig. 3.

Electron micrographs of negatively stained LDL(total), LDL(+) and LDL(−). Intact particles (left) were incubated at 82 °C in T-jump experiments as described in Fig. 2; sample aliquots were taken after 15 min (middle) or 30 min (right) of incubation. TEM was performed as described in the Experimental Section. White arrows indicate fused LDL particles. Inserts: zoomed-out views illustrate LDL aggregation. The bar size is 50 nm.
After 15 min, initial aggregation was detected in all fractions (Fig. 3, zoomed-out inserts in central panels) as evidenced by occasional large aggregates of LDL (clumps surrounded by dense regions of stain). Such aggregation is probably responsible for the increase in turbidity observed during the first 15 min of incubation. In addition, LDL(total) showed formation of smaller lipoprotein-like particles after 15 min at 82 °C, in contrast to LDL(+) that remained homogeneous in size (Fig. 3, central panel, and Supplemental Fig. S7). This suggests that the formation of smaller particles upon heating of LDL(total) at this stage could be attributed mainly to the effect of LDL(−).
After 30 min of incubation (Fig. 3, right column),a significant portion of LDL in all fractions showed rupture, i.e. loss of lipoprotein-like morphology and coalescence into lipid droplets. These observations were consistent with changes in CD at 320 nm observed in all fractions after 30 min (Fig. 2C), indicating repacking of apolar lipids upon their release from the lipoprotein core and coalescence into droplets. Interestingly, in LDL(total), the surviving particles tended to be larger than the intact particles. Again, this effect could be attributed mainly to LDL(−) that can augment both fission (seen at 15 min) and fusion (at 30 min) of LDL particles.
In summary, TEM results are in excellent agreement with the kinetic studies of LDL fractions using turbidity and near-UV CD (Figs. 1, 2). These results show that all LDL fractions undergo aggregation at relatively early stages of thermal remodeling (first 15 min at 82 °C). In addition, LDL(total) and, especially, LDL(−) show formation of larger and smaller particles due to fusion and fission, respectively, while LDL(+) remain homogeneous in size. Therefore, fusion and fission of LDL(total) can be attributed largely to the action of LDL(−). The small particles formed upon fission are probably stabilized by the exchangeable apolipoproteins, such as apoA-I, apoE, apoC-III or apoJ, that are enriched in LDL(−), as well as by the micelle-forming lipids lyso-phosphatidylcholine (lyso-PC) and NEFA whose baseline content is also elevated in LDL(−) [28,39,40].
3.5. Role of pre-aggregated LDL assessed in kinetic experiments
Our biophysical results revealed that, although LDL(−) are much less prone to precipitation upon prolonged exposure to high temperatures (black lines in Fig. 1B, C and Fig. 2A, B), they show faster thermal remodeling such as aggregation, fusion and fission at early stages (Fig. 2A, B; Fig. 3 central column, 15 min). These early stages are particularly relevant, as they mimic aspects of structural transitions in lipoproteins observed at physiologic conditions [5]. Therefore, most of our biophysical analysis is focused on these early stages of thermal transition.
The lag phase of the transition, which was observed in all LDL fractions except LDL(−) (Fig. 2), may result from different biochemical and/or physical properties of these fractions. Although the events occurring during the lag phase are not entirely clear, they probably involve accumulation of physical defects (e.g. conformational changes in apoB and formation of small LDL aggregates) as well as chemical modifications (e.g. accumulation of hydrolytic and oxidation products) that prime LDL for fusion [11]. The lag phase in other aggregation reactions, such as crystallization or fiber growth, often involves nucleation. Hence, it is possible that small LDL aggregates formed at the early stages of the transition can nucleate massive LDL aggregation, fusion and rupture at later stages. Because small LDL aggregates have been found under physiologic conditions in LDL(−) but not in LDL(+), we reasoned that the presence of such aggregates may explain the difference in the transition kinetics between LDL subfractions, in particular, the presence of the lag phase in LDL(+) but not in LDL(−) (Fig. 2A, B). To test this idea, we used LDL(+), a fraction that is free from pre-existing lipoprotein aggregates [17]. The formation of aggregated but not ruptured LDL(+) was induced by incubation for 15 min at 82 °C (Fig. 2C); the transition was stopped by rapid cooling; this was followed by a second consecutive T-jump to 82 °C from the same sample, which was monitored by turbidity (Fig. 2D, light-grey dashed line). The results clearly showed that in the second T-jump, the lag phase was largely eliminated but the transition kinetics remained unchanged and followed a similar time course as that in intact LDL(+) after 15 min of incubation at 82 °C (Fig. 2D, grey lines). Notably, this transition kinetics remained distinctly different from that of LDL(−) (Fig. 2D, black line). We conclude that, although the presence of pre-aggregated LDL may influence the early stage in LDL transition, the overall kinetics of this transition in the pre-aggregated sample remains invariant. Therefore, the differences in the transition kinetics of LDL subclasses must result from their distinct biochemical composition rather than their initial physical state (monomeric or aggregated).
3.6. Changes in the biochemical composition of LDL upon heating
To establish whether biochemical differences are responsible for the distinct structural remodeling of LDL(−) and LDL(+), we analyzed the heat-induced changes in protein and lipid content of LDL subfractions. Quantification of LDL lipids by enzymocolorimetric methods suggested small changes in lipid composition upon LDL incubation at 82 °C (Fig. 4A–C). Biochemical analysis at different time points (after 15, 30 and 90 min at 82 °C) showed a slight progressive increase in the cholesterol, triglyceride and phospholipid content in all LDL fractions, which was more pronounced in LDL(total) and LDL(+) than in LDL(−) (Fig. 4A– C). This unexpected increase was probably a consequence of particle disruption upon heating, which increased the accessibility of lipids, particularly those in the apolar core, to enzymes used for lipid measurements. This effect could explain why such an apparent increase was higher in LDL(total) and LDL(+), which are more labile to disruption, than in LDL(−).
Fig. 4.
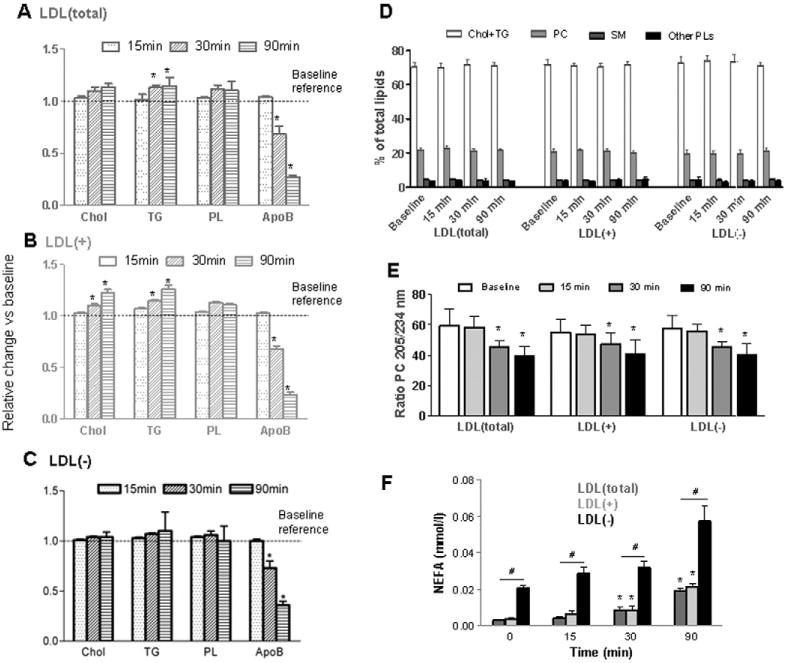
Heat-induced changes in biochemical composition of LDL subfractions. LDL(total), LDL(+) and LDL(−) were analyzed prior to heating (baseline) or after incubation for 15, 30 or 90 min at 82 °C. Relative changes (compared to the baseline) of the major protein, apoB, and major lipids are shown for (A) LDL(total), (B) LDL(+), and (C) LDL(−). (D) Quantification of cholesterol and triglycerides (Chol + TG), phosphatidylcholine (PC), sphingomyelin (SM) and other phospholipids in LDL subfractions; relative content as a fraction of total lipids is shown (see Supplemental Fig.S6 for details). (E) Phospholipid oxidation characterized by the absorbance ratio at 205 nm to 234 nm of the PC peak. (F) NEFA content in LDL subfractions. Data in A, B, C and Fare the mean ± SD of 8 independent samples. Data in D and E are the mean ± SD of 4 independent samples. See text for details of the biochemical analyses.*indicates P <0.05 vs. baseline values. # indicates P < 0.05 vs. LDL(total) or LDL(+).
The determination of LDL composition by enzymatic methods cannot detect phospholipid degradation. To ascertain whether phospholipids were degraded upon LDL heating, HPLC analysis was performed. This method separates the first peak, which includes total (free and esterified) cholesterol and triglycerides, from the peaks corresponding to PC and sphingomyelin (SM). (Supplemental Fig. S8). Fig. 4D shows the relative content of PC and SM, expressed as the fraction of total lipids. These data confirmed that major phospholipids were not extensively degraded after 90 min incubation at 82 °C. No evidence for LPC formation was observed (elution approximately at 30 min of the chromatogram, Supplemental Fig. S8).
HPLC analysis also provided information on the oxidative status upon heating. The ratio of the areas of the PC peak at 205 nm (maximum of absorbance) and 234 nm (conjugated dienes) can be used to estimate the degree of oxidation of phospholipids [17,29]. We observed a decrease of the 205/234 peak ratio in samples incubated at 82 °C, reflecting progressive oxidation during heating (Fig. 4E). Such a decrease was evident at 30 min and was very similar in all LDL subfractions.
Importantly, a significant increase in NEFA after heating LDL(total) and LDL(+) was observed. Although the baseline NEFA content was very low in these subfractions, the relative increase in NEFA after 90 min of incubation at 82 °C was approximately 600%. In comparison, the relative increase LDL(−) was <300% (Fig. 4F), mainly because LDL(−) have higher NEFA content at a baseline.
Regarding the protein moiety, we observed a decrease in the apoB content, which could be due, in part, to the loss of recognition by antibodies used to quantify apoB, but also to protein degradation (Fig. 4A– C). SDS-PAGE suggested progressive degradation of apoB upon heating, showing less intense bands after 90 min of thermal denaturation (Fig. 5, upper panels). Since similar decrease in apoB was observed in LDL(total), LDL(+) and LDL(−) (Fig. 4A–B), apoB degradation could not explain the observed differences in the denaturation behavior of these LDL fractions.
Fig. 5.
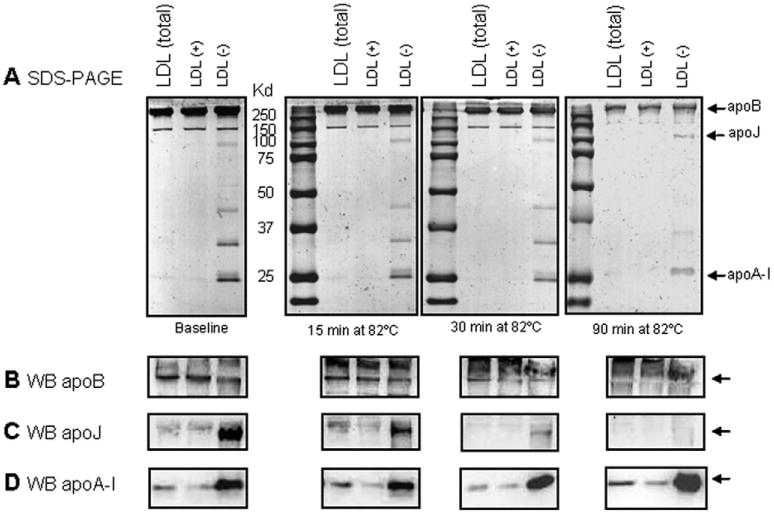
Protein degradation upon thermal denaturation of LDL. (A) SDS-PAGE (10%, Coomassie blue staining) was used to assess changes to apoB after 15, 30 and 90 min of incubation at 82 °C. Gels were run at 100 V for 1.5 h. (B, C) Following SDS-PAGE, Western blot analysis was used to detect apoB (B), apoJ (C) and apoA-I (D).
3.7. Effect of apoA-I and apoJ on LDL aggregation upon heating
The increased resistance to heat-induced disruption and precipitation of LDL(−) could potentially be due to the presence of minor apolipoproteins in this subfraction [39] which stabilize LDL. For instance, apoJ and apoA-I are known to prevent LDL aggregation, fusion and rupture [21,22]. Western blot analysis confirmed increased apoA-I and apoJ content in LDL(−) compared to LDL(total) or LDL(+) and showed that endogenous apoJ remained intact after 15 min of incubation at 82 °C, while endogenous apoA-I was largely intact even after 90 min (Fig. 5C–D and Supplemental Fig. S9). Both apolipoproteins could help to solubilize lipids and stabilize LDL surface against rupture and precipitation.
To test this idea, we first evaluated how the addition of lipid-free apoA-I influenced thermal denaturation of LDL(+) that has little endogenous apoA-I (Fig. 6A). T-jump data demonstrated that exogenous apoA-I retarded LDL aggregation and fusion and also delayed precipitation. However, LDL(+) retained the sigmoidal transition time course with its characteristic lag phase, which was not affected by apoA-I (Fig. 6A). Consequently, exogenous apoA-I increases the solubility of LDL at any stage of thermal denaturation.
Fig. 6.
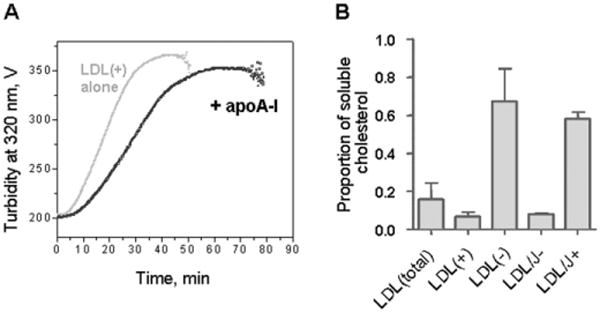
Effects of apoA-I and apoJ on thermal stability and solubility of LDL. (A) Effects of exogenous apoA-I on LDL stability. Lipid-free human apoA-I (0.2 mg/mL) was added to LDL(+) under standard conditions. The data were recorded from LDL(+) alone (grey) or from LDL(+) and apoA-I (black) during a T-jump from 25 to 82 °C by turbidity at 320 nm. (B) Effects of endogenous apoJ on LDL solubility. ApoJ-depleted (LDL/J-) and apoJ-containing LDL (LDL/J+) were obtained from LDL(total) by affinity chromatography as described in Methods. LDL subfractions were incubated at 82 °C for 90 min, were centrifuged at 14,000 rpm for 10 min at 4 °C, and cholesterol was measured in the supernatant. The results are expressed as the proportion of soluble LDL cholesterol.
Next, we evaluated how the depletion of apoJ-containing particles influences LDL solubility upon heat denaturation (Fig. 6B). LDL fractions differing in the amount of endogenous apoJ, including apoJ-containing (LDL/apoJ+) and apoJ-depleted (LDL/apoJ-) fractions separated by affinity chromatography, were incubated for 90 min at 82 °C, centrifuged at 14,000 rpm for 10 min at 4 °C, and the amount of soluble cholesterol was quantified. Particles that contained little or no apoJ, including LDL(total), LDL(+) and LDL/apoJ-, showed precipitation of >80% of their cholesterol. In contrast, particles containing endogenous apoJ, such as LDL(−) and LDL/apoJ+, showed precipitation of only 30% to 40% of their cholesterol after heating (Fig. 6). This observation supports the idea that endogenous apoJ helps confer solubility to LDL particles.
4. Discussion
LDL aggregation is a key step in the initiation of atherosclerosis. Aggregation is prerequisite for LDL fusion and formation of large lipoprotein-like particles that can further coalesce into lipid droplets characteristic of atherosclerotic lesions. These processes in vivo are driven mainly by hydrolytic modifications in apoB and lipids, which are particularly labile in LDL. Such modifications can be mediated by several factors. For example, increased expression of a number of proteases and lipases in atherosclerotic areas of the arterial wall [41–44] augments the degradation of LDL proteins and lipids in the sub-endothelial space. The degradation of apoB and phospholipids in LDL is also promoted by oxidative modifications that are thought to be extensive in atherosclerotic lesions [45]. Aggregation of LDL in vivo can be a very slow process involving many players, which is difficult to analyze in a laboratory setting. Thermal denaturation, which greatly speeds up this process, mimics key aspects of LDL aggregation, fusion and rupture in vivo (Supplemental Fig. S1), and the products of these processes are similar to LDL-derived extracellular deposits in atherosclerotic lesions. Therefore, thermal denaturation provides a useful tool to accelerate this process, dissect its steps, and determine contributions of individual factors.
Our spectroscopic and electron microscopic data show that, at the initial stages of the heat denaturation, LDL(−) aggregates and fuses faster than LDL(+) (Figs. 1–3). This observation agrees with previous reports that LDL(−) has increased susceptibility to aggregation upon other perturbations, such as intense agitation, prolonged incubation at 37 °C, or phospholipase treatment [17,18,27,29,38,46]. Our TEM and NGGE data confirm the presence of aggregated particles in LDL(−) isolated from blood. TEM analysis shows that the initial heat-induced aggregation behavior of LDL(total) is intermediate between that of LDL(+) and LDL(−) (Figs. 2, 3), suggesting that LDL(−) promotes aggregation and fusion of LDL(+) at these early stages. Collectively, these results support the idea that LDL(−) has increased susceptibility to aggregation and can initiate the aggregation of LDL(total).
In the current study we show that the main heat-induced biochemical change in LDL is the degradation of apoB. This observation supports the use of thermal denaturation as a relevant experimental model for the analysis of the effects of apoB degradation on LDL aggregation, fusion and rupture. However, since apoB proteolysis was similar in various LDL subfractions (Fig. 4A–C), it is unlikely to produce the distinct thermal remodeling observed in LDL(+) and LDL(−). Techniques of the current study cannot reliably detect specific conformational changes in apoB during thermal denaturation because far-UV CD spectra are distorted by light scattering of large aggregated particles. However, CD spectra in Fig. 1A are consistent with previous studies reporting anomalous conformation in apoB on LDL(−) [19,47,48]. This anomalous protein conformation may influence thermal denaturation of LDL(−) potentially contributing to its increased susceptibility to aggregation.
A major biochemical difference between LDL subfractions is the content of NEFA, which is higher at a baseline in LDL(−) but shows a large heat-induced increase in all fractions (Fig. 4F). NEFA are highly fusogenic, and even a few percent increase in NEFA content significantly accelerates aggregation, fusion and rupture of LDL or other lipoproteins, whereas the removal of NEFA by albumin decelerates these transitions [33]. Therefore, we propose that the initial rapid aggregation and fusion of LDL(−) and its ability to promote aggregation of LDL(+) results, at least in part, from its high baseline NEFA content.
We hypothesize that the lack of lag phase in LDL(−) at the early stage of thermal denaturation (Fig. 2) results from increased baseline content of NEFA in LDL(−). In contrast, in LDL(total) and LDL(+), whose baseline content of NEFA is very low, aggregation does not proceed until NEFA are generated upon heating, which helps explain the lag phase observed in these fractions. The exact origin of NEFA generation upon heating is unclear and could potentially result from the degradation of phospholipids, triglycerides or esterified cholesterol. Importantly, on a molar basis, the amount of NEFA at any time is much lower than that of other lipids; hence, degradation of just a few molecules of phospholipids, triglycerides and/or esterified cholesterol will significantly increase the NEFA content in LDL. In summary, lipid analyses of LDL subfractions at various stages of thermal denaturation suggest that accelerated fusion of LDL(−) observed at early stages of denaturation results, at least in part, from increased baseline content of NEFA in LDL(−).
Surprisingly, we also found that, upon prolonged incubation at high temperatures, a substantial part of aggregated LDL(−) remains soluble, whereas LDL(+) and LDL(total) precipitate (Fig. 1). This distinct behavior was not observed in previous studies that used other perturbation methods to induce aggregation of LDL(−) [17,18,27, 29,38,46]. However, the latest stages of LDL coalescence into lipid droplets were not reached in those studies. Therefore, thermal denaturation is suitable to analyze both early- and late-stage products of LDL remodeling, revealing unique aggregation properties of individual LDL subfractions.
We propose that minor LDL apolipoproteins, such as apoA-I and apoJ that are nearly absent from LDL(+) but are relatively abundant in LDL(−), help to stabilize the lipoprotein assembly against disintegration and precipitation during denaturation. Both apoA-I [26] and, to a lesser extent, apoJ are thermostable, and are significantly increased in LDL(−). The stabilizing effects of apoJ and apoA-I could be due to somewhat different mechanisms. ApoJ is an extracellular chaperone that protects proteins from stress-induced aggregation, including high temperature [49–51]. Therefore, apoJ could stabilize apoB against conformational changes that promote the heat-induced disintegration of LDL. A recent study reported that, compared to apoJ-containing LDL, apoJ-depleted LDL is more susceptible to enzyme-induced aggregation and apoB degradation by α-chemotrypsin [21]. Similarly, apoJ could slow down conformational changes and degradation of apoB and thereby stabilize LDL against thermal disruption.
The addition of apoA-I to LDL(+) retards LDL aggregation and fusion at early stages of the heat denaturation and LDL precipitation at late stages (Fig. 6). The inhibition of the heat-induced LDL aggregation by apoA-I agrees with previous studies by Khoo et al. [22] and by our team [52]; it results from the ability of apoA-I to bind to phospholipid surface and block the hydrophobic defects. Therefore, at early stages of thermal denaturation, apoA-I can protect LDL from aggregation and fusion, counterbalancing the destabilizing effects of NEFA that are progressively generated. In addition, we speculate that apoA-I and other exchangeable (water-soluble) apolipoproteins (such as apoE, apoCs, etc.) that are found in small amounts on LDL help solubilize lipids during thermal denaturation, leading to the formation of small lipoprotein-like complexes such as those observed in this (Fig. 3B, top and bottom panels) and in our previous studies [33,34]. Together, these effects strengthen the protective roles of these minor LDL apolipoproteins that collectively constitute just a few percent of the total protein content in LDL. In addition, the abnormal conformation of apoB on LDL(−) can potentially contribute to the unique behavior of this LDL subfraction during thermal denaturation.
5. Conclusions
Our results help identify key determinants for LDL aggregation, fusion and coalescence into lipid droplets, and reveal that LDL(−) presents an unique thermal denaturation behavior, with rapid non-sigmoidal initial aggregation, which is probably due to high baseline content of NEFA. Minor LDL(−) apolipoproteins, such as apoA-I and apoJ, help counterbalance this effect and protect particles from complete disintegration and coalescence into lipid droplets. Since apoA-I, apoJ and short-chain NEFA are water-soluble (exchangeable), they can potentially transfer from LDL(−) to LDL(+) and thereby influence the aggregation and precipitation of total LDL.
The physiological significance of these observations renders further investigation. It is clear that the increased initial susceptibility to aggregation and fusion is a pro-atherogenic trait of LDL(−), which reflects the property of NEFA as a potent lipoprotein fusogen [33]. This trait is partially counterbalanced by the protective effects of the exchangeable apolipoproteins, such as apoA-I and apoJ, that stabilize LDL(−) against fusion and coalescence into lipid droplets. The latter points to a fundamental anti-atherogenic role of these minor LDL apolipoproteins that help prevent the accumulation of LDL-derived cholesterol in atherosclerotic lesions.
Supplementary Material
Acknowledgments
We thank Dr. Mengxiao Lu for help at the early stages of this project. This work was supported by the Spanish Ministry of Health grant FIS/ISCIII PI13/00364 (JLS-Q) with FEDER funds, and by the National Institutes of Health grant GM067260 (OG, SJ and DLG). AR, MP-C, JO-L and JLS-Q are members of the Quality Research Group 2014 SGR 246 from Generalitat de Catalunya and of Cardiovascular Research Network (RIC, RD12/0042/0043) from FIS/ISCIII. AR is the recipient of a “Sara Borrell” post-doctoral contract (CD12/00439, Spanish Ministry of Health, FIS/ISCIII, Spain).
Abbreviations
- Apo
apolipoprotein
- α-CT
α-chymotrypsin
- CD
circular dichroism
- CE
cholesterol ester
- DAG
diacylglycerol
- GAGs
glycosaminoglycans
- LDL(−)
electronegative LDL
- LDL(+)
non-electronegative LDL
- LDL(total)
total plasma LDL containing both LDL(+) and LDL(−)
- LPC
lyso-phosphatidylcholine
- NGGE
non-denaturing polyacrylamide gradient gel electrophoresis
- NEFA
non-esterified fatty acids
- PC
phos-phatidylcholine
- PG
proteoglycans
- SM
sphingomyelin
- TEM
transmission electron microscopy
- TG
triacylglycerol
Footnotes
Transparency document: The Transparency document associated with this article can be found, in online version.
Appendix A. Supplementary data: Supplementary data to this article can be found online at http://dx. doi.org/10.1016/j.bbalip.2016.05.008.
References
- 1.Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998;9:471–474. doi: 10.1097/00041433-199810000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Williams KJ, Tabas I. Lipoprotein retention – and clues for atheroma regression. Arterioscler Thromb Vasc Biol. 2005;25:1536–1540. doi: 10.1161/01.ATV.0000174795.62387.d3. [DOI] [PubMed] [Google Scholar]
- 3.Oorni K, Pentikainen MO, Ala-Korpela M, Kovanen PT. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J Lipid Res. 2000;41:1703–1714. [PubMed] [Google Scholar]
- 4.Pentikainen MO, Oorni K, Ala-Korpela M, Kovanen PT. Modified LDL - trigger of atherosclerosis and inflammation in the arterial intima. J Intern Med. 2000;247:359–370. doi: 10.1046/j.1365-2796.2000.00655.x. [DOI] [PubMed] [Google Scholar]
- 5.Lu M, Gursky O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol Concepts. 2013;4:501–518. doi: 10.1515/bmc-2013-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hakala JK, Oorni K, Pentikainen MO, Hurt-Camejo E, Kovanen PT. Lipolysis of LDL by human secretory phospholipase A(2) induces particle fusion and enhances the retention of LDL to human aortic proteoglycans. Arterioscler Thromb Vasc Biol. 2001;21:1053–1058. doi: 10.1161/01.atv.21.6.1053. [DOI] [PubMed] [Google Scholar]
- 7.Hurt-Camejo E, Camejo G, Peilot H, Oorni K, Kovanen P. Phospholipase A(2) in vascular disease. Circ Res. 2001;89:298–304. doi: 10.1161/hh1601.095598. [DOI] [PubMed] [Google Scholar]
- 8.Lahdesmaki K, Plihtari R, Soininen P, Hurt-Camejo E, Ala-Korpela M, Oorni K, Kovanen PT. Phospholipase A(2)-modified LDL particles retain the generated hydrolytic products and are more atherogenic at acidic pH. Atherosclerosis. 2009;207:352–359. doi: 10.1016/j.atherosclerosis.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 9.Oorni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles. Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. 1998;273:29127–29134. doi: 10.1074/jbc.273.44.29127. [DOI] [PubMed] [Google Scholar]
- 10.Oorni K, Sneck M, Bromme D, Pentikainen MO, Lindstedt KA, Mayranpaa M, Aitio H, Kovanen PT. Cysteine protease cathepsin F is expressed in human atherosclerotic lesions, is secreted by cultured macrophages, and modifies low density lipoprotein particles in vitro. J Biol Chem. 2004;279:34776–34784. doi: 10.1074/jbc.M310814200. [DOI] [PubMed] [Google Scholar]
- 11.Lu M, Gantz DL, Herscovitz H, Gursky O. Kinetic analysis of thermal stability of human low density lipoproteins: a model for LDL fusion in atherogenesis. J Lipid Res. 2012;53:2175–2185. doi: 10.1194/jlr.M029629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bancells C, Benitez S, Jauhiainen M, Ordonez-Llanos J, Kovanen PT, Villegas S, Sanchez-Quesada JL, Oorni K. High binding affinity of electronegative LDL to human aortic proteoglycans depends on its aggregation level. J Lipid Res. 2009;50:446–455. doi: 10.1194/jlr.M800318-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Estruch M, Sanchez-Quesada JL, Ordonez Llanos J, Benitez S. Electronegative LDL: a circulating modified LDL with a role in inflammation. Mediat Inflamm. 2013;2013:181324. doi: 10.1155/2013/181324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez-Quesada JL, Benitez S, Ordonez-Llanos J. Electronegative low-density lipoprotein. Curr Opin Lipidol. 2004;15:329–335. doi: 10.1097/00041433-200406000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez-Quesada JL, Estruch M, Benítez S, Ordonez-Llanos J. Electronegative LDL: a useful biomarker of cardiovascular risk? Clin Lipidol. 2012;7:345–359. [Google Scholar]
- 16.Sanchez-Quesada JL, Villegas S, Ordonez-Llanos J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr Opin Lipidol. 2012;23:479–486. doi: 10.1097/MOL.0b013e328357c933. [DOI] [PubMed] [Google Scholar]
- 17.Bancells C, Villegas S, Blanco FJ, Benitez S, Gallego I, Beloki L, Perez-Cuellar M, Ordonez-Llanos J, Sanchez-Quesada JL. Aggregated electronegative low density lipoprotein in human plasma shows a high tendency toward phospholipolysis and particle fusion. J Biol Chem. 2010;285:32425–32435. doi: 10.1074/jbc.M110.139691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parasassi T, De Spirito M, Mei G, Brunelli R, Greco G, Lenzi L, Maulucci G, Nicolai E, Papi M, Arcovito G, Tosatto SC, Ursini F. Low density lipoprotein misfolding and amyloidogenesis. FASEB J. 2008;22:2350–2356. doi: 10.1096/fj.07-097774. [DOI] [PubMed] [Google Scholar]
- 19.Brunelli R, De Spirito M, Mei G, Papi M, Perrone G, Stefanutti C, Parasassi T. Misfolding of apoprotein B-100, LDL aggregation and 17-beta-estradiol in atherogenesis. Curr Med Chem. 2014;21:2276–2283. doi: 10.2174/0929867321666140120114944. [DOI] [PubMed] [Google Scholar]
- 20.Bancells C, Canals F, Benitez S, Colome N, Julve J, Ordonez-Llanos J, Sanchez-Quesada JL. Proteomic analysis of electronegative low-density lipoprotein. J Lipid Res. 2010 doi: 10.1194/jlr.M009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Bujidos M, Rull A, Gonzalez-Cura B, Perez-Cuellar M, Montoliu-Gaya L, Villegas S, Ordonez-Llanos J, Sanchez-Quesada JL. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2015;29:1688–1700. doi: 10.1096/fj.14-264036. [DOI] [PubMed] [Google Scholar]
- 22.Khoo JC, Miller E, McLoughlin P, Steinberg D. Prevention of low density lipoprotein aggregation by high density lipoprotein or apolipoprotein A-I. J Lipid Res. 1990;31:645–652. [PubMed] [Google Scholar]
- 23.Benjwal S, Verma S, Rohm KH, Gursky O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006;15:635–639. doi: 10.1110/ps.051917406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao X, Yuan S, Jayaraman S, Gursky O. Differential stability of high-density lipoprotein subclasses: effects of particle size and protein composition. J Mol Biol. 2009;387:628–638. doi: 10.1016/j.jmb.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guha M, Gursky O. Human plasma very low-density lipoproteins are stabilized by electrostatic interactions and destabilized by acidic pH. J Lipids. 2011;2011:493720. doi: 10.1155/2011/493720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gursky O. Structural stability and functional remodeling of high-density lipoproteins. FEBS Lett. 2015;589:2627–2639. doi: 10.1016/j.febslet.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez-Quesada JL, Camacho M, Anton R, Benitez S, Vila L, Ordonez-Llanos J. Electronegative LDL of FH subjects: chemical characterization and induction of chemokine release from human endothelial cells. Atherosclerosis. 2003;166:261–270. doi: 10.1016/s0021-9150(02)00374-x. [DOI] [PubMed] [Google Scholar]
- 28.Benitez S, Villegas V, Bancells C, Jorba O, Gonzalez-Sastre F, Ordonez-Llanos J, Sanchez-Quesada JL. Impaired binding affinity of electronegative low-density lipoprotein (LDL) to the LDL receptor is related to nonesterified fatty acids and lysophosphatidylcholine content. Biochemistry. 2004;43:15863–15872. doi: 10.1021/bi048825z. [DOI] [PubMed] [Google Scholar]
- 29.Bancells C, Benitez S, Villegas S, Jorba O, Ordonez-Llanos J, Sanchez-Quesada JL. Novel phospholipolytic activities associated with electronegative low-density lipoprotein are involved in increased self-aggregation. Biochemistry. 2008;47:8186–8194. doi: 10.1021/bi800537h. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez-Quesada JL, Benitez S, Otal C, Franco M, Blanco-Vaca F, Ordonez-Llanos J. Density distribution of electronegative LDL in normolipemic and hyperlipemic subjects. J Lipid Res. 2002;43:699–705. [PubMed] [Google Scholar]
- 31.Wallace BA, Mao D. Circular dichroism analyses of membrane proteins: an examination of differential light scattering and absorption flattening effects in large membrane vesicles and membrane sheets. Anal Biochem. 1984;142:317–328. doi: 10.1016/0003-2697(84)90471-8. [DOI] [PubMed] [Google Scholar]
- 32.Jayaraman S, Gantz D, Gursky O. Structural basis for thermal stability of human low-density lipoprotein. Biochemistry. 2005;44:3965–3971. doi: 10.1021/bi047493v. [DOI] [PubMed] [Google Scholar]
- 33.Jayaraman S, Gantz DL, Gursky O. Effects of phospholipase A(2) and its products on structural stability of human LDL: relevance to formation of LDL-derived lipid droplets. J Lipid Res. 2011;52:549–557. doi: 10.1194/jlr.M012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guha M, England C, Herscovitz H, Gursky O. Thermal transitions in human very-low-density lipoprotein: fusion, rupture, and dissociation of HDL-like particles. Biochemistry. 2007;46:6043–6049. doi: 10.1021/bi7001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Jayaraman S, Guha M, Wally J, Lu M, Atkinson D, Gursky O. Application of circular dichroism to lipoproteins: structure, stability and remodeling of good and bad cholesterol, Circular Dichroism: Theory and Spectroscopy. Nova Publishers. 2012:175–215. [Google Scholar]
- 36.Gursky O, Ranjana, Gantz DL. Complex of human apolipoprotein C-1 with phospholipid: thermodynamic or kinetic stability? Biochemistry. 2002;41:7373–7384. doi: 10.1021/bi025588w. [DOI] [PubMed] [Google Scholar]
- 37.Brunelli R, Balogh G, Costa G, De Spirito M, Greco G, Mei G, Nicolai E, Vigh L, Ursini F, Parasassi T. Estradiol binding prevents ApoB-100 misfolding in electronegative LDL(−) Biochemistry. 2010;49:7297–7302. doi: 10.1021/bi100715f. [DOI] [PubMed] [Google Scholar]
- 38.Greco G, Balogh G, Brunelli R, Costa G, De Spirito M, Lenzi L, Mei G, Ursini F, Parasassi T. Generation in human plasma of misfolded, aggregation-prone electronegative low density lipoprotein. Biophys J. 2009;97:628–635. doi: 10.1016/j.bpj.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bancells C, Canals F, Benitez S, Colome N, Julve J, Ordonez-Llanos J, Sanchez-Quesada JL. Proteomic analysis of electronegative low-density lipoprotein. J Lipid Res. 2010;51:3508–3515. doi: 10.1194/jlr.M009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benitez S, Sanchez-Quesada JL, Lucero L, Arcelus R, Ribas V, Jorba O, Castellvi A, Alonso E, Blanco-Vaca F, Ordonez-Llanos J. Changes in low-density lipoprotein electronegativity and oxidizability after aerobic exercise are related to the increase in associated non-esterified fatty acids. Atherosclerosis. 2002;160:223–232. doi: 10.1016/s0021-9150(01)00565-2. [DOI] [PubMed] [Google Scholar]
- 41.Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. 1999;19:2648–2658. doi: 10.1161/01.atv.19.11.2648. [DOI] [PubMed] [Google Scholar]
- 42.Jonsson-Rylander AC, Lundin S, Rosengren B, Pettersson C, Hurt-Camejo E. Role of secretory phospholipases in atherogenesis. Curr Atheroscler Rep. 2008;10:252–259. doi: 10.1007/s11883-008-0039-6. [DOI] [PubMed] [Google Scholar]
- 43.Pentikainen MO, Hyvonen MT, Oorni K, Hevonoja T, Korhonen A, Lehtonen-Smeds EM, Ala-Korpela M, Kovanen PT. Altered phospholipid-apoB-100 interactions and generation of extra membrane material in proteolysis-induced fusion of LDL particles. J Lipid Res. 2001;42:916–922. [PubMed] [Google Scholar]
- 44.Siefert SA, Sarkar R. Matrix metalloproteinases in vascular physiology and disease. Vascular. 2012;20:210–216. doi: 10.1258/vasc.2011.201202. [DOI] [PubMed] [Google Scholar]
- 45.Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med. 2001;11:93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Bujidos M, Rull A, Gonzalez-Cura B, Perez-Cuellar M, Montoliu-Gaya L, Villegas S, Ordonez-Llanos J, Sanchez-Quesada JL. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2015 doi: 10.1096/fj.14-264036. in press. [DOI] [PubMed] [Google Scholar]
- 47.Bancells C, Benitez S, Ordonez-Llanos J, Oorni K, Kovanen PT, Milne RW, Sanchez-Quesada JL. Immunochemical analysis of the electronegative LDL subfraction shows that abnormal N-terminal apolipoprotein B conformation is involved in increased binding to proteoglycans. J Biol Chem. 2011;286:1125–1133. doi: 10.1074/jbc.M110.175315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parasassi T, Bittolo-Bon G, Brunelli R, Cazzolato G, Krasnowska EK, Mei G, Sevanian A, Ursini F. Loss of apoB-100 secondary structure and conformation in hydroperoxide rich, electronegative LDL(−), Free Radic. Biol Med. 2001;31:82–89. doi: 10.1016/s0891-5849(01)00555-x. [DOI] [PubMed] [Google Scholar]
- 49.Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. 2000;25:95–98. doi: 10.1016/s0968-0004(99)01534-0. [DOI] [PubMed] [Google Scholar]
- 50.Caccamo AE, Desenzani S, Belloni L, Borghetti AF, Bettuzzi S. Nuclear clusterin accumulation during heat shock response: implications for cell survival and thermo-tolerance induction in immortalized and prostate cancer cells. J Cell Physiol. 2006;207:208–219. doi: 10.1002/jcp.20561. [DOI] [PubMed] [Google Scholar]
- 51.Humphreys DT, Carver JA, Easterbrook-Smith SB, Wilson MR. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J Biol Chem. 1999;274:6875–6881. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- 52.Bancells C, Sanchez-Quesada JL, Birkelund R, Ordonez-Llanos J, Benitez S. HDL and electronegative LDL exchange anti- and pro-inflammatory properties. J Lipid Res. 2010;51:2947–2956. doi: 10.1194/jlr.M005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.