

Abstract
RNase P is a universal enzyme that removes 5′ leader sequences from tRNA precursors. The enzyme is therefore essential for maturation of functional tRNAs and mRNA translation. RNase P represents a unique example of an enzyme that can occur either as ribonucleoprotein or as protein alone. The latter form of the enzyme, called protein-only RNase P (PRORP), is widespread in eukaryotes in which it can provide organellar or nuclear RNase P activities. Here, we have focused on Arabidopsis nuclear PRORP2 and its interaction with tRNA substrates. Affinity measurements helped assess the respective importance of individual pentatricopeptide repeat motifs in PRORP2 for RNA binding. We characterized the PRORP2 structure by X-ray crystallography and by small-angle X-ray scattering in solution as well as that of its complex with a tRNA precursor by small-angle X-ray scattering. Of note, our study reports the first structural data of a PRORP–tRNA complex. Combined with complementary biochemical and biophysical analyses, our structural data suggest that PRORP2 undergoes conformational changes to accommodate its substrate. In particular, the catalytic domain and the RNA-binding domain can move around a central hinge. Altogether, this work provides a refined model of the PRORP–tRNA complex that illustrates how protein-only RNase P enzymes specifically bind tRNA and highlights the contribution of protein dynamics to achieve this specific interaction.
Keywords: precursor tRNA (pre-tRNA), ribonuclease P (RNase P), RNA processing, small-angle X-ray scattering (SAXS), X-ray crystallography, PRORP, pentatricopeptide repeat (PPR), tRNA maturation
Introduction
Transfer RNA maturation involves many post-transcriptional steps. Among them, 5′ leader sequences are removed by an endonuclease activity called RNase P (1). This processing step was assumed to be universally performed by ribonucleoprotein (RNP)5 particles containing a ribozyme and up to 10 protein subunits until the discovery of protein-only RNase P in eukaryotes (2).
Protein-only RNase P (PRORP) enzymes were first identified in human mitochondria and plants (3, 4). Contrary to initial appreciations, PRORP enzymes are not restricted to organelles in a few species (5). They are actually widespread in eukaryotes, occurring in mitochondria and/or chloroplasts as well as in nuclei in four of five eukaryote supergroups (6). In some groups, i.e. in most Chloroplastida (Viridiplantae), Stramenopiles, and Trypanosomatida, PRORP enzymes have seemingly entirely replaced ribonucleoproteins for RNase P activity as experimentally shown for Arabidopsis thaliana (7), Chlamydomonas reinhardtii (8), and Trypanosoma brucei (9). PRORP enzymes were also characterized in the moss Physcomitrella patens (10). Intriguingly, in this species, a nuclear PRORP is not essential, although no recognizable ribonucleoprotein RNase P is present (6). Remarkably, the occurrence of PRORP and RNP RNase P appears mutually exclusive in compartments or in entire organisms (6). RNPs or PRORPs might have been retained in specific cell compartments or entire organisms because they have different substrate spectra in vivo. Alternatively, in specific clades, RNPs or PRORPs might have evolved additional functions that cannot be held by the other type of enzyme (6).
The biochemical characterization of PRORP enzymes, their structure in solution (11), and the crystal structures of mitochondrial Arabidopsis and human PRORP enzymes (12–14) have revealed two-domain enzymes. They contain a C-terminal nuclease domain belonging to the N4BP1, YacP-like nuclease (NYN) family (15) and an N-terminal pentatricopeptide repeat (PPR) domain (16, 17) believed to be responsible for RNA binding and substrate specificity. These two main domains are bridged by a central zinc-binding domain (2). A comparative kinetic analysis of RNase P activities by PRORP and RNPs has suggested that the two types of enzymes use different catalytic mechanisms (18). Remarkably, this comparative analysis also revealed that RNPs are better catalysts than PRORP enzymes at least in vitro. However, the identification of tRNA residues in contact with PRORP (11) and RNP RNase P (19) has suggested that RNPs and PRORP use a similar strategy to recognize their substrates (11). Still, how this is achieved at the protein level remained unknown, and the dynamics required for PRORP mode of action was unexplored (20).
To tackle these questions, we determined the crystal structure of Arabidopsis nuclear PRORP2 together with a SAXS model in solution and explored the role of its PPR domain for substrate binding. We found that two particular PPR motifs are required for specific tRNA recognition by PRORP2. Furthermore, we studied the PRORP2–tRNA complex in solution by SAXS to establish an interaction model. Taken together, our results identify structural features in PRORP2 important for the RNA binding process and suggest that the enzyme is flexible and undergoes conformational changes to perform its activity.
Results
Crystal and solution structure of Arabidopsis nuclear PRORP2
Crystallization conditions were extensively screened for different PRORP2 constructs, including catalytically impaired and shortened forms. Crystals were only obtained for the full-length wild-type PRORP2 (21). Their analysis led to the determination of the enzyme structure at a resolution of 3.05 Å (Fig. 1A and supplemental Table S1). As proposed based on sequence similarities with the organellar PRORP1 (2, 11), PRORP2 adopts the same characteristic Λ-shape. The N-terminal arm consists of a PPR domain made of five PPR motifs, the C-terminal arm consists of the NYN metallonuclease domain, and a bipartite zinc-binding domain (ZBD) bridges the two arms together. The first 27 residues, including the nuclear localization signal, as well as the last 20 amino acids, including the His6 affinity tag, were probably floppy in the crystal packing and could not be visualized in the electron density map. The two copies of PRORP2 present in the triclinic unit cell are very similar (root mean square deviation, 0.85 Å; supplemental Fig. S1). They also superimpose well with PRORP2 monomers (average root mean square deviation, 1.32 Å) recently described by Karasik et al. (22). As in the latter crystal structure determined concomitantly to ours in the same triclinic environment and in fairly similar crystallization conditions, we do not observe any cation bound to conserved aspartate residues in the catalytic site, although we and Karasik et al. (22) show that, similar to PRORP1, Mg2+ ions are required for catalysis (supplemental Fig. S2).
Figure 1.

Crystal and solution structures of A. thaliana nuclear PRORP2. A, side view in schematic representation illustrating the overall organization of PRORP2. The enzyme is composed of two major domains, the N-terminal PPR domain made of five PPR motifs (PPR1–5 shown in violet, red, orange, yellow, and green, respectively) and the C-terminal metallonuclease domain (in blue) linked together by a zinc-binding module (in cyan). B, bottom view highlighting the residues characterized by mutagenesis in this work. Corresponding side chains in PPR motifs (positions 5 and 35) are depicted as sticks as well as the two aspartates converted to alanines in the catalytically inactive mutant. C, solution model including flexible N- and C-terminal regions invisible by crystallography obtained under SAXS constraints. The theoretical SAXS profile is overlaid on experimental data. The Guinier plot is given in the inset. All molecular representations were prepared with PyMOL (version 1.7, Schrödinger, LLC). a.u., arbitrary units.
The full-length PRORP2 and a more compact form, PRORP2xs, were characterized in solution by SAXS as well (Fig. 1C). Their gyration radii (Rg) of respectively 35 and 32 Å derived from the Guinier analysis are in good agreement with the value calculated from the crystal structure (29.5 Å) deprived of N- and C-terminal extensions (supplemental Fig. S3). Possible conformations of PRORP2 were explored by a normal mode analysis (NMA), and the model fitting best to the SAXS data (supplemental Fig. S4) was extended to include missing N- and C-regions and refined under SAXS constraints (supplemental Fig. S5). Fig. 1C represents the resulting model showing the core of nuclear PRORP2 in solution. SAXS data on both PRORP1 and PRORP2 indicate that the structural organization of the enzyme with its central hinge between the catalytic domain and the ZBD introduces flexibility in its backbone and allows more open conformations than those observed in crystal structures (see Discussion and supplemental Fig. S4).
Defining conditions for a stable PRORP2–tRNA complex
A prerequisite for the characterization of the PRORP2–tRNA complex was to define buffer conditions compatible with various biochemical and biophysical analyses and ensuring both protein and RNA stability. The effect of the salt concentration was tested on PRORP2 catalytic activity. Initial buffer conditions contained 250 mm NaCl to increase the solubility of the enzyme, but it appeared to be detrimental to both crystallization (21) and activity. As shown in supplemental Fig. S6, the cleavage of pre-tRNA sharply decreases beyond 150 mm NaCl. The latter concentration was chosen in combination with 5% glycerol (w/v) as a good compromise to maintain both PRORP2 monodispersity in solution and its activity.
Defining a compact but well processed RNA substrate was another prerequisite, especially for SAXS or crystallography to avoid long and flexible extensions. We therefore gradually trimmed the leader sequence of our Arabidopsis pre-tRNACys(GCA) constructs from 51 down to 5 nucleotides (supplemental Fig. S7). The best compromise was obtained with the L5T0 tRNACys (i.e. with a leader sequence of 5 nucleotides, no trailer sequence, and no added CCA), which proved to be fully cleaved and showed up as a single band on a native gel. This is in agreement with a previous study (22) showing that PRORP2 preferentially binds substrates with short 5′ leaders and 3′ trailers. The L5T0 construct was selected for all subsequent analyses. In the same line, the short PRORP2xs construct was preferred over the full-length catalytic mutant to avoid long and flexible regions and to facilitate SAXS modeling (supplemental Fig. S5).
PRORP2 affinity for pre-tRNA substrates
The experimental setup defined above was used to determine the affinity of Arabidopsis PRORP2 for pre-tRNA by several biophysical methods. Hence, a catalytically inactive mutant of PRORP2xs containing two alanines in place of two aspartates at positions 421 and 422 in the NYN domain (7) was used in combination with the L5T0 tRNA substrate. Isothermal titration calorimetry (ITC) revealed a first KD value of 1 μm for the interaction of PRORP2 with tRNA with a ΔH of −3.9 104 cal·mol−1, a ΔS of −107 cal·mol−1·degree−1, and a stoichiometry of 0.4 (Fig. 2A), thus suggesting that a proportion of either of the partners would not form a complex or that one of the partners (tRNA) partially oligomerizes as seen by size exclusion chromatography (SEC) in similar conditions (data not shown). Subsequent dynamic light scattering (DLS) and SEC measurements confirmed that the sample did not aggregate during the experiment. To confirm these results, the PRORP2–tRNA complex formation was also monitored by microscale thermophoresis (MST) using a tRNACys precursor 5′-labeled with a fluorescent dye (Cy5). This revealed a KD of 1 μm with 95% confidence limits between 0.8 and 1.3 μm in triplicate experiments (Fig. 2B). Finally, the PRORP–tRNA complex was subjected to sedimentation velocity analytical ultracentrifugation (SV-AUC). Replicate analyses of titration series with a constant concentration of pre-tRNACys and increasing concentrations of PRORP2 yielded an estimated KD value of 1 μm (95% confidence limits, 0.4, 2.3 μm) (Fig. 2C) in good accordance with the values measured with MST and ITC. Altogether, results indicate a KD value of the PRORP–tRNA complex in the micromolar range. This relatively low affinity suggests a transient interaction of PRORP enzymes with tRNA precursors as expected for a maturation enzyme.
Figure 2.
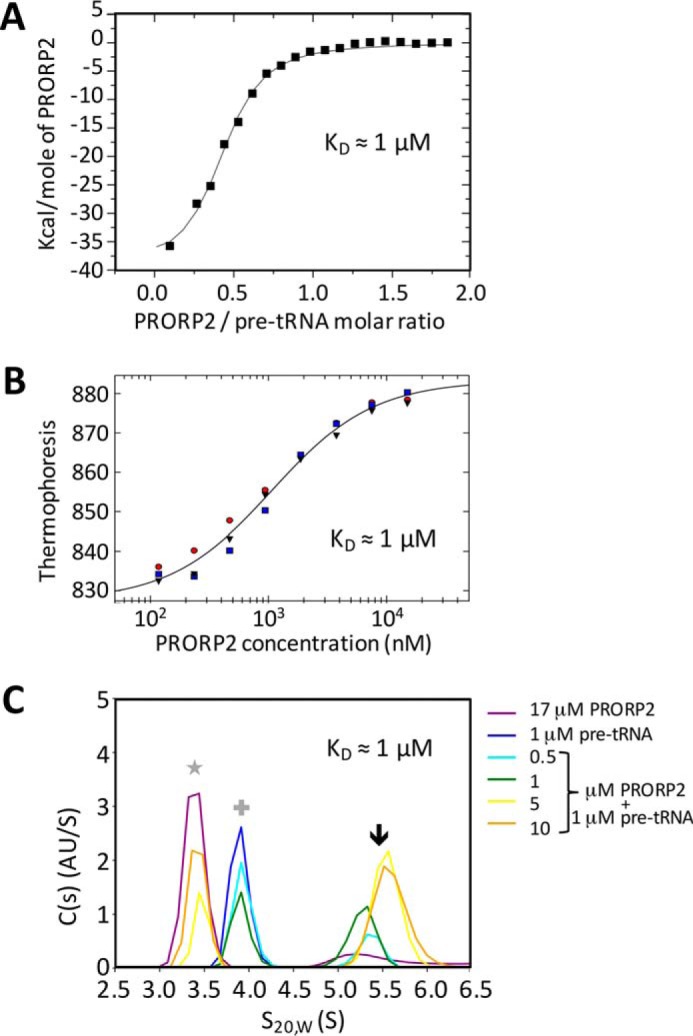
Titration of PRORP2 interaction with tRNA. A–C, the interaction of a catalytically inactive PRORP2 with an L5T0 tRNACys precursor having a 5-nucleotide-long leader sequence was monitored by ITC (A), MST (B), and AUC (C). In the ITC plot, the slope of the tangent indicates the affinity constant. MST experiments were performed with 52 nm Cy5 fluorescent RNA. For the MST plot, the normalized fluorescence (thermophoresis) of all MST traces is plotted against the concentration of PRORP2 in nm shown on a log scale. Red, blue, and gray indicate three replicate experiments. In the AUC plot, different colors indicate the particular concentrations of PRORP2 and/or RNA in the respective AUC experiments. The star indicates signals for PRORP2 alone, the cross indicates signals for RNA alone, and the arrow indicates signals for PRORP2–pre-tRNACys complexes. KD values that could be derived from the three biophysical approaches are indicated in the respective panels.
Relative importance of PPR motifs for interaction with tRNA
PRORP proteins belong to the huge family of PPR proteins. These eukaryote-specific RNA-binding proteins are involved in a wide variety of post-transcriptional processes such as RNA editing, splicing, and the maturation of transcript ends (16, 23). They are composed of ∼35-amino acid tandem repeats of degenerate primary sequences although bearing a conserved helix-turn-helix structure. The investigation of PPR protein mode of action has revealed that each PPR motif specifically interacts with a defined ribonucleotide. Some residues are particularly important to achieve this specificity, i.e. two residues located toward the start and at the end of PPR motifs, termed here positions 5 and 35, according to Cheng et al. (23). The nature of amino acids at these positions defines a combinatorial code for RNA recognition by PPR motifs (24–26). The PPR domain of Arabidopsis PRORP2 is composed of five such motifs (PPR1–PPR5) (supplemental Fig. S8). Positions 5 and 35 of motifs PPR2 and PPR3 are particularly well conserved in plants with the occurrences of a Gln or Asn at PPR2 position 5 and an Asn or Ser at PPR2 position 35 as well as a Thr at PPR3 position 5 and an Arg at PPR3 position 35 in an alignment of 100 plant PRORP sequences (6). Shorter alignments are exemplarily shown in supplemental Figs. S8 and S9. Although the “PPR code” is not yet fully understood (i.e. target RNAs cannot be predicted for many PPR proteins or motifs, and the exact involvement of other residues besides positions 5 and 35 for specific RNA binding is not entirely known), the latest resources and knowledge on PPR RNA recognition (23) enable the prediction that PRORP2 motif PPR2 would recognize a cytidine, whereas motif PPR3 would recognize a purine. In contrast, no clear prediction can be made for PPR motifs 1, 4, and 5.
Accordingly, to test the relative importance of individual PPR motifs for PRORP function, presumably for substrate recognition, PRORP2 was mutated at positions 5 and 35 of its five PPR motifs. The nature of the amino acids and mutations at these positions is indicated on supplemental Fig. S8. The five double mutants were expressed and purified to homogeneity. Solubility, stability, and structural integrity of mutants were verified by DLS and SAXS. All mutants behaved similarly to the wild-type enzyme and its catalytically inactive version in solution (supplemental Fig. S10) except PPR4 variant, which is unstable and prone to aggregation especially upon RNA removal. Therefore, the latter mutant could only be used for cleavage assays at low concentration.
Hence, the five mutant proteins were assayed for in vitro RNase P activity with two tRNACys precursors with leader sequences of either 5 or 51 nucleotides. The quantification of relative cleavages revealed that PPR mutants 1, 4, and 5 were as active as wild-type PRORP2. However, activity decreased by 45% (on average for the two tRNA substrates) in PPR2 mutant and by 91% in PPR3 mutant (Fig. 3A). This suggests that motifs PPR2 and -3 are the most important for substrate recognition by PRORP2 in accordance with comparable results obtained for the organellar PRORP1 (27).
Figure 3.

PRORP2 PPR motifs 2 and 3 are required for pre-tRNA substrate binding. A, RNase P in vitro cleavage assays were performed with Arabidopsis wild-type PRORP2 (WT) as well as with double mutants modified at positions 5 and 35 of the respective PPR motifs (PPR1–PPR5) and tRNACys precursors with either 5-nucleotide leader sequences (upper panel) or with 51-nucleotide leader sequences (lower panel). − indicates reactions with RNA alone. PRORP cleavage products were separated by 10% denaturing PAGE and quantified with ImageJ. Numbers indicate percentages of cleavage as defined by Gobert et al. (11). Values were normalized so that 100 corresponds to cleavage observed for wild-type PRORP2. The molecular weights of markers (M) are given in ribonucleotides. B, analytical ultracentrifugation sedimentation plots of catalytically inactive WT PRORP2 and PPR mutants in complex with tRNACys precursors having 5-nucleotide-long leader sequences. Different colors indicate the particular concentrations of PRORP2 isoforms and/or RNA in the respective experiments. Red dotted lines indicate signals for PRORP2 alone, blue dotted lines indicate signals for RNA alone, and purple dotted lines indicate signals for PRORP2-tRNACys complexes. KD values that could be derived for the different complexes are indicated in the respective panels.
Still, RNase P cleavage experiments represent indirect evidence for PRORP binding to tRNA. We therefore analyzed direct interaction between PPR mutants and RNA using sedimentation velocity AUC to determine which PPR motifs are indeed involved in tRNA binding. For this, the PRORP2 PPR double mutants were further mutated in the catalytic site as described above to obtain catalytically inactive proteins enabling KD measurements. The observed broad distributions of sedimenting species (between the faster tRNA component and the complex species) are characteristic of rapidly reversible systems due to dynamically associating and dissociating events during sedimentation. The weight average s-values of the fast component of the reaction boundary (swfast) isotherms were generated and analyzed to get the binding constants. The KD of PPR5 mutant interaction with tRNACys was estimated to 0.9 μm (95% confidence limits, 0.7, 1.1 μm), similar to wild-type PRORP2 (95% confidence limits, 0.6, 1.9 μm), whereas the KD values of PPR1 and PPR2 mutants were estimated to 2.2 μm (95% confidence limits, 1.7, 3.0 μm) and 2 μm (95% confidence limits, 1.4, 2.7 μm) respectively. For PPR3, no estimate of the KD was possible due to the absence of complex formation in the experimental conditions (Fig. 3B). The analysis of AUC data showed that KD variations were not due to the aggregation or misfolding of PPR mutants. No data could be obtained for PPR4 mutant because of its instability at the concentrations required for AUC (see above).
Altogether, results indicate that motif PPR3 is the most important to form the PRORP2–tRNA complex. Mutations of PPR2 motif affect both catalysis and RNA binding although to a lesser extent. PPR1 mutant is only moderately affected for RNA binding. This is probably because the mutation at position 35 introduces a negative charge that can interfere with RNA binding. Still, although PPR1 mutant does not display a loss of RNase P activity, our data suggest that the PPR1 motif is in the vicinity of the RNA in accordance with previous work showing contacts between the N-terminal extremity of PRORP and tRNA (28). In contrast, PPR motifs 4 and 5 do not seem to be involved in RNA interaction because their mutations do not alter RNA binding and cleavage.
Modeling of PRORP2–tRNA complex based on SAXS data
The conditions defined to favor a stable PRORP2–tRNA interaction were used to study the PRORP2xs–L5T0 pre-tRNA complex by SAXS. For this purpose, PRORP2xs, pre-tRNAs, and their complex were separated by analytical SEC just upstream of the SAXS cell. Because the three entities could hardly be resolved by SEC (Fig. 4A) and because of the μm affinity of PRORP for tRNA, the SEC separation was carried out in the presence of 1.5 μm free enzyme in the mobile phase to avoid complex dissociation. The complex was detected in the second part of the main scattering peak where the estimated Rg along data collection makes a plateau at ∼34 Å (Fig. 4B), i.e. a value 6% larger than that observed for PRORP2xs alone (supplemental Fig. S3). The SAXS profile and the P(r) distribution are also clearly distinct from those obtained with the isolated enzyme (supplemental Fig. S3).
Figure 4.

SAXS-based model of PRORP2 in complex with a pre-tRNA substrate. A, SEC elution profiles (Bio SEC-3/150Å HPLC column) obtained with L5T0 pre-tRNA (green chromatogram; injection, 0.2 nmol; peak elution time, 12.5 min), PRORP2xs (blue; injection, 0.9 nmol; peak elution time, 11.9 min), the complex formed during an ITC titration (orange; injection, 20 μl of ITC mixture; peak elution time, 12.0 min), and a 2:1 PRORP2xs/L5T0 mixture (red; injection, 0.6:0.3 nmol; peak elution times, 11.9 and 12.3 min). This illustrates the difficulty to stabilize the complex and to separate it from individual partners. B, evolution of X-ray scattering intensity I(0) (blue plot) and Rg (red plot) along the SEC elution of the complex in a mobile phase supplemented with PRORP2xs to minimize the dissociation of the complex. A data set of 240 SAXS images was collected in the 10–18-min window symbolized by the blue arrow. SAXS images (frames 112–130) were averaged in the region indicated by the green bar where Rg is ∼34 Å, leading to the profile shown in C. C, best solution model of the complex obtained under distance (PPR2 and -3 interacting with C56 and G18, respectively) and SAXS constraints. The theoretical SAXS profile (green curve) is overlaid on experimental data (blue dots). The Guinier plot is given in the inset. Alternative possibilities leading to higher χvalues are shown in supplemental Fig. S11. The zoom on the right highlights the proximity of residues 5 and 35 of PPR motifs 2 and 3 (analyzed by mutagenesis) with tRNA residues C56 and G18 at the corner of the tRNA (see open blue arrow). Abs., absorbance; a.u., arbitrary units.
To build a model of the PRORP2–tRNA complex, an ensemble of data previously available and determined here were considered.
Our previous footprinting analysis identified tRNA residues protected from RNase digestion and thus in contact with PRORP in the tRNA D and T loops (U16, G18, G19, and C56) (11).
These residues and others that are universally conserved among canonical tRNAs (29) were mutated. For some of them, mutations resulted in a complete loss of RNase P activity, i.e. G18, C56, and R57 (where R represents a purine) in the full-length tRNA (11) as well as C56 and R57 in a minihelix corresponding to the tRNA acceptor domain (30).
Mutations also showed that the anticodon domain of tRNAs is completely dispensable for RNase P activity (11, 30).
The PRORP2 catalytic pocket that includes Asp-421 and Asp-422 (and binds catalytic magnesium (12)) must be next to tRNA positions −1 and +1.
PPR mutagenesis and AUC data determined here suggest that PPR motifs 2 and 3 are involved in tRNA binding accordingly at the opposite side of the acceptor arm, i.e. in the D and T loops.
The PPR code predicts that motifs PPR2 and 3 should bind a cytidine and a purine, respectively. Best candidates are positions G18, G19, C56, and R57.
Taking into account these spatial criteria, a series of models of the complex was generated from the crystal structure of PRORP2 and the model of L5T0 pre-tRNACys (prepared by homology modeling using Assemble (31) and based on Escherichia coli tRNACys(GCA) crystal structure, Protein Data Bank code 1U0B (32)). These models test different combinations of PPR–nucleotide interactions. Their theoretical scattering profiles were compared with experimental SAXS data to select the best model (i.e. with best goodness of fit or lowest χ value). Because PPR3 was found to be the most important for RNA interaction, models were first built with PPR3 interacting with either G18, G19, or R57. In the models involving G19 and R57, the orientation of the tRNA does not allow an additional interaction of PPR2 with a tRNA residue. In contrast, in the model involving PPR3 interaction with G18, PPR2 is close to C56. This model of the complex displayed in Fig. 4 is the only one satisfying all distance criteria: (i) motifs PPR2 and PPR3 binding two residues in the D/T loops of tRNA and (ii) nucleotides −1 and +1 in the vicinity of metal binding aspartates. Most importantly, this model also gives the best fit with SAXS experimental data (χ of 2.4). Other combinations involving a single PPR interaction with base G19, C56, or G57 lead to a reorientation of the tRNA with respect to PRORP2 and higher χ values (ranging from 3.8 to 5.0), indicating that the shapes of these models are less representative of the objects present in solution (supplemental Fig. S11).
In our model, the backbone conformation of D/T loops was essentially kept as in the E. coli template tRNA, but the nucleobases C56 and G18 were flipped in syn conformation to point toward the protein. Indeed, in the unprocessed pre-tRNA transcript, these bases are certainly unpaired to keep D/T loops flexible and accessible to modification enzymes and thus to PPR modules as well. For instance, tertiary interactions that stabilize mature tRNAs (29), i.e. the C56–G19 or the G18–Ψ55 interaction may hardly take place before PRORP binds pre-tRNA because RNase P cleavage is one of the first steps of pre-tRNA maturation after transcription (33). Among PRORP1 lysines shown in a recent mass spectrometry study to cross-link with the tRNA (28), the only two that are conserved in PRORP2 (Lys-42 and Lys-387, which are equivalent to Lys-109 and Lys-439 in PRORP1) are close to the tRNA backbone in our SAXS model of the complex. The comparison of the crystal structure and SAXS models of PRORP2 alone or in complex with pre-tRNA suggests that the enzyme core is quite flexible with a main hinge between the zinc-binding domain and the catalytic domain. In particular, structural differences between free and complexed forms of PRORP show that major conformational changes may take place upon binding of pre-tRNA. It appears that the plasticity of PRORP2 backbone allows opening of its Λ-shape and a rotation of the catalytic domain to accommodate tRNA substrates (Fig. 5).
Figure 5.

Domain reorientation in PRORP2. A comparison of structures of PRORP2 in solution, in the crystal, and in solution in complex with a pre-tRNA substrate (left, middle, and right, respectively) highlights the movement of the catalytic domain with respect to the PPR domain and the zinc-binding domain. The three models have been superimposed according to the latter two domains and are shown in two perpendicular views, from the side and from the inner region of the Λ-shape facing the RNA substrate. In solution, the catalytic domain free and complexed with PRORP2 undergoes a 52° and 23° rotation compared with its position in the crystal structure, illustrating the flexibility of the hinge region and the structural plasticity of PRORP2. The characterization of PRORP1 in solution (see supplemental Fig. S4) shows a similar behavior and suggests that this may be a general property of PRORP enzymes.
Discussion
Since the first descriptions of protein-only RNase P (3, 4), crystal structures and biophysical studies of Arabidopsis and human PRORP have revealed the functional organization and structural diversity of these enzymes (for a review, see Schelcher et al. (20)). Initial investigations of PRORP mode of action showed which tRNA residues are in contact with PRORP (11), but the way tRNA recognition is achieved by PRORP was unknown, and no data were available on the structure of the PRORP–tRNA complex and on the dynamics of the enzyme.
Mutagenesis of PPR motifs and the model derived from SAXS data acquired on the PRORP2–tRNA complex suggest that only the PPR2 and -3 motifs of PRORP are involved in tRNA recognition. Besides PRORP, all characterized PPR proteins bind single-stranded RNA (16), and the current trend is that tandem arrays of PPR motifs specifically bind unpaired RNA bases in a sequence-specific manner (24). Because PRORP binds any tRNA of canonical structure, it was proposed that PRORP diverged from the general mode of action of PPR proteins and developed a specific RNA recognition mode based on structural features rather than on linear sequences (2, 30). Our results suggest that this is not completely the case at least for motifs PPR2 and -3 that appear to follow the canonical base recognition by PPR motifs. Indeed, PPR2 could recognize C56, and PPR3 could recognize a purine at the corner of the tRNA. This purine could be either G18 or G57, although our SAXS model indicates that G18 is closer to motif PPR3 and is thus more likely recognized by this motif when PRORP interacts with a full tRNA. However, in the context of a minihelix mimicking the acceptor arm with a less constrained single-stranded T loop region, as used by Brillante et al. (30), PPR3 might also be able to bind G57, whereas PPR2 interacts with C56.
In contrast, motifs PPR4 and -5 do not seem to participate in tRNA binding, and their function, besides maintaining the PPR architecture, remains elusive. They might be involved in the recognition of yet unidentified non-tRNA substrates of PRORP2. Alternatively, the role of these motifs might be to position PPR2/3 at the right distance from the active site of the NYN domain. The overall PRORP fold would then act as a ruler that selects the cleavage site with respect to the tRNA corner held by PPR2/3. However, although positions 5 and 35 of PPR motifs are recognized as the most important for specific RNA binding, the functional relevance of other positions such as position 2, which was also proposed to be involved in RNA interaction, for PRORP/tRNA recognition remains unexplored.
In addition to the PRORP RNA recognition process, we determined and compared the crystal and different solution models of PRORP2 to capture various conformations of PRORP and look at the conformational landscape of PRORP proteins. To explore possible domain reorientations, we performed a NMA, which is known to provide a good description of low-frequency collective motions in proteins (34). The comparison of NMA perturbed models with our SAXS data in solution suggests that both PRORP1 and PRORP2 can adopt a variety of conformations because of the presence of a hinge between the catalytic NYN domain and the central zinc-binding domain. Related movements correspond to the opening of the Λ-shape of PRORP and the rotation of its catalytic domain around the central hinge (supplemental Fig. S3). Models were further refined against SAXS data with the genetic algorithm DADIMODO and led to an excellent fit for PRORP2xs, again showing multiple compatible conformations with a reorientation of the catalytic domain (supplemental Fig. S5). The presence of long and floppy extensions in the full-length enzyme led to slightly higher goodness of fit (χ), but the trend remained the same. The superposition of the PRORP2 crystal structure and solution models alone or in complex suggests the existence of a continuum of conformations that allow the accommodation of RNA substrates and possibly with a longer helical domain than a classical tRNA acceptor arm (Fig. 5). This adaptability is exemplified by the capacity of PRORP to cleave substrates such as tRNAHis, which has an 8-base pair acceptor stem and can be processed by PRORP at positions −1 and +1 (35) or by the cleavage of tRNA-like substrates as observed for mitochondrial nad6 and orf291 mRNAs (7, 36). Overall, the intrinsic plasticity of PRORP2, which likely applies to all members of the PRORP family, may be crucial to bind RNA substrates and to release matured RNA products.
Such type of conformational adaptation has been observed in other monomeric tRNA-binding enzymes. In the case of the human mitochondrial phenylalaninyl-tRNA synthetase, the crystal structure of the isolated enzyme revealed a very compact conformation. SAXS data on the complex with tRNA indicated a large rigid-body motion of the anticodon-binding domain upon tRNA binding (37). Other examples are the bacterial elongation factor Tu and the conformational transition following GTP hydrolysis that leads to ribosome translocation and elongation factor Tu release (38); the bacterial TruB, which catalyzes pseudouridine formation at U55 in tRNA and undergoes a large rigid body displacement of its C-terminal region upon tRNA binding (39); and the archaeal O-phosphoseryl-tRNA kinase, the enzyme that phosphorylates Ser-tRNASec to produce O-phosphoseryl-tRNASec, which displays a >60-Å movement of its C-terminal domain to bind the variable region of tRNASec (40). Large conformational movements thus seem to be a general feature to modulate the recognition, binding, or release of tRNAs.
Experimental procedures
PRORP2 and tRNA purification
Expression of the full-length enzyme in the wild-type and catalytically impaired (PRORP2mut) versions as well as of PPR mutants and their purification by immobilized metal affinity chromatography and SEC were performed as described by Pinker et al. (21) A more compact and catalytically inactive form (PRORP2xs) was designed, and the region of PRORP2 cDNA coding for residues 25–516 was introduced in a pTYB1 plasmid (New England Biolabs) to produce the protein C-terminally fused to self-cleavable intein and chitin-binding domain. In both PRORP2mut and PRORP2xs, two essential aspartate residues (421 and 422) were mutated to alanines to inhibit the nucleolytic activity and facilitate the formation of stable complexes, and PPR motifs were mutated to abolish RNA interaction. Mutations were introduced with the QuikChange® Site-Directed Mutagenesis kit (Stratagene) using PCR primers designed with the manufacturer's qcprimerdesign online tool.
The expression of PRORP2xs was performed in E. coli BL21 (DE3) strain at 17 °C in 2 liters of LB medium containing ampicillin and induced with isopropyl 1-thio-β-d-galactopyranoside. The cells were harvested by centrifugation, resuspended in a buffer containing protease inhibitors (Sigma, catalogue number S8830; one tablet per 100 ml), 50 mm HEPES-Na, pH 7.5, 250 mm NaCl, 15% (w/v) glycerol, and 1 mm TCEP and were disrupted by ultrasonication. Cell debris was removed by centrifugation, and the supernatant was applied onto a 50-ml chitin affinity column (New England Biolabs) equilibrated with the same buffer including 500 mm NaCl. To trigger intein cleavage and PRORP2xs release, 50 mm DTT was added to the buffer, and the protein was incubated on the column for 16 h at 4 °C. After elution, PRORP2xs was further purified by SEC on a Superdex 200 10/300 GL column (GE Healthcare) and stored in 50 mm HEPES-Na, pH 7.5, 250 mm, NaCl, 15% (w/v) glycerol, and 1 mm TCEP. To reduce the NaCl concentration prior to complex formation, this storage buffer was exchanged by ultrafiltration or dialysis with a buffer containing 150 mm NaCl, 30 mm HEPES-Na, pH 7.5, 5% (w/v) glycerol, and 1 mm TCEP (buffer P). Sample quality was systematically assessed by SDS-PAGE and DLS as described previously (11, 21). Arabidopsis mt-tRNACys(GCA) L5T0 and L51T30 pre-tRNA cDNAs (with leader sequences of 5 or 51 nucleotides as well as trailer sequences of 0 or 30 nucleotides) were amplified from Arabidopsis seedling total cDNA, cloned in pUC19, and transcribed in vitro by T7 RNA polymerase as described by Gobert et al. (11).
Determination of PRORP2 crystal structure
Crystallization and crystallographic analysis were performed as described by Pinker et al. (21). In brief, PRORP2 in buffer P was crystallized in 2-μl batch drops at 4 °C by mixing (1:1 ratio) with a crystallant solution containing 200 mm sodium malonate, pH 6, and 20% (w/v) PEG 3350. Diffraction data were collected at 100 K with a wavelength of 1 Å using a PILATUS 2M detector on the X06DA beamline at the Swiss Light Source (SLS Synchrotron, Paul Scherrer Institute, Villigen, Switzerland). Search models for molecular replacement were derived from the structure of organellar PRORP1 (Protein Data Bank code 4G23) using MODELLER (41). A molecular replacement search was performed with Phaser and the PHENIX package (42, 43). Models of single PRORP2 monomer were split in three domains (PPR, ZBD, and NYN), and one copy of each was placed in the P1 unit cell. As the self-correlation function indicated the presence of 2-fold non-crystallographic symmetry, the second molecule was localized using a second copy of the catalytic domain. Two complete copies of PRORP2 were then refined at 3.05-Å resolution using a maximum likelihood target as implemented in PHENIX (42). Non-crystallographic symmetry constraints between the two monomers were applied during refinement as well as a translation-libration-screw-rotation (TLS) model for atomic displacement parameters. Manual inspection and rebuilding were performed with Coot (44). The final structure (Protein Data Bank code 5FT9) describes two monomers of PRORP2 including residues 28–515. N-terminal (1–27) and C-terminal (516–536) tails and two loops (312–321 and 499–514) are not visible in the electron density map. Data collection and refinement statistics are given in supplemental Table S1.
Isothermal titration calorimetry
ITC was performed on a MicroCal ITC instrument (Malvern Instruments, Malvern, UK) at 20 °C with 187 μm PRORP2 in buffer P with 5 mm MgCl2. 19 samples of 3 μl of protein solution were injected with a 120-s interval in the cell containing 280 μl of 22 μm L5T0 tRNACys precursor dialyzed in the same buffer. Data analysis was performed with MicroCal Origin7 software.
Microscale thermophoresis
MST was performed with a Monolith NT 115 instrument (NanoTemper Technologies GmbH, Munich, Germany). 10-μl aliquots of a serial dilution of PRORP2 (188 μm to 6 nm) in buffer P with 5 mm MgCl2 and 0.3 mg/ml BSA were added to 10 μl of a 100 nm solution of 5′-Cy5-labeled L5T0 tRNACys (IBA GmBH, Göttingen, Germany). The light-emitting diode laser power was set to 20%, the IR laser power was set to 40%, and the variation of fluorescence related to molecule and complex mobility was monitored at 20 °C according to the manufacturer's instructions. Data analysis was performed with Monolith MO.Affinity analysis software.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed in a Beckman Coulter ProteomeLab XL-I instrument at 20 °C and 50,000 rpm with absorbance detection. 400 μl of different ratios of PRORP2 and L5T0 tRNACys were prepared in buffer P with 5 mm MgCl2. tRNA concentration was kept constant at 0.7 μm, and protein was added to final concentrations varying from 0.25 to 10 μm. Samples of 0.7 μm tRNA alone and 10 μm PRORP2 alone were also measured. The fitting of data was performed using SEDFIT software (www.analyticalultracentrifugation.com/)6 (55) and a continuous sedimentation coefficient distribution model. The distributions obtained for each sample were integrated to determine the weight average sedimentation coefficient effective particle theory swfast as a function of protein concentrations and to generate swfast isotherms (isotherms of the fast boundary component of rapidly interacting systems based on the Gilbert-Jenkins theory (45). The isotherms were loaded into SEDPHAT for fitting with the heteroassociation model A + B ↔ AB to obtain an estimate of the KD. In the analysis, sA and sB were fixed at the experimentally determined values for PRORP2 and tRNA, respectively, whereas KAB and sAB were subject to optimization through nonlinear regression. The error surface projection analysis was exploited to determine the error intervals of the best fit KD values at a 95% confidence level. Buffer density, buffer viscosity, and protein partial specific volumes were calculated using SEDNTERP software. The software GUSSI was used to plot and integrate the sedimentation coefficient distributions and to generate the isotherms (46).
RNase P activity assays
RNase P cleavage assays were performed with three replicates using 0.5 μm transcript and 0.15 μm PRORP2 protein in buffer containing 30 mm Tris-HCl, pH 8, 40 mm NaCl, 4.5 mm MgCl2, 20 μg·ml−1 BSA, and 100 nm DTT for 15 min at 25 °C as described previously (4). RNA fragments were separated by denaturing PAGE, visualized by ethidium bromide staining, and quantified as described (4).
Small-angle X-ray scattering analysis
SAXS experiments were performed on the SWING beamline at Synchrotron SOLEIL (Saint Aubin, France) as described previously (11). In brief, samples were loaded onto a size exclusion column (Bio SEC-3 with 150-Å pore size, Agilent Technologies), and SAXS measurements were performed throughout elution. A mixture of 30 μl of 320 μm catalytically inactive PRORP2xs and 25 μl of 185 μm L5T0 pre-tRNACys in buffer P with 5 mm MgCl2 was injected in the column equilibrated with buffer P and 5 mm MgCl2 supplemented with 1.5 μm PRORP2xs to prevent dissociation of partners during elution. PRORP2 and PRORP2xs were also characterized alone in buffer P. Data processing, interpretation, and Rg evaluation over elution profiles were performed using Foxtrot (47), and data analysis (determination of Rg and dmax) was performed with the ATSAS package (48). Based on NMA with ElNemo (49), crystal structures of PRORP1 and -2 were perturbed to select models that better represented the SAXS profile of PRORP2 because of a goodness of fit calculated with CRYSOL (50, 51). Complete models of PRORP2 and PRORP2xs (including their respective N- and C-terminal extensions not described in the crystal structure) were generated under SAXS constraints using DADIMODO (52), a genetic algorithm-based refinement program. Models of PRORP2–tRNA complex were built with SASREF (53) under SAXS constraints using this PRORP2xs solution structure with a flexible hinge between the zinc-binding and catalytic domains and taking into account distance restraints between above mentioned residues in PPR2/3 modules, the catalytic domain, and nucleotides in the tRNA. SASREF models were manually inspected to remove steric clashes and adjusted in PyMOL using the SAXS plug-in SASpy (54).
Author contributions
P. G. and Cl. S. designed and coordinated the experiments. F. P., Ce. S., P. F.-M., A. G., C. B., A. T., P. R., P. G., and Cl. S. performed the experiments and analyzed the results. P. G. and Cl. S. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. E. Ennifar and C. Da Veiga for assistance and advice during ITC analysis, teams of SWING beamline (SOLEIL synchrotron, Saint-Aubin, France) and X06DA beamline (Swiss Light Source, Villigen, Switzerland) for beamtime allocation and support during data collection, and Dr. M. Meyer and B. Masquida for the collection of PRORP2 diffraction data.
This work was supported by the French CNRS, the University of Strasbourg, Agence Nationale de la Recherche (ANR) Blanc “PRO-RNase P” Research Grant ANR-11-BSV8-008-01/02 (to P. G. and Cl. S.), and the LabEx consortium “MitoCross” in the frame of the French National Program “Investissement d'Avenir” Grant ANR-11-LABX-0057_MITOCROSS. This work was also supported by the French Infrastructure for Integrated Structural Biology (FRISBI) (Grant ANR-10-INSB-05, which granted access to the resources of FRISBI and Instruct, a Landmark European Strategy Forum on Research Infrastructures project. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S11 and Table S1.
The atomic coordinates and structure factors (code 5FT9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- RNP
- ribonucleoprotein
- PRORP
- protein-only RNase P
- PPR
- pentatricopeptide repeat
- SAXS
- small-angle X-ray scattering
- NYN
- N4BP1, YacP-like nuclease
- ZBD
- zinc-binding domain
- NMA
- normal mode analysis
- ITC
- isothermal titration calorimetry
- SEC
- size exclusion chromatography
- DLS
- dynamic light scattering
- MST
- microscale thermophoresis
- AUC
- analytical ultracentrifugation
- TCEP
- tris(2-carboxyethyl)phosphine
- swfast
- weight average s-values of the fast component of the reaction boundary.
References
- 1. Altman S. (2007) A view of RNase P. Mol. Biosyst. 3, 604–607 [DOI] [PubMed] [Google Scholar]
- 2. Pinker F., Bonnard G., Gobert A., Gutmann B., Hammani K., Sauter C., Gegenheimer P. A., and Giegé P. (2013) PPR proteins shed a new light on RNase P biology. RNA Biol. 10, 1457–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holzmann J., Frank P., Löffler E., Bennett K. L., Gerner C., and Rossmanith W. (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–474 [DOI] [PubMed] [Google Scholar]
- 4. Gobert A., Gutmann B., Taschner A., Gössringer M., Holzmann J., Hartmann R. K., Rossmanith W., and Giegé P. (2010) A single Arabidopsis organellar protein has RNase P activity. Nat. Struct. Mol. Biol. 17, 740–744 [DOI] [PubMed] [Google Scholar]
- 5. Lai L. B., Vioque A., Kirsebom L. A., and Gopalan V. (2010) Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett. 584, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lechner M., Rossmanith W., Hartmann R. K., Thölken C., Gutmann B., Giegé P., and Gobert A. (2015) Distribution of ribonucleoprotein and protein-Only RNase P in Eukarya. Mol. Biol. Evol. 32, 3186–3193 [DOI] [PubMed] [Google Scholar]
- 7. Gutmann B., Gobert A., and Giegé P. (2012) PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev. 26, 1022–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonnard G., Gobert A., Arrivé M., Pinker F., Salinas-Giegé T., and Giegé P. (2016) A single gene encodes both organelles and nuclear RNase P enzymes in Chlamydomonas reinhardtii. Plant J. 87, 270–280 [DOI] [PubMed] [Google Scholar]
- 9. Taschner A., Weber C., Buzet A., Hartmann R. K., Hartig A., and Rossmanith W. (2012) Nuclear RNase P of Trypanosoma brucei: a single protein in place of the multicomponent RNA-protein complex. Cell Rep. 2, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugita C., Komura Y., Tanaka K., Kometani K., Satoh H., and Sugita M. (2014) Molecular characterization of three PRORP proteins in the moss Physcomitrella patens: nuclear PRORP protein is not essential for moss viability. PLoS One 9, e108962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gobert A., Pinker F., Fuchsbauer O., Gutmann B., Boutin R., Roblin P., Sauter C., and Giegé P. (2013) Structural insights into protein-only RNase P complexed with tRNA. Nat. Commun. 4, 1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Howard M. J., Lim W. H., Fierke C. A., and Koutmos M. (2012) Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc. Natl. Acad. Sci. U.S.A. 109, 16149–16154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reinhard L., Sridhara S., and Hällberg B. M. (2015) Structure of the nuclease subunit of human mitochondrial RNase P. Nucleic Acids Res. 43, 5664–5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li F., Liu X., Zhou W., Yang X., and Shen Y. (2015) Auto-inhibitory mechanism of the human mitochondrial RNase P protein complex. Sci. Rep. 5, 9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anantharaman V., and Aravind L. (2006) The NYN domains: novel predicted RNAses with a PIN domain-like fold. RNA Biol. 3, 18–27 [DOI] [PubMed] [Google Scholar]
- 16. Giegé P. (2013) Pentatricopeptide repeat proteins: a set of modular RNA-specific binders massively used for organelle gene expression. RNA Biol. 10, 1417–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammani K., Bonnard G., Bouchoucha A., Gobert A., Pinker F., Salinas T., and Giegé P. (2014) Helical repeats modular proteins are major players for organelle gene expression. Biochimie 100, 141–150 [DOI] [PubMed] [Google Scholar]
- 18. Pavlova L. V., Gössringer M., Weber C., Buzet A., Rossmanith W., and Hartmann R. K. (2012) tRNA processing by protein-only versus RNA-based RNase P: kinetic analysis reveals mechanistic differences. Chembiochem 13, 2270–2276 [DOI] [PubMed] [Google Scholar]
- 19. Reiter N. J., Osterman A., Torres-Larios A., Swinger K. K., Pan T., and Mondragón A. (2010) Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468, 784–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schelcher C., Sauter C., and Giegé P. (2016) Mechanistic and structural studies of protein-only RNase P compared to ribonucleoproteins reveal the two faces of the same enzymatic activity. Biomolecules 6, E30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pinker F., Giegé P., and Sauter C. (2015) Crystallization and crystallographic analysis of an Arabidopsis nuclear proteinaceous RNase P. Acta Crystallogr. F Struct. Biol. Commun. 71, 1372–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karasik A., Shanmuganathan A., Howard M. J., Fierke C. A., and Koutmos M. (2016) Nuclear protein-only ribonuclease P2 structure and biochemical characterization provide insight into the conserved properties of tRNA 5′ end processing enzymes. J. Mol. Biol. 428, 26–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng S., Gutmann B., Zhong X., Ye Y., Fisher M. F., Bai F., Castleden I., Song Y., Song B., Huang J., Liu X., Xu X., Lim B. L., Bond C. S., Yiu S. M., et al. (2016) Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 85, 532–547 [DOI] [PubMed] [Google Scholar]
- 24. Barkan A., Rojas M., Fujii S., Yap A., Chong Y. S., Bond C. S., and Small I. (2012) A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 8, e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takenaka M., Zehrmann A., Brennicke A., and Graichen K. (2013) Improved computational target site prediction for pentatricopeptide repeat RNA editing factors. PLoS One 8, e65343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yagi Y., Hayashi S., Kobayashi K., Hirayama T., and Nakamura T. (2013) Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One 8, e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imai T., Nakamura T., Maeda T., Nakayama K., Gao X., Nakashima T., Kakuta Y., and Kimura M. (2014) Pentatricopeptide repeat motifs in the processing enzyme PRORP1 in Arabidopsis thaliana play a crucial role in recognition of nucleotide bases at TpsiC loop in precursor tRNAs. Biochem. Biophys. Res. Commun. 450, 1541–1546 [DOI] [PubMed] [Google Scholar]
- 28. Chen T. H., Tanimoto A., Shkriabai N., Kvaratskhelia M., Wysocki V., and Gopalan V. (2016) Use of chemical modification and mass spectrometry to identify substrate-contacting sites in proteinaceous RNase P, a tRNA processing enzyme. Nucleic Acids Res. 44, 5344–5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giegé R. (2008) Toward a more complete view of tRNA biology. Nat. Struct. Mol. Biol. 15, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 30. Brillante N., Gößringer M., Lindenhofer D., Toth U., Rossmanith W., and Hartmann R. K. (2016) Substrate recognition and cleavage-site selection by a single-subunit protein-only RNase P. Nucleic Acids Res. 44, 2323–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jossinet F., Ludwig T. E., and Westhof E. (2010) Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 26, 2057–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hauenstein S., Zhang C. M., Hou Y. M., and Perona J. J. (2004) Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat. Struct. Mol. Biol. 11, 1134–1141 [DOI] [PubMed] [Google Scholar]
- 33. Hopper A. K., Pai D. A., and Engelke D. R. (2010) Cellular dynamics of tRNAs and their genes. FEBS Lett. 584, 310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tama F., and Sanejouand Y. H. (2001) Conformational change of proteins arising from normal mode calculations. Protein Eng. 14, 1–6 [DOI] [PubMed] [Google Scholar]
- 35. Placido A., Sieber F., Gobert A., Gallerani R., Giegé P., and Maréchal-Drouard L. (2010) Plant mitochondria use two pathways for the biogenesis of tRNAHis. Nucleic Acids Res. 38, 7711–7717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujii S., Suzuki T., Giegé P., Higashiyama T., Koizuka N., and Shikanai T. (2016) The restorer-of-fertility-like 2 pentatricopeptide repeat protein and RNase P are required for the processing of mitochondrial orf291 RNA in Arabidopsis. Plant J. 86, 504–513 [DOI] [PubMed] [Google Scholar]
- 37. Klipcan L., Moor N., Finarov I., Kessler N., Sukhanova M., and Safro M. G. (2012) Crystal structure of human mitochondrial PheRS complexed with tRNA(Phe) in the active “open” state. J. Mol. Biol. 415, 527–537 [DOI] [PubMed] [Google Scholar]
- 38. Polekhina G., Thirup S., Kjeldgaard M., Nissen P., Lippmann C., and Nyborg J. (1996) Helix unwinding in the effector region of elongation factor EF-Tu-GDP. Structure 4, 1141–1151 [DOI] [PubMed] [Google Scholar]
- 39. Pan H., Agarwalla S., Moustakas D. T., Finer-Moore J., and Stroud R. M. (2003) Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc. Natl. Acad. Sci. U.S.A. 100, 12648–12653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sherrer R. L., Araiso Y., Aldag C., Ishitani R., Ho J. M., Söll D., and Nureki O. (2011) C-terminal domain of archaeal O-phosphoseryl-tRNA kinase displays large-scale motion to bind the 7-bp D-stem of archaeal tRNA(Sec). Nucleic Acids Res. 39, 1034–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fiser A., and Sali A. (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 [DOI] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 45. Schuck P. (2010) Sedimentation patterns of rapidly reversible protein interactions. Biophys. J. 98, 2005–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brautigam C. A. (2015) Calculations and publication-quality illustrations for analytical ultracentrifugation data. Methods Enzymol. 562, 109–133 [DOI] [PubMed] [Google Scholar]
- 47. David G., and Perez J. (2009) Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 42, 892–900 [Google Scholar]
- 48. Konarev P., Petoukhov M., Volkov V., and Svergun D. (2006) ATSAS 2.1: a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suhre K., and Sanejouand Y. H. (2004) ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 51. Svergun D. I., Barberato C., and Koch M. H. J. (1995) CRYSO—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 52. Evrard G., Mareuil F., Bontems F., Sizun C., and Pérez J. (2011) DADIMODO: a program for refining the structure of multidomain proteins and complexes against small-angle scattering data and NMR-derived restraints. J. Appl. Crystallogr. 44, 1264–1271 [Google Scholar]
- 53. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Panjkovich A., and Svergun D. I. (2016) Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys. Chem. Chem. Phys. 18, 5707–5719 [DOI] [PubMed] [Google Scholar]
- 55. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.