

Abstract
Hyperactivation of Akt is associated with oncogenic changes in the growth, survival, and chemoresistance of cancer cells. The PI3K/phosphoinositide-dependent kinase (PDK) 1 pathway represents the canonical mechanism for phosphorylation of Akt at its primary activation site, Thr-308. We observed that Ca2+/calmodulin (CaM)-dependent protein kinase kinase 2 (β) (CaMKK2) is highly expressed in high-grade serous ovarian cancer, and we investigated its role in Akt activation in ovarian cancer (OVCa) cell lines (OVCAR-3, SKOV-3, and Caov-3). Knockdown or pharmacological inhibition of CaMKK2 produced phenotypes expected of Akt inhibition, including reductions in cell growth and cell viability and in the regulation of Akt downstream targets involved in G1/S transition and apoptosis. CaMKK2 knockdown or inhibition decreased Akt phosphorylation at Thr-308 and Ser-473 to extents similar to those of PDK1 knockdown or PI3K inhibition. Combined CaMKK2 and PDK1 knockdown or CaMKK and PI3K inhibition, respectively, produced additive effects on p-Akt and cell growth, consistent with direct Akt phosphorylation by CaMKK2. This conclusion was supported by the absence of effects of CaMKK2 knockdown/inhibition on alternative means of activating Akt via p-Akt Thr-450, p-PDK1 Ser-241, or p-IRS1 Ser-636/639. Recombinant CaMKK2 directly activated recombinant Akt by phosphorylation at Thr-308 in a Ca2+/CaM-dependent manner. In OVCa cells, p-Akt Thr-308 was significantly inhibited by intracellular Ca2+i chelation or CaM inhibition. Ionomycin-induced Ca2+ influx promoted p-Akt, an effect blocked by PDK1, and/or CaMKK2, siRNAs, and by PI3K and/or CaMKK inhibitors. CaMKK2 knockdown potentiated the effects of the chemotherapeutic drugs carboplatin and PX-866 to reduce proliferation and survival of OVCa cells.
Keywords: Akt PKB, calmodulin (CaM), ovarian cancer, phosphorylation, protein kinase
Introduction
High-grade serous ovarian cancer (HGSOC)2 is the most commonly diagnosed (70–80%) and aggressive subtype of ovarian cancer (OVCa). HGSOC is characterized by a high frequency of copy number alterations (CNA) and a low frequency of point mutations. CNA in HGSOC occur in genes involved in homologous recombination repair (BRCA), cell cycle (Rb and cyclins), and the PI3K/Akt (PKB) pathway (1–5). CNA of the PI3K/Akt pathway include the PI3K catalytic subunit p110α (PIK3CA) and AKT2. These CNA along with activating mutations in PIK3CA and inactivating mutations of PTEN (phosphatase and tensin homologue) are thought to drive ovarian tumorigenesis by promoting Akt hyperactivation (6).
The PI3K/Akt pathway is a major signaling network for control of the growth and survival of normal and neoplastic cells and is oncogenic for multiple cancer types, including OVCa (7, 8). PI3K synthesizes phosphatidylinositol 3,4,5-trisphosphate, which recruits Akt and phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane via their pleckstrin homology (PH) domains, resulting in PDK1 phosphorylation of Akt at its activation loop site Thr-308. Once phosphorylated at Thr-308, Akt phosphorylates SIN1 of the mechanistic target of rapamycin (mTOR) complex 2 (mTORC2), which activates mTORC2, resulting in phosphorylation of Akt at Ser-473 (9). Phosphorylation of Akt at both Thr-308 and Ser-473 is required for maximal activation. Dephosphorylation of phosphatidylinositol 3,4,5-trisphosphate by PTEN exerts a suppressive effect on the activity of the PI3K/PDK1/Akt pathway.
Akt activation results in promotion of protein translation, cell growth, and cell survival. Protein translation is mediated by Akt phosphorylation of PRAS40 (proline-rich Akt substrate 40) leading to the release of mTORC1 from an inhibited state allowing for its phosphorylation of the p70 ribosomal protein S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) (10). Akt promotes cell growth and survival by increasing cyclin D1 protein stability and gene transcription and by decreasing the transcription of pro-apoptotic genes, through the phosphorylation of glycogen synthase kinase 3β (GSK3β) and Forkhead box O3a (FoxO3a), respectively (11, 12). Increased cyclin D1/Cdk4/6 promotes G1/S phase cell cycle transition by hyperphosphorylation of the tumor suppressor Rb, thus inactivating it and allowing transit of E2F to the nucleus to promote transcription of genes required for S phase progression. In addition, Akt promotes cell survival through the inhibition of pro-apoptotic signaling cascades, which include inhibition of the executor caspases and consequent activation of poly(ADP-ribose) polymerase (PARP) through inhibition of PARP cleavage (7, 8).
The pathway leading to Akt activation is typically conceptualized with PDK1 as the sole upstream kinase activating Akt by Thr-308 phosphorylation. Thus, PDK1−/− embryonic stem (ES) cells fail to show growth factor (GF)-responsive Akt phosphorylation at Thr-308 (13). Although it is well established that PDK1 is a major upstream Akt-activating kinase, it is possible that additional kinase(s), which are not expressed developmentally at the ES cell stage, are not GF-responsive, or are overexpressed in cancer, might catalyze Akt phosphorylation.
It was previously reported that in neuroblastoma–glioma NG108 cells, Akt is phosphorylated at Thr-308 by Ca2+/calmodulin (CaM)-dependent kinase kinase (CaMKK) in response to Ca2+ influx (14). CaMKK exists as two paralogues, 1 (α) and 2 (β), with closely related structures and similar enzymatic properties (15–18). CaMKK1 and CaMKK2 activate both CaMKI and CaMKIV by phosphorylating their activation loop sites (Thr-177 and Thr-200, respectively) (16). CaMKK2 is also an upstream-activating kinase for 5′-AMP-activated kinase (AMPK) (19–21). These latter studies established the precedents that CaMKK2-catalyzed phosphorylation may be directed to a target, which is not itself Ca2+/CaM-dependent, and can occur in cells that express another upstream-activating kinase (STK11/LKB1) (22).
Akt hyperactivation is thought to be the main contributor to platinum chemotherapeutic resistance in HGSOC (23). Underscoring the importance of this pathway for OVCa progression are the multiple clinical trials of PI3K/PDK1/Akt pathway inhibitors for OVCa therapy. In this study, we observed high CaMKK2 expression in OVCa clinical specimens and probed its role in Akt activation in multiple platinum-resistant HGSOC cell lines. We report herein evidence that CaMKK2 promotes the growth and survival of platinum-resistant OVCa cells. CaMKK2 phosphorylates and activates Akt, independently of PI3K and PDK1. CaMKK2 activation of Akt is seen in many downstream targets known to mediate Akt's regulation of cell growth, proliferation, apoptosis, protein synthesis, and protein stability. Akt phosphorylation was significantly inhibited by intracellular Ca2+ (Ca2+i) chelation or CaM inhibition. Ionomycin-induced Ca2+ influx promoted Akt phosphorylation, an effect blocked by PDK1, and/or CaMKK2, siRNAs, and by PI3K, and/or CaMKK, inhibitors, results suggesting the involvement of both the PI3K/PDK1 and CaMKK pathways in mediation of Ca2+-induced Akt activation. Potentiation of the anti-proliferative and anti-survival effects of carboplatin and the PI3K inhibitor PX-866 (24) by CaMKK2 silencing suggests that CaMKK2 may be a promising therapeutic target for OVCa in combination with platinum compounds and/or PI3K/PDK1 inhibitors.
Results
CaMKK2 expression in malignant ovarian tissue
CaMKK2 is normally expressed at low levels in non-neural tissues (18) but is markedly increased in prostate cancer and has been implicated in prostate and breast oncogenesis (25–28). Specimens from OVCa patients diagnosed with high-grade serous papillary cystadenocarcinoma were subjected to IHC. CaMKK2 expression was significantly higher in malignant ovarian tissue compared with non-malignant stromal elements and showed a cytoplasmic distribution (Fig. 1, A and B). CaMKK2 also appears to be well expressed in high-grade OVCa with mucinous features (Fig. 1C).
Figure 1.

CaMKK2 expression in OVCa. A–C, representative images of CaMKK2 IHC-stained samples from three patients out of four are shown. Upper and lower panels represent images from the same patient. A and B, high-grade serous papillary ovarian cystadenocarcinoma. Top, non-malignant stromal + malignant. Bottom, malignant. C, high-grade ovarian adenocarcinoma with mucinous features showing islands of tumors floating in mucinous pools. Magnifications: A–C, top, ×20; A and B, bottom, ×40; C, bottom, ×60.
CaMKK2 regulates the growth and viability of OVCa cells
To determine the role of CaMKK2 in the regulation of growth and viability of OVCa cells, CaMKK2 knockdown in OVCAR-3 cells was conducted using siRNAs that target distinct regions of the CaMKK2 mRNA (siRNAs #1 and 2). Using either siRNA, CaMKK2 knockdown decreased the growth rate of OVCAR-3 cells, resulting in significant decreases in the number of viable cells of 54.4% (siRNA #1) and 41.6% (siRNA #2), relative to non-specific (NS) controls (Fig. 2A). Because reduction in the intracellular levels of CaMKK2 protein could exert effects apart from decreasing catalytic activity, for example by disruption of protein-protein interactions or mislocalization of proteins involved in regulating cell growth and viability, we utilized the selective CaMKK inhibitor STO-609 (29). This approach enabled the suppression of catalytic activity without altering the protein levels of CaMKK2. Consistent with the siRNA effects, STO-609 decreased cell growth in a dose-dependent fashion (Fig. 2B). Knockdown or inhibition of CaMKK2 also significantly decreased cell viability, indicating that CaMKK2 is required for optimal cell survival (Fig. 2, C and D). To confirm the results obtained with OVCAR-3 cells, we utilized another well-characterized OVCa cell line, SKOV-3 (30). OVCAR-3 and SKOV-3 cells were derived from malignant ascites of human HGSOC and resistant to cisplatin and other DNA-damaging chemotherapeutic agents. Similar to the decrease in growth achieved with STO-609 treatment of OVCAR-3 cells, CaMKK inhibition decreased SKOV-3 cell growth with a comparable IC50 value to that observed in OVCAR-3 cells (supplemental Fig. S1).
Figure 2.

CaMKK2 regulates OVCAR-3 OVCa cell growth and viability. A, CaMKK2 knockdown decreased cell growth. Cells were transfected with NS or CaMKK2 siRNAs #1 (top) and #2 (bottom), and growth of viable cells was quantified. On day 5, p < 0.01 for CaMKK2 (#1 and 2) siRNAs was relative to their respective NS siRNAs. B, CaMKK pharmacological inhibitor STO-609 decreased cell growth. Cells were treated with STO-609 (0–100 μm; 5 days), and growth rates were quantified. Values represent mean ± S.D. The IC50 value of STO-609 is shown in the inset table. C, CaMKK2 knockdown decreased cell viability. Cells were transfected with NS or CaMKK2 siRNAs (#1 or 2), and percent cell viability was quantified (5 days). Values represent mean ± S.D. relative to the respective NS siRNAs. D, treatment with STO-609 decreased cell viability. Cells were treated with STO-609 (50 μm; 5 days), and percent cell viability was quantified. Values represent mean ± S.D. relative to vehicle control. C and D, **, p < 0.01; ***, p < 0.001. A–D, n = 3–5 independent experiments.
These results suggest that the effects of silencing CaMKK2 expression or reducing its activity on cell growth may be due to its regulation of both proliferation and viability. To test this hypothesis, S phase DNA synthesis was quantified by incorporation of the thymidine analogue 5-ethynyl-2′-deoxyuridine (EdU). CaMKK2 knockdown significantly decreased EdU incorporation by 44%, confirming the inhibitory effects of CaMKK2 knockdown on proliferation (Fig. 3A).
Figure 3.

CaMKK2 knockdown or pharmacological inhibition reduces proliferation and induces apoptosis in OVCAR-3 cells. A, CaMKK2 knockdown decreased cell proliferation. Cells were transfected with NS or CaMKK2 siRNAs (#1 or 2; 3 days). Proliferation was quantified by DNA synthesis assay. B–D, CaMKK2 knockdown regulated markers of apoptosis. Cells were transfected with NS or CaMKK2 siRNAs (#1 or 2). B, caspase-3/7 activity was quantified (5 days). C, cleavage of pro-caspase was assessed after normalization to GAPDH or β-tubulin (5 days). D, cleavage of PARP was assessed after normalization to GAPDH or β-tubulin (2 days). A–D, values represent mean ± S.D. relative to their respective NS siRNAs. E, STO-609-induced apoptosis. Cells were treated with STO-609 (50 μm; 5 days), and effects on apoptosis were evaluated by two-parameter flow cytometry using FITC (caspase 3/7) and PerCP (SYTOXTM). STO-609 decreased number of viable cells (Q3) and increased the number of cells in early (Q4) and late (Q2) apoptosis. Values in the inset table represent mean ± S.D. relative to the vehicle control pertaining to that quadrant. *, p < 0.05; **, p < 0.01; ***, p < 0.001. A–E, n = 3 independent experiments.
To determine whether the CaMKK2 knockdown-induced decrease in cell viability (Fig. 2, C and D) in OVCAR-3 cells occurred by apoptosis, we assayed the activity of the executioner caspases, 3 and 7, of the apoptotic cascade. CaMKK2 knockdown increased caspase 3/7 activity by 36% (siRNA #1) and 52% (siRNA #2), indicating that cell death was apoptotic (Fig. 3B). As predicted, knockdown of CaMKK2 in OVCAR-3 cells significantly increased cleavage of pro-caspase 3 and the caspase target, PARP, consistent with an apoptotic mechanism (Fig. 3, C and D). Finally, two-parameter flow cytometry was conducted by quantifying caspase 3/7 intracellular activity and DNA binding of SYTOXTM to distinguish early from late apoptosis. Treatment of OVCAR-3 cells with STO-609 significantly increased the number of cells in both early and late apoptosis and deceased the number of viable cells compared with vehicle control (Fig. 3E). These data indicate that CaMKK2, through its catalytic activity, regulates growth and survival in OVCa cells.
CaMKK2 regulates Akt activation in OVCa cells
Because the deleterious effects produced by silencing CaMKK2 expression or by reducing its activity pharmacologically on OVCAR-3 cell growth and survival strikingly phenocopied those expected of Akt inhibition (Figs. 2 and 3 and supplemental Fig. S1), we asked whether CaMKK2 is required for optimal Akt phosphorylation. CaMKK2 knockdown in OVCAR-3 cells decreased phosphorylation of Akt at its primary activation site, Thr-308 by 65% (siRNA #1) and 78% (siRNA #2) (Fig. 4A). In addition, CaMKK2 knockdown (siRNA #2) decreased phosphorylation at the Akt secondary activation site (Ser-473) (Fig. 5B). In accord with these results, STO-609 decreased Akt phosphorylation in OVCAR-3 cells at Thr-308 and Ser-473, 65 and 40%, respectively (Fig. 4, B and C). Similarly, regulation of Akt phosphorylation at Thr-308 and Ser-473 by CaMKK2 in SKOV-3 cells and a third OVCa cell line, Caov-3 was observed (supplemental Fig. S2, A–C). Altogether, these data suggest that CaMKK2 plays a significant role in maintaining Akt phosphorylation in OVCa cells.
Figure 4.

CaMKK2 regulates Akt phosphorylation in OVCAR-3 cells. A, CaMKK2 knockdown decreased p-Akt Thr-308. Cells were transfected with NS or CaMKK2 siRNAs (#1 or 2; 3 days), and phosphorylation of Akt at Thr-308 was quantified. B and C, pharmacological inhibition of CaMKK with STO-609 decreased Akt phosphorylation at Thr-308 and Ser-473, respectively. Cells were treated with STO-609 (25 μm; 5 days), and phosphorylation of Akt at Thr-308 and Ser-473 was quantified. A–C, values were normalized to Akt total and represent mean fold-change ± S.D. relative to NS siRNA or vehicle control. **, p < 0.01; ***, p < 0.001. n = 3–4 independent experiments.
Figure 5.

CaMKK2 and PI3K/PDK1 regulate Akt phosphorylation and cell growth in an additive fashion in OVCAR-3 cells. A and B, CaMKK2 and PDK1 additively regulate Akt phosphorylation. Cells were transfected with NS, PDK1, and CaMKK2 siRNAs (#2) singly or in combination, and phosphorylation of Akt at Thr-308 and Ser-473 was quantified. C and D, CaMKK2 and PI3K additively regulate Akt phosphorylation. Cells were treated with vehicle or the pharmacological inhibitor of PI3K, PX-866 (50 nm), and vehicle or STO-609 (25 μm) singly or in combination for 4 h, and phosphorylation of Akt at Thr-308 and Ser-473 was quantified. A–D, quantification of percent decreases in p-Akt Thr-308 and Ser-473 are shown in the tables below A and C and B and D, respectively. Values were normalized to Akt total. E, PDK1 and CaMKK2 knockdowns inhibit cell growth in a greater than additive fashion. Cells were transfected with NS, PDK1, and CaMKK2 siRNAs (#2) singly or in combination, and growth of viable cells was quantified. A–E, values represent mean fold-change ± S.D. relative to the respective NS siRNA or vehicle control. *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to NS siRNA or vehicle control. +, p < 0.05; ++, p < 0.01; +++, p < 0.001 for individual siRNAs or drug treatments relative to combined siRNAs or drug treatments, respectively. n = 2–6 independent experiments.
We next addressed the issue of whether CaMKK2 regulates p-Akt independently of PDK1 by comparing the effects of silencing CaMKK2 and PDK1 singly, and in combination, on Akt phosphorylation. Silencing efficiencies were equivalent between the targets whether they were transfected singly or in combination (supplemental Table S1). We reasoned that if CaMKK2 exerted its effects indirectly on p-Akt via phosphorylation of PDK1, the almost complete absence of PDK1 in the combined knockdown should largely abolish the effect of CaMKK2 knockdown on p-Akt. Conversely, if CaMKK2 phosphorylates Akt directly and independently of PDK1, its knockdown should yield a further decrease in p-Akt when combined with PDK1 knockdown.
CaMKK2 and PDK1 single knockdowns produced roughly equivalent decrements in p-Akt Thr-308 and Ser-473 (Fig. 5, A and B). These results suggest that CaMKK2 may be comparable in importance for optimal Akt phosphorylation as its canonical activator PDK1. Consistent with independent actions of PDK1 and CaMKK2, combined knockdowns in OVCAR-3 cells produced a decrease in p-Akt Thr-308 of 81%, significantly greater than that produced by either knockdown alone and virtually identical to the 80% predicted value if the kinases phosphorylate Akt independently (Fig. 5A). Similarly, the combined PDK1 and CaMKK2 knockdown is predicted to yield a decrease in p-Akt Ser-473 of 82%, which was similar to that obtained (78%) and greater than that obtained with either PDK1 or CaMKK2 knockdown alone (although the latter comparison did not achieve statistical significance) (Fig. 5B). In concordance with results in OVCAR-3 cells, combined PDK1 and CaMKK2 knockdowns in SKOV-3 cells produced a 65% reduction in p-Akt Thr-308 corresponding closely to the predicted value of a 68% reduction, based on independent actions of CaMKK2 and PDK1 (supplemental Fig. S2D).
Activation of Akt via the PI3K/PDK1 pathway requires the generation of phosphoinositides by PI3K, which allows for membrane recruitment of Akt and PDK1 by their respective PH domains (31, 32). To determine whether CaMKK2 activity regulates Akt independently of PI3K activity, Akt phosphorylation was assessed using STO-609 and the PI3K inhibitor, PX-866. Consistent with independent actions of PI3K and CaMKK2, the combined inhibition in OVCAR-3 cells produced a decrease in p-Akt Thr-308 of 69%, significantly greater than that produced by either drug alone, and virtually identical to the 70% predicted value if the kinases regulate Akt phosphorylation independently (Fig. 5C). Similarly, the combined inhibition of PI3K and CaMKK2 led to a decrease in p-Akt Ser-473 of 80%, which was virtually identical to the 79% predicted value for independent actions, and greater than that obtained with either drug alone (Fig. 5D). Finally, the additivity of PI3K and CaMKK inhibition was shown in the third cell line examined, Caov-3 (supplemental Fig. S2E). Together, these results support a mechanism by which CaMKK2 directly phosphorylates Akt at Thr-308 independently of the PI3K/PDK1 pathway, leading to subsequent phosphorylation at Ser-473 presumably as a consequence of the activation of mTORC2.
Although these data provide strong evidence for CaMKK2 phosphorylation of Akt being independent of the activity of the PI3K/PDK1 pathway, we considered the possibility that there is a more indirect mechanism by which CaMKK2 might be required for optimal Akt activation. Three possible alternate mechanisms were considered. First, the stability of Akt has been shown to be regulated by its phosphorylation at the turn motif Thr-450 by mTORC2, to facilitate C-terminal folding, and to allow for the turn motif to interact with basic residues of the kinase domain (33). We therefore sought to evaluate whether CaMKK2 phosphorylates Akt at Thr-450 inducing conformational changes exposing Thr-308 to other kinases. However, CaMKK2 knockdown in OVCAR-3 cells did not affect p-Akt Thr-450 (Fig. 6A). The lack of involvement of Thr-450 phosphorylation is consistent with the high degree of substrate specificity of CaMKK2 for structural determinants within, but not outside of, protein kinase activation loops.
Figure 6.
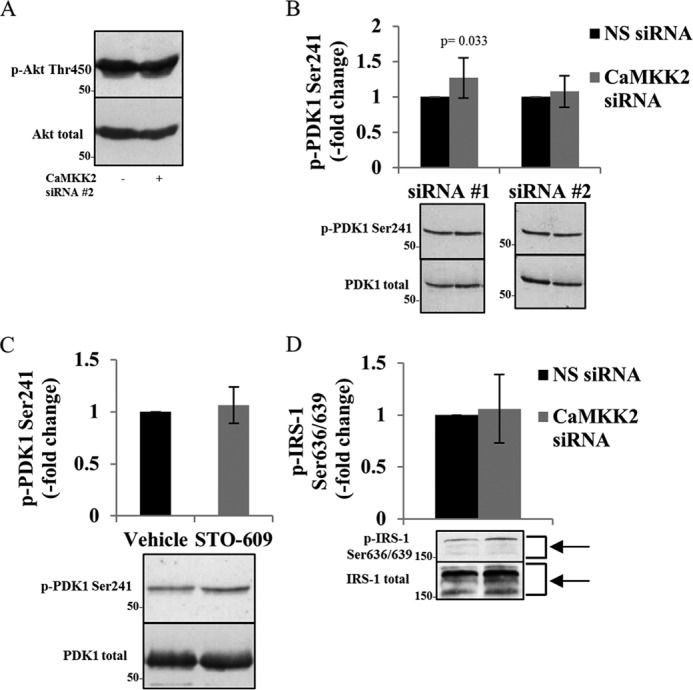
CaMKK2 knockdown or pharmacological inhibition does not affect Akt stability, PDK1-activating phosphorylation, or the mTORC1/S6K1 feedback loop in OVCAR-3 cells. A, CaMKK2 knockdown did not change p-Akt Thr-450, a marker of Akt stability. Cells were transfected with NS or CaMKK2 siRNAs (#2; 2 days), and Akt phosphorylation at Thr-450 was assessed. Akt total was used as loading control. B and C, CaMKK2 knockdown or inhibition did not reduce PDK1-activating phosphorylation. Cells were transfected with NS or CaMKK2 siRNAs (#1 or 2; 3 days) or treated with STO-609 (25 μm; 5 days), respectively, and phosphorylation of PDK1 at Ser-241 was quantified. Values were normalized to PDK1 total. D, CaMKK2 knockdown did not increase p-IRS-1 Ser-636/639. Cells were transfected with NS or CaMKK2 siRNAs (#2; 3 days), and phosphorylation of IRS-1 at Ser-636/639 was quantified. Values were normalized to IRS-1 total. B–D, values represent mean fold-change ± S.D. relative to the respective NS siRNA or vehicle control. n = 3–5 independent experiments.
A second potential mechanism is suggested by the observations that the autophosphorylation of PDK1 in its activation loop at Ser-241 is required for PDK1 activity (34, 35). The fact that this reaction is intermolecular implies that there may be conditions under which Ser-241 is accessible to an exogenous kinase. Because the only known physiological targets of CaMKK2 are protein kinase activation loop residues (36), we sought to exclude the possibility that CaMKK2 may indirectly regulate Akt by phosphorylating PDK1 at Ser-241. Knockdown or pharmacological inhibition of CaMKK2 did not decrease p-PDK1 Ser-241 (Fig. 6, B and C).
The third potential alternative mechanism involves the negative feedback loop in which Akt phosphorylation of mTORC1 leads to its activation and subsequent mTOR catalyzed phosphorylation of ribosomal protein S6 kinase β-1 (S6K1). In turn, S6K1 phosphorylation of the insulin receptor substrate-1 (IRS-1) at Ser-636/639 dampens insulin signaling upstream of Akt at the insulin receptor, resulting in decreased Akt phosphorylation (37). Thus, we also sought to exclude the possibility that CaMKK2 regulates Akt activity via inhibition of one of the components of the mTORC1/S6K1/IRS-1 negative feedback loop, thus resulting in continuous activation of Akt. Phosphorylation of IRS-1 at Ser-636/639 with CaMKK2 knockdown however was not increased, indicating that CaMKK2 does not regulate Akt phosphorylation via the mTORC1/S6K1/IRS-1 negative feedback loop (Fig. 6D).
Because a reduction in p-Akt reduces OVCa cell growth, due to impairments in cell survival and cell cycle progression, we predicted that combined PDK1 and CaMKK2 knockdowns should produce greater growth inhibition than knockdowns of either kinase alone. As shown in Fig. 5E, combined PDK1 and CaMKK2 knockdowns decreased the number of viable cells by 49%, which was significantly greater than that obtained with individual PDK1 and CaMKK2 knockdowns (16 and 26% respectively), and is greater than the predicted value for additivity of 38% (p < 0.01). This apparently greater than additive effect is consistent with evidence that PDK1 and CaMKK2 regulate proliferation via downstream targets in addition to Akt, for example S6K and CaMK1, respectively (25, 28, 38–40).
CaMKK2 directly phosphorylates, and activates, Akt in a Ca2+/CaM-dependent manner in vitro
For CaMKK2 to be considered an independent Akt regulator, CaMKK2-dependent Akt activation should be demonstrable in vitro. For this purpose, we pre-incubated recombinant CaMKK2 with recombinant WT Akt or T308A/S473A (doubly mutated, DM) Akt under phosphorylating conditions in the presence or absence of Ca2+/CaM, followed by an Akt kinase activity assay. In the presence of Ca2+/CaM, CaMKK2 activated WT Akt in a CaMKK2 concentration-dependent manner, whereas DM Akt activity was not activated, thus confirming direct activation of Akt by CaMKK2. Activation of WT Akt by CaMKK2 was greatly decreased in the absence of Ca2+/CaM, demonstrating that activation of Akt by CaMKK2 is dependent on Ca2+/CaM (Fig. 7A). We then evaluated the phosphorylation site(s) responsible for Akt activation. CaMKK2 directly phosphorylated Akt at Thr-308 but was unable to phosphorylate Akt at Ser-473 (Fig. 7B). Note that the faint band detected with the p-Akt Ser-473 antibody in the absence of CaMKK2 is explainable by the low level of phosphorylation at this site occurring endogenously during expression in Sf9 cells (see “Experimental procedures”). Because CaMKK2 did not directly phosphorylate Akt Ser-473 in vitro, the decrements in phosphoserine 473 caused by CaMKK2 inhibition and knockdown (Figs. 4C and 5, B and D; supplemental Fig. S2, B and E) are consistent with a positive feedback loop between activated Akt (p-Akt Thr-308) and SIN1/mTORC2 in OVCa cells, resulting in Akt phosphorylation at Ser-473 and full Akt activation (9).
Figure 7.

CaMKK2 activates Akt by direct phosphorylation at Thr-308 in a Ca2+/CaM-dependent fashion. CaMKK2, but not Akt, binds to Ca2+/CaM. A, CaMKK2 activated WT Akt, but not T308A/S473A (DM) Akt. Akt activation required the presence of Ca2+/CaM. Recombinant CaMKK2 and WT Akt or DM Akt were pre-incubated under phosphorylating conditions in the presence or absence of Ca2+/CaM. Akt peptide kinase activity was then assayed, and results were plotted as a function of CaMKK2 concentration. Values represent mean ± S.D. relative to 0 μm CaMKK2. *, p < 0.05; **, p < 0.01; ***, p < 0.001 for WT Akt ± Ca2+/CaM relative to DM Akt + Ca2+/CaM. +, p < 0.05; ++, p < 0.01 for WT Akt + Ca2+/CaM relative to WT Akt − Ca2+/CaM. n = 4 independent experiments. B, CaMKK2 phosphorylated Akt at Thr-308 but not at Ser-473. CaMKK2 and/or WT Akt or DM Akt were incubated under phosphorylating conditions in the presence of Ca2+/CaM. Akt phosphorylation at Thr-308 and Ser-473 was assessed. C, CaMKK2, but not Akt, bound Ca2+/CaM. The indicated amounts of recombinant GST-CaMKK2 and WT His-Akt were electrophoretically transferred onto PVDF membranes and incubated with biotinylated-CaM in the presence (Ca2+) or absence (EGTA) of Ca2+, and the membranes were immunoblotted for CaM.
Akt has been reported to bind CaM in the PH domain in a Ca2+-dependent manner, leading to the hypothesis that Ca2+/CaM binding causes recruitment to the membrane and subsequent activation by PDK1 (41, 42). Because WT Akt is activated by CaMKK2 in a Ca2+/CaM-dependent manner in vitro (Fig. 7A), we investigated which kinase was regulated by Ca2+/CaM in this assay. A CaM-binding experiment was performed on the CaMKK2 and WT Akt recombinant enzymes that were used in the in vitro kinase assay, in the presence (+Ca2+) or absence (EGTA) of Ca2+. Fig. 7C shows that CaMKK2 bound CaM in a Ca2+-dependent fashion but that Akt did not demonstrate detectable Ca2+/CaM binding. These data indicate that the Ca2+-CaM–CaMKK2 complex promotes Akt activation by direct phosphorylation at Thr-308.
We next investigated the role of Ca2+ and CaM in Akt activation in OVCAR-3 cells. The intracellular Ca2+ chelator BAPTA-AM and the CaM antagonist W-7 decreased p-Akt Thr-308, respectively (Fig. 8, A and B). In a gain-of-function approach, treatment of cells with the Ca2+-ionophore ionomycin, in the presence of Ca2+, resulted in a prominent increase in Akt phosphorylation that was reduced by W-7 (Fig. 8C). These data indicate that Ca2+ influx promotes Akt phosphorylation in a CaM-dependent manner. To determine whether the effect of ionomycin-induced Ca2+ influx to increase Akt phosphorylation required CaMKK2 and/or PDK1, the experiment illustrated in Fig. 8D was performed. As shown, both kinases appear to be required and independently act to mediate the requirement for Ca2+ influx.
Figure 8.

Akt is regulated by Ca2+ and CaM in OVCAR-3 cells. A, the intracellular Ca2+ chelator BAPTA-AM decreased p-Akt Thr-308. Cells were pre-incubated in Ca2+-free medium for 2 h and then treated with vehicle or BAPTA-AM (10 μm) in Ca2+-free medium for the times indicated, and phosphorylation of Akt at Thr-308 was quantified. B, CaM inhibitor W-7 decreased p-Akt Thr-308. Cells were treated with vehicle or W-7 (10 μm) in HBSS for the times indicated, and phosphorylation of Akt at Thr-308 was quantified. C, Ca2+ ionophore ionomycin, in the presence of Ca2+, increased p-Akt Thr-308 in a CaM-dependent manner. Cells were pre-treated with vehicle or W-7 (10 μm; 45 min) in HBSS, then treated with vehicle or ionomycin (1 μm) and Ca2+ (1 mm) for an additional 15 min in HBSS, and phosphorylation of Akt at Thr-308 was quantified. D, silencing of PDK1 and CaMKK2 decreased ionomycin-induced phosphorylation of Akt at Thr-308. Cells were transfected with NS, PDK1, and CaMKK2 siRNAs (#2) singly or in combination and then treated with ionomycin (1 μm) and Ca2+ (1 mm) for 15 min in HBSS. Phosphorylation of Akt at Thr-308 was quantified. E, pharmacological inhibition of PI3K and CaMKK activities decreased ionomycin-induced phosphorylation of Akt at Thr-308. Cells were pre-treated with vehicle or PX-866 (25 nm) and vehicle or STO-609 (50 μm) singly or in combination for 4 h in 10% FBS-containing medium, and then reated with ionomycin (1 μm) and Ca2+ (1 mm) for 15 min in HBSS, and phosphorylation of Akt at Thr-308 was quantified. A–E, values were normalized to Akt total and represent mean fold-change ± S.D. relative to the respective vehicle or vehicle + NS siRNA controls. *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to the respective vehicle or vehicle + NS siRNA control. n = 2–5 independent experiments. C, +, p < 0.05 for W-7 and ionomycin + Ca2+ relative to vehicle and ionomycin + Ca2+. D and E, ++, p < 0.01 for single kinase inhibitor relative to combined kinase inhibitors. ‡, p < 0.05; ‡‡, p < 0.01; ‡‡‡, p < 0.001 relative to ionomycin + Ca2+.
There have been several reports that Ca2+/CaM binds to the Src homology 2 domain of the regulatory 85-kDa subunit of PI3K to promote Akt activation (43, 44). To determine whether the activity of PI3K and/or that of CaMKK2 is involved in this Ca2+/CaM-mediated Akt activation, cells were treated with PX-866 and STO-609, singly or in combination, in the presence of ionomycin and Ca2+. As shown in Fig. 8E, the increase in p-Akt Thr-308 with ionomycin was blocked by both PX-866 and STO-609, suggesting that the Ca2+-dependent phosphorylation of Akt is independently mediated by both PI3K and CaMKK2. The data in Figs. 7 and 8 support a mechanism whereby Ca2+/CaM binds to and activates CaMKK2, allowing for the Ca2+-CaM–CaMKK2 complex to phosphorylate and activate Akt, and that generation of phosphoinositides by PI3K and the presence of PDK1 are also required for Ca2+-dependent Akt activation.
CaMKK2 knockdown leads to alterations in the activities of Akt downstream targets and the expression of G1/S phase cell cycle regulators in OVCa cells
We then explored the effects of CaMKK2 knockdown on the activities of a broad range of downstream Akt targets regulating protein translation, apoptosis, and cell cycle progression. Akt regulates protein translation by direct phosphorylation of PRAS40 at Thr-246, which releases the inhibition of raptor, resulting in mTORC1 phosphorylation of S6K and 4E-BP1 (45). CaMKK2 knockdown or inhibition significantly decreased p-PRAS40 Thr-246 (Fig. 9, A and B). Moreover, because the specific Akt inhibitor MK-2206 completely abolished PRAS40 phosphorylation, the reduction in PRAS40 phosphorylation by CaMKK2 knockdown confirms Akt as the mediator of this phosphorylation rather than other potential kinase targets for CaMKK2 (Fig. 9C). In further confirmation of the importance of CaMKK2-mediated Akt activation, we show that the deficit in p-PRAS40 Thr-246 induced by CaMKK2 knockdown is rescued by membrane-targeted myr-Akt (Fig. 9D).
Figure 9.

CaMKK2 knockdown or inhibition decreases phosphorylation of the Akt-specific target PRAS40 in an Akt-dependent manner in OVCAR-3 cells. A, CaMKK2 knockdown decreased p-PRAS40 Thr-246. Cells were transfected with NS or CaMKK2 siRNAs (#2; 3 days), and phosphorylation of PRAS40 at Thr-246 was quantified. B, STO-609 decreased p-PRAS40 Thr-246. Cells were treated with STO-609 (50 μm; 4 h), and phosphorylation of PRAS40 at Thr-246 was quantified. C, Akt-specific inhibitor MK-2206 completely abolished p-PRAS40 Thr-246. Cells were treated with vehicle or MK-2206 (10 μm; 2.5 h) and then treated with vehicle or STO-609 (50 μm; 4 h), singly and in combination, and phosphorylation of PRAS40 at Thr-246 was assessed. D, CaMKK2 knockdown decreased p-PRAS40 Thr-246, a phenotype that was rescued by expression of myr-Akt. Cells were transfected with NS or CaMKK2 siRNAs (#2) singly or in combination with pECE or myrAkt1, plasmids, and then treated with ionomycin (1 μm) and Ca2+ (1 mm) for 15 min in HBSS. Phosphorylation of PRAS40 at Thr-246 was quantified. A, B, and D, values were normalized to PRAS40 total and then to GAPDH. Values represent mean fold-change ± S.D. relative to NS or vehicle control or NS siRNA + pECE. *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to NS or vehicle control or to NS siRNA + pECE. D, +, p < 0.05 for CaMKK2 siRNA relative to CaMKK2 siRNA + myrAkt1. n = 3–8 independent experiments. S.E., short exposure; L.E., long exposure.
Activation of Akt promotes cell survival by inhibiting the transcription of the pro-apoptotic protein Bim, which is regulated by FoxO3a. Phosphorylation of FoxO3a at Ser-318/321 by Akt results in FoxO3a nuclear export, preventing transcription of Bim, and thereby inhibiting apoptosis (12). CaMKK2 knockdown decreased p-FoxO3a Ser-318/321 and promoted nuclear accumulation of FoxO3a (Fig. 10A). This observation is consistent with CaMKK2 knockdown or pharmacological inhibition, resulting in loss of cell viability (Fig. 2, C and D) via apoptosis (Fig. 3, B–E). Another mechanism by which Akt suppresses apoptosis is through its phosphorylation of the tumor suppressor tuberin (TSC2) at Thr-1462 resulting in its inactivation. CaMKK2 knockdown reduced p-TSC2 Thr-1462 in OVCAR-3 and SKOV-3 cells (Fig. 10B and supplemental Fig. S3A).
Figure 10.

CaMKK2 knockdown leads to alterations in the activities of Akt downstream targets and in the expression of G1/S phase cell cycle regulators in OVCAR-3 cells. A–C, CaMKK2 knockdown decreased the phosphorylation of the Akt downstream targets nuclear FoxO3a, TSC2, and GSK3β. A, cells were transfected with NS or CaMKK2 siRNA (#2; 2 days), and phosphorylation of nuclear FoxO3a at Ser-318/321 was quantified. Values were normalized to nuclear FoxO3a and then to histone H3. B, cells were transfected with NS or CaMKK2 siRNAs (#1, or 2; 3 days), and phosphorylation of TSC2 at Thr-1462 was quantified. Values were normalized to TSC2 total. C, cells were transfected with NS or CaMKK2 siRNAs (# 1 or 2; 3 days), and phosphorylation of GSK3β at Ser-9 was quantified. Values were normalized to GSK3β total. The position of p-GSK3α Ser-21 is also shown but not quantified. D–G, CaMKK2 knockdown increased the proportion of phosphorylated cyclin D1 and decreased cyclin D1 protein, protein stability, and mRNA, respectively. D, cells were transfected with NS or CaMKK2 siRNAs (#2; 3 days), and phosphorylation of cyclin D1 at Thr-286 was quantified. Values were normalized to cyclin D1 and then to β-tubulin. E, cells were transfected with NS or CaMKK2 siRNAs (#2; 2, 3 days), and cyclin D1 protein levels were quantified. Values were normalized to GAPDH. F, cells were transfected with NS or CaMKK2 siRNAs (#2; 3 days) and treated with CHX (100 μm) for the times indicated, and cyclin D1 protein stability was quantified (see under “Experimental procedures”). Values were normalized to β-tubulin. The line equations including the R2 values are displayed for each of the NS siRNA and CaMKK2 siRNA linear trend lines. G, cells were transfected with NS or CaMKK2 siRNAs (#1 or 2; 3 days), and cyclin D1 mRNA levels were quantified by qRT-PCR using two independent primer sets. Values represent mean fold-change ± S.D. relative to the respective NS siRNA with the respective primer set. H, CaMKK2 knockdown decreased the phosphorylation of the cyclin D1/Cdk4/6 downstream target Rb. Cells were transfected with NS or CaMKK2 siRNAs (#1, or 2; 3 days), and phosphorylation of Rb at Ser-807/811 was quantified. Values were normalized to Rb total. I, CaMKK2 knockdown increased the expression of the Cdk inhibitors p21 and p27. Cells were transfected with NS or CaMKK2 siRNAs (#2; 2 days), and p21 and p27 protein levels were quantified. Values were normalized to GAPDH. A–I, values represent mean fold-change ± S.D. relative to the respective NS siRNA or no CHX (F). *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to the respective NS siRNA or vehicle control. n = 3–6 independent experiments. F, +, p < 0.05 for CaMKK2 siRNA relative to NS siRNA.
Akt promotes cell growth through its positive regulation of the G1/S phase cell cycle regulator, cyclin D1. Cyclin D1, an oncogenic driver of cell growth that is tightly regulated by mitogenic stimuli, must accumulate to sufficient levels to activate Cdks 4 and 6, which phosphorylate and inactivate the tumor suppressor protein Rb and result in G1/S phase cell cycle transition (11, 46). Conversely, GSK3β negatively regulates cyclin D1 expression via phosphorylation of cyclin D1 at Thr-286 to promote its association with CRM1, thus mediating nuclear export of cyclin D1 and its subsequent proteasomal degradation (47). Akt maintains cyclin D1 levels and promotes G1/S phase cell cycle progression through the inactivation of GSK3β by its phosphorylation at Ser-9 (11). Consistent with phenotypes of Akt inhibition, CaMKK2 knockdown decreased p-GSK3β Ser-9 and, as predicted from the mechanism, resulted in a reduction in the levels of cyclin D1, with a greater proportion in the phosphorylated form (Fig. 10, C and D).
Akt mediated control of nuclear export of cyclin D1 and its proteosomal degradation, as well as effects on overall protein synthesis, predict that cyclin D1 protein is regulated by both translational and post-translational effects (11). As shown, cyclin D1 protein levels were reduced in both the cytoplasm and nucleus, and the rate of its degradation was accelerated by CaMKK2 knockdown (Fig. 10, E and F, and supplemental Fig. S3, B and C). Because the rate of cyclin D1 protein degradation was quantified using cycloheximide (CHX), we considered the potential complication that a drop in protein synthesis, as induced by CHX, results in a compensatory activation of Akt by its phosphorylation at Thr-308 (48). We replicated this phenomenon in OVCAR-3 cells (supplemental Fig. S3D). However, this effect is internally controlled in Fig. 10F because we are comparing the effect of NS siRNA and CaMKK2 siRNA both in the presence of CHX.
GSK3β not only negatively regulates cyclin D1 protein expression, but also cyclin D1 transcription by phosphorylating β-catenin to promote cytoplasmic retention and degradation of β-catenin and reducing its binding to the cyclin D1 promotor (49). CaMKK2 knockdown decreased cyclin D1 mRNA levels, confirmed using two independent qRT-PCR primer sets (Fig. 10G). To confirm that CaMKK2 knockdown reduces the levels of cyclin D1 available for activation of Cdk4/6 activities, phosphorylation of the downstream target Rb was assessed. CaMKK2 knockdown decreased phosphorylation of the cyclin D1/Cdk4/6 target Rb at Ser-807/811 by 70% (Fig. 10H).
Increased expression of the Cdk inhibitors p21 and p27 inhibit cyclin D1 by binding to, and inactivating Cdk4/6, thus preventing transition into S phase (50). Fig. 10I shows that CaMKK2 knockdown significantly increased p21 and p27 levels, complementing the decreases seen in p-Rb and cyclin D1 (Fig. 10, E, F, and H; supplemental Fig. S3, B and C). Regulation of the G1/S phase cell cycle regulators shown in Fig. 10 and supplemental Fig. S3, B and C may represent both Akt-dependent and -independent roles for CaMKK2, which is suggested by the greater than additive effect of combined PDK1 and CaMKK2 knockdowns on cell growth (Fig. 5E). Together, these data suggest that CaMKK2 is important for G1/S phase cell cycle progression and for maintaining the viability of OVCa cells. CaMKK2 has the ability to regulate downstream kinases other than Akt, three of which are well studied, CaMKI, CaMKIV, and AMPK (15–21). Thus we cannot exclude the possibility that not every phenotype observed with CaMKK2 knockdown or inhibition is necessarily attributable to Akt. Nonetheless, the broad array of Akt targets that are impacted by CaMKK2 interventions strongly suggest that flux through the CaMKK2 pathway is substantial for Akt control of growth, survival, and protein synthesis in OVCa cells.
CaMKK2 knockdown potentiates the anti-proliferative and anti-survival effects of OVCa chemotherapeutic drugs
Platinum agents, such as carboplatin, are first line therapy for OVCa; however, about 80% of patients become platinum-resistant (PR-OVCa) within the first 6 months of therapy (51). Previous studies reported that Akt amplification in PR-OVCa overcomes platinum-induced DNA damage-triggered apoptosis and that, conversely, inhibition of the PI3K/Akt pathway resensitizes PR-OVCa cells to carboplatin (23). As shown in Fig. 11, CaMKK2 knockdown potentiated the inhibitory effects of carboplatin and the PI3K inhibitor, PX-866, on OVCAR-3 cell growth and viability, respectively.
Figure 11.

CaMKK2 knockdown potentiates the inhibitory effects of carboplatin and PX-866 on OVCAR-3 cell growth and viability. A and B, left panels, OVCAR-3 cells were transfected with NS or CaMKK2 siRNAs (#2) and treated with carboplatin (0–251 μm) or PX-866 (0–50 μm), respectively, starting 1 day after transfection for an additional 5 days, and growth of viable cells was quantified. IC50 values are shown in the tables below the respective log dose-response curves. Middle and right panels, effects of the indicated doses of carboplatin and PX-866 on percent viability at two different doses. Values represent mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to NS siRNA − drug. +, p < 0.05; ++, p < 0.01; +++, p < 0.001 for CaMKK2 siRNA + drug relative to NS siRNA + drug. n = 2–6 independent experiments.
Collectively, the results of this investigation are compatible with a mechanism in which CaMKK2 responds to an elevation in Ca2+i, by phosphorylation of Akt at Thr-308 in a Ca2+/CaM-dependent and PI3K/PDK1-independent fashion. Following Ser-473 phosphorylation, fully activated Akt phosphorylates downstream targets driving cell growth and suppressing apoptosis. This model may provide both a framework for further investigation at the molecular level and study of whether CaMKK2 represents a novel and promising target for OVCa therapy in combination with carboplatin and/or PI3K inhibitors.
Discussion
The current model by which the oncogenic kinase Akt is activated is almost invariably conceptualized as a single, linear GF/PI3K/PDK1/Akt/mTORC cascade referred to here as the “canonical” pathway. Clinical trials for a variety of cancers have entailed targeting this pathway at levels up- and downstream of Akt, as well as at the level of Akt itself. The rationale for this strategy has been established by a wealth of pre-clinical and clinical data supporting the importance of Akt in human malignancies (1–9, 23, 24, 52).
However, there have been reports that Akt activity may be enhanced or maintained by non-canonical pathway(s). One such pathway involves Ca2+i/CaM as an intracellular mediator of Akt activation. It was initially reported that CaMKK, in neuroblastoma cells, is capable of activating Akt in response to NMDA-induced Ca2+ influx (14). Although this study has received relatively little experimental follow-up, there have been reports of CaMKK-dependent Akt activation in LNCaP and HEK293 cells (53, 54). In addition, studies have shown that treatment of mammary carcinoma cells with BAPTA-AM or W-7 causes significant decrements in Akt phosphorylation (41), that CaM directly binds to Akt (42), and that GF stimulation of cancer cells is capable of raising Ca2+i (55). Taken together, these lines of evidence raise the possibility that platinum-resistant HGSOC develops with the participation of the Ca2+/CaM/CaMKK pathway distinct from, but possibly in cooperation with, the canonical PI3K/PDK1 pathway.
We addressed this hypothesis by first examining the expression of CaMKK2 in OVCa. CaMKK2 expression in non-neuronal tissues is normally low (18). However, Fig. 1 shows that CaMKK2 is highly expressed in clinical specimens representative of HGSOC and mucinous OVCa, data consistent with The Cancer Genome Atlas (TCGA) (http://www.cbioportal.org/public-portal/)3 and The Human Protein Atlas (http://www.proteinatlas.org/)3 datasets reporting amplification and high protein expression respectively, of CaMKK2 in OVCa. These data suggest that CaMKK2 may be subject to selection during ovarian tumor evolution to maintain growth and survival.
IHC of HGSOC and mucinous high-grade OVCa revealed only cytoplasmic staining (Fig. 1). In previous reports, we observed that in the prostate cancer cell lines LNCaP and C4-2, dihydrotestosterone induces CaMKK2 expression and its nuclear translocation (25, 56). However, we have not to date observed in OVCa cells hormonal regulation of CaMKK2 expression by estrogen, progesterone, or dihydrotestosterone (67). Our working interpretation is that for PCa, CaMKK2 nuclear translocation is apparently related to the development of castrate resistance accompanied by increasing tumor grade and changes in the androgen receptor.
CaMKK2 knockdown in OVCa cell lines down-regulated cell growth and survival, phenotypes consistent with Akt mediation of these effects. Using recombinant, purified enzymes, we found CaMKK2 to be a regulator in vitro of Akt activation by phosphorylation at Thr-308 in its activation loop and in cell-based experiments at both Thr-308 and Ser-473. These findings extended the relevance of CaMKK control of Akt activity to a cancer cell type for which the canonical PI3K/PDK1 pathway is oncogenic. CaMKK2 and PDK1 knockdowns produced quantitatively similar reductions in Akt phosphorylation, as did CaMKK and PI3K inhibitors, highlighting the importance of CaMKK2 activity for Akt activation. These findings imply that in OVCa cells, CaMKK2 could provide significant Akt activation under conditions in which the PI3K/PDK1 pathway is targeted for inhibition.
The high level of CaMKK2 expression in clinical specimens of HGSOC suggests a possible relationship of CaMKK2 expression to tumor progression and/or drug resistance and to the state of Akt phosphorylation in ovarian tumors. To understand at the molecular and cellular levels, the rationale for CaMKK2 expression in HGSOC, we utilized OVCAR-3, SKOV-3, and Caov-3, cell lines representative of platinum-resistant HGSOC that are among the five most extensively studied in OVCa research (based on PubMed citations, 2012) (30). To assess whether the protein levels of CaMKK2 and PDK1 would be predictive of the extent of Akt phosphorylation under conditions of continuous stimulation by GFs in their basal media, we quantified their relative levels in the three OVCa cell lines. It may be concluded that there is no simple quantitative relationship between CaMKK2 (or PDK1) protein concentrations and the degree of Akt phosphorylation in these OVCa cell lines (supplemental Fig. S4). This is likely due to the variable activities of both PDK1 and CaMKK2 controlled by signaling events under specific cell culture conditions and the presence or absence of loss- or gain-of-function mutations in other pathway components such as PI3K or PTEN. Nonetheless, it is relevant that the requirement of CaMKK2 for optimal Akt phosphorylation is not unique to OVCAR-3 cells and can occur in both Caov-3 and SKOV-3 cells, the latter of which bears a PIK3CA-activating mutation (30) predicting a greater role for PDK1 in Akt-activating phosphorylation. Clearly, future in vivo studies will be required to establish mechanisms by which CaMKK2 expression is regulated by tumor progression and/or selective pressure during chemotherapy and to describe quantitatively how this expression is related to the state of activation of Akt.
The requirement of Ca2+/CaM for in vitro phosphorylation of Akt by CaMKK2 prompted an investigation into the control of Akt by Ca2+ and CaM in OVCa cells. Chelation of Ca2+i by BAPTA-AM and CaM antagonism by W-7 significantly drove down p-Akt Thr-308 under normal growth conditions, and W-7 blocked the increase in p-Akt Thr-308 produced by the Ca2+ ionophore, ionomycin (Fig. 8, A–C). These results reinforce the idea that Ca2+/CaM-stimulated Akt phosphorylation, through downstream effects on cell cycle regulators, modulates progression at G1/S, a checkpoint at which Ca2+i oscillations are known to occur (55). Importantly, ionomycin-induced Akt phosphorylation was blocked by both PDK1, and CaMKK2, siRNAs and by PI3K, and CaMKK, inhibitors, suggesting that Ca2+-mediated Akt activation is driven by both CaMKK- and PI3K/PDK1-dependent pathways (Fig. 8, D and E).
Depending on the state of the cells under study (normal versus malignant) and the tissue from which the cells are derived, there are several potential mechanisms that could account for the existence of separate CaMKK- and PI3K/PDK1-driven pathways for activating Akt. One is that Ca2+i/CaM provides an alternate route to that of GF-stimulated phosphoinositide generation for activating Akt and maintaining cellular growth and survival in pathological settings. Prior to angiogenesis, within the poorly vascularized (hypoxic) core of a tumor, there may be inadequate concentrations of GFs available for full activation of the PI3K/PDK1/Akt pathway. Cancer cells that could sustain Akt activation by Ca2+i stimulation of CaMKK2 or other means could maintain cell growth and survival, despite the lack of GFs, and acquire a selective growth advantage. Another possibility is that GFs, rather than acting as Ca2+i-independent modules, could utilize a Ca2+i-dependent pathway to regulate Akt and function in cellular environments with adequate supplies of GFs (such as the growth conditions in this study). For example, GF stimulation of PLCϵ causes increases in inositol 1,4,5-trisphosphate levels and the release of Ca2+i from endoplasmic reticulum stores, which could enhance Ca2+/CaM-dependent events such as CaMKK2 activation (57).
A final alternative is that these two pathways for Akt activation operate in concert with each other. We did not observe Ca2+-dependent CaM binding to recombinant Akt1 (Fig. 7). However, such binding may occur intracellularly as it has been reported that Ca2+-dependent CaM binding to the PH domain of Akt1 occurs and is competitive with respect to phosphoinositides (42). One can envisage a mechanism to sustain Akt activation as a response to reduced concentrations of phosphoinositides through recruitment of Ca2+/CaM/CaMKK2 to Akt forming a CaMKK2–CaM–Akt ternary complex to mediate transduction of the Ca2+i signal. Such a mechanism could explain the apparently greater than additive effects of PX-866 and STO-609 to block the ionomycin-induced Akt phosphorylation (Fig. 8E), as due to a PX-866 reduction in phosphoinositides, sensitizing Akt phosphorylation to CaMKK2 inhibition.
These hypothetical mechanisms are generally consistent with other investigations, with one possible exception. Unlike this study (Fig. 4C and 5D; supplemental Fig. S2, B and E), it was reported that p-Akt Ser-473 was unresponsive to STO-609, by which it was concluded that CaMKK activity is unrelated to Ca2+/CaM-dependent Akt phosphorylation (41). However, the STO-609 concentration utilized in this latter investigation (0.27 μm) was considerably less than in this present study, which is based on an observed STO-609 IC50 of ∼20 μm toward OVCa cell growth, and the drug effects of the latter are supported by RNAi experiments.
The results presented here raise an interesting question as to the evolutionary benefit to the cell of having a single regulatory kinase, CaMKK2, promote the activation of Akt to drive rapid growth accompanied by energy utilization but also AMPK for the purposes of energy conservation. Although AMPK has been long thought of as exerting a tumor-suppressive effect, it has also been proposed to enhance the survival of cancer cells subjected to metabolic stress, suggesting that depending on cell state, it may act either as a tumor suppressor or “contextual oncogene” (58). We propose that there is a dynamic balance between Akt promotion of rapid tumor growth and AMPK-mediated energy conservation to promote tumor cell survival under conditions of bioenergetic stress (e.g. GF deprivation), a balance that may underlie the non-linear (Gompertzian) growth of solid tumors (59). It will be of interest in future experiments to explore this hypothesized balance in which CaMKK2 may participate.
We have also considered the potential clinical implications of these results. GF receptor/PI3K and Ca2+-driven pathways for Akt activation could represent redundant means by which the tumor cell ensures continued growth and survival in adapting to changing tumor microenvironments. This implies that mutagenic alterations in the relative expression/activity levels of the various pathway components (PI3K/PDK1 versus CaMKK2) in response to tumor environmental changes and/or drug therapy may allow one or the other pathway to predominate. CaMKK2 is well expressed in HGSOC and mucinous OVCa (Fig. 1), although the relationship of its expression to chemotherapeutic resistance is unknown. PIK3CA- and AKT-activating mutations and PTEN-inactivating mutations represent less than 5% of HGSOC (60), and it has been suggested that PI3K/Akt pathway mutations/CNA are likely to represent markers of chemoresistance, for example to platinum compounds (23). The possibility that Ca2+/CaM-CaMKK2 could promote chemoresistance by signaling through Akt is consistent with data that CaM antagonists restore cisplatin sensitivity in platinum-resistant OVCa cells (61). This finding is supported by our data that the CaM inhibitor W-7 potently inhibited OVCa growth and viability (supplemental Fig. S5) and that silencing of CaMKK2 potentiated the anti-proliferative effects and reductions in viability, produced by carboplatin in OVCa cells (Fig. 11A).
Finally, in addition to resistance to clinically utilized platinum compounds, resistance to PI3K inhibitors is known to develop (62, 63). Thus, it is conceivable that inhibition of Akt activation by a combination of PI3K and CaMKK inhibitors would require lower doses and delay PI3K inhibitor resistance and/or have less toxicity in promoting apoptosis and cell cycle arrest compared with the drugs used as monotherapy, a claim supported by the data of Fig. 11.
We conclude that given the importance of the Akt pathway for OVCa progression and platinum resistance, the results of this study implicating CaMKK2 as an Akt activator suggest that development of clinically active CaMKK inhibitors may prove to be a fruitful approach to full realization of the therapeutic potential of combination therapy targeted to the oncogenic PI3K/PDK1/Akt signaling axis.
Experimental procedures
Chemicals
STO-609 was purchased from Tocris. PX-866 was purchased from Cell Signaling. W-7 and BAPTA-AM were purchased from Calbiochem. Ionomycin was purchased from Invitrogen. Carboplatin and cycloheximide were purchased from Sigma. MK-2206 was purchased from AdooQ.
Cell culture
NIH:OVCAR-3 (OVCAR-3), SK-OV-3 (SKOV-3), and Caov-3 (Caov3) cells were obtained from American Type Culture Collection (ATCC) and authenticated by ATCC by short tandem repeat DNA analysis. OVCAR-3 cells were cultured in RPMI 1640 medium supplemented with 20% FBS, 10 mm HEPES, 1 mm sodium pyruvate, 1× penicillin/streptomycin, 2 mm l-glutamine, 2.4 mg/ml d-glucose, and 1% insulin. SKOV-3 and Caov-3 cells were cultured in McCoy's 5A and DMEM high glucose media, respectively, and supplemented with 10% FBS and 1× penicillin/streptomycin. Cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
IHC
Sections of human high-grade serous papillary ovarian cystadenocarcinoma and high-grade ovarian carcinoma with mucinous features were obtained from de-identified paraffin blocks from the Department of Pathology, Roswell Park Cancer Institute. IHC was performed as described previously using an anti-CaMKK2 antibody (Sigma/Prestige, 1:500) (25).
RNA interference and transfection
siRNA sequences are listed under supplemental Experimental procedures. 18 h prior to transfection, cells were plated at densities of 3 × 105 cells/60-mm plate or 1 × 106 cells/100-mm plate in antibiotic-free medium. Cells were transfected with the following siRNAs: NS control, CaMKK2 (#1 or #2), or PDK1, siRNAs in serum-free and antibiotic-free medium using Lipofectamine 2000 (Invitrogen) as the transfection agent. After 6 h, medium was replaced with antibiotic-free serum-containing medium, and cells were grown for durations indicated in the figure legends. Combined transfection of CaMKK2 and PDK1 siRNAs utilized a sequential design. Cells were transfected twice according to the following schedule: NS control: NS siRNA day 0, NS siRNA day 2; PDK1 alone: PDK1 siRNA day 0, NS siRNA day 2; CaMKK2 alone: NS siRNA day 0, CaMKK2 siRNA day 2; combined PDK1, CaMKK2: PDK1 siRNA day 0, CaMKK2 siRNA day 2. All cultures were then grown for an additional 3 days and harvested on day 5.
Cell growth and viability
For knockdown experiments in which cell growth and viability were measured (Figs. 2, A and C, and 5E), OVCa cells were transfected with NS, CaMKK2, and PDK1, siRNAs singly or in combination as described above and in the respective legends. For carboplatin and PX-866 treatments combined with CaMKK2 knockdown (Fig. 11), OVCa cells were transfected with NS or CaMKK2 siRNAs (#2) on day 0, treated with carboplatin or vehicle (ddH2O) or with PX-866 or vehicle (<0.5% DMSO) on day 1 in 10% FBS-containing medium, and counted on day 6. For STO-609 treatments (Fig. 2 B and D; supplemental Fig. S1), cells were treated with drug or vehicle (<0.7 mm NaOH) on day 0 in medium containing one-half the concentration of FBS as their normal growth media, and counted on day 5. For W7 treatments (supplemental Fig. S5), cells were treated with drug or vehicle (<0.8% DMSO) on day 0 in 10% FBS-containing medium and counted on day 5. Floating and attached cells were collected and centrifuged at 125 × g for 6 min at room temperature. Cell pellets were resuspended, and aliquots were diluted in trypan blue (Invitrogen). Live and dead cells were counted using a hemocytometer or by automated cell counting (TC20; Bio-Rad). Percent cell viability = (number of live cells ÷ (number of live + dead cells)) × 100. Dose-response curves and IC50 values were generated using GraphPad software.
DNA synthesis assay
OVCAR-3 cells were transfected with NS or CaMKK2 siRNAs in 6-well plates for 2 days. Cells were labeled with 10 μm of the thymidine analogue EdU, placed on a rotary shaker for 2 min, and incubated for 24 h. Incorporation of EdU was measured as recommended using a Click-iT EdU microplate assay kit (Invitrogen).
Caspase 3/7 activity assay
OVCAR-3 cells were transfected with NS, or CaMKK2, siRNAs. On day 5, cells were processed, and caspase activity was determined using a caspase GLO 3/7 activity assay as per the manufacturer's modified protocol (Promega) (64).
Flow cytometry apoptosis assay
OVCAR-3 cells were treated with 50 μm STO-609 or vehicle (<0.4 mm NaOH) for 5 days. Cells were detached with trypsin, centrifuged, and resuspended in PBS. The cells were then labeled with 500 nm CellEvent caspase 3/7 green detection reagent (caspase 3/7) and 1 μm SYTOX AADvanced Dead Cell Stain (SYTOXTM) using the CellEvent caspase 3/7 Green Flow cytometry assay kit (Invitrogen) as per the manufacturer's protocol. Cells were excited with a 488-nm blue laser, using a 515/20 BP filter for caspase 3/7 (FITC) and a 695/40 BP filter for SYTOXTM (PerCP). Single cells were analyzed using a BD LSRFortessa-SORP flow cytometer and BD FACSDiva software.
Western blotting
Western blotting was performed as described previously (25) using primary antibodies listed in the supplemental Experimental procedures. Protein level intensities obtained from within the linear range of exposures were quantified after local background subtraction using Quantity One software (Bio-Rad) or Image Studio (Licor) and shown in figures with representative blots.
Treatment of cells with PX-866 and STO-609
As described in Fig. 5, C and D and supplemental Fig. S2E, OVCa cells were plated at a density of >3 × 105 cells/60-mm plate and treated with 50 nm PX-866 or vehicle (<0.03% DMSO) or 25 μm STO-609 or vehicle (<0.2 mm NaOH) singly or in combination for 4 h in media containing one-half the concentration of FBS as their normal growth media.
Akt expression and purification
His-tagged human full-length (WT) and DM (T308A/S473A) Akt1 recombinant proteins were generated from 1 liter each of Sf9 cells growing exponentially in serum-free Medium-II (Life Technologies, Inc.) at 26 °C. 60 h after infection with baculovirus at a multiplicity of infection of 10:1, Sf9 cells were collected by centrifugation and washed twice in cold PBS prior to lysis in hypotonic buffer using a high-pressure microfluidizer. Recombinant protein purification by FPLC was performed by affinity chromatography on TALONTM metal affinity columns using imidazole gradient elution followed by gel-exclusion chromatography, similar to methods described previously (65). Protein purity was estimated to be at least 90% by Coomassie staining of purified proteins on SDS-PAGE. Protein sequence and phosphorylation were investigated by mass spectrometry and Western blotting with phospho-specific antibodies. Under the growth conditions employed, less than 10% of WT Akt protein was phosphorylated, with no evidence of phosphorylation of the DM Akt protein.
In vitro Akt peptide kinase activity assay
His-tagged human full-length (WT) and DM, T308A/S473A, Akt1 recombinant proteins were pre-incubated with recombinant, baculovirus-expressed GST-tagged human full-length CaMKK2 (Life Technologies, Inc.) for 40 min at 30 °C in a pre-incubation buffer consisting of 50 mm Tris, pH 7.6, 0.5 mm DTT, 10 mm MgCl2, 200 μm ATP, 0.5 mg/ml BSA, (± 1 mm CaCl2, 1 μm CaM). Akt peptide kinase reactions were performed after addition of 25 μm Akt/SKG substrate peptide (Tocris) and [γ-32P]ATP to a final concentration of 100 μm (∼80 cpm/pmol). Peptide phosphorylation was determined as described previously (66). Reaction rates were confirmed to be linear. Nonspecific peptide kinase activity of CaMKK2 (at its respective concentrations) toward the Akt peptide substrate in the absence of Akt was subtracted as blank.
Calmodulin binding (CaM overlay)
Purified recombinant GST-CaMKK2 (87 kDa) and WT His-Akt (61 kDa) enzymes were resolved by SDS-PAGE and electrophoretically transferred to PVDF membranes, as described previously (25). Membranes were washed with T-TBS + Ca2+ (1 mm CaCl2) or T-TBS − Ca2+ (1 mm EGTA) and blocked in T-TBS 5% BSA + Ca2+ (1 mm CaCl2) or T-TBS 5% BSA − Ca2+ (1 mm EGTA). Membranes were incubated with 0.5 μg/ml biotinylated porcine brain CaM (EMD Millipore) with shaking for 30 min at room temperature. Membranes were then washed and incubated with a primary anti-CaM antibody overnight and processed further as described previously (25).
Treatment of cells with BAPTA-AM, W-7, W-7 and ionomycin + Ca2+, and PX-866/STO-609 and ionomycin + Ca2+
As described in Fig. 8, OVCAR-3 cells were plated at a density of >3 × 105 cells/60-mm plate. Cells were washed with Hanks' balanced salt solution (HBSS) (Corning). The following treatments were conducted. For BAPTA-AM, cells were incubated in Ca2+-free incomplete RPMI 1640 medium (Invitrogen) for 2 h and then treated with 10 μm BAPTA or vehicle (0.3% DMSO) in HBSS for the times indicated. For W-7, cells were treated with 10 μm W-7 or vehicle (0.08% DMSO) in HBSS for the times indicated. For W-7 and ionomycin + Ca2+, cells were pre-treated with 10 μm W-7 or vehicle for 45 min in HBSS and then treated with 1 μm ionomycin + 1 mm CaCl2 or vehicle (0.08% DMSO) in HBSS for 15 min. For PX-866/STO-609 and ionomycin + Ca2+, cells were pre-treated with 25 nm PX-866 or vehicle (0.025% DMSO) or 50 μm STO-609 or vehicle (0.2 mm NaOH) in 10% FBS-containing medium for 4 h. Cells were washed; medium was changed to HBSS, and cells were incubated for 45 min. Cells were then treated with 1 μm ionomycin, 1 mm CaCl2, or vehicle (0.08% DMSO) in HBSS for 15 min.
Treatment of cells with MK-2206 and STO-609
As described in Fig. 9C, OVCAR-3 cells were plated at a density of >4.5 × 105 cells/60-mm plate. Cells were washed with HBSS (Corning). Cells were pre-treated with 10 μm MK-2206 (AdooQ) or vehicle (0.05% DMSO) in 10% FBS-containing medium for 2.5 h and treated with 50 μm STO-609 or vehicle (0.2 mm NaOH) for an additional 4 h.
Plasmids
pECE and myr-Akt δ4-129 (myrAkt1) were the gifts from William Rutter. The control pECE was used at the same concentrations as the Akt plasmid.
CaMKK2 knockdown and expression of myrAkt1
As described in Fig. 9D, OVCAR-3 cells were transfected with NS or CaMKK2 siRNAs for 2 days. Cells were then transfected with 750 ng of pECE control, or myrAkt1, plasmids for 1 day. Cells were washed, and medium was changed to HBSS, and cells were incubated for 45 min. Cells were then treated with 1 μm ionomycin + 1 mm CaCl2 in HBSS for 15 min.
Nuclear-cytoplasmic subcellular distribution
OVCAR-3 cells were transfected with NS or CaMKK2 siRNAs, and nuclear and cytoplasmic compartments of OVCAR-3 cells were separated as described previously (24).
qRT-PCR
qRT-PCR was performed as described (25) and quantitated by the ΔΔCt method with GAPDH as the reference gene. Primer sequences are listed in supplemental Experimental procedures.
Cyclin D1 protein stability
OVCAR-3 cells were transfected as described under “RNA interference and transfection” with NS or CaMKK2 siRNAs and treated with 100 μm CHX or vehicle (ddH2O) for the times indicated in Fig. 10F. Western blotting was performed using an anti-cyclin D1 antibody. First-order exponential decay kinetics were analyzed by natural logarithmic transformation of cyclin D1 protein levels normalized to β-tubulin and represent percent cyclin D1 protein remaining relative to no CHX (0 min) for the respective siRNAs.
Statistical analysis
Statistical significance was determined by unpaired Student's t test. p values <0.05 were considered statistically significant. All results represent 2–8 independent experiments.
Author contributions
A. M. G. designed the study, performed all of its experiments, analyzed the data, and wrote the manuscript. G. A. evaluated the immunohistochemistry of ovarian cancer specimens. L. M. E. contributed to supplemental Figs. S1 and S3 and provided additional technical assistance. S. D. contributed to supplemental Fig. S5, B and C, and provided additional contributions to the design of the study. L. G. K. contributed to the design of the study. T. F. F. provided purified, wild type and mutant, recombinant Akt enzymes. A. M. E. designed the study, wrote the manuscript, and coordinated the study. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge the helpful discussions and technical assistance of Dr. Wilma Hofmann and Elaine Goldstein. We thank Dr. Joan Baizer for assistance with immunohistochemistry, Christine Willock for help with Akt protein purification, and Dr. Thomas Neubert for help with mass spectrometric analysis of purified Akt proteins.
This work was supported by Mark Diamond Research Fund SU-15-04 (to A. M. G.), University at Buffalo Foundation and Department of Defense Grant OC150368 (to A. M. E.), and by National Institutes of Health Grant OD018340 (to T. F. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S5, Table S1, and Experimental procedures.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- HGSOC
- high-grade serous ovarian cancer
- CaM
- calmodulin
- CaMKK
- CaM-dependent protein kinase kinase
- OVCa
- ovarian cancer
- CNA
- copy number alteration
- Rb
- retinoblastoma
- PARP
- poly(ADP-ribose) polymerase
- PH
- pleckstrin homology
- mTOR
- mechanistic target of rapamycin
- IHC
- immunohistochemistry
- EdU
- 5-ethynyl-2′-deoxyuridine
- DM
- double mutant
- CHX
- cycloheximide
- PR-OVCa
- platinum-resistant-OVCa
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester
- Cdk
- cyclin-dependent kinase
- S6K
- S6 kinase
- GF
- growth factor
- AMPK
- 5′-AMP-activated kinase
- NS
- non-specific
- qRT-PCR
- quantitative reverse transcription-PCR
- ddH2O
- double distilled H2O
- HBSS
- Hanks' balanced salt solution.
References
- 1. Yap T. A., Carden C. P., and Kaye S. B. (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat. Rev. Cancer 9, 167–181 [DOI] [PubMed] [Google Scholar]
- 2. Hanrahan A. J., Schultz N., Westfal M. L., Sakr R. A., Giri D. D., Scarperi S., Janakiraman M., Janikariman M., Olvera N., Stevens E. V., She Q. B., Aghajanian C., King T. A., Stanchina Ed, Spriggs D. R., et al. (2012) Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2, 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Marco C., Rinaldo N., Bruni P., Malzoni C., Zullo F., Fabiani F., Losito S., Scrima M., Marino F. Z., Franco R., Quintiero A., Agosti V., and Viglietto G. (2013) Multiple genetic alterations within the PI3K pathway are responsible for AKT activation in patients with ovarian carcinoma. PLoS ONE 8, e55362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patch A. M., Christie E. L., Etemadmoghadam D., Garsed D. W., George J., Fereday S., Nones K., Cowin P., Alsop K., Bailey P. J., Kassahn K. S., Newell F., Quinn M. C., Kazakoff S., Quek K., et al. (2015) Whole-genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494 [DOI] [PubMed] [Google Scholar]
- 5. Bast R. C. Jr., and Mills G. B. (2012) Dissecting “PI3Kness”: the complexity of personalized therapy for ovarian cancer. Cancer Discov. 2, 16–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Altomare D. A., Wang H. Q., Skele K. L., De Rienzo A., Klein-Szanto A. J., Godwin A. K., and Testa J. R. (2004) AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 23, 5853–5857 [DOI] [PubMed] [Google Scholar]
- 7. Wong K. K., Engelman J. A., and Cantley L. C. (2010) Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 20, 87–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Franke T. F. (2008) PI3K/Akt: getting it right matters. Oncogene 27, 6473–6488 [DOI] [PubMed] [Google Scholar]
- 9. Humphrey S. J., Yang G., Yang P., Fazakerley D. J., Stöckli J., Yang J. Y., and James D. E. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L., Harris T. E., and Lawrence J. C. (2008) Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 283, 15619–15627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim J. K., and Diehl J. A. (2009) Nuclear cyclin D1: an oncogenic driver in human cancer. J. Cell. Physiol. 220, 292–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X., Tang N., Hadden T. J., and Rishi A. K. (2011) Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 1813, 1978–1986 [DOI] [PubMed] [Google Scholar]
- 13. Williams M. R., Arthur J. S., Balendran A., van der Kaay J., Poli V., Cohen P., and Alessi D. R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10, 439–448 [DOI] [PubMed] [Google Scholar]
- 14. Yano S., Tokumitsu H., and Soderling T. R. (1998) Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396, 584–587 [DOI] [PubMed] [Google Scholar]
- 15. Tokumitsu H., Enslen H., and Soderling T. R. (1995) Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 270, 19320–19324 [DOI] [PubMed] [Google Scholar]
- 16. Edelman A. M., Mitchelhill K. I., Selbert M. A., Anderson K. A., Hook S. S., Stapleton D., Goldstein E. G., Means A. R., and Kemp B. E. (1996) Multiple Ca2+-calmodulin-dependent protein kinase kinases from rat brain. Purification, regulation by Ca2+-calmodulin, and partial amino acid sequence. J. Biol. Chem. 271, 10806–10810 [DOI] [PubMed] [Google Scholar]
- 17. Kitani T., Okuno S., and Fujisawa H. (1997) Molecular cloning of Ca2+/calmodulin-dependent protein kinase kinase β. J. Biochem. 122, 243–250 [DOI] [PubMed] [Google Scholar]
- 18. Anderson K. A., Means R. L., Huang Q. H., Kemp B. E., Goldstein E. G., Selbert M. A., Edelman A. M., Fremeau R. T., and Means A. R. (1998) Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase β. J. Biol. Chem. 273, 31880–31889 [DOI] [PubMed] [Google Scholar]
- 19. Hurley R. L., Anderson K. A., Franzone J. M., Kemp B. E., Means A. R., and Witters L. A. (2005) The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280, 29060–29066 [DOI] [PubMed] [Google Scholar]
- 20. Hawley S. A., Pan D. A., Mustard K. J., Ross L., Bain J., Edelman A. M., Frenguelli B. G., and Hardie D. G. (2005) Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19 [DOI] [PubMed] [Google Scholar]
- 21. Woods A., Dickerson K., Heath R., Hong S. P., Momcilovic M., Johnstone S. R., Carlson M., and Carling D. (2005) Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 [DOI] [PubMed] [Google Scholar]
- 22. Hardie D. G. (2014) AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 20, 939–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carden C. P., Stewart A., Thavasu P., Kipps E., Pope L., Crespo M., Miranda S., Attard G., Garrett M. D., Clarke P. A., Workman P., de Bono J. S., Gore M., Kaye S. B., and Banerji U. (2012) The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol. Cancer Ther. 11, 1609–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ihle N. T., Williams R., Chow S., Chew W., Berggren M. I., Paine-Murrieta G., Minion D. J., Halter R. J., Wipf P., Abraham R., Kirkpatrick L., and Powis G. (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol. Cancer Ther. 3, 763–772 [PubMed] [Google Scholar]
- 25. Karacosta L. G., Foster B. A., Azabdaftari G., Feliciano D. M., and Edelman A. M. (2012) A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 287, 24832–24843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frigo D. E., Howe M. K., Wittmann B. M., Brunner A. M., Cushman I., Wang Q., Brown M., Means A. R., and McDonnell D. P. (2011) CaM kinase kinase β-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 71, 528–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Massie C. E., Lynch A., Ramos-Montoya A., Boren J., Stark R., Fazli L., Warren A., Scott H., Madhu B., Sharma N., Bon H., Zecchini V., Smith D. M., Denicola G. M., Mathews N., et al. (2011) The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 30, 2719–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodriguez-Mora O. G., LaHair M. M., McCubrey J. A., and Franklin R. A. (2005) Calcium/calmodulin-dependent kinase I and calcium/calmodulin-dependent kinase kinase participate in the control of cell cycle progression in MCF-7 human breast cancer cells. Cancer Res. 65, 5408–5416 [DOI] [PubMed] [Google Scholar]
- 29. Tokumitsu H., Inuzuka H., Ishikawa Y., Ikeda M., Saji I., and Kobayashi R. (2002) STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 277, 15813–15818 [DOI] [PubMed] [Google Scholar]
- 30. Domcke S., Sinha R., Levine D. A., Sander C., and Schultz N. (2013) Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson K. E., Coadwell J., Stephens L. R., and Hawkins P. T. (1998) Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr. Biol. 8, 684–691 [DOI] [PubMed] [Google Scholar]
- 32. Currie R. A., Walker K. S., Gray A., Deak M., Casamayor A., Downes C. P., Cohen P., Alessi D. R., and Lucocq J. (1999) Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 337, 575–583 [PMC free article] [PubMed] [Google Scholar]
- 33. Facchinetti V., Ouyang W., Wei H., Soto N., Lazorchak A., Gould C., Lowry C., Newton A. C., Mao Y., Miao R. Q., Sessa W. C., Qin J., Zhang P., Su B., and Jacinto E. (2008) The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 27, 1932–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Casamayor A., Morrice N. A., and Alessi D. R. (1999) Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem. J. 342, 287–292 [PMC free article] [PubMed] [Google Scholar]
- 35. Wick M. J., Ramos F. J., Chen H., Quon M. J., Dong L. Q., and Liu F. (2003) Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J. Biol. Chem. 278, 42913–42919 [DOI] [PubMed] [Google Scholar]
- 36. Racioppi L., and Means A. R. (2012) Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J. Biol. Chem. 287, 31658–31665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J., Gao Z., Yin J., Quon M. J., and Ye J. (2008) S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 283, 35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anderson K. A., Noeldner P. K., Reece K., Wadzinski B. E., and Means A. R. (2004) Regulation and function of the calcium/calmodulin-dependent protein kinase IV/protein serine/threonine phosphatase 2A signaling complex. J. Biol. Chem. 279, 31708–31716 [DOI] [PubMed] [Google Scholar]
- 39. Mora A., Komander D., van Aalten D. M., and Alessi D. R. (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 15, 161–170 [DOI] [PubMed] [Google Scholar]
- 40. Kahl C. R., and Means A. R. (2004) Regulation of cyclin D1/Cdk4 complexes by calcium/calmodulin-dependent protein kinase I. J. Biol. Chem. 279, 15411–15419 [DOI] [PubMed] [Google Scholar]
- 41. Deb T. B., Coticchia C. M., and Dickson R. B. (2004) Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 279, 38903–38911 [DOI] [PubMed] [Google Scholar]
- 42. Dong B., Valencia C. A., and Liu R. (2007) Ca2+/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J. Biol. Chem. 282, 25131–25140 [DOI] [PubMed] [Google Scholar]
- 43. Joyal J. L., Burks D. J., Pons S., Matter W. F., Vlahos C. J., White M. F., and Sacks D. B. (1997) Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 272, 28183–28186 [DOI] [PubMed] [Google Scholar]
- 44. Xu J., Zhang Q. G., Li C., and Zhang G. Y. (2007) Subtoxic N-methyl-d-aspartate delayed neuronal death in ischemic brain injury through TrkB receptor- and calmodulin-mediated PI-3K/Akt pathway activation. Hippocampus 17, 525–537 [DOI] [PubMed] [Google Scholar]
- 45. Bhat M., Robichaud N., Hulea L., Sonenberg N., Pelletier J., and Topisirovic I. (2015) Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 14, 261–278 [DOI] [PubMed] [Google Scholar]
- 46. Giacinti C., and Giordano A. (2006) RB and cell cycle progression. Oncogene 25, 5220–5227 [DOI] [PubMed] [Google Scholar]
- 47. Alt J. R., Cleveland J. L., Hannink M., and Diehl J. A. (2000) Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 14, 3102–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dai C. L., Shi J., Chen Y., Iqbal K., Liu F., and Gong C. X. (2013) Inhibition of protein synthesis alters protein degradation through activation of protein kinase B (AKT). J. Biol. Chem. 288, 23875–23883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takahashi-Yanaga F., and Sasaguri T. (2008) GSK-3β regulates cyclin D1 expression: a new target for chemotherapy. Cell. Signal. 20, 581–589 [DOI] [PubMed] [Google Scholar]
- 50. Malumbres M., and Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 51. Davis A., Tinker A. V., and Friedlander M. (2014) “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol. Oncol. 133, 624–631 [DOI] [PubMed] [Google Scholar]
- 52. Cheung M., and Testa J. R. (2013) Diverse mechanisms of AKT pathway activation in human malignancy. Curr. Cancer Drug Targets 13, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen B. C., Wu W. T., Ho F. M., and Lin W. W. (2002) Inhibition of interleukin-1β-induced NF-κB activation by calcium/calmodulin-dependent protein kinase kinase occurs through Akt activation associated with interleukin-1 receptor-associated kinase phosphorylation and uncoupling of MyD88. J. Biol. Chem. 277, 24169–24179 [DOI] [PubMed] [Google Scholar]
- 54. Schmitt J. M., Smith S., Hart B., and Fletcher L. (2012) CaM kinase control of AKT and LNCaP cell survival. J. Cell. Biochem. 113, 1514–1526 [DOI] [PubMed] [Google Scholar]
- 55. Roderick H. L., and Cook S. J. (2008) Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375 [DOI] [PubMed] [Google Scholar]
- 56. Karacosta L. G., Kuroski L. A., Hofmann W. A., Azabdaftari G., Mastri M., Gocher A. M., Dai S., Hoste A. J., and Edelman A. M. (2016) Nucleoporin 62 and Ca2+/calmodulin dependent kinase kinase 2 regulate androgen receptor activity in castrate resistant prostate cancer cells. Prostate 76, 294–306 [DOI] [PubMed] [Google Scholar]
- 57. Lemmon M. A., and Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liang J., and Mills G. B. (2013) AMPK: a contextual oncogene or tumor suppressor? Cancer Res. 73, 2929–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gerlee P. (2013) The model muddle: in search of tumor growth laws. Cancer Res. 73, 2407–2411 [DOI] [PubMed] [Google Scholar]
- 60. Banerjee S., and Kaye S. B. (2013) New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin. Cancer Res. 19, 961–968 [DOI] [PubMed] [Google Scholar]
- 61. Kikuchi Y., Iwano I., Miyauchi M., Sasa H., Nagata I., and Kuki E. (1990) Restorative effects of calmodulin antagonists on reduced cisplatin uptake by cisplatin-resistant human ovarian cancer cells. Gynecol. Oncol. 39, 199–203 [DOI] [PubMed] [Google Scholar]
- 62. Muranen T., Selfors L. M., Worster D. T., Iwanicki M. P., Song L., Morales F. C., Gao S., Mills G. B., and Brugge J. S. (2012) Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rodon J., Dienstmann R., Serra V., and Tabernero J. (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 10, 143–153 [DOI] [PubMed] [Google Scholar]
- 64. Alvero A. B., Montagna M. K., and Mor G. (2008) Correlation of caspase activity and in vitro chemo-response in epithelial ovarian cancer cell lines. Methods Mol. Biol. 414, 79–82 [DOI] [PubMed] [Google Scholar]
- 65. Franke T. F. (2000) Assays for Akt. Methods Enzymol. 322, 400–410 [DOI] [PubMed] [Google Scholar]
- 66. White R. R., Kwon Y. G., Taing M., Lawrence D. S., and Edelman A. M. (1998) Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J. Biol. Chem. 273, 3166–3172 [DOI] [PubMed] [Google Scholar]
- 67. Gocher A. M. (2013) Regulation of hormone-responsive cancers by Ca2+/calmodulin-dependent protein kinase kinase 2. M.Sc. thesis, State University of New York, Buffalo [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.