

Abstract
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is the master regulator of the antioxidant response, and its function is tightly regulated at the transcriptional, translational, and post-translational levels. It is well-known that Nrf2 is regulated at the protein level by proteasomal degradation via Kelch-like ECH-associated protein 1 (Keap1), but how Nrf2 is regulated at the translational level is less clear. Here, we show that pharmacological stimulation increases Nrf2 levels by overcoming basal translational repression. We developed a novel reporter assay that enabled identification of natural compounds that induce Nrf2 translation by a mechanism independent of Keap1-mediated degradation. Apigenin, resveratrol, and piceatannol all induced Nrf2 translation. More importantly, the pharmacologically induced Nrf2 overcomes Keap1 regulation, translocates to the nucleus, and activates the antioxidant response. We conclude that translational regulation controls physiological levels of Nrf2, and this can be modulated by apigenin, resveratrol, and piceatannol. Also, targeting this mechanism with novel compounds could provide new insights into prevention and treatment of multiple diseases in which oxidative stress plays a significant role.
Keywords: antioxidant, molecular pharmacology, nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), oxidative stress, translation control
Introduction
Eukaryotic cells have evolved a highly sophisticated mechanism for cellular defense against harmful oxidative conditions. Oxidative stress is a major player in the perpetuation of the most frequent human diseases related to advanced age such as atherosclerosis (1), cancer (2), chronic obstructive pulmonary disease (3), and neurodegenerative diseases (4, 5). The antioxidant capacity of a cell is governed by a broad network of enzymes that dynamically respond to unfavorable environmental conditions for the prevention and repair of oxidative damage. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is the transcription factor that is considered the master regulator of this antioxidant response, and its function is tightly regulated at different levels, including transcription, translation, and post-translational control (6–9). A better understanding of the processes regulating the function of Nrf2 will offer the possibility of identifying novel ways to enhance its function for the treatment or prevention of diseases where oxidative stress plays a significant role. Under basal conditions, cytoplasmic Nrf2 is bound to the Kelch-like ECH-associated protein 1 (Keap1), which facilitates its proteasomal degradation after ubiquitination by the Cul3/E3 ubiquitin ligase system, which maintains the intracellular levels of Nrf2 at very low concentrations (10, 11).
Upon exposure to external stimuli, i.e. UV radiation, carcinogens, xenobiotics, etc. or internal free radical stressors, the Nrf2–Keap1 complex dissociates after post-translational modifications of reactive cysteines in Keap1. This process prevents the degradation of Nrf2 and facilitates the Nrf2 accumulation and intranuclear translocation (12). In the nucleus, Nrf2 forms a heterodimer with a small Maf protein (13) and interacts with the antioxidant-response element (ARE)3 in the promoter region of target genes, increasing the expression of the battery of protective antioxidant genes.
Newly translated Nrf2 is required to actively counteract the effect of electrophiles, rather than just inhibiting Keap1-mediated protein degradation (12, 14, 15). Recent reports also indicate that the expression of Nrf2 is regulated at the level of translation (9, 16). The translational control allows the cells to quickly respond to noxious conditions by specifically regulating temporal and spatial translation by keeping specific mRNA molecules in a repressed state. This allows their translation in response to environmental signals without requiring mRNA transcription, maturation, and nuclear export (17).
Recently, we reported the identification of a novel regulatory mechanism controlling the translation of the Nrf2 mRNA inside the open reading frame (ORF). Specifically, the terminal portion of the ORF sequence was shown to repress the basal translation. Also, we showed that it is possible to overcome this repression by creating a mutant with an alternative codon composition in this portion that does not alter the amino acid sequence of Nrf2 (18).
Recently, we got interested in identifying molecules that physiologically enhance the translation of Nrf2. Here, we report the identification of apigenin, resveratrol, and piceatannol in a library of 84 known small molecules that are antioxidants/oxidants as activators of the translation of Nrf2. Moreover, we demonstrate that an increase in Nrf2 translation promotes nuclear translocation and activation of the antioxidant response independent of Keap1.
Results
Development of a reporter system to identify compounds that promote Nrf2 translation
The mRNA sequence in the Nrf2 ORF between nucleotides 1159 and 1815 bp prevents the translation of eGFP when fused at the 3′-end. This construct used a stop codon between the two sequences to demonstrate that the inhibition of GFP translation is mediated by the mRNA sequence of Nrf2 and not by the amino acids encoded by the sequence (18). Here, we used a similar approach to generate a novel reporter system except that we used the luciferase gene instead of eGFP to fuse it to the translation regulatory portion of Nrf2 (Fig. 1A). To validate the functionality of the novel reporter, we compared the expression of the vector-containing luciferase fused to the Nrf2 regulatory element and luciferase alone. We found that the Nrf2 regulatory element inhibits more than 70% of the expression of luciferase (Fig. 1B) and also confirmed that this finding was not due to reduced levels of the Luc2–Sg3 mRNA transcript (Fig. 1C). We used this luciferase-based reporter system to screen a library containing 84 known antioxidant and pro-oxidant molecules to identify potential activators of Nrf2 translation. Only the flavonoid apigenin and the stilbene resveratrol with its metabolite piceatannol were able to promote an increase in translation of the luciferase reporter regulated by the Nrf2 translational control element (Fig. 2 and supplemental Table 1). Additionally, we observed no difference for this property if the cells were treated for 12 or 24 h (data not shown). In contrast, none of the compounds caused a similar effect on cells transfected with a construct of luciferase alone, confirming that the compounds require the 3′-sequence of Nrf2 to promote an increase in protein expression (Fig. 3A and supplemental Table 2).
Figure 1.
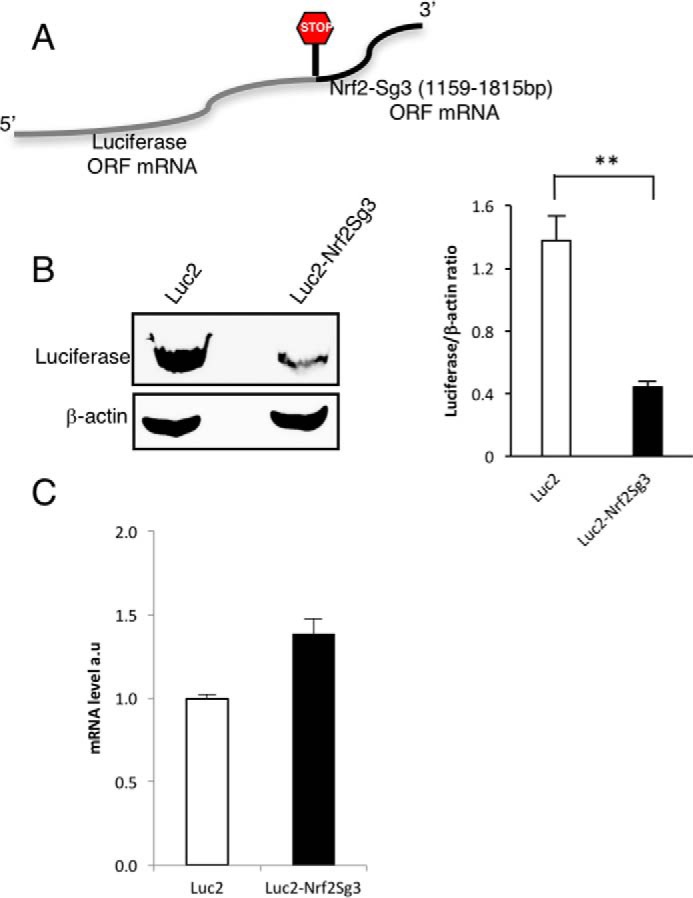
Novel reporter system to detect activators of Nrf2 translation. A, schematic representation of the composition of the recombinant transcript used to identify activators of Nrf2 translation. B, expression of luciferase in HEK293T cells was evaluated by Western blotting with or without the fusion of the 3′-region of Nrf2 as depicted in A (n = 3). The relative intensity of the bands is represented as the ratio with β-actin as a loading control. The statistical significance of the difference in the levels of expression of Luc2 versus Luc2-sg3 is indicated by **, p < 0.01. C, real-time PCR (n = 3) of the transcripts of luciferase or Luciferase-Nrf2 overexpressed in HEK293T cells.
Figure 2.
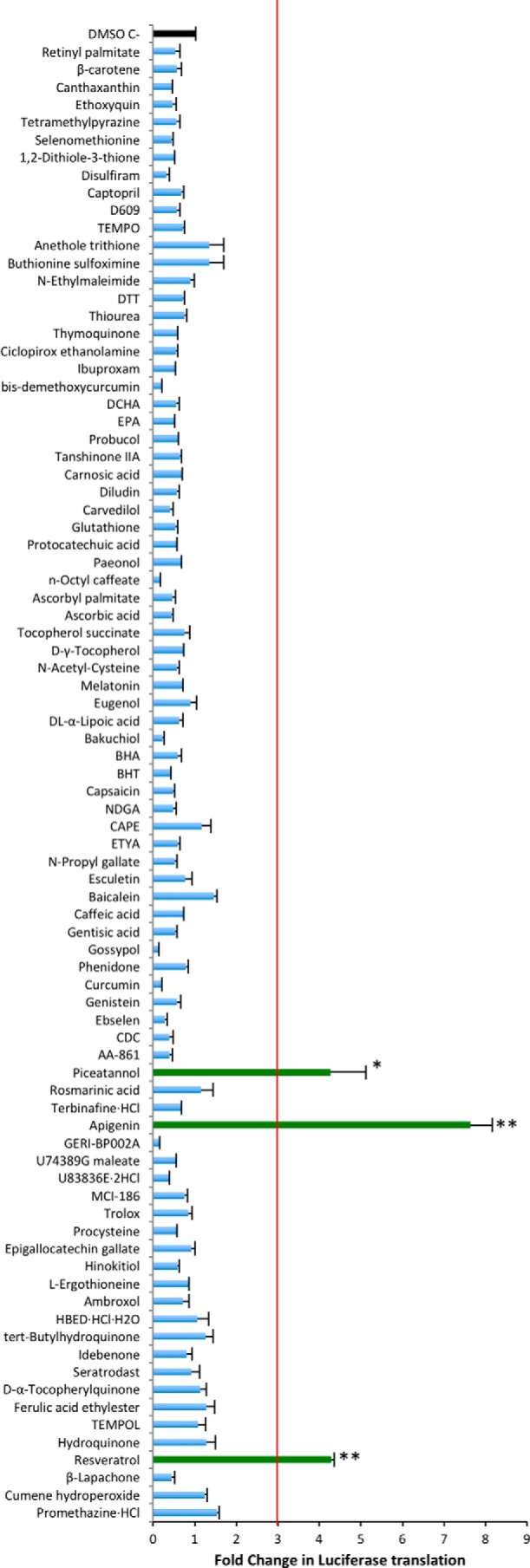
Detection of compounds that increase the expression of Luc2–Nrf2Sg3. Changes in the expression of the construct Luc2–Nrf2Sg3 were evaluated after treatment with 84 compounds from the redox library (mean ± S.E. n = 3). The statistical significance of the difference in the levels of expression of luciferase after the treatment with DMSO alone versus each of the compounds in the library is indicated by **, p < 0.0001, and *, p < 0.005. The expression was normalized against the cell viability data measured with Cell-titer Glo® after the treatment with the library. The fold change in expression was calculated against the basal expression of Luc2–Nrf2Sg3 in cells treated with DMSO only as a negative control. A secondary confirmatory assay was done for compounds that increased the expression of Luc2–Nrf2Sg3 more than three times (red line).
Figure 3.

Specificity of compound hits for increasing the translation of Luc2–Nrf2Sg3 versus Luc2 alone. A, Z-score values are plotted against the fold change (supplemental Tables 1 and 2) in the translation of either the construct Luc2–Nrf2Sg3 or Luc2 alone after the treatment with 84 compounds from the redox library. Apigenin, resveratrol, and piceatannol increased the expression of the reporter more than 3-fold and with a Z-score higher than three only in the construct containing the Nrf2–Sg3 sequence. B, chemical structures of apigenin, resveratrol, and piceatannol.
To determine the ability of apigenin, resveratrol, and piceatannol to increase Nrf2 protein expression independent of Keap1, we performed a secondary experiment using a Nrf2 construct lacking amino acids 17–32 that was resistant to Keap1-mediated degradation (19). The cells were treated for 24 h post-transfection with apigenin, resveratrol, and piceatannol. We also used the well-known inhibitors of the Keap1/Nrf2 interaction (tert-butylhydroquinone, epigallocatechin gallate, and sulforaphane) as controls. Only apigenin, resveratrol, and piceatannol were able to increase the expression of Δ17–32 Nrf2 (Fig. 4A). As expected, the known Keap1 modulators (TBHQ, EGCG, and sulforaphane) were not able to alter the expression of the Δ17–32 Nrf2 construct but were able to increase the expression of the recombinant wild-type construct (Fig. 4B). This was further confirmed by evaluating the expression of recombinant wild-type Nrf2 in a HEK293T Keap1 knock-out cell line treated with apigenin, where we show that a Keap1-independent mechanism was responsible for promoting an increase in the levels of Nrf2 (Fig. 4C). The identification and confirmation of apigenin as an inducer of the expression of Nrf2 allowed us to evaluate the quality of the reporter system by calculating the z-factor as described by Zhang et al. (20). We determined that the z-factor was 0.58, which reflects as an excellent assay for future screening studies (20).
Figure 4.

Apigenin increases the expression of Nrf2 independent of Keap-1. A, apigenin, resveratrol, and piceatannol promote an increase in the expression of the Keap1-resistant Δ17–32 Nrf2 in transfected HEK293T cells (n = 3). The statistical significance of the difference in the levels of expression of Δ17–32 Nrf2 in untreated versus treated cells is indicated by **, p < 0.01. Additionally, the effect on Nrf2 translation of TBHQ, EGCG, and sulforaphane was evaluated. B, apigenin also increases the expression of wild-type Nrf2 transfected into HEK293T cells. The effect of well-known inhibitors of the Keap1/Nrf2 interaction (TBHQ, EGCG, and sulforaphane) was also compared against apigenin (n = 3). The statistical significance of the difference in the levels of expression of wild-type Nrf2 in untreated versus treated cells is indicated by **, p < 0.01, or *, p < 0.05. C, apigenin increases the expression of recombinant WTNrf2 in transfected HEK293T cells independent of Keap1. The relative intensity of the Nrf2 band in all the Western blottings is represented as a ratio with β-actin as a loading control. The statistical significance of the difference in the levels of expression of Nrf2 in apigenin-treated versus untreated cells is indicated by **, p < 0.01.
Apigenin as a tool to study Nrf2 translation regulation
Based on the screening results, we chose to pursue the capability of apigenin to increase the activity of Nrf2 by promoting an increase in protein expression. We used Western blotting to confirm that the increase in Nrf2 expression by apigenin was dose-dependent but was independent of Keap1 (Fig. 5A). Next, we showed that apigenin actively promotes Nrf2 nuclear translocation of recombinant WTNrf2 (Fig. 5B). Additionally, we verified by Western blotting that apigenin induces the expression of endogenous Nrf2 in HepG2 cells (Fig. 5C). The HepG2 cell line is known to have a functional Nrf2 pathway (21). Thus, by using a commercial reporter cell line (ARE-HepG2), we were able to confirm the activation of the antioxidant response pathway upon treatment with apigenin in a dose-dependent manner (Fig. 5D). Interestingly enough, contrary to what has been reported for mouse skin cells (22), where apigenin was shown to increase the expression of Nrf2 we did not find an increase in transcription of the endogenous Nrf2 upon HepG2 cell stimulation with apigenin (Fig. 5E).
Figure 5.

Apigenin promotes nuclear translocation and activation of the antioxidant response. A, apigenin induced a dose-dependent increase in the expression of recombinant Keap1-resistant Δ17–32 Nrf2 in transfected HEK293T (n = 3). The statistical significance of the difference in the levels of expression of Δ17–32 Nrf2 in untreated versus treated cells is indicated by **, p < 0.01. The relative intensity of the Nrf2 band is represented as a ratio with β-actin as a loading control. B, apigenin treatment promoted translocation of recombinant WTNrf2 to the nucleus in transfected HEK293T (n = 3). The statistical significance of the difference in the levels of Nrf2 in apigenin-treated versus untreated cells is indicated by **, p < 0.01. Actin and histone 3 were used as loading controls. The relative intensity of the Nrf2 band is represented as a ratio with β-actin for the total lysate and the cytoplasmic fractions and as a ratio with histone 3 for the nuclear fractions. C, expression of the endogenous Nrf2 in HepG2 cells was evaluated after the treatment with apigenin for 12 h (n = 3). D, apigenin was a strong activator of the ARE in the ARE-HepG2 cell line. TBHQ at 50 μm was used as a positive control (C+) (mean ± S.E. n = 6). The statistical significance of the difference in the increase in Nrf2 activity after the treatment with apigenin or TBHQ versus untreated cells is indicated by **, p < 0.01, or *, p < 0.05. E, mRNA levels of the endogenous Nrf2 in HepG2 cells was evaluated by real-time PCR after 12 h of treatment with apigenin (12.5 μm) (n = 3).
To further evaluate the molecular mechanism and to determine the basis for the increase of Nrf2 after the treatment with apigenin, the increase in Nrf2 transcription, inhibition of proteasomal degradation, and increase in Nrf2 translation were studied. Nrf2 transcription studies using real-time PCR showed that apigenin does not induce changes in the transcription of the recombinant Nrf2 (Fig. 6A). The potential of apigenin to inhibit proteasomal degradation was evaluated by measuring the polyubiquitinated proteins upon treatment with either apigenin or the proteasome inhibitor MG132 as a positive control. Fig. 6B shows that apigenin was not effective in inhibiting proteasomal degradation. Additionally, we showed that apigenin does not induce the common post-translational modifications in Keap1 (Fig. 6C), which are induced by tert-butylhydroquinone or sulforaphane (23). In other words, the novel mechanism is Keap1-independent. Finally, we evaluated the possibility that the increased expression of Nrf2 upon apigenin treatment was dependent on promoting Nrf2 translation. For this, we used a technology that allows the labeling of newly synthesized proteins with a methionine analogue (5-azidohomoalanine) for the subsequent purification for detection of only the newly synthesized proteins (24, 25). With this state of the art approach, we were able to show that apigenin directly stimulates the translation of Nrf2, because newly synthesized Nrf2 was only identified in the purified fraction of cells treated for 4 h with apigenin (Fig. 6D).
Figure 6.

Apigenin activates the translation of Nrf2. A, real-time PCR indicated that apigenin did not affect the transcription of rNrf2 in HEK293T cells. B, analysis of polyubiquitinated proteins in HEK293T cells after apigenin treatment suggested that it was not an inhibitor of the proteasome (n = 3). As a positive control, the effect of the proteasome inhibitor MG132 was included in the assay. The relative intensity of the signal from the polyubiquitinated proteins is indicated as a ratio with β-actin as the loading control. The statistical significance of the difference in the levels of polyubiquitinated proteins in treated versus control cells is indicated by **, p < 0.01. C, apigenin treatment of HA-Keap1-transfected HEK293T cells did not induce post-translational modifications in Keap1 (arrow) when compared with TBHQ, a well-known inhibitor of the Nrf2 degradation mediated by Keap1 (n = 3). The relative intensity of the bands for modified Keap1 is indicated as a ratio with β-actin as a loading control. The statistical significance of the difference in the levels of modified Keap1 in treated versus untreated cells is indicated by **, p < 0.01. The arrow indicates the appearance of a slow migrating isoform of Keap1 due to cysteine disulfur bridge formation. D, apigenin activated the translation of Nrf2. An assay to isolate newly synthesized proteins was performed by following the steps indicated in the top diagram. The presence of newly synthesized recombinant Nrf2 in the eluates from cells treated or untreated with apigenin was evaluated by Western blotting (n = 3).
Activation of the AMPK pathway partially promotes Nrf2 translation
To identify potential molecular mechanism(s) for apigenin to activate Nrf2 translation, we searched the literature for a common molecular target between structurally similar apigenin, resveratrol, and piceatannol (Fig. 3B). All three compounds are known activators of the AMP-activated protein kinase (AMPK) pathway (26–30). AMPK is a central regulator of cellular energy homeostasis and primarily responds to the levels of AMP/ATP to phosphorylate its targets but also can be controlled by upstream regulatory kinases such as calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2, also known as CAMKKβ) (31) and serine/threonine protein kinase LKB1 (32). It has been shown that apigenin activates AMPK indirectly by activating its upstream regulator CAMKK2 (30). Thus, we decided to explore the requirement of AMPK or CAMKK2 for the activation of Nrf2 translation by apigenin. The cells previously transfected with the Luc2–Nrf2Sg3 reporter were treated with dorsomorphin (also known as compound C), an AMPK inhibitor (33) and STO-609, a CAMKK2 inhibitor (34). Two hours later, we added apigenin to determine whether the kinase inhibitors were able to block the translation of the Luc2–Nrf2Sg3 reporter. We were able to confirm that the inhibition of AMPK or CAMKK2 reduces the ability of apigenin to induce the translation of Nrf2 between 40 and 50%, respectively. Importantly, this reduction in activation of Nrf2 was not as a result of inhibitor toxicity (Fig. 7, A and B). To confirm the role of these two proteins in Nrf2 translational control, we decided to co-transfect an activated α subunit of AMPK and the full-length Camkk2 gene from mouse (91% identity with humans) with WTNrf2 into HEK293T cells to evaluate the expression of recombinant Nrf2. With this, we were able to show that the co-transfection of activated AMPK or Camkk2 increases the basal translation of recombinant Nrf2 (Fig. 7C).
Figure 7.

AMPK and CAMKK2 regulate Nrf2 translation. HEK293T cells transfected with the Nrf2 translation reporter (Luc2–Sg3Nrf2) were pre-treated with increasing doses of an AMPK inhibitor (A) and a CAMKK2 inhibitor (B) for 2 h before stimulation of Nrf2 translation with apigenin for 24 h. The fold change in the expression of luciferase after apigenin treatment with or without the presence of increasing doses of the inhibitor was calculated (n = 6). The statistical significance of the difference in the reduction of luciferase expression after stimulation with apigenin in cells treated with the kinase inhibitor versus non-treated cells is indicated by **, p < 0.01. The cell viability of the cells under treatment was evaluated in parallel with Cell-Titer Glo to verify the absence of the reduction in cell viability with the treatments higher than 15% (n = 6). C, HEK293T cells were co-transfected with wild-type Nrf2 and activated AMPK or Camkk2 to evaluate the effect of the kinases on the expression of Nrf2. The expression of recombinant wild-type Nrf2 was detected by Western blotting 24 h after co-transfection (n = 3). The overexpression of the recombinant kinases was verified by Western blotting with an anti-c-Myc antibody, and β-actin was used as a loading control. The relative intensity of the bands for Nrf2 is indicated as a ratio with β-actin as a loading control. The statistical significance of the difference in the levels of expression of Nrf2 in cells co-transfected with the active kinases versus untreated cells is indicated by **, p < 0.01.
Nrf2 translational regulation inside the ORF does not respond to oxidative stress
Since the discovery of Nrf2 translational control (9), most of the research in this area has centered on the regulation of Nrf2 translation by oxidative stress (9, 36). It is known that there are two different translation regulatory elements in the 5′-UTR of the Nrf2 mRNA transcript, and it has been reported that these two elements are regulated by hydrogen peroxide (36, 37). Based on this, we evaluated the ability of well-known inducers of oxidative stress to stimulate the novel reporter of Nrf2 translation regulated by the Sg3 motif (nucleotides 1159–1815 bp in the ORF). We treated HEK293T cells transfected with the Luc2–Nrf2Sg3 by increasing concentrations of hydrogen peroxide, tert-butyl hydroperoxide, and paraquat for 24 h to evaluate the luciferase expression and compare it against the effect of apigenin. None of the inducers of oxidative stress increased the translation of luciferase when compared with apigenin in the tested concentrations (Fig. 8), suggesting that oxidative stress is not required to activate the translation of Nrf2 regulated by the 3′-portion of the ORF.
Figure 8.
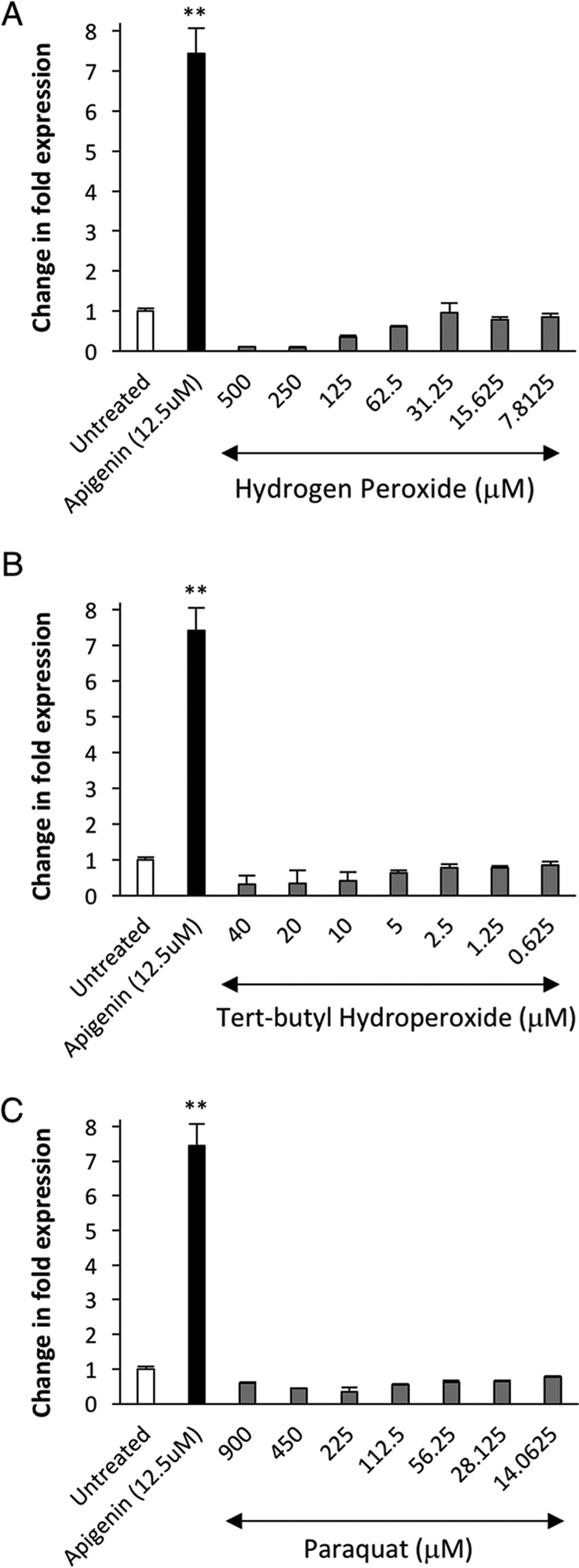
Oxidative stress does not overcome the translational repression motif that exists inside the open reading frame of Nrf2. HEK293T cells transfected with the Nrf2 translation reporter (Luc2–Sg3Nrf2) were treated 24 h after transfection with increasing doses of three inducers of oxidative stress: A, hydrogen peroxide; B, tert-butyl hydro peroxide; and C, paraquat, to evaluate the induction of luciferase translation and compared against apigenin. 24 h after the treatment with inducers of oxidative stress, the fold change in expression of luciferase was calculated against the basal expression of Luc2–Nrf2Sg3 in cells treated with DMSO only as a negative control (n = 6). The statistical significance of the difference in the levels of expression of luciferase in cells treated with apigenin or inducers of oxidative stress versus untreated cells is indicated by **, p < 0.01. The expression was normalized against the cell viability data measured with Cell-titer Glo® after the treatment. The range of concentrations for each inducer of oxidative stress was selected around known reported concentrations that induce oxidative stress in HEK293T cells (35, 73, 74).
Discussion
Nrf2 has become an important target for the treatment and/or prevention of multiple chronic diseases influenced by oxidative stress (38–42). To date, research on Nrf2 activation for therapeutic purposes has predominantly centered on interrupting the regulatory process controlled by Keap1, which promotes Nrf2 degradation (43–45). A smaller subset of reports indicates that there are other Keap1-independent mechanisms that regulate the activity of Nrf2. For example, it is known that the overexpression of the gene DPP3 can promote nuclear translocation and activation of Nrf2 without affecting Keap1 (46). Moreover, the post-translational modifications on Nrf2 by phosphorylation, ubiquitination, or acetylation can alter its activity by interfering with its cytoplasmic and nuclear localization (47, 48). Although several mechanisms are known to activate Nrf2, strategies to translate these findings into the development of novel molecules for treating or preventing chronic diseases without affecting Keap1 have not been reported.
A myriad of xenobiotics and natural compounds have been shown to have antioxidant activity in mammalian cells. Some of these molecules, such as vitamin C, are free radical scavengers with the capacity to quench reactive oxygen or nitrogen species (49). Other groups of natural compounds instead can activate the endogenous antioxidant response by promoting nuclear translocation of Nrf2. For some of these activators of Nrf2, such as sulforaphane, curcumin, epigallocatechin gallate, etc., the mechanism involves the prevention of the Keap1-mediated Nrf2 protein degradation (50). Other compounds activate Nrf2, but the mechanism of activation is poorly understood. However, it is possible that some of these natural compounds present in the human diet could increase Nrf2 translation. To test this hypothesis, we decided to create a novel reporter system to identify molecules that were able to increase the translation of Nrf2.
Our recent work on the regulation of Nrf2 at the level of translation (18) indicated that the pharmacophores that promote an increase in the translation of Nrf2 might have the potential for improving antioxidant capacity by exploiting this alternative mechanism. Therefore, we used the property of the translation regulatory element of Nrf2 to inhibit the translation of reporter genes such as eGFP and luciferase to develop a reporter system that allows for the identification of pharmacophores that induce Nrf2 translation. We selected luciferase instead of eGFP to construct a Nrf2 translation reporter because luciferase is preferred for small molecule screening with reporter assays due to the existence of multiple molecules with native fluorescence (51) that can generate false positives. Also, we are not aware of previous reports that show the capacity of small molecules to stimulate the translation of Nrf2. Thus, before committing to future vast and expensive molecular screenings, we reduced the risk for testing our novel reporter system by selecting a small library with well-known molecules that were either antioxidants or pro-oxidants. Interestingly enough, we found only three polyphenols that were able to promote an increase in the translation of the reporter (Fig. 3B). This capacity was verified in an independent assay by evaluating the changes in the expression of Nrf2 in a Keap1 knock-out cell line by Western blotting after treatment with apigenin (Fig. 4C). Also, because apigenin exerted the strongest activation of the reporter and promoted a higher expression of Nrf2, we used it as a tool to confirm that the mechanism was mediated by activation of Nrf2 protein translation (Fig. 6D).
The antioxidant and protective characteristics of apigenin have been previously reported with animal models and human studies (52–54). Also, it was known that apigenin activates the Nrf2 pathway (55, 56), but a clear understanding of the molecular mechanism for this effect was not completely understood. Here, we show that apigenin directly affects the production of Nrf2 by reversing translational repression. Additionally, our data suggest that increasing the translation of Nrf2 with small molecules is an alternative way to promote nuclear translocation of Nrf2 and induction of the antioxidant response (Fig. 9).
Figure 9.

Turning on Nrf2 by activating its translation. The translation of Nrf2 is repressed under basal circumstances by a sequence contained inside the open reading frame between nucleotides 1159 and 1815 bp (red portion). The translation of Nrf2 can be stimulated by treating the cell with apigenin (step 1). This process promotes an increase in the levels of Nrf2 (step 2) and its nuclear translocation (step 3), where together with other proteins such as small Maf proteins, they recognize the ARE in the promoter of their target genes. This promotes the transcription of the genes needed for activating the antioxidant response (step 4).
Previous studies have reported the presence of translation regulatory elements in the 5′- and 3′-UTR of the mRNA transcript of Nrf2. In particular, these regulatory elements present in the 5′-UTR respond to hydrogen peroxide to increase the translation of Nrf2 (36, 37). In contrast, the 3′-region of the mRNA of Nrf2 allows the regulation of Nrf2 by microRNAs (57, 58). More than likely, this type of regulation is more tissue-specific and is likely to facilitate the control of the levels of the Nrf2 transcript in specific tissues or cell types that express those microRNAs. We have previously described a new type of regulation of Nrf2 translation that occurs inside the ORF (18). In this study, we show that this regulatory element does not respond to classical Keap1 regulators or to well-known inducers of oxidative stress such as hydrogen peroxide, tert-butyl hydroperoxide, or paraquat.
The molecular mechanism that allows the 3′-portion of the ORF of Nrf2 to regulate its translation it is not well-understood. Previously, we reported that the mechanism is dependent on the mRNA sequence but not in the amino acids encoded by the nucleotides 1159–1815 bp. The identification of apigenin as a potent inducer of Nrf2 translation adds another set of questions to this puzzle. For example, how is apigenin able to increase the translation of Nrf2? Based on what has been found in other genes regulated at the level of translation (17, 59), we speculate that unknown proteins or RNA molecules are interacting with the 3′-portion of the mRNA ORF of Nrf2 to regulate its expression according to environmental conditions. Apigenin might prevent or enhance the interaction of these trans-regulatory factors directly or indirectly with the mRNA of Nrf2. An example of this type of mechanism was reported for the P53 gene in keratinocytes, where it was found that apigenin enhanced the translation of P53 by promoting the interaction of the HUR protein with the mRNA of P53 (60). Additionally, there is a possibility that other indirect mechanisms induced by apigenin, such as kinase activation, could induce the translation of Nrf2. Here, we have shown that the AMPK kinase and CAMKK2 kinase are important for a full activation of the translation of Nrf2 mediated by apigenin. Significantly, the role of these kinases in regulating the translation of Nrf2 was confirmed by co-transfection experiments that show partial restoration of Nrf2 translation in the absence of apigenin (Fig. 7C). Interestingly, the co-expression of CAMKK2 with Nrf2 led to a much stronger activation of Nrf2 translation than the co-transfection with activated AMPK. This finding is in agreement with the reports that indicated that CAMKK2 is a direct target of apigenin (30) and resveratrol (61) and that the activation of AMPK is secondary because AMPK is a downstream effector of CAMKK2 (31). It is known that CAMKK2 has two additional downstream effectors, calcium/calmodulin-dependent protein kinase I and IV (CAMKI and CAMKIV) (62). Interestingly, CAMKIV has been reported to be directly implicated in protein translation regulation. Specifically, liver cancer cells that overexpress CAMKK2 display higher levels of activated CAMKIV, and this stimulates protein translation by causing phosphorylation of multiple targets of the mechanistic target of rapamycin pathway implicated in translation regulation (63). Thus, the higher expression of Nrf2 when co-expressed with CAMKK2 instead of AMPK can be explained by the synergistic activation of two downstream effectors (CAMKIV and AMPK) that directly affect protein translational control. Finally, we show that the activation of Nrf2 translation by recombinant CAMKK2 did not reach the expression levels induced by apigenin alone, indicating that there might be other proteins that are targeted by apigenin to induce the translation of Nrf2.
In conclusion, our findings suggest an alternative mechanism for maintaining physiological levels of Nrf2 through translational control inside the ORF. We believe that this is complementary to the fast degradation of Nrf2 mediated by Keap1 to efficiently respond to different types of environmental stimuli that might be harmful to the cell. Therefore, although the Keap1 regulatory process would be efficient in responding to electrophilic stressors, the novel mechanism based on translational control seems to respond to a specific type of xenobiotics or natural bioactive compounds. It has been established that the function of Nrf2 is central to respond to multiple types of harmful stimuli (64, 65) and that there are Keap1-independent mechanisms that can regulate the activity of Nrf2 (66–68). Thus, this suggests that eukaryotic cells have evolved multiple sensing mechanisms for different types of stressors to regulate one central transcription factor to respond to environmental stress. Our findings raise further questions centered around these natural bioactive compounds as follows: Does the translation of Nrf2 respond to a metabolite synthesized in mammalian cells or does it only respond to xenobiotic or phytochemical molecules? Finding the answers will advance the development of novel activators of Nrf2 for their potential use in the prevention and treatment of multiple diseases where oxidative stress plays a major role.
Experimental procedures
Reagents
All the reagents used were of the highest purity available. Apigenin and piceatannol were obtained from Santa Cruz Biotechnology. Trans-resveratrol, sulforaphane, dorsomorphin (AMPK inhibitor), STO-609 (CAMKK2 inhibitor), and epigallocatechin gallate were purchased from Cayman Biochemicals. tert-Butyl hydroperoxide, paraquat, and hydrogen peroxide were obtained from Sigma. The Screen-WellTM REDOX library (Enzo Life Sciences) that contains 84 compounds with defined pro-oxidant or antioxidant activity was used to identify activators of Nrf2 translation.
DNA constructs
The DNA constructs for WTNrf2 (this construct encodes the protein referred to as rNrf2) and Nrf2Δ17–32 were cloned in the plasmid PLEX-MCS (Thermo Fisher Scientific), and the required oligos and the sequence of the mutant Nrf2 were previously described (18). The plasmid containing the HA-Keap1 gene was previously described (69) and was obtained from Addgene (plasmid no. 21556). The constructs Luc2 and Luc2–Nrf2Sg3 were cloned in the PLEX vector, which was previously digested with BamHI and AgeI (Thermo Fisher Scientific) using the Gibson assembly method (70). For generating the construct Luc2–Nrf2Sg3, the luciferase2 gene was amplified from the plasmid PGL4 (Promega) using the primers forward 5′-ATA GAA GAC ACC GAC TCT ACT AGA GGA TCC GCC GCC ACC ATG GAA GAT GCC AAA AAC ATT AA-3′ and reverse 5′-TGG TGT TTT AGG ACC ATT CTG TTT GAC ACT TTA TTA CAC GGC GAT CTT GC-3′, and the NRF2 segment 1159–1815 bp was amplified from the clone WTNrf2 by using the primer set forward 5′-A GTG TCA AAC AGA ATG GTC CTA AA-3′ and reverse 5′-GTT GGC GCA GCA GCC GGG GCA GCA ACC GGT GTT TTT CTT AAC ATC TGG CTT CTT-3′. Both fragments contained recombination sites that are underlined, and two stop codons were included between the two fragments. After PCR purification, these two PCR fragments were cloned into the previously digested PLEX-MCS by using the Gibson assembly mix (New England Biolabs) following the manufacturer's recommendations. To generate the construct containing only Luc2, we used the same methodology except that the luciferase2 gene was amplified with the reverse oligo 5′-GTT GGC GCA GCA GCC GGG GCA GCA ACC GGT TTA TTA CAC GGC GAT CTT GC-3′ in combination with the forward oligo described above. The plasmid containing the gene for the c-Myc-tagged mouse calcium/calmodulin-dependent protein kinase kinase 2 (Camkk2) was obtained from Origene Technologies (MR208650). The DNA construct to overexpress a constitutively active form of the AMPK α2 protein (gene symbol PRKAA2) was constructed by cloning the first 936 nucleotides of the open reading frame, as described previously (71), into the mammalian vector pHTC (Promega catalogue G7711) previously digested with XhoI using the Gibson assembly method (70). The PRKAA2 construct was built as a synthetic gblock by IDT DNA Technologies and contained a c-Myc C-terminal tag.
The authenticity of the constructs described here was confirmed by sequencing at Genewiz Inc., and all the primers were synthesized by IDT DNA Technologies.
Cells and transfection
HEK293T cells were obtained from ATCC and were grown in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum. HepG2 cells were also obtained from ATCC and were cultured in minimal essential medium supplemented with 10% fetal bovine serum. ARE-HepG2 cells were obtained from BPS Bioscience and were grown under similar conditions as the HepG2 cells described above. All recombinant plasmids were extracted using the Pureyield Maxiprep system (Promega) and were transfected into HEK293T by using Jetprime (Polyplus) by following the manufacturers' recommendations without modifications. The CRISPR/Cas9 system for genomic modification was used to develop a HEK293T Keap1 knock-out cell line (HEK293T-Keap KO) with vectors obtained from Santa Cruz Biotechnology (catalogue no. sc-400190 and sc-400190-HDR). For this, a plasmid co-expressing Cas9 and a Keap1-targeting sgRNA was co-transfected into HEK293T cells with a donor plasmid containing a puromycin resistance gene and red fluorescent protein. 72 h after transfection, positive cells were selected with puromycin following the manufacturer's recommendations. Single clones were isolated by limited dilution, and the knock-out of Keap1 was confirmed by Western blotting.
Antibodies and protein lysates
The antibody for Strep II tag was a mouse monoclonal from Genescript (catalogue no. A01742-40); monoclonal anti Nrf2 was from Cell Signaling (catalogue no. 12721P); monoclonal anti Keap1 was from Origene (catalogue no. TA502059); monoclonal anti-histone 3 was from Active Motif (catalogue no. 39763); and monoclonal anti-luciferase, monoclonal anti-ubiquitin, monoclonal anti-β-actin, anti-HA tag, and anti-c-Myc tag were from Santa Cruz Biotechnology (catalogue no. sc-57603, sc-8017, sc-47778 HRP, sc-73294, and sc-40, respectively). Cytoplasmic and nuclear fractions of HEK293T cells were obtained following cellular lysis in a hypotonic buffer (0.1× PBS, 0.001% Triton X-100, and protease inhibitor mixture (Thermo Fisher Scientific)). Briefly, hypotonic lysis buffer (200 μl) was added to the HEK293T cell pellet of 2 × 106 cells. The resulting suspension was agitated by pipetting and was placed on ice for 15 min. Following incubation, the suspension was vortexed for 1 min and centrifuged at 16,000 × g for 30 min. The supernatant containing the cytoplasmic fraction was recovered, and the pellet containing the nuclei was washed twice with PBS and lysed with MPER Lysis buffer (Pierce) under sonication for 30 s, followed by centrifugation to isolate a soluble nuclear lysate. The protein concentration of both fractions was obtained by following the Bradford assay (Bio-Rad).
Real-time PCR
The expression of recombinant Nrf2, Luc2, and Luc2–Nrf2Sg3 was evaluated in HEK293T cells by RT-PCR. The expression of the endogenous Nrf2 (NFE2L2) was assessed in HepG2 cells by the same technique. HEK293T cells were plated on 6-well plates and were independently transfected with WTNrf2, Luc2, and Luc2–Nrf2Sg3. All the cells were harvested after 24 h. Untransfected HepG2 cells or transfected HEK293T cells with WTNrf2 were treated with or without apigenin for 12 h to quantitate the concentration of endogenous or recombinant Nrf2 post-treatment, respectively. After harvesting the cells, the total cellular RNA was extracted using RNAzol (Molecular Research Center Inc.) and was treated with DNase I (Promega) following the manufacturer's recommendations. 500 ng of RNA from each treatment was used to set up a one-step real-time quantitation assay using the TaqMan RNA-to-CT one-step kit (Applied Biosystems). The RNA quantitation was conducted in Step One Plus® real-time PCR system (Applied Biosystems), and the PCR cycles proceeded as follows: initial reverse transcription for 15 min at 48 °C, followed by 10 min at 95 °C for activating Taq polymerase, followed by 40 cycles of 15 s at 95 °C, and 1 min at 60 °C. All the reactions were done in triplicates. The Primetime® qPCR probe and oligos used for the reaction were made by IDT DNA Technologies and targeted a unique sequence located in the recombinant constructs in the region that contained the Strep II tag sequence. The sequences were as follows: probe with FAM and TAMRA 5′-FAM-ATT GAG GAT GGG ACC AAT TGC TGC-TAMRA-3′; Primer1 5′-GGA AGT ACA GGT TCT CGC TG-3′; Primer2 5′-CAG TTC GAG AAG GGC GG-3′. For detecting the endogenous Nrf2, the following Primetime® qPCR probe was used: 5′-FAM-ATC CAT GTC CCT TGA CAG CAC AGA AG-TAMRA-3′; Primer1 5′-GTC TTT GGA TTT AGC GTT TCA GA-3′; Primer2 5′-TCT TGC CTC CAA AGT ATG TCA A-3′. To guarantee that equal amounts of RNA were added to the one-step real-time PCR mixture, the expression of β-actin was also quantitated in a parallel reaction by using the following probe and oligos: Probe 5′-FAM-TCA TCC ATG GTG AGC TGG CGG-TAMRA-3′; Primer1 5′-CCT TGC ACA TGC CGG AG-3′; Primer2 5′-ACA GAG CCT CGC CTT TG-3′. The threshold cycle (CT), which inversely correlates with the target mRNA level, was measured as the cycle number at which the reporter fluorescent emission appeared above the background threshold. Data analysis was performed with Step-one software as described in the manufacturer's instructions (Applied Biosystems).
Luciferase reporter assay for identifying Nrf2 translation activators or inhibitors
HEK293T cells were transfected with the plasmid containing the reporter Luc2–Nrf2Sg3 or a vector containing the luciferase gene alone with HTS-jetPEI (Polyplus), as per the manufacturer's recommendations. The cells were seeded into white 96-well plates (Greiner Bio-one) at a concentration of 1 × 104 cells per well in a final media volume of 100 μl. To identify Nrf2 translation activators, we used a compound library containing 84 known antioxidant/pro-oxidant molecules. Twenty four hours after seeding, the compounds in the Screen-WellTM REDOX library (Enzo Life Sciences) were transferred to the culture plates to a final concentration of 10 μm by using a Janus automated work station (PerkinElmer Life Sciences) and incubated at 37 °C, 5% CO2. Twelve hours after treatment, the One-Glo® luciferase assay system (Promega) was used as recommended by the manufacturer. The luciferase activity was measured using a Glomax luminometer (Promega) following the manufacturer's setup for the One-Glo® luciferase assay system. To study the role of the AMPK pathway in the translation of Nrf2 two kinase inhibitors (dorsomorphin, an AMPK inhibitor, and STO-609, a CAMKK2 inhibitor) were added to HEK293T previously transfected with the reporter Luc2–Nrf2Sg3. The kinase inhibitors were added 24 h after transfection, and 2 h later the cells were treated with apigenin (12.5 μm). Twenty four hours later, the luciferase activity was detected by adding the One-Glo® luciferase assay system (Promega). To determine the cell viability, replicate plates were made, and the cell viability was measured by quantifying ATP with CellTiter®-Glo (Promega) using the standard protocol and expressed as percent viability compared with vehicle-treated cells. To study the role of three inducers of oxidative stress (tert-butyl hydroperoxide, paraquat, and hydrogen peroxide) in the translation of Nrf2, we treated HEK293T previously transfected with the reporter Luc2–Nrf2Sg3 with increasing concentrations of each of the oxidants for 24 h in white 96-well plates followed by luciferase activity determination with the One-Glo® luciferase assay system (Promega).
Luciferase reporter assay for detecting the activation of the antioxidant-response element in ARE-HepG2 cells
ARE-HepG2 cells (BPS Bioscience) was seeded into white 96-well plates (Greiner Bio-one) at a concentration of 1 × 104 cells per well in a final media volume of 100 μl. Twenty four hours after seeding, the cells were treated for 12 h with increasing concentrations of apigenin or tert-butylhydroquinone as a positive control (50 μm). For detecting the luciferase expression after the treatment, the One-Glo® luciferase assay system (Promega) with a Glomax luminometer (Promega) was used as indicated above.
Selective isolation and detection of the newly synthesized proteins
A modified version of the method of Dieterich et al. (24, 25) was used to label and isolated newly synthesized proteins upon treatment with apigenin. For this, we used HEK293T cells previously transfected (24 h) with wild-type Nrf2. For labeling, we replaced regular DMEM with pre-warmed DMEM without methionine (MP Bio) with l-azidohomoalanine at 4 mm for incorporation into newly synthesized proteins. Subsequently, we treated the cells with or without apigenin at 12.5 μm for 4 h. After harvesting the cells, these were lysed, and the proteins were quantified with the Bradford assay (Bio-Rad). To isolate newly synthesized proteins, they were biotinylated by using biotin alkyne (Life Technologies, Inc.) with the Click-it® protein reaction buffer (Life Technologies, Inc.) that specifically biotinylated the proteins where l-azidohomoalanine was incorporated. The isolation of the biotinylated proteins was performed with a purification column containing CaptAvidinTM (Life Technologies, Inc.) as recommended by the manufacturer. The eluates from treated or untreated cells were concentrated by using an Amicon Ultra (Millipore) centrifugal filter with a cutoff of 3 kDa. The concentrated eluates were separated by SDS-PAGE and processed for Western blotting for the detection of Nrf2.
Statistical analysis
All figures in this work are representative of at least three independent experiments. Differences between control and treated samples in which two groups were compared were analyzed by t test analysis. The comparison of multiple groups was performed by using one-way analysis of variance with Dunnett's test in Graphpad Prism version 6. p values <0.05 were considered statistically significant. The Z score is the number of standard deviations from the mean for the expression of luciferase for each readout value (n = 3) after the treatment with compounds from the redox library and was calculated as described by Malo et al. (72). The Z-factor for determining the quality of the novel assay to discover inducers of Nrf2 translation was done as described by Zhang et al. (20) by using the readout values of the expression of Luc2–Nrf2Sg3 treated with DMSO (n = 8) or apigenin (n = 8) as follows: Z factor = 1 − (3S.D.(apigenin) + 3SD(DMSO)/mean(apigenin) − mean(DMSO)), S.D. = standard deviation.
Author contributions
O. P. L. conceived the project, designed the experiments, performed the research, analyzed the data, and wrote the paper. C. A. B. performed real-time PCR and subcellular fractionations. S. M. designed the experiments, analyzed the data, and wrote the paper.
Supplementary Material
This work was supported in part by National Institutes of Health Grants R01AI064017, R03AG051095, and R03DK105267-01. The authors declare that they have a conflict of interest due to a patent application generated with partial data from this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2.
- ARE
- antioxidant-response element
- AMPK
- AMP-activated protein kinase
- eGFP
- enhanced GFP
- CAMK
- calcium/calmodulin-dependent protein kinase
- TBHQ
- tert-butylhydroquinone
- EGCG
- epigallocatechin gallate
- oligo
- oligonucleotide
- qPCR
- quantitative PCR
- FAM
- 6-carboxyfluorescein
- TAMRA
- 6-carboxy-N,N,N′,N′-tetramethylrhodamine.
References
- 1. Kim Y. W., and Byzova T. V. (2014) Oxidative stress in angiogenesis and vascular disease. Blood 123, 625–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorrini C., Harris I. S., and Mak T. W. (2013) Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 [DOI] [PubMed] [Google Scholar]
- 3. Kirkham P. A., and Barnes P. J. (2013) Oxidative stress in COPD. Chest 144, 266–273 [DOI] [PubMed] [Google Scholar]
- 4. Butterfield D. A., Swomley A. M., and Sultana R. (2013) Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox Signal. 19, 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Niranjan R. (2014) The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson's disease: focus on astrocytes. Mol. Neurobiol. 49, 28–38 [DOI] [PubMed] [Google Scholar]
- 6. Cullinan S. B., Zhang D., Hannink M., Arvisais E., Kaufman R. J., and Diehl J. A. (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., et al. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang H. C., Nguyen T., and Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774 [DOI] [PubMed] [Google Scholar]
- 9. Purdom-Dickinson S. E., Sheveleva E. V., Sun H., and Chen Q. M. (2007) Translational control of Nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 72, 1074–1081 [DOI] [PubMed] [Google Scholar]
- 10. Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J. D., and Yamamoto M. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi A., Kang M. I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., and Yamamoto M. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate for proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kobayashi A., Kang M. I., Watai Y., Tong K. I., Shibata T., Uchida K., and Yamamoto M. (2006) Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity if Keap1. Mol. Cell. Biol. 26, 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., and Nabeshima Y. (1997) An Nrf2 small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322 [DOI] [PubMed] [Google Scholar]
- 14. Itoh K., Tong K. I., and Yamamoto M. (2004) Molecular mechanism activating Nrf2–Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 36, 1208–1213 [DOI] [PubMed] [Google Scholar]
- 15. Itoh K., Wakabayashi N., Katoh Y., Ishii T., O'Connor T., and Yamamoto M. (2003) Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391 [DOI] [PubMed] [Google Scholar]
- 16. Shay K. P., Michels A. J., Li W., Kong A.-N., and Hagen T. M. (2012) Cap-independent Nrf2 translation is part of a lipoic acid-stimulated detoxification stress response. Biochim. Biophys. Acta 1823, 1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gebauer F., and Hentze M. W. (2004) Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez-Leal O., Barrero C. A., and Merali S. (2013) Translational control of Nrf2 within the open reading frame. Biochem. Biophys. Res. Commun. 437, 134–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McMahon M., Thomas N., Itoh K., Yamamoto M., and Hayes J. D. (2004) Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 279, 31556–31567 [DOI] [PubMed] [Google Scholar]
- 20. Zhang J. H., Chung T. D., and Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 21. Kanazawa K., Uehara M., Yanagitani H., and Hashimoto T. (2006) Bioavailable flavonoids to suppress the formation of 8-OHdG in HepG2 cells. Arch. Biochem. Biophys. 455, 197–203 [DOI] [PubMed] [Google Scholar]
- 22. Paredes-Gonzalez X., Fuentes F., Su Z. Y., and Kong A. N. (2014) Apigenin reactivates Nrf2 anti-oxidative stress signaling in mouse skin epidermal JB6 P+ cells through epigenetics modifications. AAPS J. 16, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang D. D., and Hannink M. (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., and Schuman E. M. (2007) Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2, 532–540 [DOI] [PubMed] [Google Scholar]
- 25. Dieterich D. C., Link A. J., Graumann J., Tirrell D. A., and Schuman E. M. (2006) Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. U.S.A. 103, 9482–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baur J. A., Pearson K. J., Price N. L., Jamieson H. A., Lerin C., Kalra A., Prabhu V. V., Allard J. S., Lopez-Lluch G., Lewis K., Pistell P. J., Poosala S., Becker K. G., Boss O., Gwinn D., et al. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dasgupta B., and Milbrandt J. (2007) Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. U.S.A. 104, 7217–7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Minakawa M., Miura Y., and Yagasaki K. (2012) Piceatannol, a resveratrol derivative, promotes glucose uptake through glucose transporter 4 translocation to plasma membrane in L6 myocytes and suppresses blood glucose levels in type 2 diabetic model db/db mice. Biochem. Biophys. Res. Commun. 422, 469–475 [DOI] [PubMed] [Google Scholar]
- 29. Ono M., and Fujimori K. (2011) Antiadipogenic effect of dietary apigenin through activation of AMPK in 3T3-L1 cells. J. Agric. Food Chem. 59, 13346–13352 [DOI] [PubMed] [Google Scholar]
- 30. Tong X., Smith K. A., and Pelling J. C. (2012) Apigenin, a chemopreventive bioflavonoid, induces AMP-activated protein kinase activation in human keratinocytes. Mol. Carcinog. 51, 268–279 [DOI] [PubMed] [Google Scholar]
- 31. Woods A., Dickerson K., Heath R., Hong S. P., Momcilovic M., Johnstone S. R., Carlson M., and Carling D. (2005) Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 [DOI] [PubMed] [Google Scholar]
- 32. Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., and Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 33. Vucicevic L., Misirkic M., Janjetovic K., Vilimanovich U., Sudar E., Isenovic E., Prica M., Harhaji-Trajkovic L., Kravic-Stevovic T., Bumbasirevic V., and Trajkovic V. (2011) Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 7, 40–50 [DOI] [PubMed] [Google Scholar]
- 34. Tokumitsu H., Inuzuka H., Ishikawa Y., Ikeda M., Saji I., and Kobayashi R. (2002) STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 277, 15813–15818 [DOI] [PubMed] [Google Scholar]
- 35. Shin S. Y., Kim C. G., Jho E. H., Rho M. S., Kim Y. S., Kim Y. H., and Lee Y. H. (2004) Hydrogen peroxide negatively modulates Wnt signaling through downregulation of β-catenin. Cancer Lett. 212, 225–231 [DOI] [PubMed] [Google Scholar]
- 36. Li W., Thakor N., Xu E. Y., Huang Y., Chen C., Yu R., Holcik M., and Kong A. N. (2010) An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 38, 778–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee S. C., Zhang J., Strom J., Yang D., Dinh T. N., Kappeler K., and Chen Q. M. (2017) G-Quadruplex in the NRF2 mRNA 5′ untranslated region regulates de novo NRF2 protein translation under oxidative stress. Mol. Cell. Biol. 37, e00122–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boutten A., Goven D., Boczkowski J., and Bonay M. (2010) Oxidative stress targets in pulmonary emphysema: focus on the Nrf2 pathway. Expert Opin. Ther. Targets 14, 329–346 [DOI] [PubMed] [Google Scholar]
- 39. de Vries H. E., Witte M., Hondius D., Rozemuller A. J., Drukarch B., Hoozemans J., and van Horssen J. (2008) Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 45, 1375–1383 [DOI] [PubMed] [Google Scholar]
- 40. Jung K. A., and Kwak M. K. (2010) The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules 15, 7266–7291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li J., Ichikawa T., Janicki J. S., and Cui T. (2009) Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin. Ther. Targets 13, 785–794 [DOI] [PubMed] [Google Scholar]
- 42. Ruiz S., Pergola P. E., Zager R. A., and Vaziri N. D. (2013) Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 83, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Devling T. W., Lindsay C. D., McLellan L. I., McMahon M., and Hayes J. D. (2005) Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc. Natl. Acad. Sci. U.S.A. 102, 7280–7285A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dinkova-Kostova A. T., Liby K. T., Stephenson K. K., Holtzclaw W. D., Gao X., Suh N., Williams C., Risingsong R., Honda T., Gribble G. W., Sporn M. B., and Talalay P. (2005) Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. U.S.A. 102, 4584–4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao X., and Talalay P. (2004) Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc. Natl. Acad. Sci. U.S.A. 101, 10446–10451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y., Kern J. T., Walker J. R., Johnson J. A., Schultz P. G., and Luesch H. (2007) A genomic screen for activators of the antioxidant response element. Proc. Natl. Acad. Sci. U.S.A. 104, 5205–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma Q. (2013) Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chowdhry S., Zhang Y., McMahon M., Sutherland C., Cuadrado A., and Hayes J. D. (2013) Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32, 3765–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carr A. C., McCall M. R., and Frei B. (2000) Oxidation of LDL by myeloperoxidase and reactive nitrogen species–reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 20, 1716–1723 [DOI] [PubMed] [Google Scholar]
- 50. Chen C., and Kong A. N. (2005) Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol. Sci. 26, 318–326 [DOI] [PubMed] [Google Scholar]
- 51. Simeonov A., Jadhav A., Thomas C. J., Wang Y., Huang R., Southall N. T., Shinn P., Smith J., Austin C. P., Auld D. S., and Inglese J. (2008) Fluorescence spectroscopic profiling of compound libraries. J. Med. Chem. 51, 2363–2371 [DOI] [PubMed] [Google Scholar]
- 52. Jeyabal P. V., Syed M. B., Venkataraman M., Sambandham J. K., and Sakthisekaran D. (2005) Apigenin inhibits oxidative stress-induced macromolecular damage in N-nitrosodiethylamine (NDEA)-induced hepatocellular carcinogenesis in Wistar albino rats. Mol. Carcinog. 44, 11–20 [DOI] [PubMed] [Google Scholar]
- 53. Nielsen S. E., Young J. F., Daneshvar B., Lauridsen S. T., Knuthsen P., Sandström B., and Dragsted L. O. (1999) Effect of parsley (Petroselinum crispum) intake on urinary apigenin excretion, blood antioxidant enzymes and biomarkers for oxidative stress in human subjects. Br. J. Nutr. 81, 447–455 [DOI] [PubMed] [Google Scholar]
- 54. Singh J. P., Selvendiran K., Banu S. M., Padmavathi R., and Sakthisekaran D. (2004) Protective role of apigenin on the status of lipid peroxidation and antioxidant defense against hepatocarcinogenesis in Wistar albino rats. Phytomedicine 11, 309–314 [DOI] [PubMed] [Google Scholar]
- 55. Huang C.-S., Lii C.-K., Lin A.-H., Yeh Y.-W., Yao H.-T., Li C.-C., Wang T.-S., and Chen H.-W. (2013) Protection by chrysin, apigenin, and luteolin against oxidative stress is mediated by the Nrf2-dependent up-regulation of heme oxygenase 1 and glutamate cysteine ligase in rat primary hepatocytes. Arch. Toxicol. 87, 167–178 [DOI] [PubMed] [Google Scholar]
- 56. Kode A., Rajendrasozhan S., Caito S., Yang S.-R., Megson I. L., and Rahman I. (2008) Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L478–L488 [DOI] [PubMed] [Google Scholar]
- 57. Singh B., Ronghe A. M., Chatterjee A., Bhat N. K., and Bhat H. K. (2013) MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis 34, 1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang M., Yao Y., Eades G., Zhang Y., and Zhou Q. (2011) MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 129, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perez-Leal O., Barrero C. A., Clarkson A. B., Casero R. A. Jr., and Merali S. (2012) Polyamine-regulated translation of spermidine/spermine-N1-acetyltransferase. Mol. Cell. Biol. 32, 1453–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tong X., and Pelling J. C. (2009) Enhancement of p53 expression in keratinocytes by the bioflavonoid apigenin is associated with RNA-binding protein HuR. Mol. Carcinog. 48, 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vingtdeux V., Giliberto L., Zhao H., Chandakkar P., Wu Q., Simon J. E., Janle E. M., Lobo J., Ferruzzi M. G., Davies P., and Marambaud P. (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J. Biol. Chem. 285, 9100–9113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marcelo K. L., Means A. R., and York B. (2016) The Ca2+/calmodulin/CaMKK2 axis: Nature's metabolic CaMshaft. Trends Endocrinol. Metab. 27, 706–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin F., Marcelo K. L., Rajapakshe K., Coarfa C., Dean A., Wilganowski N., Robinson H., Sevick E., Bissig K. D., Goldie L. C., Means A. R., and York B. (2015) The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 62, 505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jennings P., Limonciel A., Felice L., and Leonard M. O. (2013) An overview of transcriptional regulation in response to toxicological insult. Arch. Toxicol. 87, 49–72 [DOI] [PubMed] [Google Scholar]
- 65. Niture S. K., Khatri R., and Jaiswal A. K. (2014) Regulation of Nrf2-an update. Free Radic. Biol. Med. 66, 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kensler T. W., Wakabayashi N., and Biswal S. (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116 [DOI] [PubMed] [Google Scholar]
- 67. Qin S., and Hou D. X. (2016) Multiple regulations of Keap1/Nrf2 system by dietary phytochemicals. Mol. Nutr. Food Res. 60, 1731–1755 [DOI] [PubMed] [Google Scholar]
- 68. Tebay L. E., Robertson H., Durant S. T., Vitale S. R., Penning T. M., Dinkova-Kostova A. T., and Hayes J. D. (2015) Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 88, 108–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Furukawa M., and Xiong Y. (2005) BTB protein keap1 targets antioxidant transcription factor nrf2 for ubiquitination by the cullin 3-Roc1 ligase. Mol. Cell. Biol. 25, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 71. Foretz M., Ancellin N., Andreelli F., Saintillan Y., Grondin P., Kahn A., Thorens B., Vaulont S., and Viollet B. (2005) Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54, 1331–1339 [DOI] [PubMed] [Google Scholar]
- 72. Malo N., Hanley J. A., Cerquozzi S., Pelletier J., and Nadon R. (2006) Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 24, 167–175 [DOI] [PubMed] [Google Scholar]
- 73. Guzy R. D., Hoyos B., Robin E., Chen H., Liu L., Mansfield K. D., Simon M. C., Hammerling U., and Schumacker P. T. (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408 [DOI] [PubMed] [Google Scholar]
- 74. Park E. K., Mak S. K., Kultz D., and Hammock B. D. (2008) Determination of cytotoxicity of nephrotoxins on murine and human kidney cell lines. J. Environ. Sci. Health 43, 71–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.