

Abstract
There is currently great interest in human serine racemase, the enzyme responsible for producing the NMDA co-agonist d-serine. Reported correlation of d-serine levels with disorders including Alzheimer's disease, ALS, and ischemic brain damage (elevated d-serine) and schizophrenia (reduced d-serine) has further piqued this interest. Reported here is a structure/activity relationship study of position Ser84, the putative re-face base. In the most extreme case of functional reprogramming, the S84D mutant displays a dramatic reversal of β-elimination substrate specificity in favor of l-serine over the normally preferred l-serine-O-sulfate (∼1200-fold change in kcat/Km ratios) and l (l-THA; ∼5000-fold change in kcat/Km ratios) alternative substrates. On the other hand, the S84T (which performs l-Ser racemization activity), S84A (good kcat but high Km for l-THA elimination), and S84N mutants (nearly WT efficiency for l-Ser elimination) displayed intermediate activity, all showing a preference for the anionic substrates, but generally attenuated compared with the native enzyme. Inhibition studies with l-erythro-β-hydroxyaspartate follow this trend, with both WT serine racemase and the S84N mutant being competitively inhibited, with Ki = 31 ± 1.5 μm and 1.5 ± 0.1 mm, respectively, and the S84D being inert to inhibition. Computational modeling pointed to a key role for residue Arg-135 in binding and properly positioning the l-THA and l-serine-O-sulfate substrates and the l-erythro-β-hydroxyaspartate inhibitor. Examination of available sequence data suggests that Arg-135 may have originated for l-THA-like β-elimination function in earlier evolutionary variants, and examination of available structural data suggests that a Ser84-H2O-Lys114 hydrogen-bonding network in human serine racemase lowers the pKa of the Ser84 re-face base.
Keywords: enzyme kinetics, enzyme mechanism, molecular modeling, pyridoxal phosphate, site-directed mutagenesis
Introduction
The discovery of d-serine in the brain and its importance in modulating NMDA receptor activity provided the first bona fide example of a d-amino acid in human biology. The quest to uncover the source of this d-serine led to the identification of mammalian serine racemase (1). Wolosker et al. (1) successfully cloned human serine racemase (hSR)3 at the turn of the millennium. The observation that all d-serine apparently originates in l-serine added another significant branch to the complex metabolic network associated with l-serine and an important new signaling function for the amino acid (Fig. 1).
Figure 1.

l-Serine as a central metabolite. l-Serine plays a central role in biology, from one-carbon metabolism to transsulfuration, phospholipid/phosphoprotein function, and d-serine biosynthesis.
To be sure, l-serine already was known to possess an array of physiological functions, including serving as both the source of one-carbon equivalents in N5,N10-methylenetetrahydrofolate (utilized for DNA synthesis; i.e. installation of the 5-methyl group in the uracil ring to give the thymine base) and of the neurotransmitter and NMDAR co-agonist, glycine, through the action of a single pyridoxal phosphate (PLP) enzyme, serine hydroxymethyltransferase. l-Serine also serves a central role in maintaining redox homeostasis, because all glutathione equivalents originate in the l-serine backbone, with the sulfur atom from dietary methionine being installed at the β-carbon through the sequential action of two additional PLP-dependent enzymes, cystathionine β-synthase (CBS) and cystathionine eliminase (also known as cystathionine γ-lyase). l-Serine also serves as an important constituent of the phospholipidome and is one of three constituent amino acids of proteins (along with l-threonine and l-tyrosine) that underpin the phosphoproteome, as controlled by the action of protein phosphoserine kinase phosphatases.
From the point of view of neuronal signaling, both d-serine and glycine serve as co-agonists of the NMDA receptor (Fig. 2), binding at the “glycine site” but with the observation that d-serine is a more potent agonist than glycine itself, showing efficacy at several orders of magnitude lower concentration in a seminal study by Ascher and co-workers (2) in a rat hypoglossal motoneuron system (2–4). Whereas it had previously been thought that d-serine is biosynthesized by SR in astroglial cells, more recent evidence indicates that l-serine produced in the astroglia from 3-phosphoglycerate is actively shuttled to the neurons where SR is present and converts the l-serine to d-serine (5, 6).
Figure 2.

hSR: physiological role and 3D structure. A, biosynthesis of the neuromodulators H2S and d-serine and their postulated roles in stimulation of the NMDA receptor (schematic); B, homology model for hSR based upon Protein Data Bank entries 3L6B (recombinant modified human; internal aldimine) and 2ZPU (S. pombe; substrate-modified internal aldimine). Blue, C-terminal PDZ domain (interacts with GRIP-1 and PICK-1); lavender, structural dication (Mg2+, Ca2+ or Mn2+; salmon, palmitoylation site (Thr227); red helical region, putative nitrosylation site (Cys113); green, ATP-binding site; red loop region, putative phosphorylation site (Thr71).
As is illustrated in Fig. 2A, d-serine generated in the presynaptic neuron serves as stimulatory co-agonist of the postsynaptic NMDA receptor, acting in concert with the primary agonist, l-glutamate. Of particular interest to our laboratory (7–13), the gaseous neurotransmitter, H2S, produced by another PLP enzyme (CBS) in the brain also elicits an NMDA excitatory response (14), potentially via an adenylate cyclase-cAMP-dependent protein kinase–mediated mechanism (15). Indeed, both SR and CBS have emerged as potential targets for ischemic stroke, because there is evidence that both d-serine (16, 17) and H2S (18) promote neuronal infarction following such a stroke event. Elevated d-serine levels have also been associated with Alzheimer's disease (19) and ALS (20), suggesting that SR may emerge as a potential target for neurodegenerative disease. On the other hand, low d-serine levels (i.e. SR hypofunction) have been correlated with schizophrenia (21–23). In contrast to these examples of SR dysregulation, in the healthy brain, basal d-serine signaling is essential for synaptic efficiency and long-term potentiation associated with learning and memory (24).
The hSR enzyme is known to be activated allosterically by ATP and requires a divalent cation (Mg2+, Mn2+) for activity (Fig. 2B) (25–27). The enzyme is reported to be post-translationally modified by phosphorylation (28), palmitoylation (29), and nitrosylation (30). SR levels can be modulated by ubiquitin tagging for proteasomal degradation (31). The C-terminal PDZ domain is important in protein–protein interactions, with PICK-1 (32, 33), GRIP-1 (34), and PSD-95 (35), for example. X-ray crystallographic structures of the Schizosaccharomyces pombe (36), maize (37), mouse (38), and rat and human (39) SR enzymes are available.
Mammalian SR has a type II β-eliminase fold reminiscent of the classical PLP-dependent enzyme, tryptophan synthase. Accordingly, it is perhaps not surprising that the enzyme catalyzes both the β-elimination of l-serine and its racemization to d-serine (Fig. 3). Mechanistically, a dual-base mechanism has been proposed, whereby Lys56 serves as the si-face base (40), α-deprotonating an appropriately oriented external aldimine of l-serine, giving rise to a common, cofactor-stabilized carbanionic intermediate (41). Subsequent re-face protonation by the putative re-face base, Ser84 (42), leads to the d-serine racemization product, whereas expulsion of the (presumably protonated) β-OH leaving group leads to pyruvate, the β-elimination product. l-Serine-O-sulfate (l-SOS) and l-threo-β-hydroxyaspartate (l-THA) are known to serve as very efficient alternative substrates for this latter β-elimination manifold (43). This overall mechanism is consistent with recent QM/MM calculations (44).
Figure 3.

Mechanism of serine racemase. Shown is the racemization manifold versus the β-elimination manifold. Note that both carbanionic and fully delocalized quinonoid intermediate pathways can be considered.
Results
We set out to examine the mechanism of hSR, with a particular focus on the influence of the putative re-face base upon reactivity. The assignment of Ser84 as the re-face base itself raises a key mechanistic question, namely how can this residue have an appropriately low pKa to perform this general acid/base function? Careful examination of available structural information suggests that Lys114 may serve to (de)protonate Ser84 through a hydrogen bond network involving an essential water molecule (Fig. 4A). This putative Ser84-Wat372-Lys114 hydrogen bond network appears to resemble the Ser-cis-Ser-Lys catalytic triad (Fig. 4B) (45–48) that is typically seen in the amidase signature enzyme family that includes peptide amidases (49–51) and fatty acid amide hydrolases (52–54) as well as some β-lactamases (55) and a recently described hydrazidase enzyme (56). To our knowledge, this model for serine acidification has not previously been proposed for a PLP enzyme.
Figure 4.
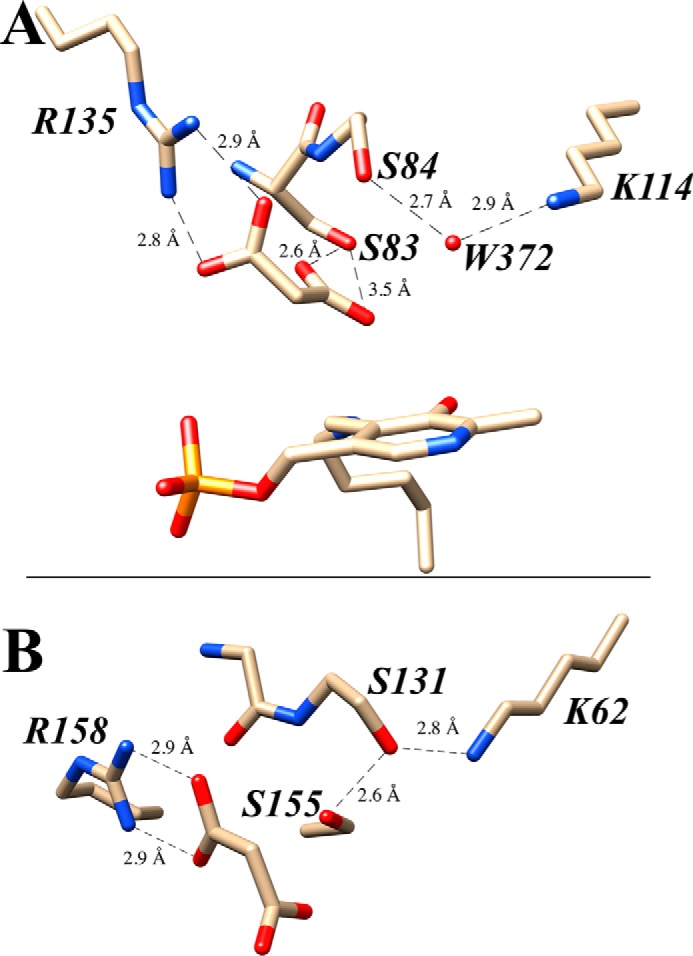
Acidity of Ser84: Ser-(OH2)-Lys versus Ser-(cis-Ser)-Lys triad. A, proposed hydrogen-bonding network involving the re-face base, Ser84, and the neighboring Wat372 molecule and Lys114 residue. B, comparison with the Ser155–cis-Ser131–Lys62 catalytic triad in the amidase signature family of enzymes. Crystal structures 3L6B (hSR; A) and 1OCL (malonamidase; B) are used in this image; both structures contain a bound malonate as inhibitor (A) and product (B), respectively.
To facilitate experimental studies, it was found that improved hSR solubility could be achieved by expressing the protein as an N-terminal maltose-binding protein (MBP) fusion construct (MBP–hSR). Literature reports of heterologous SR expression indicate that these efforts have been plagued with difficulty, leading to low yields of active enzyme, ranging from 1 to 2 mg/liter of culture, as summarized in Fig. 5 (40, 43, 57) and confirmed in our hands with both the N-terminal His- and GST-tagged constructs. However, the MBP–hSR construct reproducibly gave ∼15 mg of purified fusion protein. This is largely attributed to improved solubility. Pixel densitometric gel analysis demonstrates that whereas His–hSR showed only 7% soluble hSR protein, the MBP–hSR fusion is estimated to give 48% of the protein in the supernatant, a nearly 7-fold increase in solubility. The 15 mg of MBP–hSR translates to ∼6.4 mg of hSR versus the 1–2 mg obtained for His–hSR, a significant improvement. Removal of the tag via factor Xa digestion followed by ATP column purification resulted in a doubling of specific activity, as expected.
Figure 5.

Use of the MBP–hSR construct for expression and purification. A, purification gel comparison of the MBP–hSR and His6–hSR constructs by Coomassie Blue staining and Western blotting. B, removal of the MBP tag. C, comparison of kinetic properties of the MBP–hSR with hSR values reported in the literature (40, 43, 57). D, comparison of MBP–hSR specific activity with that of other reported SR constructs (1, 43, 57, 60, 75).
Native PAGE experiments are indicative of a dimeric structure for the hSR–MBP fusion protein, consistent with previous reports and crystal structures for the SR (39). Interestingly, gel filtration (Sephacryl S-200) shows an apparent molecular mass of 247 Da for the new construct, suggestive of an oligomeric composition of 3.1. This may be reflective of an equilibrium between a dimeric and tetrameric form of MBP–hSR under the conditions of the gel filtration experiment. This notion would be consistent with a recent observation by Mozzarelli and co-workers (58) that hSR is capable of forming an active tetramer in the presence of ATP and the divalent cation (Ca2+ or Mg2+) normally associated with the protein.
Active site mutants/kinetic evaluation
We next chose to examine the influence of re-face base on enzyme function. In previous work by Wolosker and co-workers (40), mutagenesis of residues 151–154 led to a reversal of functional preference with l-serine as the substrate. As noted (Fig. 6), normally mammalian serine racemase favors l-serine elimination activity over l-serine racemase activity, in a ratio of ∼4:1. Their most notable mutant, Q154D, displayed a dramatic reversal of these activities to a ratio of 1:3.
Figure 6.

Molecular modeling results: WT versus S84D. Relaxation of the hSR homology model from Protein Data Bank entries 3L6B and 2ZPU (Fig. 2) with GROMACS version 4, followed by molecular docking of the l-THA-PLP-external aldimine (Autodock version 4) leads to the active site structures shown, both schematically (A) and in 3D (B). Arg135 is projected to have an important role in l-THA substrate positioning in wild-type hSR (lavender). On the other hand, Arg135 is engaged in a new salt bridge with Asp84 (green) in the S84D mutant, making it unavailable for l-THA or l-SOS substrate positioning.
Focusing on the active site, previous reports had identified Ser84 as the re-face base in homologous enzymes belonging to S. pombe (42) and slime mold (Dictyostelium discoideum) (59). In both cases, the re-face Ser to Ala mutant was constructed and resulted in loss of function for racemization. Utilizing the MBP–hSR platform, we generated the corresponding mammalian SR S84A mutant, in addition to the S84D, S84N, and S84T variants. There had been one earlier report of the former construct, but the activity of this mutant was only studied with the natural substrate, l-serine (60). The rationale was to examine changes in hydrogen bond donor ability, charge, and sterics for putative re-face base by studying the behavior of these mutants across four assays: racemization and β-elimination reaction across a battery of three substrates.
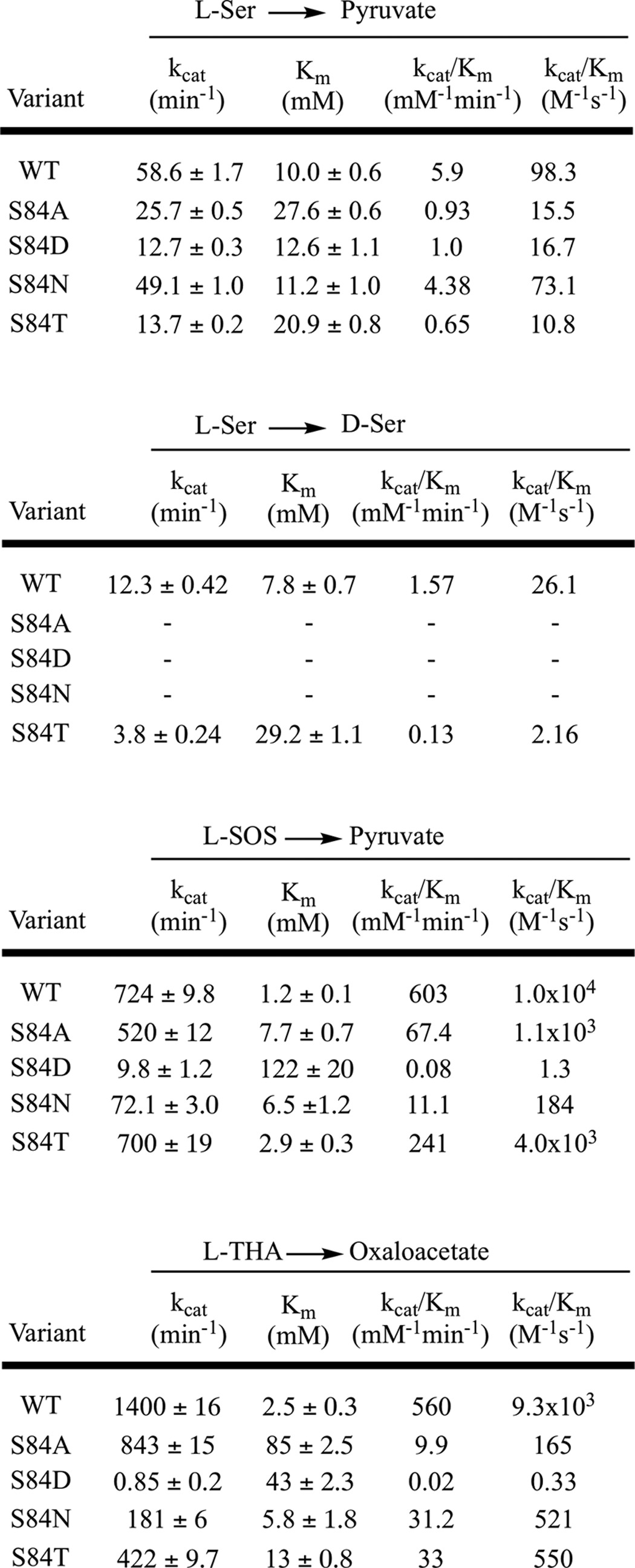
The results are summarized in Table 1. Compared with the wild-type hSR, the S84D mutant shows the most dramatic difference in substrate preference as measured by the catalytic efficiency (i.e. measured kcat/Km value). Whereas the charged substrates l-SOS and l-THA are highly favored in wild-type SR, S84D shows a dramatic reversal of this preference. In the most pronounced case, for l-THA compared with l-serine, this preference changes from 100:1 in the wild-type to 1:50 (see Table 1). This represents a 5000-fold change in substrate preference. A similar ∼1200-fold change is observed for l-SOS. It is important to note that this was not a “mutate-and-kill” effect, because the catalytic efficiency of l-serine only decreased ∼6-fold in the S84D mutant. That PLP enzymes can exhibit significant catalytic promiscuity even with subtle changes has recently been highlighted by Patrick and co-workers (61) in detailing the promiscuous alanine racemase activity seen with mutant cystathionine-β-lyase, both in Escherichia coli.
Table 1.
Kinetic inventory of hSR variants across the major enzymatic reaction manifolds
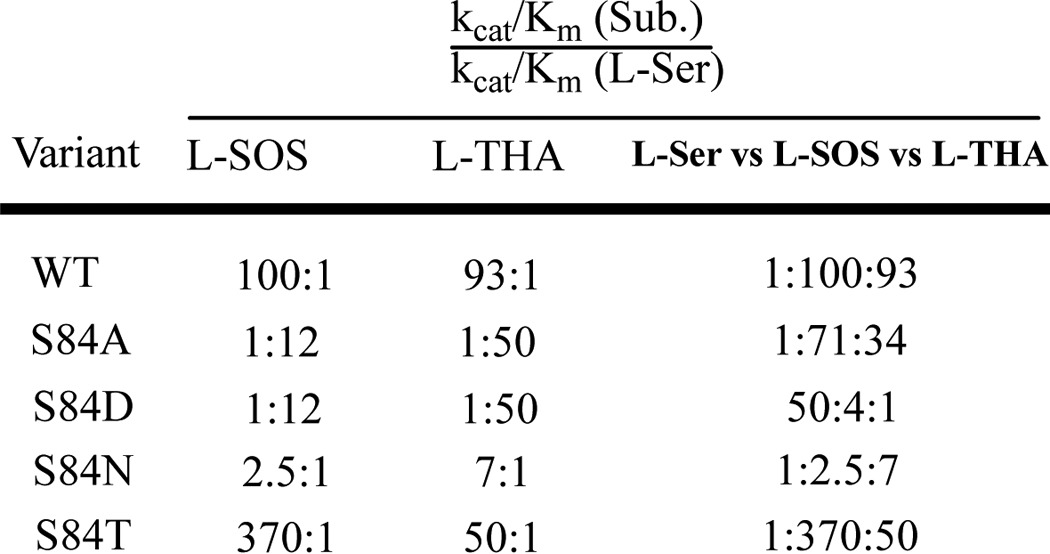
The S84N mutant shows intermediate behavior, exhibiting neither the strong preference for charged substrates seen in the wild type nor the inverted substrate preference seen in the S84D mutant. Specifically, the S84N mutant shows a modest 2.5:1 preference in l-SOS over l-serine and a 7:1 preference of l-THA over l-serine for β-elimination (Table 2). Although unable to catalyze l-serine racemization, this mutant is also the most fit mutant in catalyzing l-serine elimination, displaying ∼75% of the catalytic efficiency of the native enzyme.
Table 2.
Relative catalytic efficiencies of substrates as a function of hSR variant
The S84A mutant also loses l-Ser racemization completely and has attenuated l-Ser elimination activity (>6-fold drop in catalytic efficiency, almost equally due to kcat and Km effects), and although this mutant displays quite respectable kcat values for the charged substrates, it pays a significant penalty in Km for l-THA (34-fold).
Whereas the alterations in substrate profile seen with the S84D mutant were dramatic, the S84T mutant displayed a more nuanced change in substrate preference that is also useful in considering the hSR mechanism. Thus, S84T-hSR retains the native hSR preference for charged substrates, but that preference is now selective for l-SOS over l-THA. In other words, the β-elimination of l-THA is now favored only 35-fold versus the β-elimination of l-serine compared with the original 100-fold, whereas l-SOS is now eliminated some 350-fold more efficiently than l-serine. Expressed differently, but perhaps more succinctly, whereas the kcat for l-SOS elimination is unchanged for the S84T mutant, the corresponding kcat values for l-serine and l-THA elimination suffer a 4–5-fold penalty with this subtle mutation in re-face base structure. Thus, the S84T-hSR is able to discriminate between these two charged substrates, showing an order of magnitude higher catalytic efficiency for processing l-SOS versus l-THA, whereas no such preference is seen in wild-type enzyme.
Molecular dynamics/docking studies
These kinetic results inspired us to employ molecular dynamics simulation and substrate docking experiments with the goal of potentially shedding light on the origins of these observations on substrate preference. See “Experimental procedures” for details on how the homology model was constructed and how the molecular dockings were carried out and analyzed. All three elimination substrates were docked to both wild-type hSR and the S84D mutant. Displayed in Fig. 6 is the structure of l-THA-derived external aldimine docked into the wild-type hSR active site. The β-carboxylate of the substrate is engaged in a salt bridge interaction with Arg135. This is somewhat reminiscent of the published crystal structures of SR with bound malonate inhibitor, wherein one carboxylate of the malonate appears to be similarly engaged (39). However, unlike l-THA, malonate is, of course, not covalently engaged with the PLP cofactor at all.
When l-THA is docked into the active site of the S84D-hSR mutant, the results are much different. What we observe is Arg135 now interacting with the newly mutated S84D. The new re-face base, Asp84, has “hijacked” Arg135, forming an intramolecular salt bridge, which effectively renders Arg135 unavailable to assist with the binding and proper positioning of the charged substrates, l-SOS and l-THA. As can be seen in Fig. 6, this leads to a significant distortion of the substrate in the active site. This is illustrated with an overlaid image of the two structures (WT in lavender and S84D mutant in green).
The wild-type hSR enzyme is represented by purple residues, whereas the S84D mutant is shown in green. The external aldimine formed between l-THA and PLP is shown in silver. The interaction between the substrate and wild-type Arg135 is again displayed. However, when Ser84 is converted to Asp84 (green), it becomes quite apparent how far Arg135 has now migrated in the active site. This key active site residue no longer engages the substrate; rather, it interacts solely with Asp84.
How might this simple mutation lead to the kinetic differences that we observe for the three hSR β-elimination substrates? During the course of the docking studies, we noticed a large change in substrate orientation within the active site. These orientations were examined closely with an eye toward how PLP enzymes operate normally, according to the Dunathan hypothesis (62). Looking more closely at the docking results for the external aldimine complexes of all three substrates, a large difference was observed in the behavior of l-serine compared with that of its charged counterparts.
In Fig. 7 (left column), a typical example of the preferred docking orientation of each of the three substrates in the wild-type SR active site is shown. For all three cases, the substrate is aligned with a proper Dunathan orientation of the α-C–H bond to be broken. In other words, for all three cases, the α-C–H is nearly parallel with the π-system of the cofactor imine. In this case, Arg135 is not only important for substrate binding/recognition; it appears to also be vital to substrate positioning for catalysis.
Figure 7.

Molecular modeling to examine stereoelectronics in external aldimines for WT- and S84D-hSR. Molecular docking (Autodock version 4) results imply that whereas the l-Ser, l-SOS, and l-THA β-elimination substrates show proper Dunathan alignment in their respective external aldimines for WT-hSR, this alignment is significantly altered in the S84D mutant.
For the mutant S84D, we no longer see proper orientation. Whereas l-serine remains relatively unchanged in position, l-SOS and l-THA exhibit a substantial rotation about the key C4′-N-Cα-H dihedral angle. This non-Dunathan orientation is consistent with the dramatic decrease in β-elimination activity for these substrates with the S84D mutant, in good agreement with the experimental kinetic results. This apparent importance of Arg135 raises interesting questions about the evolutionary history of serine racemase, as will be discussed below.
In an effort to understand the intermediate substrate preferences of the S84N mutant, molecular docking was also undertaken here. In nearly all docked structures, the new asparagine 84 residue was seen to interact with aspartate 238, probably through hydrogen bonding. Perhaps because residue 84 is no longer able to contribute to the α-carboxylate binding site, one sees a major cluster of conformers in which Arg135 is now engaged with the α-carboxylate.
Indeed with the charged substrates, l-SOS and l-THA, whereas in the WT enzyme these substrates appear to be “locked” into position for catalysis through electrostatic pairing with Arg135 (Figs. 6 and 7), in the S84N mutant, molecular docking identifies two nearly equally populated clusters of conformers, the aforementioned Arg135–α-carboxylate cluster (Fig. 8, left) and a second cluster of conformers in which the Arg135–side chain interaction is retained (Fig. 8, right). Inspection of the C4′-N-Cα-H dihedral angle for the individual members of each of these major clusters gives a dihedral angle range of 140–190° for the Arg135–α-carboxylate cluster and of 80–105° for the Arg135–side chain cluster. The latter dihedral angle window appears to be in the stereoelectronically allowed range for Dunathan-compliant α-deprotonation. Representative examples of members of each cluster for both the bound l-SOS-external aldimine (top half) and the bound l-THA-external aldimine are depicted in Fig. 8. Overall, this Arg135-toggle model for substrate binding for the S84N mutant is consistent with the modest preference for these charged substrates displayed.
Figure 8.

Molecular modeling suggests two types of bound conformations for l-SOS and l-THA in S84N-hSR. Molecular docking (Autodock version 4) results show two clusters of bound external aldimine conformers for the l-SOS and l-THA substrates with the S84N mutant. The conformers on the left exhibit an Arg135–α-carboxylate salt bridge and are non-Dunathan-aligned, whereas in conformers on the right, Arg135 is engaged with the charged side chain, leading to proper alignment for α-deprotonation.
Overviewing this set of hSR mutants, then, in light of these docking results, one sees that WT enzyme appears to permit a sort of three-point binding interaction with Ser83, Ser84 (α-carboxylate), and Arg135 (charged side chains). Mutation to S84N appears to remove residue Asn84 from the binding pocket as it becomes engaged with Asp238. This appears to drive an Arg135-toggle in binding both anionic groups in the charged substrates. Mutation to S84D removes both residue Asp84 and Arg135 because they are predicted to combine to form a salt bridge. Whereas these in silico models are consistent with the relaxed preference for l-SOS and l-THA seen in the S84N mutant and with the dramatic reversal of substrate preference seen in the S84D mutant, they remain to be tested by structural biology studies in the future.
Probing the active site with inhibitors
That said, we next set out to undertake complementary experiments to provide additional data with which to evaluate this interesting hSR binding model. Namely, given the importance ascribed to Arg135, particularly in binding and positioning the charged substrates, l-SOS and l-THA, in this model, it seemed prudent to challenge this array of hSR active sites with a battery of inhibitors. A set of four inhibitor candidates was chosen: (i) malonate, (ii) l-aspartate β-hydroxamate (l-ABH), (iii) l-erythro-β-hydroxyaspartate (l-EHA), and (iv) aminooxyacetate (AOAA). Three of these inhibitors bear anionic side chains that might be expected to engage Arg135, particularly in light of the substrate binding model being put forward here. The β-hydroxamate in l-ABH might also be expected to interact with Arg135, although probably not as strongly. Only for malonate is crystal structure information available (39), and indeed such an interaction with Arg135 is seen (Fig. 4).
All experiments were conducted in competition with l-serine, the native substrate. The first two hSR inhibitors showed dramatically different behavior with WT-hSR versus the S84N and S84D mutants (Fig. 9 and Table 3). Malonate displayed competitive inhibition with Ki = 65 ± 3.2 μm (reported Ki = 27–71 μm (38, 43, 57)). l-ABH also inhibited WT-hSR competitively, with Ki = 155 ± 11.2 μm (reported Ki of 93 μm (63)) but showed no inhibition of either the S84N or the S84D mutant. As a positive control, the oxime-forming global PLP-dependent enzyme inactivator, AOAA (1), was tested and displayed potent inactivation of WT-hSR as well as the S84N and S84D mutants (Table 3).
Figure 9.

Competitive inhibition of WT-hSR with malonate and l-ABH. A, Lineweaver–Burk plot for WT-hSR inhibition with malonate; B, secondary plot for these data. C, Lineweaver–Burk plot for WT-hSR inhibition with l-ABH; D, secondary plot.
Table 3.
Inhibitor profile across the hSR mutant array
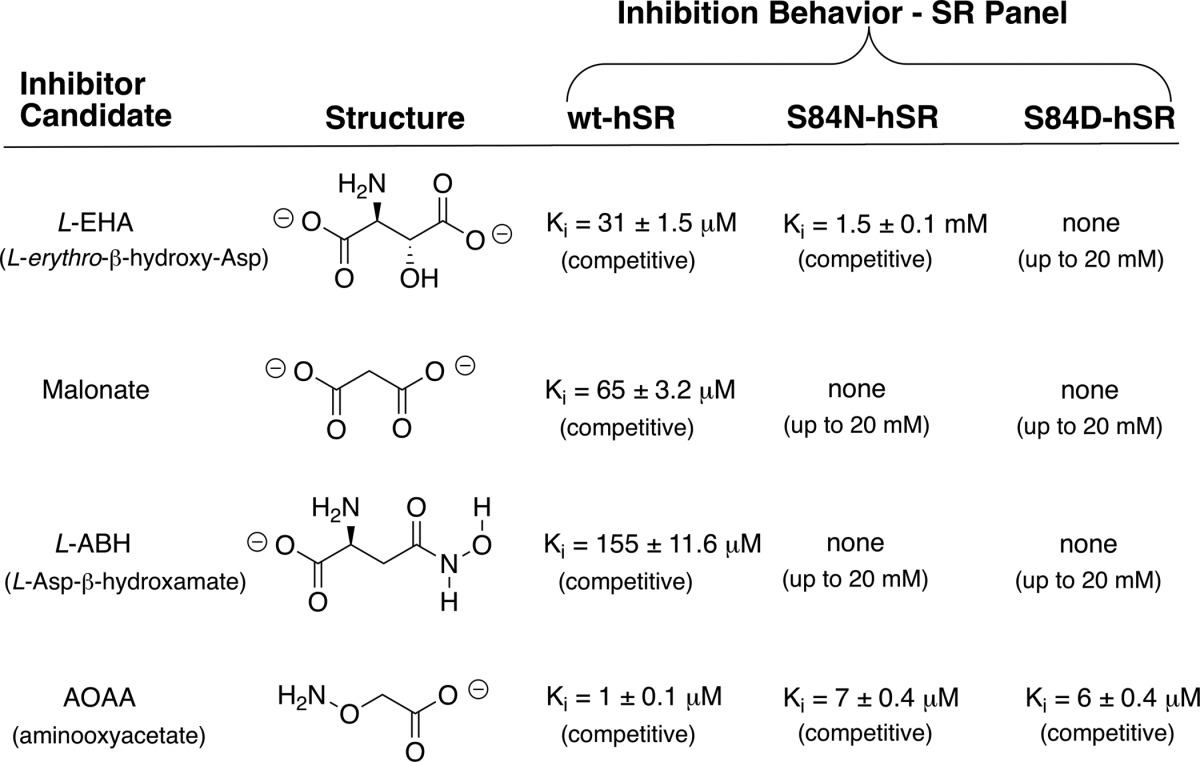
l-EHA exhibited competitive inhibition kinetics with Ki = 31 ± 1.5 μm for WT-hSR in our hands (reported Ki of 11–43 μm (43, 57)). Upon mutation of the active-site Ser84 to Asn, however, a pronounced 50-fold decrease in inhibition potency was observed as Ki increased to 1.5 mm. Even more extreme, the S84D mutant showed no inhibition with l-EHA at concentrations up to 20 mm. (Fig. 10 and Table 3).
Figure 10.

l-EHA inhibition steady-state kinetic results for WT- and S84N-hSR. A, Lineweaver–Burk plot for WT-hSR inhibition with l-EHA; B, secondary plot for these data. C, Lineweaver–Burk plot for S84N-hSR inhibition with l-EHA; D, secondary plot.
Discussion
The effectiveness of all four inhibitors with native hSR and the inability of all but the universal PLP inactivator, AOAA, to inhibit the S84D mutant is a striking contrast and is consistent with the Arg135-charged side chain binding model that emerged from the earlier substrate scan/molecular modeling. Much as in the β-elimination substrate studies, the S84N mutant again displayed behavior that is intermediate between WT-hSR and S84D-hSR in the inhibition studies. Namely, in contrast to the S84D mutant, the S84N mutant is inhibited by l-EHA (Ki = 1.5 ± 0.1 mm) but considerably less well than WT-hSR (Ki = 31 ± 1.5 μm). These results are in line with the details of the molecular modeling that emerged earlier whereby it was predicted that Arg135 would be fully available for side chain carboxylate binding in WT-hSR (Fig. 6), partially available in the S84N mutant (Fig. 8) and unavailable in the S84D mutant (Fig. 6). Indeed, molecular modeling of the putative l-EHA-external aldimine with both WT-hSR and S84N hSR indicates that these active sites are also capable of engaging the β-carboxylate of l-EHA in a salt bridge with Arg135 (Fig. 11), consistent with the ability of this compound to inhibit both enzymes.
Figure 11.

l-EHA inhibition molecular modeling. Shown are docking results (Autodock version 4) for the inhibitor l-erythro-β-hydroxyasparate (A) in the wild-type hSR active site and in the S84N-hSR active site (B).
This study also raises interesting questions from the observation of the S84T mutant, particularly its ability to retain all hSR functions and yet to discriminate between the l-SOS (preferred) and l-THA substrates. Recent investigations into the mechanisms of the related PLP-dependent β-eliminases Drosophila CBS (64) and tryptophan synthase (65) may provide guidance here. Both of these enzymes also catalyze the β-elimination of PLP-aldimine–bound l-serine along the normal reaction coordinate, for ll-cystathionine and l-tryptophan biosynthesis, respectively. In the Drosophila CBS study (64), the authors claim to observe a carbanionic intermediate that is generated upon l-serine deprotonation. It is argued that this species is an incompletely delocalized, PLP-stabilized α-carbanion, as opposed to a fully delocalized quinonoid intermediate. The authors argue that both the si-face lysine ammonium ion and a re-face serine residue are of central importance in stabilizing this key mechanistic intermediate.
Mueller and Dunn (65) have recently described a similar carbanionic intermediate for the archetypical β-replacement enzyme, tryptophan synthase, utilizing a new biophysical method that combines solid-state NMR, X-ray crystallography, and computational chemistry. Here too, the active site lysine ammonium ion is thought to stabilize the negative charge in this intermediate. It is, of course, possible that a similar carbanionic intermediate forms along the reaction coordinate for hSR. It may be that replacement of a β-H with a β-methyl group (S84T) results in a subtle repositioning of this residue, rendering it less efficient at stabilizing the developing negative charge in the enzyme-bound substrate aldimine upon α-deprotonation.
Indeed, it might well be the case that a stepwise mechanism involving initial rate-limiting substrate deprotonation takes place for the l-serine and l-THA hSR substrates, but not for l-SOS. The latter substrate, by virtue of having a good leaving group in sulfate, could presumably undergo a concerted β-elimination without the need to stabilize a discreet α-carbanionic PLP-substrate intermediate. This explanation is consistent with the observation that the hSR S84T mutant more effectively catalyzes the β-elimination of l-SOS as compared with that of l-THA, in contrast to WT-hSR. However, this is still quite speculative at this juncture. The nature of the PLP-centered intermediate in hSR catalysis has remained elusive heretofore, so future experiments will be needed to evaluate this notion. It may be possible to shed light on this hypothesis, at least indirectly, through the sort of Mueller/Dunn solid-state NMR/X-ray studies described above or through successful co-crystallization of hSR with known inhibitor l-EHA.
Evolutionary considerations
To examine the potential evolutionary implications of the favorable l-THA elimination kinetics observed here, we decided to construct a phylogenetic tree of various type II β-eliminases acting on l-serine (Fig. 12) (66). The blue dots represent enzymes known to racemize serine. What we observe is that only recently do we see serine racemase activity. Furthermore, an apparent ancestor is shared with enzymes that are annotated as l-THA dehydratases from Saccharomyces cerevisiae (67) and Pseudomonas sp. T62 (68). Very recently, such activity has been observed in a Caenorhabditis elegans enzyme as well (69). Looking more closely at the sequences of a range of proteins in this broad β-eliminase family, we observe the following trends. First, Ser84 is conserved in serine racemases and l-THA dehydratases. Perhaps most striking, Arg135 is also conserved among these examples while being absent in all other type II dehydratases (Fig. 13). These results are consistent with the substrate preferences (i.e. l-SOS and l-THA as preferred β-elimination substrates over l-Ser (Tables 1 and 2)) observed for the wild-type serine racemase. This also suggests that perhaps serine racemase is still early on in its evolution of function (see Fig. 12).
Figure 12.

Phylogenetic tree of various fold type II eliminases. Enzymes annotated as SR are marked with blue dots; enzymes annotated as having CBS activity are given lavender triangles.
Figure 13.

Alignment of fold type II enzymes. Arg135 is conserved among enzymes showing l-THA eliminase activity consistent with the functional role of this residue posited herein. h, human; m, mouse; r, rat; at, Arabidopsis thaliana; sp, S. pombe; hv, Hordeum vulgare; pa, Pseudomonas aeruginosa; pi, Pyrobaculum islandicum; sc, S. cerivisiae; p, Pseudomonas T62; ec, E. coli; s, Salmonella.
Conclusions
Since the discovery of d-serine as a potent co-agonist of the NMDA receptor, there has been growing interest in the enzyme responsible for its biosynthesis, serine racemase. Its implication in various disease states has further raised interest in developing selective inhibitors (ischemic stroke and potentially Alzheimer's disease and ALS) or stimulators (schizophrenia) of this enzyme. A new expression construct for hSR is reported here, namely the MBP–hSR fusion protein, that yields higher titers of soluble enzyme and serves as an excellent platform to study active-site mutants. Characterization of these mutants has led to a new understanding of important residues in the mechanism of serine racemase. Specifically, these complementary “enzyme/substrate mutation” studies focusing on position 84 in the enzyme and the three principal hSR β-elimination substrates have raised mechanistic hypotheses for hSR function. Studies with the hSR S84D and S84N mutants suggest an important mechanistic role for Arg135 in substrate positioning for charged β-elimination substrates that is consistent with the Dunathan hypothesis. Indeed, these observations, along with sequence alignment studies point to Arg135 as a key residue that probably was selected for in this β-eliminase family to optimize the β-elimination of l-THA (conserved in all annotated l-THA dehydratases).
Phylogenetic analysis supports the notion that Arg135 is historically important for l-THA eliminase activity. But where would organisms experience evolutionary selection pressure to evolve l-THA eliminase activity? Recently, Katane et al. (69) have speculated that C. elegans may have evolved its T01H8.2 protein, an apparent l-THA dehydratase, as an important defense mechanism. Namely, microorganisms such as Athrinium phaeospermum and Streptomyces are known to produce l-THA, and this amino acid is known to inhibit l-glutamate transport. The thinking is that C. elegans might well feed on such microorganisms and take advantage of this evolved l-THA dehydratase activity to detoxify ingested l-THA, and whereas this may be the evolutionary origin of residue Arg135 in hSR, it may well be possible to leverage this vestigial ligand-binding ability in future hSR inhibitor design studies.
Experimental procedures
Recombinant His–hSR
Human serine racemase cDNA was purchased from Origene and amplified using the following primers (IDT): 5′-CCT TCT TGC TAG CTG TGC TCA GTA TTG CAT C-3′ (forward) and 5′-CAC GCG CTC GAG AAT TCC CAC CAT TTC-3′ (reverse) for the NheI and XhoI restriction sites, respectively. Gene Runner software was used to design the primers, and the 10198-0118 PCR Reagent System (Invitrogen) was utilized. The cDNA was inserted into the pGEM-T cloning vector (Promega) and used to transform DH5α-derived E. coli made competent by the calcium chloride method. Plasmids from overnight cultures were extracted using a plasmid miniprep spin kit (Qiagen) and verified by 1% agarose gel as well as sequencing with T7 and T7term primers. After double digestion, the fragment was inserted into the pET-28c plasmid. BL21(DE3)-pLysS E. coli was used for expression.
Starter cultures of 3-ml volume, grown overnight, were used to inoculate 1 liter of Luria–Bertani broth (Difco), supplemented with an additional 5 g/liter yeast extract. Cells were grown at 37 °C and 250 rpm. When the cells reached an A600 of 0.8, the temperature was reduced to 25 °C, and the cells were induced with 0.1–0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 18 h at 300 rpm. Cells were pelleted by centrifugation for 15 min at 10,000 × g and stored at −80 °C.
Purification of His–hSR
Cells were resuspended in assay Buffer A (200 mm TEA, 150 mm KCl, 10 mm DTT, 5 mm MgCl2, 2.5 mm ATP, and 50 μm PLP, pH 8.0) at a volume of 1 ml/g of wet cell mass. Cells were disrupted by sonication on ice for 5 cycles (1 min on/1 min off) and centrifuged for 15 min at 15,000 × g. The supernatant contained modest levels of hSR and no visible SDS-PAGE band, whereas the pellet, when solubilized in the same volume of Buffer B (10 mm Tris, 100 mm sodium phosphate, 8 m urea, pH 7) as the crude supernatant, exhibited a very intense band at ∼37 kDa. To verify that this band was insoluble His-tagged hSR protein, the solubilized pellet fraction was applied to a nickel-nitrilotriacetic acid column. The protein did bind and was successfully eluted, with decreasing pH, yielding a single band on SDS-PAGE that matched the expected molecular weight for hSR.
Recombinant GST–hSR
Human serine racemase cDNA was purchased from Origene (catalog no. TC11289) and amplified using the primers (IDT) 5′-CGT TGC GGA TCC ATG TGT GCT CAG TAT TGC-3′ (forward) and 5′-CAC GCG CTC GAG AAT TCC CAC CAT TTC-3′ (reverse) for restriction sites BamHI and XhoI, respectively. The cDNA was double-digested and inserted into the pGEX-4T1 expression vector (GE Healthcare) and used to transform DH5α-derived E. coli made competent by the calcium chloride method. Plasmids from overnight cultures were extracted using a plasmid miniprep spin kit (Qiagen) and used to transform BL21(DE3)pLysS E. coli for expression.
Purification of GST–hSR
Cells were resuspended in assay Buffer A at a volume of 1 ml/g of wet cell mass. Cells were disrupted by sonication on ice for five cycles (1 min on/1 min off) and centrifuged for 15 min at 15,000 × g. The supernatant displayed more units of activity per liter of culture compared with the His–hSR construct but did not yield a visible band by SDS-PAGE. When the pellet was solubilized in Buffer B, there did exist a strong band at the expected molecular weight of the GST–hSR construct. SDS-PAGE indicated that most of the expressed protein was relegated to the insoluble pellet, so further purification was not attempted.
Recombinant MBP–hSR
The protein was designed to bear the N-terminal MBP tag present in the pMAL-c2X (New England Biolabs) vector. The following primers were employed in this system (IDT): 5′-CGT TGC GGA TCC ATG TGT GCT CAG TAT TGC-3′ (forward) and 5′-CAC CTA GTC GAC AAT TCC CAC CAT TTC C-3′ (reverse), for restriction sites BamHI and SalI, respectively. The respective hSR PCR products were ligated into the respective vectors and used to transform our DH5α-competent cells. The amplified plasmids were then analyzed and used to transform BL21(DE3)pLysS E. coli strain for expression.
Purification of hSR
Cells were resuspended in assay Buffer A at a volume of 1 ml/g of wet cell mass. Cells were disrupted by sonication and collected by centrifugation (15,000 × g). The supernatant was mixed with prebalanced amylose resin (New England Biolabs) and shaken at 4 °C for 1 h. The slurry was centrifuged 15 min at 5000 × g and decanted. The resin was applied to a column of the same resin, washed with 3 column volumes of Buffer C (50 mm Tris, 150 mm KCl, 10 mm β-mercaptoethanol, 15 μm PLP, pH 8.0), and eluted with a linear gradient (0–10 mm) of maltose. MBP–hSR was observed to elute at 3–5 mm maltose. Fractions were pooled by activity and UV280 absorbance. Pooled fractions were then concentrated (Amicon cell, 30-kDa membrane) to a final concentration of 20 mg/ml further clarified by centrifugation (15 min, 15,000 × g, 4 °C). Protein was flash-frozen in 50% (v/v) and stored at −80 °C.
To obtain tag-free hSR, the MBP tag of pMAL-c2X was cleaved by factor Xa (New England Biolabs), leaving a 6-mer (ISEFGS) N-terminal overhang immediately preceding the initial methionine of hSR. Digestion took place in Buffer D (20 mm Tris, 50 mm NaCl, 0.2 mm β-mercaptoethanol, and 10 μm PLP). After overnight digestion, the solution was applied to a 3-ml column of C-8–linked ATP-agarose (Sigma), washed with 3 column volumes of Buffer D, and eluted with 2.5 mm ATP in Buffer D.
β-Elimination activity assay
β-Elimination of l-SOS to form pyruvate was conducted under the following assay conditions: 200 mm TEA, 150 mm KCl, 5 mm MgCl2, 2.5 mm ATP, 50 μm PLP, 0.15 units of lactate dehydrogenase, 0.24 mm NADH, and 10 mm l-serine-O-sulfate. Activity was monitored by observing the decrease in absorbance at 340 nm.
l-Serine-O-sulfate β-elimination
A Shimadzu UV-2101PC spectrophotometer equipped with a 6-cell changer was used to monitor the decrease in NADH concentration as pyruvate produced from l-serine-O-sulfate elimination was reduced by lactate dehydrogenase. Assay conditions were as follows: 37 °C, pH 8, 200 mm TEA, 150 mm KCl, 5 mm MgCl2, 2.5 mm ATP, 50 μm PLP, 0.24 mm NADH, and 0.15 units of lactate dehydrogenase. Each point represents the average of three experiments. The kinetic parameters for MBP–hSR (79.5 kDa) were estimated by least squares hyperbolic fitting to the Michaelis–Menten equation.
l-Serine elimination
l-Serine elimination activity was evaluated by the same assay described for l-SOS elimination, because both reactions give pyruvate as the hSR product.
l-threo-β-Hydroxyaspartate β-elimination
l-THA elimination was monitored by the formation of oxaloacetate. Oxaloacetate formation was measured by reduction with malate dehydrogenase and NADH under the same conditions described for l-SOS.
l-Serine racemization
To obtain racemization data, d-amino acid oxidase from porcine kidney (Sigma) was used to oxidize d-serine to 3-hydroxypyruvate and H2O2. The resulting H2O2 was reduced in the presence of horseradish peroxidase and Amplex Red to generate resorufin that can be monitored continuously at 570 nm. Racemization was also monitored via a time point assay by derivatizing the products with Marfey's reagent (1-fluoro-2,4-dinitrophenyl-5-l-alanine) and resolved using reversed-phase HPLC with glycine as an internal standard.
Inhibition studies
Wild-type hSR and the S84N and S84D mutants were probed for inhibition by malonate, l-EHA, l-ABH, and AOAA. Utilizing the l-serine elimination assay at various l-serine concentrations (5, 10, 15, and 20 mm) and a battery of inhibitor concentrations, Ki values were determined following standard steady-state kinetic analysis. Wild-type hSR was examined with all inhibitors (except AOAA) at inhibitor concentrations of 65, 125, 312, 625, and 1250 μm. For the S84N mutant (aside from AOAA), inhibition was seen only with l-erythro-β-hydroxyaspartate upon incubation at 2.5, 5, 7.5, and 10 mm concentrations. The S84D mutant showed no inhibition up to 20 mm concentration with malonate, l-EHA, and l-ABH. The WT-hSR and S84N and S84D mutants were all effectively inhibited by AOAA, which was tested at 1, 2, 3, 4, 5, and 10 μm. Activity was measured by observing the decrease in absorbance at 340 nm associated with the consumption of NADH in the coupled lactate dehydrogenase reaction converting pyruvate to l-lactate.
Oligomeric state determination
Native PAGE and gel filtration analysis were used to determine the oligomeric state of the MBP–hSR construct. For native gels, molecular weight markers from GE Healthcare (High MW) were used. Electrophoresis was run using 4% stacking, 9% resolving discontinuous polyacrylamide minigels at constant current (40 mA) under flow-cooling in a Hoeffer Mighty Small II apparatus. A calibration curve (Rf versus log Mr) was constructed for the standards. The primary MBP–hSR band displayed an Rf value of 0.397, corresponding to an apparent molecular mass of 185 kDa, which corresponds to a dimer (2.3 monomeric units) based on the calculated monomeric molecular mass of 79.6 kDa. A similar analysis of the His6–hSR construct by native PAGE showed an apparent molecular mass of 81.5 kDa, again corresponding to a dimeric structure (2.1 monomeric units). Gel filtration analysis employing a GE Healthcare S-200 High-Prep Sephacryl HS column with BioLogic DuoFlow software was utilized. A calibration curve was constructed using the GE Healthcare high molecular weight gel filtration calibration kit and revealed an apparent molecular mass of 247 Da for the new construct, suggestive of an oligomeric composition of 3.1 (possible dimer–tetramer equilibration (58)).
Homology modeling
To construct a hSR homology model, the 340-amino acid human serine racemase protein sequence (NCBI accession number NP_068766) was BLASTed against the NCBI structural database (70). A multiple-sequence alignment was performed on hSR, 3L6B, 1WTC, and 2ZPU, using the ClustalW algorithm (71). This alignment forms the basis for construction the hSR model by MODELLER (72).
ATP, PLP, Ca2+, and Mg2+ were copied into the homology model from the template structures. PLP is bound to all template structures in a similar orientation. Of these, PLP from 3L6B was chosen for the hSR model. 1WTC contains a bound ATP/Mg2+ analogue that was copied into the hSR model and energy-minimized so as to represent bound ATP.
Molecular dynamics and docking experiments
All MD simulations were performed using the GROMACS software package (73). Before each run, the structure was solvated in a water box using the spc216 water model. Box dimensions were set so that the hSR model was no closer than 1 nm to the edge of the box. Simulations were performed using the GROMOS96 force field, periodic boundary conditions, standard temperature-coupling schemes, and the particle-mesh Ewald method for determining long-range electrostatics. Each MD run was preceded by a 1000-step steepest descent energy minimization and a 20-ps position-restrained MD simulation. Each full MD simulation was performed for 2 ns at 300 K, using 2-fs time steps.
The first simulation contained Ca2+, ATP/Mg2+, and non-covalently bound PLP. This setup was chosen to allow the active-site lysine (Lys56) to reorient itself to a position that would not obstruct external aldimine docking. Following the initial 2-ns MD simulation, the hSR structure was energy-minimized and prepared for docking. Autodock version 4 was used to dock the external aldimine of l-serine, l-serine-O-sulfate, l-threo-β-hydroxyaspartate, and l-erythro-β-hydroxyaspartate into the hSR active site (74).
Author contributions
D. B. B., D. L. N., G. A. A., M. L. B., and D. L. G. designed the experiments. D. L. N., G. A. A., M. L. B., and D. L. G. performed the experiments. D. B. B., D. L. N., G. A. A., M. L. B., and D. L. G. reviewed and analyzed the results, contributed to writing the manuscript, and approved the final version.
Acknowledgment
Facilities in this work were supported by a Grant-in-Aid from the National Institutes of Health Grant RR016544.
This work was supported by American Heart Association Grant-in-Aid 16GRNT313400012. This work was also supported by the IR/D (Individual Research and Development) program associated with the appointment of D. B. B. at the National Science Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- hSR
- human serine racemase
- SR
- serine racemase
- TEA
- triethanolamine
- Wat
- water molecule
- PLP
- pyridoxal phosphate
- CBS
- cystathionine β-synthase
- l-SOS
- l-serine-O-sulfate
- l-THA
- l-threo-β-hydroxyaspartate
- MBP
- maltose-binding protein
- l-ABH
- l-aspartate β-hydroxamate
- l-EHA
- l-erythro-β-hydroxyaspartate
- AOAA
- aminooxyacetate
- MD
- molecular dynamics.
References
- 1. Wolosker H., Sheth K. N., Takahashi M., Mothet J. P., Brady R. O. Jr., Ferris C. D., and Snyder S. H. (1999) Purification of serine racemase: Biosynthesis of the neuromodulator d-serine. Proc. Natl. Acad. Sci. U.S.A. 96, 721–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berger A. J., Dieudonné S., and Ascher P. (1998) Glycine uptake governs glycine site occupancy at NMDA receptors of excitatory synapses. J. Neurophysiol. 80, 3336–3340 [DOI] [PubMed] [Google Scholar]
- 3. Mothet J.-P., Parent A. T., Wolosker H., Brady R. O. Jr., Linden D. J., Ferris C. D., Rogawski M. A., and Snyder S. H. (2000) d-Serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4926–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wolosker H. (2007) NMDA receptor regulation by d-serine: New findings and perspectives. Mol. Neurobiol. 36, 152–164 [DOI] [PubMed] [Google Scholar]
- 5. Ishiwata S., Umino A., Balu D. T., Coyle J. T., and Nishikawa T. (2015) Neuronal serine racemase regulates extracellular d-serine levels in the adult mouse hippocampus. J. Neural Transm. 122, 1099–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wolosker H., Balu D. T., and Coyle J. T. (2016) The rise and fall of the d-serine-mediated gliotransmission hypothesis. Trends Neurosci. 39, 712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berkowitz D. B., Karukurichi K. R., de la Salud-Bea R., Nelson D. L., and McCune C. D. (2008) Use of fluorinated functionality in enzyme inhibitor development: mechanistic and analytical advantages. J. Fluor. Chem. 129, 731–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karukurichi K. R., de la Salud-Bea R., Jahng W. J., and Berkowitz D. B. (2007) Examination of the new α-(2′Z-fluoro)vinyl trigger with lysine decarboxylase: the absolute stereochemistry dictates the reaction course. J. Am. Chem. Soc. 129, 258–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berkowitz D. B., Wu B., and Li H. (2006) A formal [3,3]-sigmatropic rearrangement route to quaternary α-vinyl amino acids: use of allylic N-PMP trifluoroacetimidates. Org. Lett. 8, 971–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berkowitz D. B., de la Salud-Bea R., and Jahng W.-J. (2004) Synthesis of quaternary amino acids bearing a (2′Z)-fluorovinyl α-branch: potential PLP enzyme inactivators. Org. Lett. 6, 1821–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berkowitz D. B., McFadden J. M., Chisowa E., and Semerad C. L. (2000) Organoselenium-based entry into versatile, α-(2-tributylstannyl)vinyl amino acids in scalemic form: a new route to vinyl stannanes. J. Am. Chem. Soc. 122, 11031–11032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berkowitz D. B., and Smith M. K. (1996) A convenient synthesis of l-α-vinylglycine from l-homoserine lactone. Synthesis 1, 39–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berkowitz D. B., Jahng W.-J., and Pedersen M. L. (1996) α-Vinyllysine and α-vinylarginine are time-dependent inhibitors of their cognate decarboxylases. Bioorg. Med. Chem. Lett. 6, 2151–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan B. H., Wong P. T., and Bian J. S. (2010) Hydrogen sulfide: a novel signaling molecule in the central nervous system. Neurochem. Int. 56, 3–10 [DOI] [PubMed] [Google Scholar]
- 15. Kimura H. (2000) Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267, 129–133 [DOI] [PubMed] [Google Scholar]
- 16. Mustafa A. K., Ahmad A. S., Zeynalov E., Gazi S. K., Sikka G., Ehmsen J. T., Barrow R. K., Coyle J. T., Snyder S. H., and Doré S. (2010) Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J. Neurosci. 30, 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abe T., Suzuki M., Sasabe J., Takahashi S., Unekawa M., Mashima K., Iizumi T., Hamase K., Konno R., Aiso S., and Suzuki N. (2014) Cellular origin and regulation of d- and l-serine in in vitro and in vivo models of cerebral ischemia. J. Cereb. Blood Flow Metab. 34, 1928–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCune C. D., Chan S. J., Beio M. L., Shen W., Chung W. J., Szczesniak L. M., Chai C., Koh S. Q., Wong P. T. H., and Berkowitz D. B. (2016) “Zipped synthesis” by cross-metathesis provides a cystathionine β-synthase inhibitor that attenuates cellular H2S levels and reduces neuronal infarction in a rat ischemic stroke model. ACS Cent. Sci. 2, 242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Madeira C., Lourenco M. V., Vargas-Lopes C., Suemoto C. K., Brandão C. O., Reis T., Leite R. E. P., Laks J., Jacob-Filho W., Pasqualucci C. A., Grinberg L. T., Ferreira S. T., and Panizzutti R. (2015) d-Serine levels in Alzheimer's disease: implications for novel biomarker development. Transl. Psychiatry 5, e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paul P., and de Belleroche J. (2014) The role of d-serine and glycine as co-agonists of NMDA receptors in motor neuron degeneration and amyotrophic lateral sclerosis (ALS). Front. Synaptic Neurosci. 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia M., Zhu S., Shevelkin A., Ross C. A., and Pletnikov M. (2016) Disc1, astrocytes and neuronal maturation: a possible mechanistic link with implications for mental disorders. J. Neurochem. 138, 518–524 [DOI] [PubMed] [Google Scholar]
- 22. Van der Auwera S., Teumer A., Hertel J., Homuth G., Völker U., Lucht M. J., Degenhardt F., Schulze T., Rietschel M., Nöthen M. M., John U., Nauck M., and Grabe H. J. (2016) The inverse link between genetic risk for schizophrenia and migraine through NMDA (N-methyl-d-aspartate) receptor activation via d-serine. Eur. Neuropsychopharmacol. 26, 1507–1515 [DOI] [PubMed] [Google Scholar]
- 23. Puhl M. D., Mintzopoulos D., Jensen J. E., Gillis T. E., Konopaske G. T., Kaufman M. J., and Coyle J. T. (2015) In vivo magnetic resonance studies reveal neuroanatomical and neurochemical abnormalities in the serine racemase knockout mouse model of schizophrenia. Neurobiol. Dis. 73, 269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panatier A., Theodosis D. T., Mothet J.-P., Touquet B., Pollegioni L., Poulain D. A., and Oliet S. H. R. (2006) Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784 [DOI] [PubMed] [Google Scholar]
- 25. Marchetti M., Bruno S., Campanini B., Peracchi A., Mai N., and Mozzarelli A. (2013) ATP binding to human serine racemase is cooperative and modulated by glycine. FEBS J. 280, 5853–5863 [DOI] [PubMed] [Google Scholar]
- 26. Neidle A., and Dunlop D. S. (2002) Allosteric regulation of mouse brain serine racemase. Neurochem. Res. 27, 1719–1724 [DOI] [PubMed] [Google Scholar]
- 27. De Miranda J., Panizzutti R., Foltyn V. N., and Wolosker H. (2002) Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-d-aspartate (NMDA) receptor coagonist d-serine. Proc. Natl. Acad. Sci. U.S.A. 99, 14542–14547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foltyn V. N., Zehl M., Dikopoltsev E., Jensen O. N., and Wolosker H. (2010) Phosphorylation of mouse serine racemase regulates d-serine synthesis. FEBS Lett. 584, 2937–2941 [DOI] [PubMed] [Google Scholar]
- 29. Balan L., Foltyn V. N., Zehl M., Dumin E., Dikopoltsev E., Knoh D., Ohno Y., Kihara A., Jensen O. N., Radzishevsky I. S., and Wolosker H. (2009) Feedback inactivation of d-serine synthesis by NMDA receptor-elicited translocation of serine racemase to the membrane. Proc. Natl. Acad. Sci. U.S.A. 106, 7589–7594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shoji K., Mariotto S., Ciampa A. R., and Suzuki H. (2006) Regulation of serine racemase activity by d-serine and nitric oxide in human glioblastoma cells. Neurosci. Lett. 392, 75–78 [DOI] [PubMed] [Google Scholar]
- 31. Dumin E., Bendikov I., Foltyn V. N., Misumi Y., Ikehara Y., Kartvelishvily E., and Wolosker H. (2006) Modulation of d-serine levels via ubiquitin-dependent proteasomal degradation of serine racemase. J. Biol. Chem. 281, 20291–20302 [DOI] [PubMed] [Google Scholar]
- 32. Fujii K., Maeda K., Hikida T., Mustafa A. K., Balkissoon R., Xia J., Yamada T., Ozeki Y., Kawahara R., Okawa M., Huganir R. L., Ujike H., Snyder S. H., and Sawa A. (2006) Serine racemase binds to PICK1: potential relevance to schizophrenia. Mol. Psychiatry 11, 150–157 [DOI] [PubMed] [Google Scholar]
- 33. Dev K. K., and Henley J. M. (2006) The schizophrenic faces of PICK1. Trends Pharmacol. Sci. 27, 574–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baumgart F., Mancheño J. M., and Rodríguez-Crespo I. (2007) Insights into the activation of brain serine racemase by the multi-PDZ domain glutamate receptor interacting protein, divalent cations and ATP. FEBS J. 274, 4561–4571 [DOI] [PubMed] [Google Scholar]
- 35. Ma T. M., Paul B. D., Fu C., Hu S., Zhu H., Blackshaw S., Wolosker H., and Snyder S. H. (2014) Serine racemase regulated by binding to stargazin and PSD-95. J. Biol. Chem. 289, 29631–29641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamauchi T., Goto M., Wu H.-Y., Uo T., Yoshimura T., Mihara H., Kurihara T., Miyahara I., Hirotsu K., and Esaki N. (2009) Serine racemase with catalytically active lysinoalanyl residue. J. Biochem. 145, 421–424 [DOI] [PubMed] [Google Scholar]
- 37. Zou L., Song Y., Wang C., Sun J., Wang L., Cheng B., and Fan J. (2016) Crystal structure of maize serine racemase with pyridoxal 5′-phosphate. Acta Crystallogr. F Struct. Biol. Commun. 72, 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vorlová B., Nachtigallová D., Jirásková-Vaníčková J., Ajani H., Jansa P., Rezáč J., Fanfrlík J., Otyepka M., Hobza P., Konvalinka J., and Lepšík M. (2015) Malonate-based inhibitors of mammalian serine racemase: kinetic characterization and structure-based computational study. Eur. J. Med. Chem. 89, 189–197 [DOI] [PubMed] [Google Scholar]
- 39. Smith M. A., Mack V., Ebneth A., Moraes I., Felicetti B., Wood M., Schonfeld D., Mather O., Cesura A., and Barker J. (2010) The structure of mammalian serine racemase: evidence for conformational changes upon inhibitor binding. J. Biol. Chem. 285, 12873–12881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Foltyn V. N., Bendikov I., De Miranda J., Panizzutti R., Dumin E., Shleper M., Li P., Toney M. D., Kartvelishvily E., and Wolosker H. (2005) Serine racemase modulates intracellular d-serine levels through an α,β-elimination activity. J. Biol. Chem. 280, 1754–1763 [DOI] [PubMed] [Google Scholar]
- 41. Strísovský K., Jirásková J., Barinka C., Majer P., Rojas C., Slusher B. S., and Konvalinka J. (2003) Mouse brain serine racemase catalyzes specific elimination of l-serine to pyruvate. FEBS Lett. 535, 44–48 [DOI] [PubMed] [Google Scholar]
- 42. Goto M., Yamauchi T., Kamiya N., Miyahara I., Yoshimura T., Mihara H., Kurihara T., Hirotsu K., and Esaki N. (2009) Crystal structure of a homolog of mammalian serine racemase from Schizosaccharomyces pombe. J. Biol. Chem. 284, 25944–25952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strísovský K., Jirásková J., Mikulová A., Rulísek L., and Konvalinka J. (2005) Dual substrate and reaction specificity in mouse serine racemase: identification of high-affinity dicarboxylate substrate and inhibitors and analysis of the β-eliminase activity. Biochemistry 44, 13091–13100 [DOI] [PubMed] [Google Scholar]
- 44. Nitoker N., and Major D. T. (2015) Understanding the reaction mechanism and intermediate stabilization in mammalian serine racemase using multiscale quantum-classical simulations. Biochemistry 54, 516–527 [DOI] [PubMed] [Google Scholar]
- 45. Cerqueira NMFSA, Moorthy H., Fernandes P. A., and Ramos M. J. (2017) The mechanism of the Ser-(cis)Ser-Lys catalytic triad of peptide amidases. Phys. Chem. Chem. Phys. 19, 12343–12354 [DOI] [PubMed] [Google Scholar]
- 46. Valiña A. L. B., Mazumder-Shivakumar D., and Bruice T. C. (2004) Probing the Ser-Ser-Lys catalytic triad mechanism of peptide amidase: computational studies of the ground state, transition state, and intermediate. Biochemistry 43, 15657–15672 [DOI] [PubMed] [Google Scholar]
- 47. Shin S., Lee T.-H., Ha N.-C., Koo H. M., Kim S.-Y., Lee H.-S., Kim Y. S., and Oh B.-H. (2002) Structure of malonamidase E2 reveals a novel Ser-cis-Ser-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature. EMBO J. 21, 2509–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Labahn J., Neumann S., Büldt G., Kula M.-R., and Granzin J. (2002) An alternative mechanism for amidase signature enzymes. J. Mol. Biol. 322, 1053–1064 [DOI] [PubMed] [Google Scholar]
- 49. Wu Z.-M., Zheng R.-C., and Zheng Y.-G. (2017) Identification and characterization of a novel amidase signature family amidase from Parvibaculum lavamentivorans zjb14001. Protein Expr. Purif. 129, 60–68 [DOI] [PubMed] [Google Scholar]
- 50. Lee S., Park E.-H., Ko H.-J., Bang W. G., Kim H.-Y., Kim K. H., and Choi I.-G. (2015) Crystal structure analysis of a bacterial aryl acylamidase belonging to the amidase signature enzyme family. Biochem. Biophys. Res. Commun. 467, 268–274 [DOI] [PubMed] [Google Scholar]
- 51. Neu D., Lehmann T., Elleuche S., and Pollmann S. (2007) Arabidopsis amidase 1, a member of the amidase signature family. FEBS J. 274, 3440–3451 [DOI] [PubMed] [Google Scholar]
- 52. Mileni M., Kamtekar S., Wood D. C., Benson T. E., Cravatt B. F., and Stevens R. C. (2010) Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 400, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McKinney M. K., and Cravatt B. F. (2005) Structure and function of fatty acid amide hydrolase. Annu. Rev. Biochem. 74, 411–432 [DOI] [PubMed] [Google Scholar]
- 54. McKinney M. K., and Cravatt B. F. (2003) Evidence for distinct roles in catalysis for residues of the serine-serine-lysine catalytic triad of fatty acid amide hydrolase. J. Biol. Chem. 278, 37393–37399 [DOI] [PubMed] [Google Scholar]
- 55. Pratt R. F., and McLeish M. J. (2010) Structural relationship between the active sites of β-lactam-recognizing and amidase signature enzymes: convergent evolution? Biochemistry 49, 9688–9697 [DOI] [PubMed] [Google Scholar]
- 56. Akiyama T., Ishii M., Takuwa A., Oinuma K.-I., Sasaki Y., Takaya N., and Yajima S. (2017) Structural basis of the substrate recognition of hydrazidase isolated from Microbacterium sp. strain hm58-2, which catalyzes acylhydrazide compounds as its sole carbon source. Biochem. Biophys. Res. Commun. 482, 1007–1012 [DOI] [PubMed] [Google Scholar]
- 57. Hoffman H. E., Jirásková J., Ingr M., Zvelebil M., and Konvalinka J. (2009) Recombinant human serine racemase: enzymologic characterization and comparison with its mouse ortholog. Protein Expr. Purif. 63, 62–67 [DOI] [PubMed] [Google Scholar]
- 58. Bruno S., Margiotta M., Marchesani F., Paredi G., Orlandi V., Faggiano S., Ronda L., Campanini B., and Mozzarelli A. (2017) Magnesium and calcium ions differentially affect human serine racemase activity and modulate its quaternary equilibrium toward a tetrameric form. Biochim. Biophys. Acta 1865, 381–387 [DOI] [PubMed] [Google Scholar]
- 59. Ito T., Maekawa M., Hayashi S., Goto M., Hemmi H., and Yoshimura T. (2013) Catalytic mechanism of serine racemase from. Dictyostelium discoideum. Amino Acids 44, 1073–1084 [DOI] [PubMed] [Google Scholar]
- 60. Wang C.-Y., Ku S. C., Lee C.-C., and Wang A. H. J. (2012) Modulating the function of human serine racemase and human serine dehydratase by protein engineering. Protein Eng. Des. Sel. 25, 741–749 [DOI] [PubMed] [Google Scholar]
- 61. Soo V. W. C., Yosaatmadja Y., Squire C. J., and Patrick W. M. (2016) Mechanistic and evolutionary insights from the reciprocal promiscuity of two pyridoxal phosphate-dependent enzymes. J. Biol. Chem. 291, 19873–19887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dunathan H. C. (1966) Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc. Natl. Acad. Sci. U.S.A. 55, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hoffman H. E., Jirásková J., Cígler P., Sanda M., Schraml J., and Konvalinka J. (2009) Hydroxamic acids as a novel family of serine racemase inhibitors: mechanistic analysis reveals different modes of interaction with the pyridoxal-5′-phosphate cofactor. J. Med. Chem. 52, 6032–6041 [DOI] [PubMed] [Google Scholar]
- 64. Koutmos M., Kabil O., Smith J. L., and Banerjee R. (2010) Structural basis for substrate activation and regulation by cystathionine β-synthase (CBS) domains in cystathionine β-synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 20958–20963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Caulkins B. G., Young R. P., Kudla R. A., Yang C., Bittbauer T. J., Bastin B., Hilario E., Fan L., Marsella M. J., Dunn M. F., and Mueller L. J. (2016) NMR crystallography of a carbanionic intermediate in tryptophan synthase: chemical structure, tautomerization, and reaction specificity. J. Am. Chem. Soc. 138, 15214–15226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) Mega6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wada M., Nakamori S., and Takagi H. (2003) Serine racemase homologue of Saccharomyces cerevisiae has l-threo-3-hydroxyaspartate dehydratase activity. FEMS Microbiol. Lett. 225, 189–193 [DOI] [PubMed] [Google Scholar]
- 68. Murakami T., Maeda T., Yokota A., and Wada M. (2009) Gene cloning and expression of pyridoxal 5′-phosphate-dependent l-threo-3-hydroxyaspartate dehydratase from Pseudomonas sp. T62, and characterization of the recombinant enzyme. J. Biochem. 145, 661–668 [DOI] [PubMed] [Google Scholar]
- 69. Katane M., Saitoh Y., Uchiyama K., Nakayama K., Saitoh Y., Miyamoto T., Sekine M., Uda K., and Homma H. (2016) Characterization of a homologue of mammalian serine racemase from Caenorhabditis elegans: the enzyme is not critical for the metabolism of serine in vivo. Genes Cells 21, 966–977 [DOI] [PubMed] [Google Scholar]
- 70. McGinnis S., and Madden T. L. (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thompson J. D., Higgins D. G., and Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sali A, and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 73. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 74. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) Autodock4 and Autodocktools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Panizzutti R., De Miranda J., Ribeiro C. S., Engelender S., and Wolosker H. (2001) A new strategy to decrease N-methyl-d-aspartate (NMDA) receptor coactivation: inhibition of d-serine synthesis by converting serine racemase into an eliminase. Proc. Natl. Acad. Sci. U.S.A. 98, 5294–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]