

Abstract
Characterization of the second nucleotide-binding domain (NBD2) of the cystic fibrosis transmembrane conductance regulator (CFTR) has lagged behind research into the NBD1 domain, in part because NBD1 contains the F508del mutation, which is the dominant cause of cystic fibrosis. Research on NBD2 has also been hampered by the overall instability of the domain and the difficulty of producing reagents. Nonetheless, multiple disease-causing mutations reside in NBD2, and the domain is critical for CFTR function, because channel gating involves NBD1/NBD2 dimerization, and NBD2 contains the catalytically active ATPase site in CFTR. Recognizing the paucity of structural and biophysical data on NBD2, here we have defined a bioinformatics-based method for manually identifying stabilizing substitutions in NBD2, and we used an iterative process of screening single substitutions against thermal melting points to both produce minimally mutated stable constructs and individually characterize mutations. We present a range of stable constructs with minimal mutations to help inform further research on NBD2. We have used this stabilized background to study the effects of NBD2 mutations identified in cystic fibrosis (CF) patients, demonstrating that mutants such as N1303K and G1349D are characterized by lower stability, as shown previously for some NBD1 mutations, suggesting a potential role for NBD2 instability in the pathology of CF.
Keywords: ABC transporter, cystic fibrosis transmembrane conductance regulator (CFTR), protein engineering, protein misfolding, protein stability
Introduction
Cystic fibrosis (CF)2 is an autosomal recessive genetic disorder caused by loss-of-function mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) (1), an integral membrane protein that consists of two transmembrane domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2), and an intrinsically disordered regulatory (R) region (1–3). CFTR functions as a passive chloride channel (4), with expression and gating activity regulated by phosphorylation of the R region (5–7) and nucleotide binding and hydrolysis, coupled to NBD dimerization (8–11).
CFTR is the only known channel in the ATP-binding cassette (ABC) transporter superfamily (12, 13), a set of homologous proteins that all share ancestry in at least their NBDs. This family has a consistent functional mechanism, in which ATP-binding events occurring in two NBDs allow them to interact across a highly conserved dimerization interface, with the binding, dimerization, hydrolysis, and finally dissociation of the NBDs being allosterically coupled to motion of the TMDs (14). Within the ABC superfamily, the TMDs are structurally diverse, and the mechanisms for coupling to the NBDs are not universally conserved but, in transporters with CFTR-like TMDs, coupling is mediated through NBD interactions with helical extensions of the TMDs known as the intracellular domains (ICDs) (15–19).
NBD1 of CFTR has been more intensively studied than NBD2, in part because it contains the Phe-508 residue that is frequently deleted in CF patients. Deletion of Phe-508 is known to destabilize the isolated NBD1 (20, 21), reduce its ability to form NBD dimers (22), and also reduce intramolecular interactions between NBD1 and the ICDs (23, 24), resulting in global destabilization of CFTR. Two disease-causing NBD2 mutations, N1303K and p.Ile1234_Arg1239del, which are known to hinder processing of CFTR (25, 26), suggest a similar destabilization mechanism involving NBD2. Another well-studied NBD1 mutation is the G551D channel-gating mutation, which matures with similar efficiency as WT but does not gate efficiently. The G1244E, S1251N, S1255P, and G1349D mutations in NBD2 also do not disrupt CFTR maturation and are considered to be gating mutations (27). Inopportunely, the isolated NBD2 domain has proven to be even more difficult to work with than NBD1, hampering study of these NBD2 mutations by biophysical measurements. As a result, the mechanism by which pathogenic NBD2 mutations cause disease is less well understood than for analogous NBD1 mutations.
Previous attempts to stabilize NBD2 constructs capitalized on the relationship between sequence consensus and stability primarily by taking mutations from non-human CFTR sequences or other close homologs in combination with rational design. A range of constructs was produced and screened, ultimately resulting in a crystal structure (PDB code 3GD7) (28) of a construct in which the conserved C-terminal residues of the NBD2 were replaced with a portion of MalK, an unrelated protein domain used to stabilize NBD2. Despite this success, these constructs have been difficult to work with for determination of binding interactions, functional measurements, or NMR studies.
We decided to identify potential NBD2-stabilizing mutations by analyzing a comprehensive set of sequences from the ABC transporter superfamily to measure broader trends in ancestral sequence conservation across structurally similar but functionally diverse homologs, capturing data on essential functions that are not unique or specific to CFTR, and defining the consensus sequence of their shared structure. As a general rule in protein design, mutations toward the consensus sequence of a given fold are often stabilizing (29–32), but identifying back-to-consensus mutations requires a diverse population of sequences to avoid phylogenic bias (33, 34). Thus, consensus-based methods benefit when functionally diverse but structurally similar proteins can be included in the analysis.
Incorporating functionally diverse homologs from the greater ABC transporter family into our analysis requires an understanding of how these sequences are related to each other. The 48 human members of the ABC transporter superfamily can be divided into distinct clades defined as ABC subfamilies, with CFTR being the 7th member of the C subfamily (ABCC7) (13). All 12 of these subfamily C genes encode a full transporter (or channel) containing homologs of the four folded domains found in CFTR, arranged in the order TMD1-NBD1-TMD2-NBD2. The next closest clade is subfamily B, which consists of four full transporters and seven half-transporters, the latter composed of single TMD-NBD sets that can homo- or heterodimerize to form a full transporter.
The common ancestor of both subfamilies is thought to have coded for a half-transporter, with full transporters arising from multiple domain duplication events. The full transporters in subfamily B originate from a more recent domain duplication event than the one in subfamily C (13). This is relevant to sequence analysis because it means that the conservation seen in half-transporters can become asymmetric in full transporters, with functions that are only required in one domain being lost in the symmetrical partner. This asymmetry is a common feature of many full-length ABC proteins, a notable example being the degenerate ATP-binding site in CFTR NBD1. Conservation in half-channels/transporters then provides a control for identifying this asymmetric divergence.
The most recent ancestral events separating CFTR from the other members of subfamily C were the loss of transporter activity and the insertion of the ∼200-residue-long R region between NBD1 and TMD2. This intrinsically disordered phosphoregulatory region is thought to have originated from a stretch of non-coding DNA (35). Consequently, it has no homology to any other human proteins, and the effects its interdomain interactions have on the evolution of the folded domains of CFTR are unique to the gene.
ABC subfamilies B and C all still share the same basic set of folded domains, and maintaining the structural stability of those domains is one of the consistent selective pressures involved in defining the consensus sequence. However, protein stability is a complex phenotype for which there is typically no fitness benefit in going beyond the marginal stability a protein requires (36, 37). This means mutations that increase stability are not always universally conserved. Relative effects on stability do have fitness benefits, and they do impart a bias toward conservation, so stabilizing residues are more likely to be found within the more common identities found at a given position.
In this study, we describe a method to identify stabilizing mutations using a large-scale alignment of ABC transporters, including calculations of the energetic contribution of point mutations and structural analysis, which we applied to the NBD2 domain of CFTR. By combining our mutations with previously identified mutations, we successfully developed a range of stable and soluble NBD2 constructs with minimal mutations from wild type. Introducing these mutations into full-length CFTR demonstrated that the stabilizing mutations have a minimal effect on efficient processing of CFTR, confirming that NBD2 containing these mutations is a useful reagent for biophysical characterization. The critical stabilizing mutation S1359A was also shown to not alter the gating properties of CFTR. The new mutations allowed us to measure the thermal stability of NBD2 and the effect of disease-causing mutations on stability. Our results demonstrate that some NBD2 disease-causing mutations likely contribute to CF disease by destabilizing the isolated NBD2 domain, including some mutations that do not appear to affect the maturation of full-length CFTR. Other disease-causing mutations, which have previously been shown to be gating mutations, have a minimal effect on NBD2 stability.
Results
We undertook a bioinformatics assessment of sequence conservation in CFTR and ABC transporters with homologous transmembrane domains (subfamilies C and B) to characterize the sequence conservation and consensus properties of their shared fold, with the primary goal of providing statistical tools for predicting the effect a given mutation has both on the stability of CFTR and on potential CFTR-specific functions. Many techniques exist for predicting stabilizing mutations (38–43), but our goal was also an understanding of wild-type CFTR behavior and allosteric control of NBD dimerization. This calls for tools designed to identify a small population of mutations that are unlikely to interfere with CFTR-specific functions. To this end, we developed a back-to-consensus scoring approach that identifies consensus positions and applied this tool to the production of a stabilized NBD2 reagent for in vitro characterization of NBD2.
Consensus definition
To define the ABC(C/B) consensus sequence, we extracted a comprehensive list of complete sequences from the NCBI non-redundant sequence database (44), screening against partial sequences and clustering by their relationships to the 23 discrete human ABC(C/B) transporters, as described under “Experimental procedures.” All ABC(C) and 7/11 of the ABC(B) associated sequences have duplicate copies of the shared NBD fold, so to enable comparison of conservation patterns by position (NBD1 versus NBD2), we then extracted half-transporter/channel alignments, containing just one TMD-NBD pair, from every full transporter/channel group, and we re-aligned our full set of ABC(C/B) half-transporters against each half-channel set of CFTR sequences.
By comparing the two NBDs in CFTR separately to other ABC transporters, we were able to measure both how often a given residue identity occurs simultaneously in the two NBDs (symmetric conservation) and how often a given residue occurs in at least one of the NBDs but not necessarily the other (asymmetric conservation). These measurements are intended to separate invariant residues, likely to be involved in basic functions common to all NBDs, from residues involved in functions that are only required in one NBD, and which can be lost after a domain duplication event. By treating three residue type pairs as identical (Asp = Glu, Arg = Lys, and Ser = Thr) and selecting residues that show high conservation in at least one NBD, we isolated 36 residue positions that show evidence of being essential within all ABC(C/B) sequences, being consistently identified in at least 95% of the sequences within every subset of ABC(C/B) sequences. Of these positions, 9 are symmetrically conserved in both halves of all full transporters/channels, and the remaining 27 show varying degrees of asymmetric conservation, with 22 showing <90% identity at the second position in ABC(C) sequences, and 9 of those showing less than 25% identity. These 36 positions are summarized in Fig. 1A.
Figure 1.

ABC(C/B) sequence conservation profiles. Sequence profiles in weblogo (65, 66) format are shown for the 36 highly conserved ABC(C/B) positions in A and for a selection of additional positions discussed in this study in B. Each column of the sequence data shows amino acid frequencies from a different subset of the data, from left to right, ABC(B) half-transporters, NBD1 of ABC(B) full transporters, NBD2 of ABC(B) full transporters, NBD1 of ABC(C) transporters, NBD2 of ABC(C) transporters, NBD1 of CFTR, and NBD2 of CFTR. Amino acid frequencies are shown by the relative size of the letter, with the consensus identity at the top. Rows are labeled with functional or motif annotations and with the CFTR mutations occurring at each position; with CF-causing mutants from the CFTR2 data set shown in red, CFTR2 mutations that do not cause CF shown in black, and stabilizing mutations found either in the literature or in this study shown in blue. C, Asymmetric Conservation Bias in NBD1 and NBD2. Structural representation of CFTR NBD2 (from PDB code 3GD7 at 2.6 Å resolution). The helical subdomain is pink with helix 5 in brown; the α-β subdomain is blue, and motifs have been highlighted with the signature sequence in brown; the Walker A motif in red; the Walker B motif in green, and the Q loop in yellow. Positions where highly conserved residues can be lost in one NBD and that have a tendency to be lost in the same NBD when comparing paralogous sequences from subfamily C and/or subfamily B are identified. Positions biased toward being lost in NBD1 are noted by red spheres, clustering primarily around the ATP-binding site, and those biased toward being lost in NBD2 are noted in blue, clustering primarily around the helix-5 signature sequence.
Further inspection of these selected residues reveals that 20 of the 36 exist within or are adjacent to known and well-defined motifs, including the Walker A, Walker B, and the signature sequence. Of the remaining 16 there are five positions with clear structural relevance, with three conserved glycine positions found in tight turns between secondary structure elements, and two conserved helix caps. A further five of the 16 occur within the binding pocket for the ICD, specifically to the residues known as the intracellular loops (ICLs) or coupling helices, leaving only six positions for which the potential function is not as easily explained. The five highly conserved consensus positions in the ICL-binding pocket point to the importance of this region in interactions with the ICDs.
Disease relevance and stability
To determine whether disease-causing mutations were more likely to be associated with changes at the 36 broadly conserved residues, we took the list of 17 known NBD-specific CF-causing missense mutations from CFTR2 (45), counting how often disease-causing substitutions occur at positions that are conserved in all ABC(C/B) homologs. We observed that 71% of the CF-causing mutations occur at one of the 36 positions identified, which is 4.6 times higher than the 14% expected if the distribution were random. Furthermore, when comparing sequence conservation of other ABC transporters at each position, we find that 16 of 17 CF-causing mutations are either substitutions not found in other ABC transporters (e.g. V520F, S1255P, L467P, and A455E) or represent moves away from one of the 36 broadly conserved identities. The lone exception, I1234V, is a special case, because that mutation (c.3700A→G) causes aberrant splicing resulting in six amino acids being deleted from the NBD2 domain (p.Ile1234_Arg1239del) (26, 46).
We also observed that four known CFTR NBD1-stabilizing mutations, S492P, S495P, A534P, and I539T, all occur at positions where human CFTR deviates from the consensus found for other ABC(C/B) domains, and they occur at positions where the NBD1 and NBD2 consensus diverged in an asymmetric fashion after their domain duplication events (Fig. 1B).
Asymmetrical conservation bias
One of the potential causes for asymmetric conservation in full transporters/channels, relative to half-transporters/channels, is a change in the required stability of the individual domains, given that each half of the fold has the potential to act as a folding scaffold to stabilize the other. With this in mind, we extracted statistics on asymmetric conservation from the 16 non-overlapping sets of sequences we associated with each human full-length transporter/channel, comparing the relationship between the global conservation of consensus identity with loss of consensus at NBD1 and NBD2, focusing on relative bias both between independent sets of sequences and, overall, between ABC(B) and ABC(C) full transporters/channels. The full transporters found in these two families appear to originate from independent domain duplication events, so observing the same bias in both families supports the hypothesis that the observed bias results from similar functional or structural constraints on the residues, rather than their phylogenic relationships.
To determine the patterns of NBD1 versus NBD2 conservation bias, we measured consensus sequence frequencies independently for each of the four ABC(B) full transporters and 11 non-CFTR ABC(C) full transporters, calculating the rate of observing at least one NBD matching the consensus for these 15 non-overlapping sets of sequences and measuring both the conservation and reliability of the consensus sequence by the number of independent ABC(C/B) full transporter/channel sets with ≥98% match to the consensus. This cutoff was chosen to identify invariant consensus sequences with room for sequencing errors. We then counted how often the consensus residue can be lost at each NBD (accounting for error by requiring at least 10 observations per set), and we defined the bias at each residue position by the number of ABC(C) full transporters/channels showing loss of consensus in NBD1 minus the number showing loss of consensus in NBD2.
In comparing the patterns of conservation and bias, we observe that ABC(C) and ABC(B) full transporters/channels show similar bias toward losing consensus residues (supplemental Fig. S1), with loss in NBD1 occurring primarily in the ATP-binding site, with losses observed in Walker A sites (corresponding to positions 458–466 in NBD1 versus 1244–1252 in NBD2), the ATP-binding aromatic side chain at Trp-401 (versus NBD2 position 1219), and the region of the Q-loop that is proximal to the binding pocket (Fig. 1C). In contrast, residues biased to being lost in NBD2 primarily occur in the vicinity of the signature sequence (corresponding to positions 1346–1350 in NBD2 versus 548–552 in NBD1), which in the context of a head-to-tail dimer puts them in contact with NBD1's ATP-binding site, such that the pattern across both NBDs can be described as a loss of consensus that is biased toward the first ATP-binding pocket.
This bias appears to have arisen independently over two distinct duplication events and matches what is observed in CFTR (Fig. 1A). The observation that the first NBD to fold is the one more likely to lose catalytic activity is consistent with the hypothesis that NBD1 can act as a scaffold for stabilizing NBD2 as it folds, where a loss of ATP hydrolysis may result in an NBD1 that remains in an ATP-bound and dimer-compatible state while forming interactions with a partially folded NBD2. This suggests that NBD2 may have lost independent stability.
Mutation screening
Based on this possibility that the sequence divergence between NBD1 and NBD2 represents a stabilizing role for NBD1 in NBD2 folding, with NBD1 more likely to have a bias toward independent stability, we defined our “back-to-consensus” score function. This score is derived from the conservation of the substituted residue in any NBD of any ABC(C/B) transporter sequence, selectively up-weighting CFTR conservation and NBD1 conservation by treating CFTR NBD frequencies independently and then subtracting the overall conservation observed for the WT CFTR identity. The latter avoids residues for which NBD2 already matches the consensus. This is summarized in Equation 1,
| (Eq. 1) |
where X is the residue in human CFTR; Y is the substitution; and CN1, CN2, and A are the residue probabilities at a given sequence position for CFTR NBD1, CFTR NBD2, and either half of any non-CFTR ABC transporter in subfamilies B and C, respectively. To find potential stabilizing mutations within NBD2, this equation was used to score every possible NBD2 mutation, generating a ranked list of candidate mutations.
We had two goals, the first being to test whether or not these back-to-consensus mutations have a stabilizing effect, and the second being to produce a soluble NBD2 construct to be used for biophysical studies. These included testing the potential destabilization of disease-causing mutations and exploring interactions with cytosolic binding partners, such as NBD1 and the R region. Thus, we utilized two additional criteria for selecting candidate mutations. The first was distance from potential interaction surfaces, as assessed by visual inspection of the NBD2 structure (PDB code 3GD7). Thus, for example, mutations directly in the canonical NBD1/NBD2 interface were avoided. The second criterion was our ability to identify a potential rational basis for stabilization, either by manual inspection of crystal structures or by in silico analysis of the mutation using free energy of unfolding (ΔΔG) prediction.
For the ΔΔG prediction, we desired mutations that could be computationally predicted as either stabilizing or neutral, given the crystal structure, and for this analysis we used the Rosetta3.4 ΔΔG prediction with the fixed-backbone protocol (47) to model all possible substitutions from NBD2 wild type into each unique chain of the structure. The current generation of ΔΔG predictors can have large prediction errors, so rather than incorporating the predictions into the back-to-consensus score, these values were used simply to inform our manual analysis, with unreasonable predictions being largely ignored.
Selected mutations
We selected a total of four single-site substitutions with high back-to-consensus scores for screening: S1255L, K1292D, S1359A, and K1334G (supplemental Table S1). Fig. 2A shows these positions in the context of the NBD2 structure. Of these, S1359A was the most obvious choice, with the second highest return-to-consensus score and a stabilizing ΔΔG prediction of −1.5 kcal/mol. During manual inspection of the crystal structure, we also noted that the Ser-1359 residue, rather than being solvated, forms a hydrogen bond with Gln-1291 in the Q-loop, which appears to pull Gln-1291 away from the ATP-binding pocket, potentially reducing the affinity of NBD2 for ATP (Fig. 2B). Comparison of the glutamine position in the ATP-bound state of CFTR NBD2 (Gln-1291) and the multidrug resistance protein 1 (MRP1) NBD1 (PDB code 2CBZ and Gln-713) (Fig. 2C) (48) shows Gln-713, the homologous position to CFTR's Gln-1291, pointing to the upper left into the ATP-binding pocket. This makes room for a network of water-mediated contacts between helix five, residues in or adjacent to the Q-loop, and the Walker B motif, interactions that, in CFTR, are occluded by Gln-1291 when hydrogen-bonded to Ser-1359. Most of these contacts are lost in the CFTR NBD2 structure, suggesting a larger network of water-mediated interactions involving Arg-1258 and Gln-1352 that might be recovered if the hydrogen bond with Ser-1359 is disrupted by mutation and Gln-1291 is able to flip into a canonical position.
Figure 2.

Deviations from wild-type versus structure. A, positions in CFTR NBD2 of relevance to this study have been noted with spheres, with solubilizing mutations from the SGX 5sol background construct in black (five positions), our selection of new solubilizing mutations in blue (four positions), and positions of CF-causing mutations in red (four positions). Notably, N1303K is the sole CF-causing NBD2 substitution that is not in or adjacent to one of the named sequence motifs. B, expanded view of the polar contacts (yellow lines) and water molecules (small red spheres if found in PDB and red star if predicted by structure) that connect Asn-1303, helix 5, and the Q-loop. In this structure, Ser-1359 contacts the Gln-1291 directly, pulling it away from the ATP-binding pocket. C, homologous view for the NBD1 of Multidrug Resistance Protein 1 (PDB code 2CBZ at 1.7 Å resolution). The Ser-1359 analog in this structure, Ala-781, fits the ABC(C/B) consensus sequence. With alanine at this position, we see Gln-713, the Gln-1291 equivalent, extending out away from helix 5 and into the ATP-binding pocket. This change appears to open up space for a much larger network of water-mediated polar contacts underlying the Q-loop, and it coincides with a structural rearrangement enabling direct contact between helix 5 (through Gln-774) to a backbone oxygen adjacent to the Walker B motif. (Structural representations are colored as in Fig. 1.)
The remaining three mutations, which had more moderate back-to-consensus scores, were chosen due to their distance from the dimerization interface. K1334G is a return-to-ABC consensus, while also having a stabilizing ΔΔG prediction, making it a fairly obvious choice as a stabilizing mutation. K1292D and K1292E both had positive back-to-consensus scores. This position is the residue after Gln in the Q-loop, and it could play a role in ATP binding or dimerization, supporting testing of the K1292D mutation. S1255L represents a return to the ABC(C) consensus specifically for NBD2, and it was chosen because Leu also matches the identity found at NBD1 in CFTR, despite the fact that S1255M had a better overall score and ΔΔG prediction.
First round of mutation
A previously generated NBD2 construct, SGX 5sol (described under “Experimental procedures”), which is more soluble than wild type and was developed during the course of producing the NBD2 crystal structure (PDB code 3GD7), was used as the background for an initial round of mutagenesis. Two point mutations, K1292D and S1359A, were tested that we suspected would greatly improve NBD2 stability. Expression levels improved for both mutations (with the rough order S1359A > K1292D > SGX5 sol), but attempts to measure melting transitions (Tm values) using the SYPRO-Orange binding assay showed inconsistent melting transitions below 40 °C (supplemental Table S1 and supplemental Fig. S2), consistent with aggregation.
We then screened using different ATP/Mg2+ concentrations looking for conditions that might reduce aggregation, and we found that for the highest expression construct, S1359A, increasing the ATP/Mg2+ concentrations to 10 mm produced a consistent Tm value of 40 °C using the SYPRO-Orange binding assay, whereas the other constructs remained aggregation-prone (supplemental Fig. S2). Although the other constructs did not show clear melting transitions at higher ATP, the apparent transitions did appear to increase, albeit with all transitions remaining below 40 °C. This suggested that ATP's stabilizing effect is general across constructs and that reliable observation of a two-state melting transition is only possible when NBD2 reaches a stability threshold.
Sequential stabilization screens
Using the Tm value from SYPRO-Orange assays as a direct readout of construct stability, we undertook four additional rounds of single-point mutagenesis, using the most stable construct in each round as the background for the next round of point mutations. Starting in the third round we also introduced point mutations to revert the SGX 5sol background back to wild type. This was followed with another two rounds of mutagenesis to test the effects of each SGX 5sol mutation and provide a stable construct as close as possible to wild type with an intact catalytic site. The progression of average observed Tm values for both addition and reversion as well as a full description of the mutations attempted and constructs tested in each round is described in supplemental Tables S1 and S2. The three additional back-to-consensus mutations, S1255L, K1292D, and K1334G, enhanced stability between 2 and 4 °C. These, together with S1359A and the SGX 5sol mutations, yielded the most stable construct, 9sol, with a final SYPRO-Orange measured Tm value of 49 °C, ∼10 °C higher than SGX 5sol (supplemental Table S2).
Buffer conditions were varied between rounds (as described under “Experimental procedures”) in an attempt to address potential buffer effects on aggregation during purification, with SYPRO-Orange ΔTm measurements controlled internally during each round by comparison with a defined background. To address buffer effects on Tm measurements, we retested select constructs in subsequent rounds to confirm the stabilizing effects of the mutation in the new buffer conditions. Tm value differences associated with changes in buffer conditions ranged from 0 to 3 °C (supplemental Table S1). Of note, the ΔTm of point mutations relative to each other were largely consistent between rounds despite differing buffers, suggesting the mutation effects are typically buffer independent, with the H1402A mutation in the ATP-binding site being a notable lone exception. We note this is similar to the published observation that F508del has a consistent effect on NBD1 stability independent of additional background mutations (20).
During the course of the screens, we also observed that mutations dramatically decreasing expression (Table S1) almost always returned the protein to an aggregation-prone state where SYPRO-Orange assays result in large number of inconsistent transitions rather than a single defined Tm (as in supplemental Fig. S2). In some instances, we were able to recover the ability to measure a discrete SYPRO-Orange Tm by repeating the same destabilizing mutation in a higher Tm value background. These results provide evidence that loss of the ability to measure consistent SYPRO-Orange ΔTm values reflects significant destabilization of the protein.
Of the SGX 5sol mutations, only H1402A, Q1411D, and L1436D were found to have a large effect on protein stability, whereas Q1280E and Y1307N were marginally stabilizing and destabilizing, respectively (supplemental Table S2). Of these mutations, the functional role of only H1402A is known. It results in the loss of a catalytic histidine involved in ATP hydrolysis and potentially increases stability by maintaining the bound ATP. Ignoring this catalytic residue, two of the other four SGX 5sol mutations scored highly in our back-to-consensus function, satisfying the criteria we used to select potential stabilizing mutations. Q1411D had the highest back-to-consensus score of all the mutants tested (supplemental Table S1), and in the end it was the only stabilizing mutation for which we were not able to obtain a ΔTm value, because reintroduction of Gln at this position rendered constructs too unstable to test both times it was attempted. Q1280E and Y1307N had marginal back-to-consensus values, in line with their marginal effects on stability.
Structural characterization
We purified 15N-labeled samples of our most stable construct, with a total of nine mutations from wild type (9sol), five inherited from the SGX 5sol background, and we recorded NMR experiments. The spectral quality of this construct was dramatically improved relative to the SGX 5sol construct (Fig. 3). The observed chemical shifts clearly indicate that NBD2 is folded in solution. Peaks with reasonable signal-to-noise were observed for ∼90% of the backbone N–H bonds. The spectral differences between these two constructs are minor indicating that both share the same fold as expected. Evidence of chemical exchange broadening is apparent, indicating that NBD2 is dynamic on an intermediate time-scale regime, exchanging between different states, potentially including various self-associated states.
Figure 3.
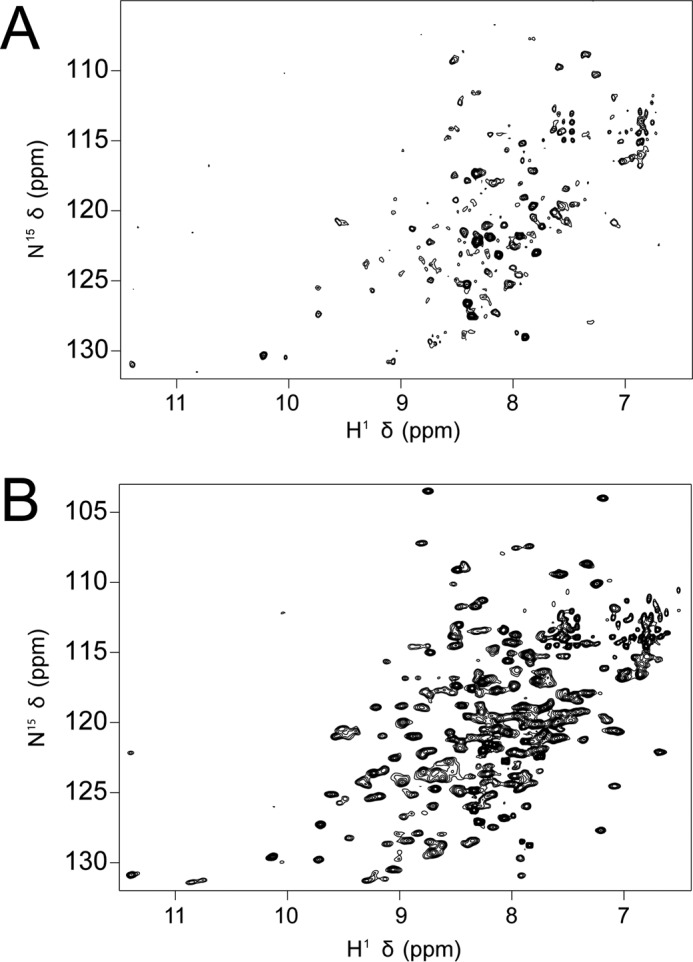
NMR spectra. NMR HSQC spectra were obtained for the SGX 5sol NBD2 background construct (A) and 9sol NBD2, our highest stability final construct (B). There is a 20-fold protein concentration difference between A and B, because the SGX 5sol construct was not soluble enough to produce an NMR sample with the concentration typically required. Thus, these spectra represent the best spectra we could produce using each construct, although they are not directly comparable. The SGX 5sol background has intense resolved peaks primarily in the disordered region of the protein, consistent with an aggregation-prone sample. In contrast, the 9sol sample has a larger number of dispersed peaks with a greater degree of uniformity in peak intensity, indicating predominantly monodisperse, folded protein, with peaks corresponding to the vast majority of resonances expected.
Expression and processing of CFTR NBD2 mutants
CFTR was transiently expressed in HEK293T cells and subsequently analyzed by Western blotting to assess the effects of these NBD2 mutations on full-length protein folding and trafficking. The wild-type and F508del proteins expressed robustly, showing differences in glycosylation state consistent with their differential subcellular localization (Fig. 4). The wild-type protein was detected as both the band B and band C forms, consistent with protein localizing in the endoplasmic reticulum and throughout the secretory and endosomal pathways. In contrast, the F508del protein showed only the band B form, demonstrating its loss of processing in the secretory pathway beyond the endoplasmic reticulum.
Figure 4.

Expression of CFTR with NBD2-stabilizing mutations. Western blottings of WT and F508del CFTR, with and without the S1359A NBD2-stabilizing mutation and other stabilizing mutations, are shown at the top of the figure. Following expression for 48–60 h, the lysates were analyzed by Western blotting using the mAb 660 α-CFTR antibody. The upper band in the WT samples corresponds to band C, and the lower band corresponds to band B. Tubulin (Tub) bands are shown as a loading control.
The NBD2-stabilizing mutations were introduced into both the wild-type and F508del backgrounds to assess how they might alter CFTR trafficking. The S1359A mutant had no observable effects on CFTR trafficking in either the wild-type or F508del backgrounds (Fig. 4). The trafficking efficiency of the wild-type protein did not appear to be adversely affected by the S1359A mutation. Similarly, the S1359A mutant did not restore or increase the trafficking of the F508del CFTR protein, as detected by Western blotting. The other NBD2 mutants were similarly assessed for their effects on wild-type and F508del trafficking in the presence and absence of the S1359A mutant. The S1255L, K1334G, and Q1411D mutants had no appreciable effect on the trafficking of the wild-type or F508del proteins either with or without the S1359A mutation. Similarly, the combination of mutations S1255L/K1334G or S1255L/K1334G/S1359A had no observable effects on either wild-type or F508del trafficking. In contrast, the K1292D mutant showed a decrease in trafficking efficiency in the wild type and wild-type/S1359A backgrounds. The introduction of the K1292D mutation decreased the quantity of band C protein relative to the observed band B. These data suggest that the K1292D mutant decreases the efficiency of CFTR processing in the secretory pathway and/or increases the rate of fully mature protein degradation.
Effect of S1359A in full-length CFTR
To confirm that the critical S1359A mutation does not interfere with WT channel function, this mutation was incorporated into full-length CFTR for characterization. S1359A did not appear to alter the expression of CFTR as judged by the mature protein band detected in a Western blotting after SDS-PAGE of microsomal membranes isolated from HEK293 cells in which the variant was transiently expressed (Fig. 5A). Both the electrophoretic mobilities and intensities of the major mature CFTR protein bands were very similar in cells expressing this variant and the wild-type protein. The minor more rapidly migrating immature band was more evident in the mutant than the wild-type sample, reflecting the fact that the former was being expressed transiently and the latter stably. Functionally, S1359A CFTR behaved virtually identically to the wild-type channel with a unitary conductance of 12.3 picosiemens and an open probability of 0.64 at 30 °C (Fig. 5B). Unaltered gating kinetics is indicated by the dwell-time histograms below the channel tracing showing that the channel mean open and closed times also were similar to those of the wild type under these conditions (11).
Figure 5.

Effect of S1359A on full-length CFTR. A, detection of S1359A and wild-type CFTR proteins in microsomal membranes of transfected HEK293 cells. 10 μg of total membrane protein were resolved by SDS-PAGE (7.5% acrylamide) and electroblotted onto a nitrocellulose membrane that was probed with primary mAb 596. B, single-channel recording after fusion of S1359A membrane vesicles with a planar lipid bilayer. The unitary conductance and open probability are indicated above the recording, and the amplitude histogram is shown to the left. Dwell time histograms below the recording were used to estimate mean closed and open times.
Stability of NBD2 disease-causing mutations
Given the observation that stabilized backgrounds allowed us to measure SYPRO-Orange ΔTm values for strongly destabilizing substitutions, we tested the effects of NBD2 missense substitutions observed in CF patients in the context of our most stable background. Eight mutations in the CFTR2 database were analyzed. G1244E, S1251N, S1255P, G1349D, and N1303K are classified as CF-causing mutations. D1270N is classified as being a mutation of varying clinical consequence. S1235R is classified as a non-CF-causing mutation. I1234V is classified as being CF-causing, but the missense substitution is likely not responsible, as the mutation (c.3700A→G) causes aberrant splicing, resulting in a six-residue deletion (p.Ile1234_Arg1239del) (26, 46).
Protein yield levels were recorded during purification of mutant NBD2 constructs expressed in bacteria. Relative to the solubilized control, the disease-relevant substitutions all showed a decrease in protein yields, whereas the sole non-CF-causing mutation in CFTR2, S1235R, had no effect on yield (supplemental Table 1). Similarly, I1234V had no effect on yield, but subsequent testing of the disease-relevant aberrantly spliced construct, p.Ile1234_Arg1239del, showed a severe loss of yield. Overall, the three most poorly expressed mutants were p.Ile1234_Arg1239del, D1270N, and N1303K. Notably, N1303K and p.Ile1234_Arg1239del are the only mutations confirmed to inhibit full maturation of CFTR to band C status (26, 49). During purification of isolated NBD2 on gel filtration columns, our low stability construct samples typically had components that eluted early, but ran at the expected molecular weight of NBD2 on SDS-polyacrylamide gels, consistent with a higher degree of NBD2 aggregation in these samples. Overall, higher stability constructs had a lower ratio of aggregate peak relative to monomer peak in gel filtration chromatography (data not shown).
The stabilities of these disease-causing mutants within the context of the NBD2 9sol construct were also measured using both differential scanning calorimetry (DSC) (Figs. 6 and 7A and Table 1) and circular dichroism (CD) spectroscopy (Fig. 7B and Table 2). Results from the two techniques were very similar, suggesting that both monitor the same thermal unfolding process. For these experiments, NBD2 was purified until only a single band was visible on Coomassie-stained SDS-polyacrylamide gels. We subjected the NBD2 9sol background construct to thermal melts at different ATP concentrations, because we suspected that ATP stabilizes NBD2. Indeed, increasing the ATP concentration from 2 to 10 mm enhanced stability by greater than 3 °C (Fig. 6A and Table 1). We next tested the protein concentration dependence of the NBD2 9sol construct. Plotting stability versus NBD2 concentration suggests either a small increase in stability with increasing concentration or no effect (Fig. 6B), with increased noise observed at lower concentrations hindering definitive interpretation.
Figure 6.

Effect of ATP concentration and protein concentration on NBD2 stability. Thermal melts were performed by DSC on the purified 9sol NBD2 in a buffer containing 20 mm NaPO4, 150 mm NaCl, 10% glycerol, 5 mm BME, 4 mm MgCl2, and ATP. A, effect of varying ATP concentrations on NBD2 stability, measured at 20 μm protein. B, effect of varying NBD2 concentrations on stability, measured at 10 mm ATP.
Figure 7.

Stability of disease-causing mutants. Thermal melts were performed on the purified 9sol NBD2 as background and three disease-causing mutant variants in buffer containing 20 mm NaPO4, 150 mm NaCl, 10% glycerol, 5 mm BME, 4 mm MgCl2 and ATP, G1244E, S1251N, and N1303K. A, thermal melt assays using DSC at two different ATP concentrations, with 10 mm in black and 4 mm in red. B, normalized thermal melts of the same set of proteins measured by CD at a wavelength of 230 nm in the same buffer as used for the DSC experiments. Protein concentration was 0.5 mg/ml (18 μm), and ATP concentration was 10 mm. Circles represent data points, and the red line represents the fit obtained by linear regression as described previously (63).
Table 1.
Thermal stability of human NBD2 constructs determined by DSC
| NBD2 construct | [ATP] | [NBD2] | Tma |
|---|---|---|---|
| mm | μm | ||
| 9solb | 9.6 ± 0.5 | 5.5 ± 0.5 | 55.6 ± 0.8 |
| 9sol | 9.5 ± 0.4 | 10.3 ± 0.7 | 55.9 ± 0.9 |
| 9sol | 10.2 ± 0.4 | 22 ± 2 | 56.6 ± 0.8 |
| 9sol | 10.0 ± 0.2 | 43 ± 4 | 56.7 ± 1.1 |
| 9sol | 9.7 ± 0.3 | 68 ± 3 | 56.8 ± 0.3 |
| 9sol | 9.2 | 95 | 57.1 |
| 9sol | 10.2 | 161 | 57.2 |
| 9sol | 9.3 | 190 | 57.3 |
| 9sol | 2.0 ± 0.2 | 24 ± 4 | 52.6 ± 0.2 |
| 9sol | 4.0 ± 0.2 | 24 ± 6 | 54.0 ± 0.1 |
| 9sol | 7.0 ± 0.2 | 22 ± 2 | 54.9 ± 0.1 |
| 9sol + N1303K | 4.0 | 9.4 | 48.1 |
| 9sol + N1303K | 10 | 9.7 | 49.5 |
| 9sol + S1251N | 4.2 | 9.4 | 54.4 |
| 9sol + S1251N | 10 | 9.8 | 54.2 |
| 9sol + G1244E | 4.1 | 11 | 55.4 |
| 9sol + G1244E | 10 | 11 | 55.2 |
| 9sol + A1402H (reverse H1402A mutation in 9sol) | 3.9 | 27 | 50.3 |
| 9sol + G1349D | 4.3 | 3.5 | 51.3 |
| 9sol + S1235R | 4.2 | 3.6 | 53.5 |
| 9sol + I1234V | 4.5 | 14 | 53.3 |
a Tm is defined as the temperature at the maximum in the DSC heat capacity profile, determined after subtraction of buffer baseline and progressive baseline.
b 9sol mutations are as follows: S1255L, Q1280E, K1292D, Y1307N, K1334G, S1359A, H1402A, Q1411D, and L1436D.
Table 2.
Thermal stability of human NBD2 constructs determined by CD spectroscopy
| NBD2 construct | Tma |
|---|---|
| 9sol | 53.5 ± 0.4 |
| 9sol + G1244E | 55.6 ± 0.6 |
| 9sol + S1251N | 53.0 ± 1.1 |
| 9sol + N1303K | 49.4 ± 0.7 |
| 9sol + p.Ile1234_Arg1239del | No transition observed |
a Experiments were measured at a protein concentration of 17 μm (0.5 mg/ml) in the same buffer as used in the DSC experiments with 4 mm ATP. Experiments were recorded three times with the standard deviations of the repeats indicated. The curves were fit to obtain a Tm value as described in Ref. 63.
In agreement with a SYPRO-Orange-monitored thermal melt, no thermal melt transition was observed for p.Ile1234_Arg1239del mutant using CD spectroscopy, supporting the hypothesis that this mutant form of NBD2 is extremely unstable. Next to p.Ile1234_Arg1239del, N1303K was the most destabilizing mutation. DSC and CD experiments indicate that N1303K reduced NBD2 stability by ∼6–7 °C (compare results at 10 mm ATP and 10 μm NBD2) and ∼4 °C, respectively. Restoring His-1402 to the 9sol construct (which has the H1402A mutation) also led to a decrease in stability, potentially due to its effects on ATP binding and the potential recovery of catalytic activity, rather than an inherent stability loss, as H1402A is expected to abrogate NBD2 ATPase activity. G1349D reduced the stability of NBD2 by ∼3 °C. The remaining tested mutations, including I1234V, S1235R, G1244E, and S1251N, had a very modest effect on stability or slightly enhanced stability (e.g. G1244E). Furthermore, analysis indicates that increasing ATP concentration stabilized the 9sol and the 9sol + N1303K but did not stabilize 9sol + S1251N nor 9sol + G1244E (Table 1).
Back-to-consensus score evaluation
Taking the full range of mutations tested over the course of the stabilization screens, including the back-to-consensus mutations, the SGX 5sol reversion to wild-type mutations, and the CFTR2 mutants, we evaluated the back-to-consensus score in Equation 1. Mutations were divided into categories based on their effect on protein yield or stability of the isolated NBD2. Comparing the back-to-consensus score (supplemental Table S1) to these categories confirmed that the score is capable of separating mutations into largely discrete groups, in which all of the destabilizing (ΔTm <1) or low yield (<70% of background expression) mutations diverge from the consensus, and all but one of the stabilizing (ΔTm >1) mutations represent a return to the ABC(C/B) consensus (Fig. 8A). The one exception to this rule is H1402A, which had a highly negative score. This exception is readily explained because the mutation was incorporated into the original SGX 5sol construct because it is known to interfere with ATP hydrolysis.
Figure 8.

Return-to-consensus score post-validation. Back-to-consensus score value ranges for different categories of substitution mutations in both NBD1 (red) and NBD2 (blue), shown as standard box plots (points show statistical outliers, whiskers show lower and upper extremes, and box shows lower quartile, median, and upper quartile) with values on the x axis, and outliers labeled by their identity. A, comparison of mutations whose expression and stability were tested in isolated NBD2 during this study, with potential processing mutants defined as either < −1 °C ΔTm or a ≥70% drop in expression, neutral mutations having ΔTm values in the range of −1 to 1, and stabilizing mutations having either >1 °C ΔTm or, in the case of Gln-1411, cannot be removed from the construct without a ≥70% drop in expression. Overall, the score matches effects on stability, with the one outlier, H1402A, being a special case known to stabilize by loss of a conserved function in ATP hydrolysis. B, comparison of score ranges by disease associations, splitting substitutions into four categories based on the CFTR1, CFTR2, and SNP databases. Mutations known to cause CF consistently have the lowest scores; those associated with the less severe disease CBAVD have intermediate but still negative scores, and the three conclusive non-disease-causing mutations in the CFTR2 database have neutral or slightly positive scores, showing that the general progression of disease association correlates with deviation from the general ABC(C/B) consensus. The SNP database covers the full range of potential scores with a median at 0, being predominantly neutral with regard to the consensus.
We extended this analysis by scoring missense mutations found in the human population, mining the CFTR1, CFTR2, and SNP databases to produce four categories of substitution. First, we took the CF-causing and not the disease-associated mutations from the CFTR2 database. Second, to expand the number of missense mutations and to look for mutations of intermediate severity, we mined the CFTR1 database looking for patients with one severe allele (F508del or N1303K) and a second missense mutation of interest. To identify mutations of intermediate severity, we specifically looked for patients who were denoted as having congenital bilateral absence of the vas deferens (CBAVD) but were not indicated to have CF. Although this manner of identifying missense mutations of intermediate severity is likely less reliable than those mutations identified as CF-causing in the CFTR2 database, together they are likely to be informative with regard to back-to-consensus score ranges. Finally, for a broader picture of mutations found in the human population, we identified missense substitutions found in NBD1 and NBD2 within the SNP database.
In Fig. 8B we show that the back-to-consensus score separates the disease relevance annotated categories by the severity of the disease. CFTR2 database CF-causing mutations have the lowest possible scores, representing deviations from residues that are absolutely conserved throughout ABC subfamilies C and B. Similarly, the three known non-disease-associated mutations from the CFTR2 database had neutral or slightly positive scores, being effectively within the consensus. Intermediate mutations, those observed in patients reported to have CBAVD but not annotated as having CF, tended to have intermediate scores. These trends held true through both NBD1 and NBD2, with similar score values in each domain, suggesting a general correlation between the severity of disease phenotypes and the degree to which the mutations involved deviate from the ABC transporter consensus.
The SNP database, by comparison, has an average score close to zero, or within consensus, with the majority of the set falling within the same range as the CBAVD and neutral sets. This larger dataset also contains a wider range of outliers, having a range that effectively covers the entire spread of the score function. In this case, the back-to-consensus score predicts that some of these SNPs are likely to be rare disease-causing mutations not yet represented in the CFTR2 database, while predicting that others are stabilizing human variants that may be able to suppress CF disease severity. Supporting this idea, the human SNP with the highest back-to-consensus score is I539T, a mutation that has already been shown to stabilize NBD1 (50) and improve processing of full-length CFTR but not channel function (51).
Comparison with automated approach
The stabilizing mutation predictions made during this study involved significant manual analysis and elements of rational design, suited to our goal of both minimizing the number of mutants in the final construct and testing the functional relevance of consensus mutations at specific positions in CFTR. Automated methods for predicting stabilized constructs using consensus data and structural modeling are available, and we assessed the impact of our manual approach by comparing it with automated predictions made by the PROSS server (43), which similarly combines sequence conservation with modeling against the Rosetta/Talaris energy function (52). Using the SGX background NBD2 sequence as input, this server produces seven construct suggestions ranging from 7 to 39 mutations, with a total of 43 individual substitutions suggested over the full set (supplemental Table S3). This set of 43 includes two of the mutations we found, K1292D and S1359A, but only in the context of at least 15 and 29 other substitutions, respectively. This approach moves farther away from the wild-type sequence and increases the risk of including destabilizing mutations, as seen by the reversion of the stabilizing H1402A mutation being suggested in four of the seven constructs. Thus, our manual, iterative approach can generate stabilizing substitutions within a more limited set of mutations that is closer to wild type and also provides evolutionary and functional insights.
Discussion
In this work, we identified consensus residues in ABC(C) and ABC(B) transporters through a comprehensive analysis of these sequence families. This allowed us to develop a back-to-consensus score function used to identify four novel stabilizing mutations in NBD2. Incorporating these mutations, we developed multiple stable NBD2 constructs suitable for biophysical and structural characterization. Manual analysis of ABC transporter structures and Rosetta energy calculations from the NBD2 crystal structure significantly contributed to deciding which well-scored mutants to attempt, making the method difficult to implement in full-length CFTR for which we have no high-resolution structure. Nonetheless, the results for previously studied and disease-causing mutations independently confirm a clear ability to predict relative expression yields or stability as well as being correlated with disease liability. Thus, although this bioinformatics method is enhanced by the availability of a high-resolution structure, the approach is highly promising as a method for identifying (de)stabilizing mutations within a minimal number of candidates based on sequence analyses alone.
The back-to-consensus score, which was calculated across a range of functionally diverse genes to identify stabilizing residues, can highlight both stabilizing mutations and mutations for which conserved functions have been lost, the latter often implicated in disease. To avoid reverting any CFTR-specific losses of conserved ABC transporter function that may exist in the NBD1/NBD2 interface, we chose to ignore residues buried in the canonical interface and those involved in the ATP-binding pockets, thus favoring residue positions that affect stability over function. In contrast, most of the disease-causing mutations analyzed, including G1244E, S1251N, S1255P, and G1349D, are integral to the ATP-binding pockets (part of Walker A, helix 1, or signature sequence) and are known to allow CFTR to mature normally, being considered primarily gating mutants (27, 49, 53, 54). Although these mutations are associated with functional rather than stability defects, there is some overlap, because G1349D also destabilizes NBD2.
We identified a core of 36 NBD positions that have essential conservation in at least one-half of ABC(C/B) sequences, and none of those essential residues in the ABC(C/B) consensus have been lost from both halves of CFTR. Conserved positions that can be lost in an asymmetric fashion in full transporters showed similar patterns of asymmetric loss in CFTR, suggesting that NBD divergence at these core positions is unlikely to involve a loss-of-function specific to CFTR. However, divergence for gain-of-function specific to CFTR could still have occurred at any of the residue positions outside of this core, and back-to-consensus mutations with intermediate or low positive scores, such as S1255L and K1292D, could still cause the loss of some unknown CFTR-specific function. Interestingly, none of the disease-causing mutations in NBD2 that we explored occur at a residue position for which conservation and sequence identity are specific to CFTR; indeed, all six instead represent deviations from the ABC(C/B) consensus, so it is less likely that their effect is due to a loss-of-function specific to CFTR.
The NBD domains are the most highly conserved regions of ABC transporters/channels, which facilitated our prediction of stabilizing mutations. The higher degree of variability in the TMDs and ICDs and the lack, at the time of our analysis, of a high-resolution full-length CFTR structure made it impossible to predict the effect NBD-stabilizing mutations near domain interface regions would have on the stability of full-length CFTR. From a mechanistic standpoint, analysis of the NBD2 crystal structure indicates that Ser-1359 pulls Gln-1291 away from its normal position where it helps to chelate Mg-ATP. This led us to postulate that the Ser-1359/Gln-1291 interaction destabilizes NBD2 by reducing its affinity for stabilizing ATP. We further postulate that S1359A would eliminate the Ser-1359/Gln-1291 interaction thus restoring normal Mg-ATP binding and enhancing NBD2 stability. Although S1359A does stabilize the isolated NBD2, CFTR expression, maturation, and activity studies indicate that the mutation has almost no effect on the stability or activity of full-length CFTR. In full-length CFTR, interaction between this region of NBD2 and the ICDs may lead to a conformational rearrangement that corrects this deficit. Analogously, the K1292D mutation, which enhances NBD2 stability in our SYPRO-Orange assay, appears to reduce maturation of CFTR through the secretory pathway. Lys-1292, which is part of the Q-loop, may also affect NBD dimerization and NBD/ICD interactions. Thus, although we were successful in predicting mutations that stabilize isolated NBD2, predicting how these mutations behave in full-length CFTR is complicated by the higher order association of individual domains in the native quaternary structure.
The disease-causing mutations that we analyzed had variable effects on stability, suggesting some are primarily functional mutations. Two out of six, N1303K and G1349D, did have reproducible stability deficits in isolated NBD2, by a variety of experimental techniques. N1303K is a severe disease-causing mutation that is known to impair maturation of CFTR, analogous to the manner in which F508del destabilization of NBD1 impairs CFTR maturation. Interestingly, like F508del, N1303K interacts with the C terminus of the Q-loop, leading us to speculate that, like F508del (22), N1303K may impair NBD dimerization, by altering the conformation of the Q-loop ensemble. Future work will examine N1303K effects on NBD dimerization.
Unlike N1303K, which is peripheral to the dimer interface, G1349D is central to the NBD dimer interface as part of the NBD2 signature sequence. Gly-1349 is in the corresponding position to Gly-551 in NBD1, but unlike Gly-551 it contributes to a non-hydrolytic ATP-binding pocket. Still, G1349D appears to be primarily a functional mutation and does not impair CFTR maturation (49, 53, 54), despite its impact on NBD2 stability. The destabilization of isolated NBD2 by G1349D could be explained by electrostatic repulsion between Asp-1349 and Asp-1377, but this may be mitigated by inter-domain interactions in full-length CFTR. Alternatively, the mild destabilization of NBD2 may not be sufficient to inhibit CFTR maturation, but it may destabilize inter-domain interactions sufficiently to cause the observed moderate gating defect observed for this mutation (53).
Interestingly, isolated NBD2 seems to be less stable than NBD1. Our most stable mutant NBD2 construct is 4 °C less stable than the WT NBD1 ΔRIΔRE construct as measured by DSC, albeit under different buffer conditions, and it should be noted that the regulatory insertion is known to destabilize NBD1 (20). Although the greater instability of NBD2 might lead one to assume that it is the weakest link in CFTR maturation, mutations that result in pathological maturation defects are more common in NBD1 (F508del, R560T, A559T, L467P, S492F, Y569D, A561E, and R560S) (55) than in NBD2 (N1303K and p.Ile1234_Arg1239del), especially in terms of gene prevalence. This points to cooperative folding, in which interactions with NBD1 support NBD2 stability, and mutations in NBD1 have the double effect of destabilizing both. In support of this, expression of CFTR with the severe processing mutation N1303K can be improved by NBD1 suppressor mutations (56), although band C/band B ratios are not improved. Suppressor mutation rescue and temperature rescue of F508del are both reduced in the absence of NBD2 (56–58). These data provide evidence for the model in which an unstable NBD2 requires a stable, dimer-competent NBD1 to achieve a stable conformation and highlight defective NBD1/NBD2 interaction for the F508del mutation (22). This hypothesis, together with the absence of NBD/ICD interactions, may explain the discrepancy between mutational effects on isolated NBD2 and full-length CFTR.
The relative instability of wild-type CFTR NBD2 raises the possibility that some of the departures from consensus that we have, in effect, reverted originally diverged because of functional selection for lower stability, for example, through mutations that affected the regulation of protein expression by a selective decrease in stability. We were not able to obtain a clear melting transition for wild-type NBD2, but the stabilized SGX NBD2 construct we used as our starting point melts below 40 °C despite having five mutations whose stabilizing effects, if combined linearly, would be greater than 5 °C, suggesting that wild-type NBD2, expressed as an isolated domain, has a Tm value significantly below human body temperature. In this context, the low stability of NBD2, and the likely essential role of its interactions in maintaining its fold, could stem from a specific regulatory function, with the ultimate maturation of functional channels is controlled in part by the effects of NBD2-stabilizing interactions, potentially including the phosphorylation state of the R region, the ATP-binding state of NBD1, and the presence of unknown cytosolic binding partners or chaperones.
Experimental procedures
Bioinformatic analysis
8457 non-redundant sequences were obtained by BLAST (59) against each of the 23 human ABC subfamily C and B members. These were then filtered down further using the USEARCH clustering method (60) with a 99% sequence identity threshold to further eliminate redundant sequences, with the longest sequences being kept, leaving a final set of 2681 sequences. The sequences were then assigned to the closest human homolog by BLAST score, with assignment of CFTR sequences confirmed by the presence of the regulatory region, and each assigned set was independently aligned to CFTR using MUSCLE (61). For the purpose of determining sequence consensus, half-transporter/channel alignments were extracted, and scripts were developed for extracting statistics from alignments to either or both halves. This half-transporter/channel approach increases the number of comparisons that can be made by alignment to CFTR, resulting in 29,736 unique individual sequence alignments to CFTR spread across 74 multiple sequence alignment files. Separating the alignment tasks in this fashion served primarily to reduce the number of completely independent insertions and deletions considered per alignment, eliminating some issues expected when attempting to align multiple insertions that have no homology to one another aside from their general location in the folded structure.
ΔΔG prediction
Predicted changes in free energy of unfolding (ΔΔG) values upon introduction of single-point mutations were obtained using Rosetta3.4 (62) with the fixed backbone protocol (47) on each chain of the NBD2 crystal structure (PDB code 3GD7) (28). Data from sequence positions with non-wild-type amino acids in the crystal structure were discarded, and the median value for each mutation at all remaining positions was retained.
Construction of mutants
We generated a background construct based on a previously described “5sol” construct that was designed by SGX Pharmaceuticals and used to solve a crystal structure of NBD2 (PDB code 3GD7). Our background construct consists of CFTR residues 1193–1445 with five point mutations (Q1280E, Y1307N, H1402A, Q1411D, and L1436D) in the Invitrogen pET-Sumo vector. Individual point mutations were introduced by quick-change mutagenesis (ACGT Corp.) and confirmed by sequencing.
Protein purification
NBD2 protein was expressed in Escherichia coli BL21DE3 in either LB or M9 media supplemented with kanamycin and grown overnight at 16 °C following induction. Following lysis, NBD2 was purified first by His tag binding to nickel resin and then by gel filtration using an S75-Superdex column. The buffer conditions for lysis and gel filtration consisted of a 20 mm NaPO4, pH 7.5, 150 mm Na+ (<150 mm Cl−), and 10% glycerol, with 2 mm ATP/MgCl2 used in the first round of mutagenesis and 10 mm ATP/MgCl2 in subsequent rounds. 50 mm arginine was used in rounds 1, 3, and 6. Reducing agent varied between rounds (either 5 mm BME or 2 mm DTT).
The effects of buffer conditions, growth conditions, reagent batch quality, and sample degradation over time were controlled within each individual round of mutagenesis. Cultures for each construct within a round of mutants were grown and purified by Ni2+ resin simultaneously. All buffers for each round were prepared as a single common batch, and gel filtration was done for each construct in series over a period no longer than 2–4 days. Purity of the gel filtration fractions was assessed by SDS-PAGE, with a consistent single fraction taken based on overall purity. The yield of NBD2 protein was measured by quantifying SDS-PAGE band volumes for matched gel filtration fractions for each mutant within a set using a Bio-Rad Gel Doc EZ Imager.
SYPRO-Orange thermal melts
Melting points were measured at protein concentrations between 10 and 30 μm, as measured by Bradford assay, with the concentrations being equal within a set. The midpoints of thermal melting transitions (Tm values) were measured by adding 15 μl of protein samples to 5 μl of 4× SYPRO-Orange (in gel filtration buffer) followed by a dye-binding thermal melt assay in a Bio-Rad CFX RT-PCR machine using a gradient from 20 to 80 °C over 80 min in 0.5 °C over 57.5-s increments (40 s + plate read). Each sample was tested with a minimum of three replicates, and the median was accepted as the Tm, where over the course of the experiment 78% of individual replicates were observed to match the median and 98% matched within one step size, giving a measurement error of ±0.5 °C.
Circular dichroism measurements
Thermal melts of NBD2 were performed on a JASCO J-810 spectropolarimeter equipped with Peltier thermoregulation. The buffer, which was the same buffer as used for the DSC experiments, was 20 mm NaPO4, 150 mm NaCl, 10% glycerol, 5 mm BME, 4 mm MgCl2, and 4 mm ATP. Scans were performed with 0.5 mg/ml protein, as measured by Bradford assay, with a scan rate of 2 °C/min. Data were fit as described in Ref. 63.
Differential scanning calorimetry
DSC was conducted with a VPCapillary DSC System (MicroCal Inc, GE Healthcare) using a scan rate of 2 °C/min. A buffer-only heat capacity curve was subtracted from the protein curve, and data were analyzed with the instrument's software. Buffer was identical to that used for the CD measurements with varying protein, ATP, and MgCl2 concentrations as noted under “Results.”
NMR acquisition
HSQC spectra were obtained on a Varian Innova 500 MHz spectrometer equipped with a triple resonance probe. Conditions for the SGX 5sol sample were as follows: 320 transients, 15 °C, 0.047 mm protein, 10% D2O, 4% glycerol, 20 mm HEPES, pH 7.6, 150 mm NaCl, 5 mm MgCl2, 0.5 mm P-ATP, 0.05% NaN3, 0.5 mm benzamidine, and 2 mm BME. Those for the 9sol sample were as follows: 256 transients, 25 °C, 1 mm protein, 10% D2O, 2% glycerol, 50 mm Tris, pH 7.5, 50 mm ATP/Mg2+, 40 mm Na+, 80 mm Cl−, 0.25 mm NaN3, and 2 mm BME.
Cell culture, full-length CFTR expression, and Western blotting
HEK293T cells (ATCC) were routinely cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin and streptomycin (Life Technologies, Inc.) at 37 °C in a 5% CO2 atmosphere. Expression of CFTR was accomplished using pCMV-CFTR expression plasmids transiently transfected using XtremeGene (Roche Applied Science) following the manufacturer's protocols. Proteins were expressed for 48–60 h; cells were lysed in RIPA lysis buffer (Millipore) and clarified by centrifugation at 15,000 relative centrifugal force (RCF) at 4 °C. The lysates were separated using Tris-glycine SDS-PAGE and subsequently transferred to PVDF membrane (Millipore). The CFTR protein was either detected using the monoclonal 660 α-CFTR antibody (University of North Carolina/Cystic Fibrosis Foundation antibody resource) and visualized using HRP-conjugated secondary antibody (Thermo Fisher Scientific) and GE Healthcare (Fig. 4) or detected with mAb596 and visualized with secondary goat anti-mouse IgG-IR800 using Odyssey infrared fluorescent imager (Licor Corp.) (Fig. 5).
Single channel analysis of S1359A in full-length CFTR
Planar lipid bilayers were prepared by painting a 0.2-mm hole drilled in a Teflon cup with a phospholipid solution in n-decane containing a 3:1 mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (Avanti Polar Lipids, Alabaster, AL). The lipid bilayer separated 1.0 ml of solution in the Teflon cup (cis side) from 5.0 ml of a solution in an outer glass chamber (trans side). Both chambers were magnetically stirred and thermally insulated. Heating and temperature control were established by a temperature control system TC2BIP (Cell Micro Controls, Norfolk, VA). The CFTR ion channels were transferred into the preformed lipid bilayer by spontaneous fusion of CFTR containing membrane vesicles. The membrane vesicles were prepared from HEK293 cells stably expressing CFTR construct as described previously (64). The expression of the CFTR protein was confirmed by immunoblotting. Membrane vesicles were phosphorylated by incubation with 50 nm PKA catalytic subunit (Promega Corp., Madison, WI) in phosphorylation buffer (10 mm HEPES, pH 7.2, 0.5 mm EGTA, 5 mm ATP, 5 mm MgCl2, and 250 mm sucrose) for 15 min at room temperature. To maintain uniform orientation and functional activity of CFTR channels transferred into the bilayer, 5 mm MgATP, 50 nm PKA, and membrane vesicles were added in the cis compartment only. Single-channel currents were recorded at +30 °C in symmetrical salt solution (300 mm Tris/HCl, pH 7.2, 3 mm MgCl2, and 1 mm EGTA) under voltage-clamp conditions at −75 mV using an Axopatch 200B amplifier (Molecular Devices, LLC, Sunnyvale, CA). The membrane voltage difference of −75 mV is the voltage difference between cis and trans (ground) compartments. For analysis, the single-channel current was digitized by Digidata 1322 (Molecular Devices, LLC, Sunnyvale, CA) with a sampling rate of 500 Hz and analyzed using pCLAMP 9.2 software (Molecular Devices, LLC, Sunnyvale, CA). Origin 7.5 software (Origin Lab, Northampton, MA) was used to fit all-point histograms by multi-peak Gaussians. Single-channel current was defined as the distance between peaks on the fitting curve and used for the calculation of the single-channel conductance. The single-channel open probability (Po) was calculated as a ratio of the area under the peak for the open state to the total area under both peaks.
Comparison with automated predictor
We performed a retrospective analysis comparing our manual approach for identifying potential stabilizing mutants to results from the PROSS server (43), an automated predictor based on similar sequence consensus and Rosetta energy terms (52). As input for PROSS, we used the SGX 5sol construct sequence and the PDB structure 3GD7. The PROSS server provided seven construct suggestions as output containing a total of 43 potential stabilizing mutations, which were used solely as a point of comparison with our predictions.
Author contributions
R. V. M. and J. D. F.-K. designed the research. R. V. M., P. A. C., H. L., Z. Y., Q. Z., A. A. A., J. E. D., and P. H. T. performed the research. R. V. M., P. A. C., Z. Y., Q. Z., A. A. A., J. E. D., P. H. T., J. R. R., C. G. B., and J. D. F.-K. analyzed the data. R. V. M., P. A. C., A. A. A., P. H. T., J. R. R., C. G. B., and J. D. F.-K. wrote the paper.
Supplementary Material
Acknowledgments
We appreciate the contributions of Jianli An in support of early DSC studies for this paper. We thank the Structural and Biophysical Core Facility at the Hospital for Sick Children for use of their resources.
This work was supported by grants from the Cystic Fibrosis Foundation Therapeutics (to J. D. F.-K., P. H. T., C. G. B., and J. R. R.), from Cystic Fibrosis Canada (to J. D. F.-K.), and from the National Institutes of Health (to P. H. T. and J. R. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1 and S2 and Tables S1–S3.
- CF
- cystic fibrosis
- CFTR
- CF transmembrane conductance regulator
- NBD
- nucleotide-binding domain
- TMD
- transmembrane domain
- ABC
- ATP binding cassette
- ICD
- intracellular domain
- ICL
- intracellular loop
- PDB
- Protein Data Bank
- SNP
- single nucleotide polymorphism
- CBAVD
- congenital bilateral absence of the vas deferens
- DSC
- differential scanning calorimetry
- P-ATP
- N6-(2-phenylethyl)-ATP
- BME
- β-mercaptoethanol.
References
- 1. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., and Chou J. L. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 2. Hyde S. C., Emsley P., Hartshorn M. J., Mimmack M. M., Gileadi U., Pearce S. R., Gallagher M. P., Gill D. R., Hubbard R. E., and Higgins C. F. (1990) Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 346, 362–365 [DOI] [PubMed] [Google Scholar]
- 3. Ostedgaard L. S., Baldursson O., Vermeer D. W., Welsh M. J., and Robertson A. D. (2000) A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc. Natl. Acad. Sci. U.S.A. 97, 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., and Welsh M. J. (1991) Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253, 202–205 [DOI] [PubMed] [Google Scholar]
- 5. Winter M. C., and Welsh M. J. (1997) Stimulation of CFTR activity by its phosphorylated R domain. Nature 389, 294–296 [DOI] [PubMed] [Google Scholar]
- 6. Cheng S. H., Rich D. P., Marshall J., Gregory R. J., Welsh M. J., and Smith A. E. (1991) Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 66, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 7. Liang X., Da Paula A. C., Bozóky Z., Zhang H., Bertrand C. A., Peters K. W., Forman-Kay J. D., and Frizzell R. A. (2012) Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis. Mol. Biol. Cell 23, 996–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vergani P., Lockless S. W., Nairn A. C., and Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berger A. L., Ikuma M., and Welsh M. J. (2005) Normal gating of CFTR requires ATP binding to both nucleotide-binding domains and hydrolysis at the second nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 102, 455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aleksandrov L., Aleksandrov A. A., Chang X. B., and Riordan J. R. (2002) The first nucleotide-binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem. 277, 15419–15425 [DOI] [PubMed] [Google Scholar]
- 11. Aleksandrov A. A., Cui L., and Riordan J. R. (2009) Relationship between nucleotide binding and ion channel gating in cystic fibrosis transmembrane conductance regulator. J. Physiol. 587, 2875–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hughes A. L. (1994) Evolution of the ATP-binding-cassette transmembrane transporters of vertebrates. Mol. Biol. Evol. 11, 899–910 [DOI] [PubMed] [Google Scholar]
- 13. Dean M., Rzhetsky A., and Allikmets R. (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166 [DOI] [PubMed] [Google Scholar]
- 14. Jones P. M., and George A. M. (1999) Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett. 179, 187–202 [DOI] [PubMed] [Google Scholar]
- 15. Dawson R. J., and Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 16. Jordan I. K., Kota K. C., Cui G., Thompson C. H., and McCarty N. A. (2008) Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 18865–18870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He L., Aleksandrov A. A., Serohijos A. W., Hegedus T., Aleksandrov L. A., Cui L., Dokholyan N. V., and Riordan J. R. (2008) Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J. Biol. Chem. 283, 26383–26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dalton J., Kalid O., Schushan M., Ben-Tal N., and Villà-Freixa J. (2012) New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J. Chem. Inf. Model 52, 1842–1853 [DOI] [PubMed] [Google Scholar]
- 19. Mornon J. P., Lehn P., and Callebaut I. (2009) Molecular models of the open and closed states of the whole human CFTR protein. Cell. Mol. Life Sci. 66, 3469–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Protasevich I., Yang Z., Wang C., Atwell S., Zhao X., Emtage S., Wetmore D., Hunt J. F., and Brouillette C. G. (2010) Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 19, 1917–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang C., Protasevich I., Yang Z., Seehausen D., Skalak T., Zhao X., Atwell S., Spencer Emtage J., Wetmore D. R., Brouillette C. G., and Hunt J. F. (2010) Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 19, 1932–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chong P. A., Farber P. J., Vernon R. M., Hudson R. P., Mittermaier A. K., and Forman-Kay J. D. (2015) Deletion of phenylalanine 508 in the first nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator increases conformational exchange and inhibits dimerization. J. Biol. Chem. 290, 22862–22878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rabeh W. M., Bossard F., Xu H., Okiyoneda T., Bagdany M., Mulvihill C. M., Du K., di Bernardo S., Liu Y., Konermann L., Roldan A., and Lukacs G. L. (2012) Correction of both NBD1 energetics and domain interface is required to restore ΔPhe-508 CFTR folding and function. Cell 148, 150–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mendoza J. L., Schmidt A., Li Q., Nuvaga E., Barrett T., Bridges R. J., Feranchak A. P., Brautigam C. A., and Thomas P. J. (2012) Requirements for efficient correction of ΔPhe-508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cui L., Aleksandrov L., Chang X. B., Hou Y. X., He L., Hegedus T., Gentzsch M., Aleksandrov A., Balch W. E., and Riordan J. R. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 365, 981–994 [DOI] [PubMed] [Google Scholar]
- 26. Molinski S. V., Gonska T., Huan L. J., Baskin B., Janahi I. A., Ray P. N., and Bear C. E. (2014) Genetic, cell biological, and clinical interrogation of the CFTR mutation c.3700 A>G (p.Ile1234Val) informs strategies for future medical intervention. Genet. Med. 16, 625–632 [DOI] [PubMed] [Google Scholar]
- 27. Yu H., Burton B., Huang C. J., Worley J., Cao D., Johnson J. P. Jr, Urrutia A., Joubran J., Seepersaud S., Sussky K., Hoffman B. J., and Van Goor F. (2012) Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros 11, 237–245 [DOI] [PubMed] [Google Scholar]
- 28. Zhao X., Conners K., Emtage S., Lu F., and Atwell S. (2008) The crystal structure of the second nucleotide-binding domain (NBD2) of CFTR suggests NBD2 subdomain movements are involved in channel opening. Pediatr. Pulmonol. 43, Suppl. 31, 205 [Google Scholar]
- 29. Steipe B., Schiller B., Plückthun A., and Steinbacher S. (1994) Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240, 188–192 [DOI] [PubMed] [Google Scholar]
- 30. Amin N., Liu A. D., Ramer S., Aehle W., Meijer D., Metin M., Wong S., Gualfetti P., and Schellenberger V. (2004) Construction of stabilized proteins by combinatorial consensus mutagenesis. Protein Eng. Des. Sel. 17, 787–793 [DOI] [PubMed] [Google Scholar]
- 31. Lehmann M., and Wyss M. (2001) Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr. Opin. Biotechnol. 12, 371–375 [DOI] [PubMed] [Google Scholar]
- 32. Lehmann M., Kostrewa D., Wyss M., Brugger R., D'Arcy A., Pasamontes L., and van Loon A. P. (2000) From DNA sequence to improved functionality: using protein sequence comparisons to rapidly design a thermostable consensus phytase. Protein Eng. 13, 49–57 [DOI] [PubMed] [Google Scholar]
- 33. Jäckel C., Bloom J. D., Kast P., Arnold F. H., and Hilvert D. (2010) Consensus protein design without phylogenetic bias. J. Mol. Biol. 399, 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bloom J. D., and Glassman M. J. (2009) Inferring stabilizing mutations from protein phylogenies: application to influenza hemagglutinin. PLoS Comput. Biol. 5, e1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sebastian A., Rishishwar L., Wang J., Bernard K. F., Conley A. B., McCarty N. A., and Jordan I. K. (2013) Origin and evolution of the cystic fibrosis transmembrane regulator protein R domain. Gene 523, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldstein R. A. (2011) The evolution and evolutionary consequences of marginal thermostability in proteins. Proteins 79, 1396–1407 [DOI] [PubMed] [Google Scholar]
- 37. Taverna D. M., and Goldstein R. A. (2002) Why are proteins marginally stable? Proteins 46, 105–109 [DOI] [PubMed] [Google Scholar]
- 38. Dehouck Y., Grosfils A., Folch B., Gilis D., Bogaerts P., and Rooman M. (2009) Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 25, 2537–2543 [DOI] [PubMed] [Google Scholar]
- 39. Wainreb G., Wolf L., Ashkenazy H., Dehouck Y., and Ben-Tal N. (2011) Protein stability: a single recorded mutation aids in predicting the effects of other mutations in the same amino acid site. Bioinformatics 27, 3286–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guerois R., Nielsen J. E., and Serrano L. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387 [DOI] [PubMed] [Google Scholar]
- 41. Tian J., Wu N., Chu X., and Fan Y. (2010) Predicting changes in protein thermostability brought about by single- or multi-site mutations. BMC Bioinformatics 11, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laimer J., Hofer H., Fritz M., Wegenkittl S., and Lackner P. (2015) MAESTRO–multi-agent stability prediction upon point mutations. BMC Bioinformatics 16, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldenzweig A., Goldsmith M., Hill S. E., Gertman O., Laurino P., Ashani Y., Dym O., Unger T., Albeck S., Prilusky J., Lieberman R. L., Aharoni A., Silman I., Sussman J. L., Tawfik D. S., and Fleishman S. J. (2016) Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 63, 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. NCBI Resource Coordinators (2014) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 42, D7–D17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sosnay P. R., Siklosi K. R., Van Goor F., Kaniecki K., Yu H., Sharma N., Ramalho A. S., Amaral M. D., Dorfman R., Zielenski J., Masica D. L., Karchin R., Millen L., Thomas P. J., Patrinos G. P., et al. (2013) Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 45, 1160–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramalho A. S., Clarke L. A., Sousa M., Felicio V., Barreto C., Lopes C., and Amaral M. D. (2016) Comparative ex vivo, in vitro and in silico analyses of a CFTR splicing mutation: importance of functional studies to establish disease liability of mutations. J. Cyst. Fibros 15, 21–33 [DOI] [PubMed] [Google Scholar]
- 47. Kellogg E. H., Leaver-Fay A., and Baker D. (2011) Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79, 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ramaen O., Leulliot N., Sizun C., Ulryck N., Pamlard O., Lallemand J. Y., Tilbeurgh Hv., and Jacquet E. (2006) Structure of the human multidrug resistance protein 1 nucleotide-binding domain 1 bound to Mg2+/ATP reveals a non-productive catalytic site. J. Mol. Biol. 359, 940–949 [DOI] [PubMed] [Google Scholar]
- 49. Gregory R. J., Rich D. P., Cheng S. H., Souza D. W., Paul S., Manavalan P., Anderson M. P., Welsh M. J., and Smith A. E. (1991) Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol. Cell. Biol. 11, 3886–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He L., Aleksandrov A. A., An J., Cui L., Yang Z., Brouillette C. G., and Riordan J. R. (2015) Restoration of NBD1 thermal stability is necessary and sufficient to correct Phe-508 CFTR folding and assembly. J. Mol. Biol. 427, 106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Da Paula A. C., Sousa M., Xu Z., Dawson E. S., Boyd A. C., Sheppard D. N., and Amaral M. D. (2010) Folding and rescue of a cystic fibrosis transmembrane conductance regulator trafficking mutant identified using human-murine chimeric proteins. J. Biol. Chem. 285, 27033–27044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O'Meara M. J., Leaver-Fay A., Tyka M. D., Stein A., Houlihan K., DiMaio F., Bradley P., Kortemme T., Baker D., Snoeyink J., and Kuhlman B. (2015) Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 11, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bompadre S. G., Sohma Y., Li M., and Hwang T. C. (2007) G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J. Gen. Physiol. 129, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Melin P., Thoreau V., Norez C., Bilan F., Kitzis A., and Becq F. (2004) The cystic fibrosis mutation G1349D within the signature motif LSHGH of NBD2 abolishes the activation of CFTR chloride channels by genistein. Biochem. Pharmacol. 67, 2187–2196 [DOI] [PubMed] [Google Scholar]
- 55. Van Goor F., Yu H., Burton B., and Hoffman B. J. (2014) Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros 13, 29–36 [DOI] [PubMed] [Google Scholar]
- 56. He L., Aleksandrov L. A., Cui L., Jensen T. J., Nesbitt K. L., and Riordan J. R. (2010) Restoration of domain folding and interdomain assembly by second-site suppressors of the ΔPhe-508 mutation in CFTR. FASEB J. 24, 3103–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Du K., and Lukacs G. L. (2009) Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 20, 1903–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rosser M. F., Grove D. E., Chen L., and Cyr D. M. (2008) Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: folding defects caused by deletion of Phe-508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Mol. Biol. Cell 19, 4570–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Edgar R. C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 [DOI] [PubMed] [Google Scholar]
- 61. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fersht A. (1998) Structure and Mechanism in Protein Science, pp. 508–523, W.H. Freeman & Co., New York [Google Scholar]
- 64. Aleksandrov A. A., Kota P., Aleksandrov L. A., He L., Jensen T., Cui L., Gentzsch M., Dokholyan N. V., and Riordan J. R. (2010) Regulatory insertion removal restores maturation, stability and function of ΔPhe-508 CFTR. J. Mol. Biol. 401, 194–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schneider T. D., and Stephens R. M. (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.