

Abstract
The visual photo-transduction cascade is a prototypical G protein–coupled receptor (GPCR) signaling system, in which light-activated rhodopsin (Rho*) is the GPCR catalyzing the exchange of GDP for GTP on the heterotrimeric G protein transducin (GT). This results in the dissociation of GT into its component αT–GTP and β1γ1 subunit complex. Structural information for the Rho*–GT complex will be essential for understanding the molecular mechanism of visual photo-transduction. Moreover, it will shed light on how GPCRs selectively couple to and activate their G protein signaling partners. Here, we report on the preparation of a stable detergent-solubilized complex between Rho* and a heterotrimer (GT*) comprising a GαT/Gαi1 chimera (αT*) and β1γ1. The complex was formed on native rod outer segment membranes upon light activation, solubilized in lauryl maltose neopentyl glycol, and purified with a combination of affinity and size-exclusion chromatography. We found that the complex is fully functional and that the stoichiometry of Rho* to GαT* is 1:1. The molecular weight of the complex was calculated from small-angle X-ray scattering data and was in good agreement with a model consisting of one Rho* and one GT*. The complex was visualized by negative-stain electron microscopy, which revealed an architecture similar to that of the β2-adrenergic receptor–GS complex, including a flexible αT* helical domain. The stability and high yield of the purified complex should allow for further efforts toward obtaining a high-resolution structure of this important signaling complex.
Keywords: 7-helix receptor, G protein-coupled receptor (GPCR), rhodopsin, signal transduction, structural biology, EM, G-protein, SAXS, photo-transduction, transducin
Introduction
G protein–coupled receptors (GPCRs),3 the largest family of transmembrane proteins, are the targets for nearly 50% of all pharmaceutical drugs (1). These receptors modulate cellular responses to a vast array of extracellular signals through the activation of heterotrimeric G proteins, with the active state being defined as the complex that forms between the agonist-bound (or light-stimulated) GPCR and nucleotide-free G protein (2). Attempts to obtain structural information for GPCRs, and especially for signaling-active GPCR–G protein complexes, have garnered a great deal of interest, both as a means to better understand the underlying mechanisms by which this important family of receptors mediates a wide range of biological outcomes and as a critical step in the design of more selective and effective drug treatments. Recent technological advancements, including novel protein engineering (3), in meso crystallization (4), and micro-focus beamlines at synchrotron facilities (5), have ushered in significant progress in the determination of high-resolution GPCR structures. However, only a few of those structures are of activated GPCRs, and thus far, only two GPCR–G protein complex structures have been solved, namely that of the β2-adrenergic receptor–GS protein complex (6) and the calcitonin receptor–GS protein complex (7). Furthermore, virtually all of these structures, with the exception of rhodopsin, have been obtained with GPCRs that were heavily modified so as to facilitate crystallization. Therefore, to help fully understand the mechanisms of GPCR-mediated G protein activation, it will be important to obtain structural information of active complexes formed between native receptors and their different G protein partners.
Rhodopsin, the photoreceptor responsible for dim light vision, is a prototypical member of the GPCR superfamily. Absorption of a single photon by rhodopsin activates many transducin (GT) molecules (the subunits designated as αT, β1, and γ1) in less than a second, generating GTP-bound αT subunits that activate the effector enzyme, cGMP phosphodiesterase (8). The photo-transduction system offers certain advantages for obtaining structural insights into GPCR signaling, as its principal components can be purified from native tissue in large quantities. As a result, rhodopsin represents the first and only GPCR for which X-ray crystal structures (9–13) have been solved in the native form.
The determination of an X-ray crystal structure of an activated rhodopsin–transducin complex not only is essential to providing a comprehensive picture of photo-transduction, but also for obtaining a better understanding of how the specificity between GPCRs and G proteins is achieved, as well as establishing the stoichiometry of the GPCR–G protein complex in the signaling-active state. To facilitate future crystallographic studies, here we describe a milligram-scale purification of an active rhodopsin–G protein complex formed with native light-activated rhodopsin (Rho*) and a GαT/Gαi1 chimera (αT*) together with the retinal β1 and γ1 subunits (GT*). The resulting complex is stable and homogeneous, and the stoichiometry between Rho* and GT* is 1:1, as determined by both UV-visible spectroscopy and radiolabeled nucleotide binding. Small angle X-ray scattering (SAXS) studies conducted with the purified Rho*–GT* complex further confirm that the complex is monodisperse, and molecular weight values calculated based on SAXS data suggest that the complex is composed of a monomeric Rho* bound to one GT* molecule. Negative-stain electron microscopy (EM) analyses of the complex confirm its homogeneity and reveal an overall architecture that is reminiscent of both EM (14) and crystallographic (6) results of the β2-adrenergic receptor–GS protein complex.
Results
Detergent selection for Rho*–GT* complex purification
To purify the Rho*–GT* complex, a suitable detergent is needed not only to extract the complex from its native rod outer segment membrane but also to maintain its stability. The first crystal structures of rhodopsin were solved using receptor solubilized in short-chain detergents, such as nonylglucoside (9), C8E4 (10), and octyl glucoside (11–13). Although these detergents can maintain the stability of dark inactivated rhodopsin for several days, the complex formed with light-activated rhodopsin and transducin cannot survive the extraction process and dissociates quickly upon solubilization. Therefore, we set out to select a detergent in which the Rho*–GT* complex would remain stably associated.
A fluorescence assay, monitoring the change in the intrinsic tryptophan fluorescence of αT* upon nucleotide exchange (15), was utilized to examine the rhodopsin-catalyzed nucleotide exchange activity in various detergents. When αT* exchanges GDP for GTPγS, a non-hydrolyzable GTP analog, there is an increase in the intrinsic tryptophan fluorescence due to a conformational change in the switch II region, one of three regions of αT* that change conformation upon GTP binding. Therefore, the rate of Rho*-catalyzed nucleotide exchange can be used to assay the activity of Rho* under various detergent conditions. Commercially available detergents from the maltoside and maltose neopentyl glycol (16) families were tested. Most of these detergents were able to maintain the Rho*-catalyzed nucleotide exchange activity at higher concentrations than their corresponding critical micelle concentration (i.e. 2× CMC), with a rate constant of ∼3 min−1 (Fig. 1, A and B). There is a clear dependence of Rho* activity on the length of the detergent hydrophobic chain. However, when using detergent concentrations above or below 2× CMC, the Rho*-stimulated nucleotide exchange activity in most of the tested detergents decreased significantly (Fig. 1C, see dodecyl maltoside), suggesting that these detergents are not well-suited for extracting the complex from rod outer segment (ROS) membranes, for which an initial high detergent concentration (typically around 1% w/v) would be required. Of the various detergents tested, only LMNG (lauryl maltose neopentyl glycol) was able to maintain high Rho*-stimulated nucleotide exchange activity at various concentrations (Fig. 1D) and was therefore selected to be used for Rho*–GT* complex purification.
Figure 1.

Rho*-catalyzed nucleotide exchange activity of αT* in various detergents. A, tryptophan fluorescence emission profiles of Rho*-catalyzed nucleotide exchange in maltoside detergents at a concentration of 2× CMC (OM, octyl maltoside; NM, nonyl maltoside; DM, decyl maltoside; UM, undecyl maltoside; DDM, dodecyl maltoside). Inset, rate constants obtained from single exponential fits of the data (n = 3). The respective rate constants are as follows: OM, 0.96 ± 0.13 min−1; NM, 2.81 ± 0.21 min−1; DM, 3.83 ± 0.01 min−1; UM, 2.60 ± 0.67 min−1; DDM, 1.52 ± 0.17 min−1. B, tryptophan fluorescence emission profiles of Rho*-catalyzed nucleotide exchange in maltose neopentyl glycol detergents at a concentration of 2× CMC (OMNG, octyl maltose neopentyl glycol; DMNG, decyl maltose neopentyl glycol; LMNG, lauryl maltose neopentyl glycol). Inset, rate constants obtained from single exponential fits of the data (n = 3). The respective rate constants as follows: OMNG, 1.83 ± 0.52 min−1; DMNG, 3.12 ± 0.56 min−1; LMNG, 2.64 ± 0.68 min−1. C, tryptophan fluorescence emission profiles of Rho*-catalyzed nucleotide exchange in DDM at various detergent concentrations (CMC = 0.0087% w/v). D, tryptophan fluorescence emission profiles of Rho*-catalyzed nucleotide exchange in LMNG at various detergent concentrations (CMC = 0.001% w/v).
Purification of the Rho*–GT* complex
The purification scheme for the Rho*–GT* complex is illustrated in Fig. 2A. The Coomassie Blue-stained SDS-PAGE profiles for the proteins at each step of the purification scheme are shown in Fig. 2B.
Figure 2.

Purification of the Rho*–GT* complex. A, purification scheme (see “Experimental procedures”). B, SDS-polyacrylamide gel of the purification process. (This is representative of more than 20 repetitions of the purification process.)
The complex was formed by first mixing αT*, a chimeric protein composed of an αT backbone with a stretch of residues from αi1, with β1γ1 in a 1.1:1 molar ratio. The αT* subunit was expressed in Escherichia coli with an N-terminal His6 tag and purified by nickel-nitrilotriacetic acid chromatography followed by ion-exchange and size-exclusion chromatography. The β1γ1 complex was purified from bovine retina. The resulting GT* heterotrimer was then mixed with urea-washed bovine ROS membranes containing native Rho in its dark state. Because of the high density of Rho in ROS, a significant amount of Rho may not be accessible to GT* upon light activation. Therefore, a substantial excess of Rho was used to assemble the Rho*–GT* complex (typically seven Rho per GT*). The mixture was subjected to illumination at 4 °C, resulting in the light activation of Rho (Rho*) and the induction of complex formation on the native ROS membranes. The mixture was then centrifuged, and as a result, the Rho*–GT* complex and excess Rho* were in the pellet, whereas the GDP released during GT activation, together with the unbound αT* subunit, remained in the supernatant.
The pellet was solubilized with buffer containing the detergent LMNG and applied to a nickel-Sepharose column. Because of the His6 tag present at the N terminus of αT*, the Rho*–GT* complex remained bound to the column and was thus separated from free Rho*. The Rho*–GT* complex was eluted from the column with imidazole and further purified by size-exclusion chromatography. The Rho*–GT* complex eluted as a single symmetrical peak (Fig. 3A). The yield of a typical purification process was about 80% based on the amount of β1γ1 complex used.
Figure 3.

Stoichiometry determination and SEC profiles of the purified Rho*–GT* complex. A, SEC profiles of the purified Rho*–GT* complex (red) and its dissociation upon the addition of GTPγS (green). (This is representative of more than 10 similar sets of SEC profiles.) B, UV-visible spectrum of purified complex. (This is representative of more than 20 similar spectra of the purified complex.) C, ratio of Rho* to GT* in the Rho*–GT* complex was determined by a [35S]GTPγS binding assay and the extinction coefficient for the chromophore retinal in Rho*. (This experiment was repeated three times.) D, SEC profiles of the Rho*–GT* complex after 1 day (blue) and 7 days (orange). (This is representative of more than 20 similar sets of SEC profiles.)
Stoichiometry, activity, and stability of the Rho*–GT* complex
Bovine Rho has been shown to form arrays of dimers in ROS membranes (17), and there have been questions raised regarding whether the stoichiometry between Rho* and GT* in the signaling complex is 1:1 or 2:1. The UV-visible spectrum of the purified complex reveals two peaks at 280 and 380 nm (Fig. 3B), corresponding to the absorbance of the protein moiety, and that of the unprotonated all-trans-retinal in the meta-II Rho* state, respectively. The A280 nm/A380 nm ratio is 3.7, which agrees with a 1:1 stoichiometry based on the reported extinction coefficients for the protein components (18–20) (a 2:1 stoichiometry would result in a A280 nm/A380 nm ratio of 2.58).
Additionally, the stoichiometry between Rho* and GT* was determined with a radio-nucleotide filter binding assay, in which the purified Rho*–GT* complex was incubated with [35S]GTPγS and then applied to a nitrocellulose filter. The amount of GT* present in the complex was estimated based on its nucleotide binding capacity, i.e. the amount of bound [35S]GTPγS, and was compared with the amount of Rho* calculated from the 380-nm absorbance. The results showed that for 1 mol of Rho*, there was 0.784 ± 0.003 mol of GTPγS present, supporting the idea of a 1:1 Rho*–GT* complex (Fig. 3B).
The purified Rho*–GT complex was fully active as it dissociated upon the addition of GTPγS, a non-hydrolysable analog of GTP (Fig. 3A). The complex was stable at 4 °C in the dark for at least 7 days, as it remained nearly completely intact with less than 5% dissociation (Fig. 3D).
SAXS analysis of Rho*–GT* complex
To further study the overall conformation of the active state of the Rho*–GT* complex, SAXS data were collected over a concentration range of the purified complex, with corrections for the background scattering being obtained by subtracting the scattering due to the SEC buffer from the scattering profiles of the Rho*–GT* samples (Fig. 4A). The Guinier regions (q*Rg <1.3) of these curves are linear (Fig. 4B), confirming that the Rho*–GT* complex is monodisperse. All the parameters calculated from the SAXS scattering profiles are summarized in Fig. 4C. The radius of gyration (Rg) values for the Rho*–GT* complex calculated from the Guinier plots range from 41.7 to 43.9 Å, which agree well with those calculated from pair distribution functions (P(r)) using GNOM (21) (41.9–43.9 Å). These results demonstrate that the value for Rg is concentration-independent. The molecular mass of the Rho*–GT* complex was calculated using both the particle mass determination method with ScÅtter software developed by Rambo et al. (22) and SAXS molecular mass developed by Fischer et al. (23). The results from ScÅtter (129.6–143.8 kDa) and SAXS molecular mass (127.4–128.9 kDa) agree well with each other and confirm that the complex is composed of 1 Rho* and 1 GT*, which gives a molecular mass value of 125 kDa. The P(r) curve (Fig. 4D) calculated from GNOM is skewed to the right, indicating an elongated overall particle shape. The cross-sectional Rg (Rc) values calculated using ScÅtter software range from 32.5 to 34.9 Å, which are about 9 Å smaller than the Rg values, confirming the existence of an elongated shape for the Rho*–GT* complex.
Figure 4.

SAXS data and analyses of the Rho*–GT* complex. A, SAXS scattering profiles from protein concentrations of 1, 0.5, and 0.25 mg/ml (colored cyan, purple, and red, respectively). (Each curve shown is the average of 10 scattering curves.) B, Guinier plots for q*Rg <1.3 region of the scattering curve. C, SAXS parameters and calculation results. D, pair distribution function calculated from 1 mg/ml scattering profile.
SAXS-based modeling of Rho*–GT* complex
The P(r) curve (Dmax = 128 Å) calculated from the scattering profile for the 1 mg/ml Rho*–GT* sample was used for generating ab initio models of the complex. Two different algorithms, DAMMIN (24) and GASBOR (25), were used. The resulting envelopes obtained using each method agree well with each other. Both revealed a small bulge protruding out from the bottom half of the envelopes, indicating the position of the helical domain of αT* (Fig. 5, A and B). A structural model of the Rho*–GT* complex, using the crystal structure of the β2AR–GS complex (PDB 3SN6) as a template, was generated to fit the envelopes. There is significant discrepancy between the structural model and ab initio envelopes at the Rho* region, which can be attributed to the existence of a detergent micelle around the receptor in solution, and as a result, the model fits poorly to the experimental data with χ2 = 24.2. To improve the quality of the structural model, a detergent corona shaped as an elliptical torus was built around Rho* using the Memprot program developed by Pérez and Koutsioubas (26). The resulting micelle model is composed of 78 LMNG detergent tails and 74 detergent heads, and its dimensions are shown in Fig. 5C. After incorporating the detergent corona into the complex model, the χ2 value decreased to 3.27, indicating a significant improvement of fitness to the experimental data. In addition, the position of the helical domain was further optimized with the program CORAL (26), and the χ2 value of the model improved to 1.83 (Fig. 5, D and E). The helical domain position in the resulting model resides between the close and open positions as indicated from X-ray crystal structures (PDB 1GOT and 3SN6, respectively) (Fig. 5E).
Figure 5.
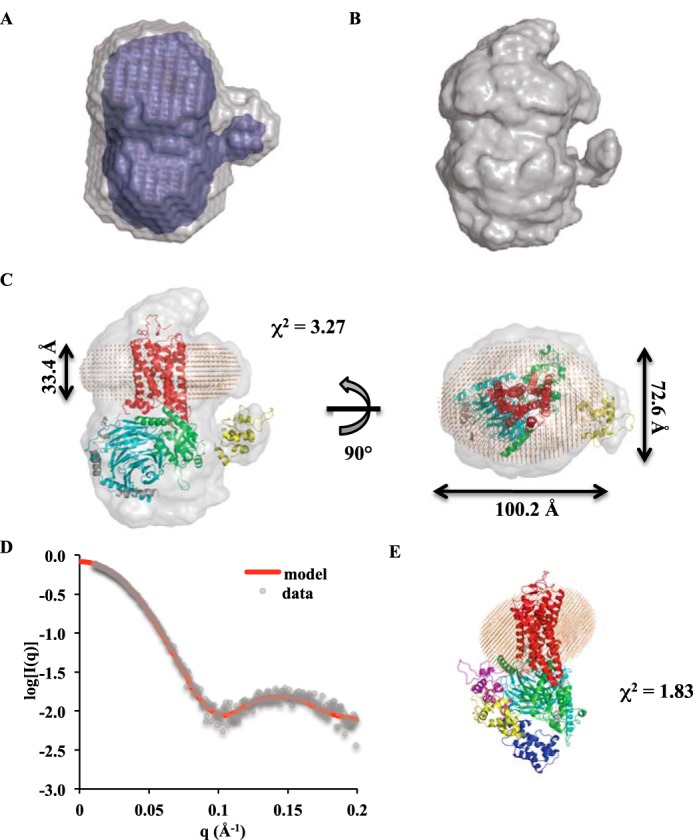
Ab initio envelopes and model of the Rho*–GT* complex. A, DAMMIN envelopes (averaged and filtered envelopes are colored in gray and blue, respectively). B, GASBOR envelope. C, superimposition of the Rho*–GT* complex model with the GASBOR envelope. Rho* is in red; the αT* GTPase domain is in green; the αT* helical domain is in yellow; β1 is in cyan; γ1 is in gray; and detergents are shown as orange beads. D, theoretical scattering profile of the Rho*–GT* complex model (red line) overlaid with experimental data (gray dots). E, position of the αT* helical domain (yellow) in the model compared with open (purple) and closed (blue) positions from X-ray crystal structures.
Electron microscopy characterization of negatively stained Rho*–GT*complex
EM visualization of the negatively stained complex samples showed a monodisperse particle population (Fig. 6A). Reference-free alignment and classification of particle projections revealed class averages with an overall density similar to that of the β2-adrenergic receptor–GS protein complex (β2AR-GS) (Fig. 6B) (14). A central oval density represents Rho* in a detergent micelle with a small protruding density often observed on top corresponding to the N terminus of Rho*. At the bottom of Rho*, two major densities representing the GT* heterotrimer are clearly visible. One of the two densities has extensive contact with the receptor density and shows an additional small globular density in various orientations in several class average images. This is similar to what was observed for the β2AR–GS complex (14) and represents the Ras-like domain of the αT* subunit, with a flexible helical domain in the Rho*–GT* complex. The other density therefore corresponds to β1γ1.
Figure 6.

EM 2D projection analysis of the Rho*–GT* complex. A, raw EM image of the detergent-solubilized Rho*–GT* complex embedded in negative stain. B, representative EM class averages of the Rho*–GT* complex (the positions of the αΤ* helical domain are indicated by an arrow). The schematic model represents the conformations reflected by the EM averages, depicting the variable positioning of the helical domain (the position of the detergent micelle is indicated by gray shaded arcs and labeled with m).
Discussion
Structural information obtained from a variety of approaches will be invaluable in shedding light on how the photoreceptor Rho engages and activates its signaling partner, the G protein GT. Such analyses will also provide insights with broad relevance toward understanding the underlying mechanisms by which GPCRs demonstrate remarkable specificity for their G protein targets, as well as whether different GPCRs use common or distinct mechanisms in catalyzing the G protein activation event. Recently, there have been several reports of the purification of detergent-solubilized Rho*–GT complexes, in which either native (28, 29) or recombinant Rho (30, 31) was utilized. Our approach toward obtaining an active Rho*–G protein complex differs from these previous efforts in two major aspects. First, rather than detergent-solubilizing and purifying Rho prior to allowing it to associate with a purified G protein, the Rho*–G protein complex was obtained by directly light-activating the urea-washed ROS membranes in the presence of the heterotrimeric and chimeric G protein GT*. This allowed the formation of a Rho*–GT* complex within a native membrane environment. The subsequent pelleting step readily removed the released GDP, which may otherwise destabilize the complex. Second, instead of using either the glycan linkages or the C terminus of Rho as purification handles, we utilized the N-terminal His tag on the recombinant αT* subunit to achieve a simple and efficient one-step purification of the detergent-solubilized Rho*–GT* complex. The resulting complex dissociated with the addition of GTPγS, demonstrating that the complex isolated in this manner is fully active.
Near-infrared light-scattering studies of ROS membranes in the early 1980s by Kuhn et al. (32) suggested a 1:1 stoichiometric association of GT with Rho*. However, atomic force microscopy images of ROS membranes revealed that Rho exists in the form of dimeric arrays in its native environment (17). Recent studies using native rhodopsin solubilized from ROS with detergent for complex formation (28, 29) argued for the existence of a pentameric complex consisting of two Rho* molecules and one GT heterotrimer as the minimal functioning unit. While in these settings, it may appear that Rho* can form dimers, the important question is what stoichiometry of Rho* and GT is required to achieve full activation. Based on our studies of a signaling-active Rho*–GT* complex, as analyzed by UV-visible spectroscopy and radioactive nucleotide binding, as well as by SAXS and EM, we conclude that the minimal unit necessary for full activation is 1 Rho* and 1 G protein. This conclusion is supported by recent studies using rhodopsin-embedded nanodiscs, which have shown that one Rho* is sufficient for coupling to and activating GT (33–34). Moreover, based on nanodisc reconstitution, monomeric rhodopsin has been shown to be sufficient for phosphorylation by G protein–coupled receptor kinase and interaction with arrestin (35–36). The recent X-ray structure of a rhodopsin–arrestin complex (37) further illustrated that rhodopsin binds arrestin in a 1:1 stoichiometry. Similarly, the β2AR has been demonstrated to interact with Gs in a monomeric manner (6, 14, 38). Therefore, several lines of evidence now point to class A GPCR monomers being sufficient for promoting their downstream signals.
In addition to the limited stoichiometry necessary to achieve a signaling-active complex, another question of interest is the positioning of the helical domain of the Gα subunit in the receptor–G protein complex. The first high-resolution structure of a G protein α subunit, for GTPγS-bound αT (39), revealed that the nucleotide is buried inside a cleft formed by the Ras-like domain and helical domain, and it was postulated that an activated receptor must induce an opening of the cleft to allow for nucleotide exchange. The first direct evidence of this inter-domain opening was provided by double electron-electron resonance experiments (40), in which distance increases as large as 20 Å between the Ras-like and helical domains of the Giα subunit were observed upon binding to light-activated ROS. The crystal structure of the β2AR–GS complex (6) shows a very dramatic 127° rotation of the GSα helical domain, which results in the opening of the nucleotide-binding pocket. Our SAXS model of the Rho*–GT* complex shows that the helical domain of αT* adopts a similar open position in solution. However, the opening is not as dramatic as displayed in the β2AR–GS complex crystal structure. Negative-stain EM studies of the β2AR–GS complex (14) reveal the flexible positioning of the GSα helical domain and suggest an ensemble of open helical domain positions that exist under physiological conditions. Because a similarly flexible helical domain is also observed in the negative-stain EM images of the Rho*–GT* complex, it is most likely that a displacement of the helical domain is a universal mechanism of receptor-mediated nucleotide exchange for different families of heterotrimeric G proteins.
In conclusion, here we describe procedures for isolating a Rho*–G protein complex with a very high yield that is amenable to a variety of types of biochemical and structural analyses. Given the availability of a number of interesting mutants of αT*, which mimic different stages in the activation event, we should soon be in a position to address fundamentally important questions regarding how different members of the GPCR family engage their G protein signaling partners and induce the necessary structural changes to drive G protein-mediated signal propagation.
Experimental procedures
Materials
Frozen dark-adapted bovine retinae were purchased from the W. L. Lawson Co. (Lincoln, NE). Detergents were from Anatrace, and all other chemicals were obtained from Sigma.
Purification of retinal proteins
Urea-washed rod outer segment (UROS) membranes were isolated as described (41), flash-frozen, and stored at −80 °C in HMN buffer (20 mm HEPES, pH 7.5, 2 mm MgCl2, 100 mm NaCl, 1 mm DTT) at a concentration of 300 μm. UROS membranes were used as the source for Rho* in Rho*–GT* complex formation. The β1γ1 subunit complex was purified essentially as described previously (42). Bovine retinae were exposed to light and subjected to sucrose gradient ultracentrifugation to prepare purified ROS membranes. After a series of isotonic and hypotonic washes, 100 μm GTP was added to release GT subunits from the membrane. β1γ1 was separated from αT through a 5-ml HiTrap Blue HP (GE Healthcare) column, and the resulting β1γ1 complex was further purified by anion-exchange chromatography through a 5-ml HiTrap Q HP (GE Healthcare) column, using Buffer A (20 mm HEPES, pH 7.5, 2 mm MgCl2, 1 mm DTT, 10% glycerol) and Buffer B (Buffer A + 1 m NaCl) to form the gradient. The β1γ1 complex typically elutes at 100 mm NaCl and was concentrated to 20 μm, flash-frozen, and stored at −80 °C.
Expression and purification of αT*
An αT/αi1 chimeric construct designated as pHis6Chi8 was obtained from Dr. Heidi Hamm (Vanderbilt University) (43), in which αT residues 215–295 were replaced with the corresponding residues from αi1, and a His6 tag was introduced at the N terminus. In addition, residues 244 and 247 were changed back to the original amino acids in αT, resulting in the αT* construct. The αT* subunit can undergo Rho*-catalyzed nucleotide exchange and activate the effector enzyme, phosphodiesterase, in a similar manner as retinal αT. αT* was expressed in BL21(DE3)-competent cells and purified as described previously (18). The protein was concentrated to 20 μm in HMN buffer with the addition of 10% glycerol, flash-frozen, and stored at −80 °C.
Detergent selection for Rho*–GT* complex purification
Fluorescence measurements were carried out with a Varian eclipse spectrofluorimeter. UROSs light-activated (Rho*) by incubation on ice under ambient light for 5 min. Rho*-catalyzed nucleotide exchange on the αT* subunit in detergents was monitored by premixing 5 nm Rho* and 300 nm β1γ1 in HMN buffer with 50 μm GTPγS and different detergents at various concentrations, monitoring tryptophan fluorescence (excitation, 300 nm; emission, 345 nm) in real time upon the addition of 300 nm αT*. All kinetic traces were corrected for the fluorescence from Rho* and β1γ1, and the data were fitted to single exponential Equation 1,
| (Eq. 1) |
where F is the fluorescence signal at any time t; F0 is the fluorescence signal at time t = 0; F∞ is the fluorescence signal at time t = ∞, and kobs is the observed rate constant.
Rhodopsin–GT* complex formation and purification
The G protein heterotrimer GT* was formed by mixing 22 nmol of αT* with 20 nmol of β1γ1 and then incubated on ice for 5 min. The Rho*–GT* complex was formed on ROS membranes by mixing GT* with UROSs containing 140 nmol of Rho and illuminating the mixture under a halogen lamp covered with a UV-absorbing glass and a 495-nm long-pass filter at 4 °C for 30 min. The suspension was aliquoted into 1.5-ml Eppendorf tubes and centrifuged at 14,000 × g for 30 min. From here on, all subsequent steps were carried out in the dark under dim red light. The supernatant was discarded, and the pellets containing the Rho*–GT* complex and excess Rho* were resuspended in 3 ml of HMN buffer + 1% (w/v) LMNG. The mixture was incubated at 4 °C with rocking for 1 h to allow for complete solubilization. HMN buffer (12 ml) was then added to lower the LMNG concentration to 0.2%, and the sample was further incubated at 4 °C with rocking for 1 h. Solubilized complex was loaded onto a 1-ml HisTrap FF column (GE Healthcare) pre-equilibrated with HMN buffer + 0.02% LMNG. The Rho*–GT* complex was eluted from the column with an imidazole gradient in HMN buffer + 0.003% LMNG, as a single peak at ∼100 mm imidazole. Peak fractions were pooled and concentrated with an Amicon Ultra-0.5 100-kDa MWCO concentrator (EMD Millipore) to 500 μl and injected onto a Superdex 200 10/300 GL column (GE Healthcare), pre-equilibrated with HMN buffer + 0.003% LMNG. Peak fractions were pooled and concentrated with an Amicon Ultra-0.5 100-kDa MWCO concentrator (EMD Millipore) to 50 μl, resulting in a complex concentration of ∼40 mg/ml.
Determination of the stoichiometry of the Rho*–GT* complex
The stoichiometry of Rho* and GT* within the complex was determined using two methods, UV-visible absorption spectroscopy and a [35S]GTPγS binding assay. For stoichiometry determinations using UV-visible spectroscopy, a 100-μl sample of the purified Rho*–GT* complex was examined in a quartz cuvette with a 1 cm path length, using a Beckman DU600 UV-visible spectrophotometer scanning from 240 to 700 nm at 240 nm/min. The ratio for Rho* to GT* within the complex was determined from the A280 nm/A380 nm ratio, using the following extinction coefficients: Rho* (ϵ280 nm = 61,800 m−1 cm−1; ϵ380 nm = 42,000 m−1 cm−1) (19, 20); αT* (ϵ280 nm = 35,870 m−1 cm−1), and β1γ1 (ϵ280 nm = 57,400 m−1 cm−1). The extinction coefficients for αT* and β1γ1 were calculated based on protein sequence using the ExPASy ProParam tool (44). Therefore, a 1:1 Rho*–GT* stoichiometry would result in an A280 nm/A380 nm value of 3.69, whereas a 2:1 Rho*–GT* stoichiometry would yield a A280 nm/A380 nm value of 2.58.
For stoichiometry determinations using a [35S]GTPγS binding assay, purified Rho*–GT* complex was incubated with 50 μm [35S]GTPγS for 10 min on ice in 20 μl of HMN buffer + 0.003% LMNG. The solution was then applied to prewet nitrocellulose filters (Schleicher & Schuell, pore size 0.45 μm) on a suction manifold. The filters were washed twice with HMN buffer, added to scintillation liquid (30% LSC Scintisafe Mixture), and counted in a scintillation counter (LS6500 multipurpose scintillation counter). The calculated amount of bound [35S]GTPγS was used as an estimate for the amount of GT* in the complex and was compared with the amount of Rho* calculated from A380 nm, using the extinction coefficient of Meta II rhodopsin, ϵ380 nm = 42,000 m−1 cm−1.
SAXS data collection and processing
SAXS data were collected at the G1 station of the Cornell High Energy Synchrotron Source (CHESS). G1 operated with an energy of 9.86 keV and provided a flux of 3 × 1011 photons/s for a 250 × 250-μm beam. Purified Rho*–GT* complex was eluted through a Superdex 200 5/150 GL column (GE Healthcare), pre-equilibrated with HMN buffer without detergent, immediately prior to the collection of SAXS data. The center peak fraction was collected, and a concentration series, 0.25, 0.5, and 1 mg/ml, was prepared in HMN buffer and kept on ice. Samples were centrifuged at 14,000 × g for 10 min before being loaded into the SAXS sample exposure window. Samples were exposed for 30 s with oscillation. Ten datasets were collected for each sample for possible radiation damage detection, and undamaged exposures were averaged. Buffer measurements with HMN buffer were conducted in-between each sample measurement. Data reduction and background subtraction were done with RAW data reduction software (45). The program ScÅtter (22) was used to obtain the Guinier plot and to calculate the radius of gyration (Rg) and I(0) values. Particle distance distribution P(r) was calculated using GNOM (20).
SAXS-based modeling of the Rho*–GT* complex
Ab initio models were calculated using two different programs, DAMMIN (24) and GASBOR (25). In DAMMIN calculations, 17 envelopes were generated (average χ2 = 0.90), and the results were averaged using DAMAVER (46) to produce the averaged and filtered envelopes. None of the 17 DAMMIN envelopes was rejected during the DAMAVER calculation, and the mean normalized special discrepancy value was 0.482 ± 0.053, indicating the model is of good quality. In addition to DAMMIN, GASBOR calculations were performed to fit the intensity in reciprocal space. The resulting envelope fits well to the experimental curve with χ2 = 0.895. A structural model was built using the X-ray crystal structure of the β2AR–GS complex (PDB code 3SN6) as a template. In this model, the X-ray crystal structures of meta-rhodopsin II (PDB code 3PXO), and the β1γ1 complex from the GT heterotrimer (PDB code 1GOT), were used for Rho* and β1γ1, respectively. A homology model of the αT* subunit was built based on the αS subunit from the X-ray crystal structure of the β2AR–GS complex (PDB code 3SN6) using the SWISS-MODEL server (44). The program Memprot (26) was used to model the LMNG detergent micelle around Rho*, in which a coarse-grained fitting algorithm was used to add detergent molecules by assuming an elliptical model for the detergent corona. Electron density values of 0.28 and 0.52 e·Å−3 were used for the hydrophobic and hydrophilic portions of the LMNG detergent molecule, respectively. The position of the helical domain (residues 57–176 in αT*) was optimized using the program CORAL (27), in which the linker regions (residues 51–56 and 177–181) connecting the helical domain to the GTPase domain were set to be random flexible loops. The resulting structural model was aligned with the envelopes using the SUPCOMB program (47).
Specimen preparation and EM imaging of negative-stained samples
Purified Rho*–GT* complex was prepared for electron microscopy using the conventional negative-staining protocol (48) and imaged at room temperature with a Tecnai T12 electron microscope operated at 120 kV, using low-dose procedures. Images were recorded at a magnification of ×71,138 and a defocus value of ∼1.5 μm on a Gatan US4000 CCD camera.
Two-dimensional classifications of the Rho*–GT* complex
All images were binned (2 × 2 pixels) to obtain a pixel size of 4.16 Å on the specimen level. Particles were manually excised using e2boxer (49) (part of the EMAN2 software suite). Two-dimensional reference-free alignment and classification of particle projections were performed using ISAC (50). A total of 9303 projections of Rho*–GT* were subjected to ISAC, producing 134 classes consistent over two-way matching and accounting for 6774 particle projections.
Author contributions
Y. G., G. W., and S. R. performed the experiments; Y. G., G. W., R. A. C., G. S., and S. R. analyzed the data; and Y. G., J. W. E., R. A. C., G. S., and S. R. wrote the manuscript.
Acknowledgments
We thank Cindy Westmiller for expert secretarial assistance. The MacCHESS facility was the recipient of National Institutes of Health Grant GM103485.
This work was supported by National Institutes of Health Grants GM047458 and DK090165. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- GPCR
- G protein–coupled receptor
- Rho
- rhodopsin
- SAXS
- small angle X-ray scattering
- LMNG
- lauryl maltose neopentyl glycol
- GTPγS
- guanosine-5′-O-(3-thiotriphosphate)
- ROS
- rod outer segment
- PDB
- Protein Data Bank
- SEC
- size-exclusion chromatography
- β2AR
- β2-adrenergic receptor
- UROS
- urea-washed rod outer segment.
References
- 1. Ma P., and Zemmel R. (2002) Value of novelty? Nat. Rev. Drug Discov. 1, 571–572 [DOI] [PubMed] [Google Scholar]
- 2. De Lean A., Stadel J. M., and Lefkowitz R. J. (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J. Biol. Chem. 255, 7108–7117 [PubMed] [Google Scholar]
- 3. Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Yao X. J., Weis W. I., Stevens R. C., and Kobilka B. K. (2007) GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 4. Caffrey M., and Cherezov V. (2009) Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu S., and Fischetti R. F. (2007) Design and performance of a compact collimator on GM/CA-CAT at the advanced photon source. Proc. SPIE. 6665, 1–8 [Google Scholar]
- 6. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liang Y. L., Khoshouei M., Radjainia M., Zhang Y., Glukhova A., Tarrasch J., Thal D. M., Furness S. G. B., Christopoulos G., Coudrat T., Danev R., Baumeister W., Miller L. J., Christopoulos A., Kobilka B. K., Wootten D., Skiniotis G., and Sexton P. M. (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stryer L. (1991) Visual excitation and recovery. J. Biol. Chem. 266, 10711–10714 [PubMed] [Google Scholar]
- 9. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., and Miyano M. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 10. Li J., Edwards P. C., Burghammer M., Villa C., and Schertler G. F. (2004) Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 343, 1409–1438 [DOI] [PubMed] [Google Scholar]
- 11. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., and Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 12. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., and Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 13. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H. W., Hofmann K. P., and Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 [DOI] [PubMed] [Google Scholar]
- 14. Westfield G. H., Rasmussen S. G., Su M., Dutta S., DeVree B. T., Chung K. Y., Calinski D., Velez-Ruiz G., Oleskie A. N., Pardon E., Chae P. S., Liu T., Li S., Woods V. L. Jr., Steyaert J., et al. (2011) Structural flexibility of the Gαs α-helical domain in the β2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci. U.S.A. 108, 16086–16091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Phillips W. J., and Cerione R. A. (1988) The intrinsic fluorescence of the α subunit of transducin. J. Biol. Chem. 263, 15498–15505 [PubMed] [Google Scholar]
- 16. Chae P. S., Rasmussen S. G., Rana R. R., Gotfryd K., Chandra R., Goren M. A., Kruse A. C., Nurva S., Loland C. J., Pierre Y., Drew D., Popot J. L., Picot D., Fox B. G., Guan L., et al. (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fotiadis D., Liang Y., Filipek S., Saperstein D. A., Engel A., and Palczewski K. (2003) Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature 421, 127–128 [DOI] [PubMed] [Google Scholar]
- 18. Majumdar S., Ramachandran S., and Cerione R. A. (2006) Perturbing the linker regions of the α-subunit of transducin. J. Biol. Chem. 281, 9219–922616469737 [Google Scholar]
- 19. Lin S. W., and Sakmar T. P. (1996) Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry 35, 11149–11159 [DOI] [PubMed] [Google Scholar]
- 20. Matthews R. G., Hubbard R., Brown P. K., and Wald G. (1963) Tautomeric forms of metarhodopsin. J. Gen. Physiol. 47, 215–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 [Google Scholar]
- 22. Rambo R. P., and Tainer J. A. (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer H., de Oliveira Neto M., Napolitano H. B., Polikarpov I., and Craievich A. F. (2010) Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Cryst. 43, 101–109 [Google Scholar]
- 24. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Svergun D. I., Petoukhov M. V., and Koch M. H. (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pérez J., and Koutsioubas A. (2015) Memprot: a program to model the detergent corona around a membrane protein based on SEC-SAXS data. Acta Crystallogr. D Biol. Crystallogr. 71, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jastrzebska B., Ringler P., Lodowski D. T., Moiseenkova-Bell V., Golczak M., Müller S. A., Palczewski K., and Engel A. (2011) Rhodopsin-transducin heteropentamer: three-dimensional structure and biochemical characterization. J. Struct. Biol. 176, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jastrzebska B., Ringler P., Palczewski K., and Engel A. (2013) The rhodopsin–transducin complex houses two distinct rhodopsin molecules. J. Struct. Biol. 182, 164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xie G., D'Antona A. M., Edwards P. C., Fransen M., Standfuss J., Schertler G. F., and Oprian D. D. (2011) Preparation of an activated rhodopsin/transducin complex using a constitutively active mutant of rhodopsin. Biochemistry 50, 10399–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maeda S., Sun D., Singhal A., Foggetta M., Schmid G., Standfuss J., Hennig M., Dawson R. J., Veprintsev D. B., and Schertler G. F. (2014) Crystallization scale preparation of a stable GPCR signaling complex between constitutively active rhodopsin and G-protein. PLoS ONE 9, e98714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kühn H., Bennett N., Michel-Villaz M., and Chabre M. (1981) Interactions between photoexcited rhodopsin and GTP-binding protein: kinetic and stoichiometric analyses from light-scattering changes. Proc. Natl. Acad. Sci. U.S.A. 78, 6873–6877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bayburt T. H., Leitz A. J., Xie G., Oprian D. D., and Sligar S. G. (2007) Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J. Biol. Chem. 282, 14875–14881 [DOI] [PubMed] [Google Scholar]
- 34. Whorton M. R., Jastrzebska B., Park P. S., Fotiadis D., Engel A., Palczewski K., and Sunahara R. K. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 283, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bayburt T. H., Vishnivetskiy S. A., McLean M. A., Morizumi T., Huang C. C., Tesmer J. J., Ernst O. P., Sligar S. G., and Gurevich V. V. (2011) Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J. Biol. Chem. 286, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vishnivetskiy S. A., Ostermaier M. K., Singhal A., Panneels V., Homan K. T., Glukhova A., Sligar S. G., Tesmer J. J., Schertler G. F., Standfuss J., and Gurevich V. V. (2013) Constitutively active rhodopsin mutants causing night blindness are effectively phosphorylated by GRKs but differ in arrestin-1 binding. Cell. Signal. 25, 2155–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H., Zhang C., et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., and Sunahara R. K. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Noel J. P., Hamm H. E., and Sigler P. B. (1993) The 2.2 Å crystal structure of transducin-α complexed with GTPγS. Nature 366, 654–663 [DOI] [PubMed] [Google Scholar]
- 40. Van Eps N., Preininger A. M., Alexander N., Kaya A. I., Meier S., Meiler J., Hamm H. E., and Hubbell W. L. (2011) Interaction of a G protein with an activated receptor opens the interdomain interface in the α subunit. Proc. Natl. Acad. Sci. U.S.A. 108, 9420–9424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Min K. C., Gravina S. A., and Sakmar T. P. (2000) Reconstitution of the vertebrate visual cascade using recombinant transducin purified from Sf9 cells. Protein Expr. Purif. 20, 514–526 [DOI] [PubMed] [Google Scholar]
- 42. Ramachandran S., and Cerione R. A. (2011) A dominant-negative Gα mutant that traps a stable rhodopsin-Gα-GTP-βγ complex. J. Biol. Chem. 286, 12702–12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skiba N. P., Bae H., and Hamm H. E. (1996) Mapping of effector binding sites of transducin α-subunit using Gαt/Gαi1 chimeras. J. Biol. Chem. 271, 413–424 [DOI] [PubMed] [Google Scholar]
- 44. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Inc., Totowa, NJ [Google Scholar]
- 45. Nielsen S. S., Noergaard Toft K., Snakenborg D., Jeppesen M. G., Jacobsen J. K., Vestergaard B., Kutter J. P., and Arleth L. (2009) BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Cryst. 42, 959–964 [Google Scholar]
- 46. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 34, 33–41 [Google Scholar]
- 48. Peisley A., and Skiniotis G. (2015) 2D projection analysis of GPCR complexes by negative statin electron microscopy. Methods Mol. Biol. 1335, 29–38 [DOI] [PubMed] [Google Scholar]
- 49. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 [DOI] [PubMed] [Google Scholar]
- 50. Yang Z., Fang J., Chittuluru J., Asturias F. J., and Penczek P. A. (2012) Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 20, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]