

Abstract
Tyrosine hydroxylase (TH) catalyzes the conversion of l-tyrosine into l-DOPA, which is the rate-limiting step in the synthesis of catecholamines, such as dopamine, in dopaminergergic neurons. Low dopamine levels and death of the dopaminergic neurons are hallmarks of Parkinson's disease (PD), where α-synuclein is also a key player. TH is highly regulated, notably by phosphorylation of several Ser/Thr residues in the N-terminal tail. However, the functional role of TH phosphorylation at the Ser-31 site (THSer(P)-31) remains unclear. Here, we report that THSer(P)-31 co-distributes with the Golgi complex and synaptic-like vesicles in rat and human dopaminergic cells. We also found that the TH microsomal fraction content decreases after inhibition of cyclin-dependent kinase 5 (Cdk5) and ERK1/2. The cellular distribution of an overexpressed phospho-null mutant, TH1-S31A, was restricted to the soma of neuroblastoma cells, with decreased association with the microsomal fraction, whereas a phospho-mimic mutant, TH1-S31E, was distributed throughout the soma and neurites. TH1-S31E associated with vesicular monoamine transporter 2 (VMAT2) and α-synuclein in neuroblastoma cells, and endogenous THSer(P)-31 was detected in VMAT2– and α-synuclein–immunoprecipitated mouse brain samples. Microtubule disruption or co-transfection with α-synuclein A53T, a PD-associated mutation, caused TH1-S31E accumulation in the cell soma. Our results indicate that Ser-31 phosphorylation may regulate TH subcellular localization by enabling its transport along microtubules, notably toward the projection terminals. These findings disclose a new mechanism of TH regulation by phosphorylation and reveal its interaction with key players in PD, opening up new research avenues for better understanding dopamine synthesis in physiological and pathological states.
Keywords: confocal microscopy, dopamine, Parkinson disease, phosphorylation, vesicles, axonal transport, tyrosine hydroxylase phosphorylated at Ser-31, tyrosine hydroxylase
Introduction
Catecholamine neurotransmission disturbances are hallmarks of neurodegenerative and neuropsychiatric diseases, such as Parkinson's disease (PD),2 Alzheimer's disease, l-DOPA-responsive dystonia, or tyrosine hydroxylase deficiency (1–4). Catecholamines are stored in vesicles that originate at the Golgi complex (GC) and are transported through the classical secretory pathway (5). GC fragmentation, loss of vesicle integrity, and dysfunctional vesicular transport have emerged as pathomechanisms involved in neurodegenerative disorders, notably PD (6–8). A key enzyme in the synthesis of catecholamine neurotransmitters is tyrosine hydroxylase (TH), which catalyzes the hydroxylation of l-tyrosine into l-DOPA (l-3,4-dihydroxyphenylalanine), the rate-limiting step in the synthesis of dopamine (DA). DA is then pumped and loaded into vesicles by the vesicular monoamine transporter 2 (VMAT2) (9). The TH gene encodes one protein isoform in lower mammals, but alternative splicing gives four in humans (TH1–TH4) (10, 11). Rat TH is homologous to the TH1 human isoform, the most abundant isoform in humans together with TH2.
TH activity is controlled by catecholamine feedback inhibition and phosphorylation of its N-terminal residues 8, 19, 31, and 40 (THSer(P)-8, THSer(P)-19, THSer(P)-31, and THSer(P)-40) by different kinases (12, 13). THSer(P)-19 induces the high-affinity binding to 14-3-3 proteins, which increases TH activity and stability (14). Ser-19 increases the phosphorylation at Ser-40 in a hierarchical manner, leading to increased activity (13). On the other hand, binding of 14-3-3 to THSer(P)-19 decreases Ser-40 phosphorylation (15). THSer(P)-40 releases TH from catecholamine feedback inhibition, increasing the activity 20-fold (16). The functional implications of THSer(P)-8 are currently lacking, and a comprehensive understanding of the physiological role of THSer(P)-31 also remains unclear. Cdk5 and ERK1/2 phosphorylate TH (human isoforms 1, 3, and 4 and rodent TH) at Ser-31 (17–19), which increases TH activity about 2-fold in vitro (20) and in situ (21). In 2006, Lehman et al. (22) reported that for human TH1, phosphorylation at Ser-31 also produced a 9-fold increase in the rate of phosphorylation at Ser-40. THSer(P)-31 is abundant at the axonal terminals of dopaminergic neurons (23), and it has been reported to increase about 3-fold in NGF-stimulated PC12 cells, whereas THSer(P)-40 is unaffected by NGF (24). Moreover, THSer(P)-31 increases TH stability in cells (25), and it may increase TH activity regardless of THSer(P)-40 under depolarizing conditions (16, 21). In the terminal fields of the central nervous system, there is a greater DA and l-DOPA content, coinciding with greater THSer(P)-31 compared with somatodendritic compartments, whereas no consistent differences in THSer(P)-40 have been reported (23, 26). Under experimental PD conditions, THSer(P)-31 stoichiometry is decreased in the striatum but increased in the substantia nigra; this pattern is also shown by DA per remaining TH; however, total amounts of TH and of THSer(P)-40 decreased in both brain areas (27). Therefore, THSer(P)-31 may affect TH activity within the range of phosphorylation stoichiometries normally seen in the central nervous system and be independent of Ser-40 phosphorylation differences. Despite the body of results, the physiological role of Ser-31 phosphorylation is still not fully understood. TH is characterized as a largely soluble and cytoplasmic tetrameric protein; however, its physiological association with membranes (28) has recently been proven to involve binding to partners, such as VMAT2 (9) and Hsc70 (29). However, the mechanism whereby TH is targeted to the synaptic vesicles still remains elusive. In this work, we investigated the functional role of Ser-31 phosphorylation, and our results indicate the association of THSer(P)-31 with the GC and the vesicular transport pathway, through VMAT2 and α-synuclein (α-syn). α-Syn is a synaptic vesicle associated-protein and a culprit in the development of PD (30). TH and α-syn are known to interact (31), but the association of both proteins at synaptic vesicles and coordinated microtubular transport is presented here for the first time. Furthermore, our results position THSer(P)-31 at sites and processes disturbed in PD and other neurodegenerative disorders, such as loss of vesicle integrity, defective vesicle trafficking, and GC fragmentation, and may contribute to explain the early loss of striatal TH and DA that characterize PD (32).
Results
THSer(P)-31 localizes to the Golgi complex and synaptic-like vesicles
The cellular distribution of the different phosphorylated forms of TH at Ser-19, Ser-31, and Ser-40 was investigated by immunofluorescence using specific antibodies. These antibodies have been used in previous studies (26, 27, 33). Western blots of HEK293 cell extracts expressing either recombinant human V5-TH1-WT or the corresponding phospho-null (Ser → Ala) and phospho-mimicking (Ser → Glu) mutations for Ser(P)-19, Ser(P)-31, and Ser(P)-40, show low reactivity for the corresponding Ser → Ala protein for the three antibodies (Fig. 1, A–C). THSer(P)-31 and THSer(P)-40 antibodies react both with the WT form (Fig. 1, B and C), phosphorylated at the corresponding site, and also with the phospho-mimicking mutants, whereas the THSer(P)-19 antibody only reacts with Ser-19–phosphorylated WT (Fig. 1A). In undifferentiated PC12Adh (from ATCC) cells permeabilized with Triton X-100, THSer(P)-19 showed a nuclear distribution, and THSer(P)-31 as well as THSer(P)-40 showed a cytosolic distribution (data not shown), consistent with recent results (34).
Figure 1.

Distribution of TH phosphorylated forms. A–C, HEK293 cells were transfected with the WT or the phospho-null V5-TH1-S19A, V5-TH1-S31A, and V5-TH1-S40A or the phospho-mimicking V5-TH1-S19E, V5-TH1-S31E, and V5-TH1-S40E constructs. Phosphorylation at Ser-19, Ser-31, or Ser-40 was detected by Western blotting, loading equal amounts of total protein. Total TH (THt), V5 (transfection control), and GAPDH (loading control) detection was also carried out. D, immunofluorescence (top panels) of THSer(P)-31 in PC12* cells treated or not with R/S to inhibit Ser-31 phosphorylation and corresponding Western blot analysis (bottom panel), where total TH and GAPDH were also detected as controls. Arrows, perinuclear signal. E, immunofluorescence of total TH, THSer(P)-19, and THSer(P)-40 in PC12* cells treated or not with R/S. F, cellular distribution of THSer(P)-31 in PC12Adh. Arrows, perinuclear signal. G, cellular distribution of THSer(P)-31 in human pluripotent induced dopaminergic neurons (iCell DopaNeurons). Arrows, perinuclear signal. H, THSer(P)-31 distribution in PC12* stimulated with 50 ng/ml 2.5S NGF for 48 h. Insets, enlarged numbered areas. In all images, 10-μm scale bars are shown, and all nuclei are stained with DAPI (blue). I, whole-lysate Western blot of PC12* and PC12Adh cell lines stimulated or not with NGF and detection of dopamine-related marker proteins such as DOPA β-hydroxylase (DBH), chromogranin A (ChrA), total TH, THSer(P)-31, and vesicular monoamine transporter 2 (VMAT2). Equal amounts of protein were loaded as shown in the total protein loading represented in the right panel.
To preserve TH interaction with partner proteins, we used saponin, a mild permeabilization agent, which allowed us to visualize THSer(P)-31 as punctate structures enriched in the perinuclear region, notably observed in the immense majority of PC12* cells (Fig. 1D). PC12* is an in-house clone derived from Ref. 35, which was chosen for its elevated responsiveness to NGF, leading to neurite abundance and stability (35), and has been used previously in signaling studies (36). Specific pharmacological inhibition of Cdk5 and ERK1/2 by roscovitine and SL327 (R/S) treatment (37, 38) abolished the THSer(P)-31 signal both in Western blotting and immunofluorescence (Fig. 1D), indicating that the perinuclear signal is specific. This distribution was clearly distinct from total TH, THSer(P)-19, and THSer(P)-40 (Fig. 1E, top panels), and these signals were not affected by the R/S inhibition of Cdk5 and ERK1/2 (Fig. 1E, bottom panels). Moreover, in 45% of PC12Adh cells (n = 130) and 35% of iCell DopaNeurons (human dopaminergic pluripotent cells) (n = 50), the perinuclear enrichment was also observed (Fig. 1, F and G). Because stimulation with NGF increases THSer(P)-31 (24), we investigated its effect on THSer(P)-31 distribution. Interestingly, NGF treatment led to a punctate THSer(P)-31 signal throughout the cell soma and neurites of PC12* (Fig. 1H), similar to the distribution of THSer(P)-31 in PC12Adh and iCell DopaNeurons (Fig. 1, F and G). It is well established that PC12 can give rise to specific phenotypes as described previously in depth (39–41) and illustrated here when profiling different proteins of the dopamine pathway in the presence or absence of NGF stimulation by Western blotting (Fig. 1I), where we observe that THSer(P)-31 content increases in PC12Adh by 10% after NGF treatment, whereas it increases by 2-fold in PC12*, in agreement with previous studies (24).
THSer(P)-31 signal in PC12* resembled a GC pattern, and its co-detection with the GC marker GM130 showed co-distribution in PC12Adh, iCell DopaNeurons, and PC12* cells (Fig. 2, A–C). Moreover, disassembly of the GC with brefeldin A abolished the THSer(P)-31 signal (Fig. 2C). To further investigate the punctate signal, we co-detected THSer(P)-31 and synaptic-like vesicle marker synaptotagmin I (sytI), which showed a certain signal overlapping in NGF-stimulated and unstimulated PC12Adh (Fig. 2D). Similarly, THSer(P)-31 in iCell DopaNeurons also co-distributed with sytI (Fig. 2E). Notably, fluorescence signals of immunolabeled VMAT2 and THSer(P)-31 highly overlapped in iCell DopaNeurons (Fig. 2E). Our results thus indicate that THSer(P)-31 associates with synaptic-like vesicles in rat PC12Adh cells and in a human dopaminergic neuronal model.
Figure 2.

Endogenous THSer(P)-31 co-distribution with Golgi complex and vesicle markers. A, co-detection of THSer(P)-31 (green) with the Golgi marker GM130 (red) in PC12Adh (maximum projection of the confocal stack is shown) and 3D rendering of each signal and the corresponding co-localization channel in yellow. Arrows, perinuclear signal. B, co-detection of THSer(P)-31 (green) with the Golgi marker GM130 (red) in DopaNeurons treated or not with R/S. Arrows, perinuclear signal. C, GC disruption in PC12* by 30 min 5 μg/ml brefeldin A (BFA) incubation and subsequent reassembly by drug washout. Untreated samples are presented as control. D, THSer(P)-31 (green) co-detection with synaptotagmin I (sytI; red) in PC12Adh cells without (top) and with (bottom) NGF treatment. E, THSer(P)-31 co-detection of sytI (top) and VMAT2 (vesicular monoamine transporter 2; bottom) in iCell DopaNeurons. For D and E, pixel height in the surface plot represents the pixel intensity in the confocal plane. In all images, nuclei are stained with DAPI, and 10-μm scale bars are shown.
Phosphorylation of TH at Ser-31 is necessary for its association with vesicles
To determine whether the association of TH to vesicles is dependent on phosphorylation at Ser-31, we treated cell cultures with Cdk5 and ERK1/2 kinase inhibitors (combined roscovitine and SL327 treatment) and isolated the microsomal fractions (enriched in endoplasmic reticulum, GC, endosomes, and vesicles among other membranous cellular structures). We compared total TH levels of treated versus untreated cells to determine whether inhibition of phosphorylation at Ser-31 caused a decrease in the amount of total TH present in the microsomal fraction. Our results show that the treatment led to a large decrease of total TH in the microsomal fractions of PC12* (Fig. 3A), suggesting that phosphorylation at Ser-31 is needed for TH to associate with vesicles. Actually, a strong decrease of the signal from THSer(P)-31 was also observed in the microsomal fraction (Fig. 3A) in agreement with Ref. 25 and confirming that the treatment led to the inhibition of phosphorylation at Ser-31.
Figure 3.

Requirement of TH phosphorylation at Ser-31 for interaction with vesicles. A, relative amount of total TH and THSer(P)-31 in PC12* microsomal fractionation analyzed by Western blotting after inhibition of Ser-31 phosphorylation with 50 μm roscovitine and 50 μm SL327 (R/S). Bars, average relative amount of TH or THSer(P)-31 compared with the untreated samples (t test was used for statistical sample comparison; ***, p < 0.001; data are represented as mean ± S.D. (error bars); n = 3). Representative blots are shown. B, relative amounts of markers to determine the vesicle biogenesis status in microsomal fraction treated or not with R/S. The markers analyzed were GM130 for GC, clathrin, and transferrin receptor (TfR) for endosomes; VMAT2 for dopamine-vesicles; and SPC25 (signal peptidase complex 25) for endoplasmic reticulum. Equal amounts of treated and untreated samples were loaded, as shown in the total protein loading (right). C, relative amount of recombinant V5-TH1-WT, V5-TH1-S31A, or V5-TH1-S31E overexpressed in microsomal fractions of neuroblastoma cells. Microsomal fractions were analyzed by Western blotting using the V5 tag for detection, and mutant THs were compared with WT (representative blots are shown). Bars, relative amount of the WT construct compared with the mutant proteins (t test was used for statistical sample comparison; *, p < 0.05; data are represented as mean ± S.D.; n = 3). D, relative amount of recombinant V5-TH1-S31E overexpressed in microsomal fractions of neuroblastoma cells treated or not with R/S. Microsomal fractions were analyzed by Western blotting using the V5 tag for detection, and treated samples were compared with controls (representative blots are shown). Bars, relative amount of the control samples compared with the treated samples (t test was used for statistical sample comparison, although no significant differences were found; data are represented as mean ± S.D; n = 3).
However, to rule out the possibility that the decreased observed was due an impairment of vesicle biogenesis caused by the kinase inhibition, we tested the effect of the treatment on different microsomal markers. Specifically, we treated PC12Adh cells with or without roscovitine and SL327 and detected GC marker (GM130), endoplasmic reticulum marker (SPC25), endosome markers (clathrin and transferrin receptor), and dopamine-containing vesicle marker (VMAT2). The treatment did not lead to decreases in the signal of any of these markers (Fig. 3B).
To further confirm the role of THSer(P)-31 in this vesicle association, we performed Western blotting of cell fraction of neuroblastoma cells expressing V5-TH1-S31A and V5-TH1-S31E, which showed that V5-TH1-S31A was decreased in the microsomal fraction by 20% when compared with the wild type, whereas V5-TH1-S31E was increased by 20% (Fig. 3C). To rule out possible artifacts, we also treated V5-TH1-S31E–transfected cells with the kinase inhibitors, and then we compared the amounts of V5-TH1-S31E in the microsomal fractions, without finding significant differences (Fig. 3D). These results reinforce the specificity of the Ser-31 phosphorylation for TH vesicle association. However, compared with the large effect of the chemical inhibition of Ser-31 phosphorylation (Fig. 3A), the effect of the phospho-null mutation was less pronounced. This may be explained by hetero-oligomerization of recombinant and endogenous monomers, as has been shown in other cases (42, 43), which would lead to a vesicular interaction of oligomers formed of phospho-null and THSer(P)-31 subunits. The phospho-variant data are consistent with the results from the chemical inhibition of Cdk5 and ERK1/2 and rule out the possibility that the observed effect would be due to either nonspecific effects of Cdk5 and ERK1/2 on other proteins directly or indirectly involved in THSer(P)-31 association with vesicles. Again, these results show that TH needs to be phosphorylated at Ser-31 to bind to vesicles.
THSer(P)-31 associates with VMAT2 and α-syn
Because TH is able to interact with negatively charged membranes (44), we investigated whether phosphorylation at Ser-31 enhances this association. To study the binding of TH to membranes, we used chromaffin vesicle membranes that had been previously trypsinized to remove any proteins that could be potential partners. Surface plasmon resonance studies using purified recombinant, non-phosphorylated human TH1 showed a certain degree of interaction with membranes (Fig. 4A), as expected (44). However, TH1 phosphorylated at Ser-31 in vitro showed no difference in the interaction with trypsinized chromaffin-vesicle membranes when compared with the non-phosphorylated TH1 (Fig. 4A). It is thus unlikely that the THSer(P)-31-driven interaction involves a direct binding of the enzyme with the vesicular membrane, and the enzyme may rather interact with vesicular membrane proteins. Indeed, Torres and co-workers (9) have reported that TH can interact with the integral vesicular membrane protein VMAT2, and our immunofluorescence images of THSer(P)-31 and VMAT2 in iCell DopaNeurons showed co-distribution (Fig. 2E). To further investigate in situ whether THSer(P)-31 binds to vesicle proteins, we performed a proximity ligation assay (PLA), which is able to detect even weak and transient protein–protein interactions (45) with anti-V5 and either anti-VMAT2 or anti-α-syn in neuroblastoma cells expressing the phospho-variants (and GFP as transfection control). Appropriate PLA-positive and -negative controls were performed in all experiments (data not shown). PLA signals for the pair V5-TH1-S31E/VMAT2 were clearly obtained. However, for the pair V5-TH1-S31A/VMAT2, they were nearly imperceptible (Fig. 4B). Similarly, V5-TH1-S31A/α-syn signals were significantly decreased compared with the V5-TH1-S31E/α-syn pair (19 ± 20 versus 47 ± 24; n = 15; p < 0.05) (Fig. 4C).
Figure 4.

THSer(P)-31 interacts with VMAT2 and α-synuclein. A, surface plasmon resonance of TH (12.5 μm; black trace) and THSer(P)-31 (12.5 μm; blue trace) binding to trypsinized chromaffin vesicle membranes (trCVM). Inset, concentration dependence of the response of TH (○) and THSer(P)-31 (●) binding to trCGM. B and C, PLA (signals shown in red) between V5 and either VMAT2 or α-syn antibodies in neuroblastoma cells expressing either V5-TH1-S31A or V5-TH1-S31E. Maximal projections of the whole-cell height are shown. Cells were co-transfected with GFP as a transfection control (green), and nuclei were stained with DAPI (blue). Scale bars, 10 μm. D, immunoprecipitation of THSer(P)-31, VMAT2, and α-syn from whole-mouse brain lysate and immunoblot against THSer(P)-31, VMAT2, and total TH are shown. All proteins were also detected in lysates incubated with only beads (right lane) or the different IgGs shown. THSer(P)-31 was detected in the input, as expected.
PLA results were confirmed by immunoprecipitation of THSer(P)-31, VMAT2, and α-syn from whole-brain extracts from mice, followed by immunoblotting against THSer(P)-31, total TH, and VMAT2 (Fig. 4D). Immunoblotting against α-syn did not render conclusive results, but taken together with the positive results obtained using PLA, our data suggest that the THSer(P)-31–α-syn association is weak and/or transient because PLA allows the detection of this type of interaction, as reported previously (45). Appropriate negative controls using only beads or the different IgG were performed in parallel (Fig. 4D). Our data show the presence of THSer(P)-31 in both samples, indicating that THSer(P)-31 associates with VMAT2 and α-syn.
THSer(P)-31 is transported along the microtubules
Association of THSer(P)-31 with VMAT2 and α-syn may drive the anterograde transport of TH from GC to neurite terminals. Therefore, we sought to investigate the localization and redistribution of exogenous TH phospho-mutants upon disruption of the microtubule network. First, detection of the V5 tag of recombinant V5-TH1-S31A and V5-TH1-S31E mutants showed that V5-TH1-S31A accumulates mainly in the soma of the neuroblastoma cells, whereas V5-TH1-S31E is distributed throughout the soma and neurites (Fig. 5A). The depolymerization of the microtubule network of neuroblastoma cells by nocodazole led to a decreased of V5-TH1-S31E signal in neurites compared with control samples (Fig. 5B), and drug washout recovered the V5-TH1-S31E signal in neurites. However, V5-TH1-S31A localization did not change upon microtubule disassembly (Fig. 5C). To further investigate THSer(P)-31 transport in DopaNeuron cells, we first inhibited TH phosphorylation at Ser-31 using Cdk5 inhibitors (R/S) to create an initial stage with the lowest phosphorylation levels possible, and then we depolymerized the microtubules with nocodazole. Afterward, we performed a drug washout to allow the phosphorylation of TH at Ser-31 and reassembly of the microtubule network. All samples were stained for THSer(P)-31 and tubulin to monitor the treatment and were treated in parallel, with data acquisition and image processing performed under the same conditions. As expected, R/S and nocodazole treatment (0 min of washout) showed a disrupted microtubule pattern compared with the controls, as well as a significantly decreased THSer(P)-31 signal both for the somas (34.99 ± 11.7 versus 22.81 ± 7.1; p < 0.001; n = 50) and for the neurites (18.85 ± 4.57 versus 8.22 ± 2.9; p < 0.001; n = 50) (Fig. 5, D and E). At the initial time points of the drug washout (5 and 15 min), increased THSer(P)-31 fluorescence was measured in the soma and, to a lesser extent in the neurites, the recovery rate of the signal being 2.2-fold faster in the somas than in the neurites. At the 15–30-min time lapse, an inversion of this trend was measured. At the final 30-min time point, fluorescence signals in somas and neurites were comparable with the controls with no statistical differences (somas: 32.95 ± 17.76 versus 34.99 ± 11.7; p > 0.4; n = 50; neurites: 15.60 ± 12.49 versus 18.86 ± 4.57; p > 0.07; n = 50). Therefore, our data show that THSer(P)-31 signal increases first in the somas and then gradually in the distal parts of the neurites. Thus, our results show that THSer(P)-31 trafficking depends on microtubule integrity for its transport to the neurite extensions in a human dopaminergic cell line.
Figure 5.

Transport of THSer(P)-31 to neurite extensions. A, distribution of V5-TH1-S31A– and V5-TH1-S31E–expressing neuroblastoma detected by V5 staining (red). Cellular membranes were stained using WGA (green). Confocal planes are presented as well as the maximum intensity projection (max. projection) of the V5 signal stack of confocal planes. Nuclei were stained with DAPI (blue). Arrows, presence (white) or absence (yellow) of V5 signal in neurites of V5-positive cells (intense red signal in the soma). B and C, detection of V5 tag in neuroblastoma cells expressing V5-TH1-S31E (B) or V5-TH1-S31A (C) after microtubule depolymerization by a cold shock and nocodazole treatment, followed by 30 min of drug washout. Control cells were processed in parallel but were not subjected to nocodazole. All samples were stained for V5 (red), tubulin (cyan), and WGA to mark the cells membranes (green) and DAPI to stain the nucleus. Maximum intensity projection of the stack of confocal planes (max. proj.) is presented. Arrows, presence (white) or absence (yellow) of V5 signal in neurites of V5-positive cells (intense red signal in the soma). D, immunofluorescence of iCell DopaNeurons detecting THSer(P)-31 (green) and tubulin (red) in samples treated first with roscovitine/SL327 for inhibition of phosphorylation of TH at Ser-31 and then subjected to a cold shock nocodazole treatment (R/S+ Nocod) for microtubule disassembly before allowing drug washout. Blue and white arrows, control levels of THSer(P)-31 fluorescence in somas and neurites, respectively. Gray and yellow arrows, low levels/absence of THSer(P)-31 fluorescence in somas and neurites, respectively. Intermediate fluorescence levels are indicated with the corresponding striped arrows. In all cases, maximal projections comprehending the whole cell height are shown. E, quantification of the THSer(P)-31 signal of DopaNeuron somas or neurites in control (untreated) samples as well as in samples treated with the R/S+Nocod and subjected to drug washout. Data are shown as average ± S.D. (error bars) (in all cases n = 50), and the changes in fluorescence per time (arbitrary units (AU)/min) are indicated below each pair of time points. ***, p < 0.001. For all confocal images, 10-μm scale bars are shown.
The A53T mutation of α-syn (α-syn-A53T) is associated with autosomal dominant forms of PD (30), and it has been shown to hinder axonal transport in rat neurons and in neuroblastoma cells by fragmenting the GC and aggregating microtubuli (47, 48). Therefore, we studied whether overexpression of His-α-syn-A53T resulted in impaired distribution of V5-TH1-S31E. We co-transfected neuroblastoma cells with V5-TH1-S31E and one of the following plasmids: GFP, His-α-syn-WT, or His-α-syn-A53T, all of which are under the same promotor, CMV. To avoid comparing samples transfected only with one plasmid with samples transfected with two constructs, we used the V5-TH1-S31E/GFP co-transfection as our control and reference. We stained the cell membranes with WGA to identify the neurites of the cells. Cells transfected with V5-TH1-S31E/GFP showed the V5 signal along the whole neurite, including the more distal parts (Fig. 6A). However, cells transfected with V5-TH1-S31E and His-α-syn-WT, and especially His-α-syn-A53T, showed intense V5 signal in the cell soma but weaker signal at the distal ends of the neurites (Fig. 6A). We then quantified the intensity of the signal along the neurite length from cells transfected with V5-TH1-S31E/GFP, V5-TH1-S31E/His-α-syn-WT, or V5-TH1-S31E/His-α-syn-A53T, and we integrated the area below the plot profile. Comparing the most distal 20 μm of the neurites, we found that the signal was significantly reduced in V5-TH1-S31E/His-α-syn-WT (p < 0.05) and especially in V5-TH1-S31E/His-α-syn-A53T (p < 0.0001) samples (Fig. 6B). Signal intensity in arbitrary units was as follows: TH1-S31E/GFP = 17.4 ± 8.1 (n = 24); V5-TH1-S31E/His-α-syn-WT = 12.2 ± 6.7 (n = 16); V5-TH1-S31E/His-α-syn-A53T = 6.9 ± 4.2 (n = 22). Our results indicate that overexpression of wild-type and especially mutant α-syn can affect the axonal transport of TH.
Figure 6.
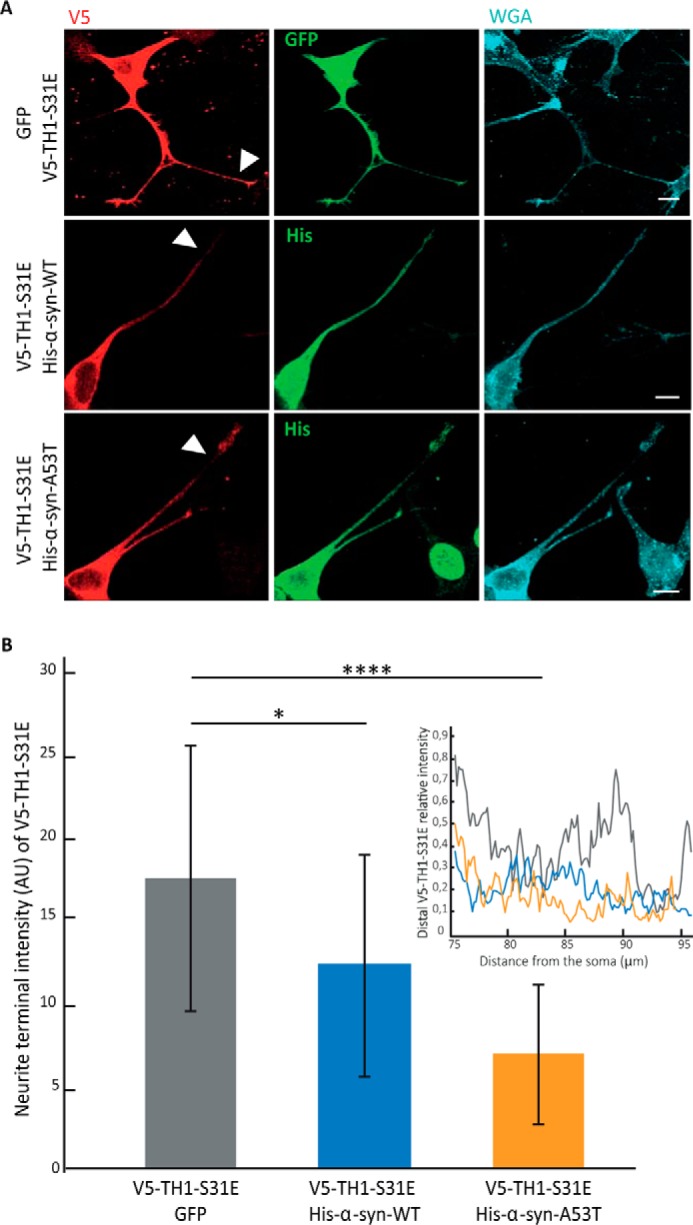
Effect of overexpression of α-synuclein on V5-TH1-S31E in neuroblastoma neurites. A, immunofluorescence of neuroblastoma cells co-transfected with V5-TH1-S31E (red) and one of the plasmids GFP, His-α-syn WT, or His-α-syn-A53T (green) and membranes stained with WGA (cyan). B, quantification of the signal of neurites from cells co-transfected with V5-TH1-S31E/GFP V5-TH1-S31E/His-α-syn-WT or V5-TH1-S31E/His-α-syn-A53T. Data are shown as average ± S.D. (error bars) and are expressed in arbitrary units (AU) (left). Representative plot profiles are shown (right). For all confocal images, 10-μm scale bars are shown. *, p < 0.05 and ****, p < 0.0001.
Influence of phosphorylation at Ser-31 on Ser-19 and Ser-40 sites
To better understand the previously reported multisite and hierarchical phosphorylation events in TH (13, 16, 20, 22), we studied whether phosphorylation at Ser-31 can modulate phosphorylation at Ser-19 and Ser-40, which are regulated by different signaling pathways. PLAs allow the detection of two epitopes that are in close vicinity (<100 nm (49)) and therefore were used to detect the co-existence of phosphorylation at two different sites. Anti-V5 together with anti-THSer(P)-19 or anti-THSer(P)-40 antibodies in neuroblastoma cells expressing V5-TH1-S31A developed significantly more PLA signals for both Ser-19 and Ser-40 phosphorylation compared with cells expressing V5-TH1-S31E (708 ± 442 versus 302 ± 102, n = 12, p < 0.05 for THSer(P)-19; 1036 ± 449 versus 56 ± 65, n = 10, p < 0.005 for THSer(P)-40) (Fig. 7, A and B). The difference in co-distribution of Ser-19 and Ser-40 phosphorylation between V5-TH1-S31E– and V5-TH1-S31A–transfected cells was corroborated by Western blot analyses (Fig. 7C). Cells expressing V5-TH1-S31A treated or not with R/S did not show significant differences in levels of phosphorylation (Fig. 7D), further supporting our conclusions.
Figure 7.

Effect of THSer(P)-31 on the phosphorylation of THSer(P)-19 and THSer(P)-40. A and B, PLAs (signals shown in red) between V5 and THSer(P)-19 (A) or THSer(P)-40 (B) antibodies in neuroblastoma cells expressing either V5-TH1-S31A and V5-TH1-S31E. Cells were co-transfected with soluble GFP (green) as a positive transfection control, and nuclei were stained with DAPI (blue). Scale bars, 10 μm. C, THSer(P)-19 or THSer(P)-40 detection using Western blotting of whole lysates of neuroblastoma cells expressing V5-TH1-S31A or V5-TH1-S31E. Bars, relative amount of TH (t test was used for statistical sample comparison; *, p < 0.05; **, p < 0.01; data are represented as mean ± S.D. (error bars); n = 3). Representative Western blots are shown. D, THSer(P)-19 or THSer(P)-40 detection using Western blotting of whole lysates of neuroblastoma cells expressing V5-TH1-S31A treated or not with R/S. Bars, relative amount of TH (data represented as mean ± S.D.; n = 3).
Taken together, our data demonstrate that phosphorylation at Ser-31 regulates TH association with vesicles and thus its transport to neurite terminals through interaction with VMAT2 and α-syn.
Discussion
TH has been described essentially as a cytosolic protein, but its membrane-associated form has recently been attracting interest (9, 29), although the regulation of the binding mechanisms remains largely unknown. Here we show that Ser-31 phosphorylation of TH regulates its association with the GC and synaptic-like vesicles. Our data also indicate that TH1Ser(P)-31 interacts, directly or indirectly, with VMAT2 and α-syn in striatal brain isolates and in cellular models. However, we observed little co-localization of TH1-S31A with α-syn and VMAT2, whereas this was prominent for the TH1-S31E mutant. It has been reported previously that α-syn interacts with, and negatively regulates, TH through activation of PP2A phosphatase, which dephosphorylates THSer(P)-40 and inactivates TH (50). In addition, it has also been shown that VMAT1/2 can be inhibited by α-syn (51). However, this work assigns to Ser-31 phosphorylation the role of controlling the association of TH to both α-syn and VMAT2. Interestingly, phosphorylation also appears to regulate the vesicular interaction of synthesizing enzymes for other neurotransmitters, such as acetylcholine and GABA. Thus, the synthesizing enzymes choline acetyltransferase and glutamate decarboxylase 65 have been found to bind to their corresponding vesicular transporter (vesicular acetylcholine transporter and vesicular GABA transporter) (52, 53), and this association seems to be phosphorylation-dependent for both glutamate decarboxylase 65 (53) and choline acetyltransferase (54). Our results thus contribute to the identification of the dopaminergic system and TH to comply with a growing class of neurotransmitter-synthesizing enzymes that couple to their transporter in a phosphorylation-dependent manner. This association may ensure an efficient packaging of the neurotransmitters into the synaptic vesicle and a proper trafficking and subcellular localization of neurotransmitter synthesis.
Based on the previous reports showing an inhibitory effect of α-syn on TH activity (50, 55), the engagement of α-syn in a vesicular complex might ensure that TH is transported most probably in an inhibited state to its destination (see below). It would also be likely that this functional transport complex contains additional regulatory or stabilizing proteins, such as Hsc70, which also has been described to interact with both VMAT2 and TH (29, 56). The DOPA decarboxylase enzyme that catalyzes the next step in DA synthesis after TH also interacts with TH, Hsc70, and VMAT2 (9, 29).
The GC enrichment of TH that we observed indicates that it associates with VMAT2 and α-syn during the vesicle formation, because VMAT2 is an integral protein that is sorted into the vesicles in the GC (57) and α-syn has been localized at the GC and dopamine-containing vesicles (58, 59), reinforcing the early stage association of these proteins. Furthermore, our data suggest that in vivo TH phosphorylated at Ser-31 may be transported from the GC in the cell soma to the terminals by the anterograde axonal transport. Early reports already proved fast axonal transport for TH, consistent with its association with vesicles (60–62), and additional transport of TH mRNA has more recently been described (61). Moreover, recent results from our laboratory using a knock-in mouse bearing a destabilizing TH mutation have shown the importance of axonal transport of TH for proper distribution of TH in striatal terminals (63).
Effects of multisite phosphorylation events have been reported for TH, and phosphorylation at Ser-31 has been reported to stimulate the in vitro phosphorylation of adjacent Ser-40 sites (22). Moreover, upon stimulation of bovine chromaffin cells, increased Ser-40 phosphorylation was observed when co-stimulating the Ser-31–targeting ERK1/2 pathways (22), but this stimulatory effect was not observed in the presence of dopamine (22). No studies have so far looked into these hierarchical effects on phosphorylation in the brain, but our results in neuroblastoma cells suggest that localization and interaction partners of TH play a role in the phosphorylation of the different sites. Interestingly, less Ser-19 and Ser-40 phosphorylation was observed for the phospho-mimicking TH1-S31E mutant, suggesting that conformational constraints and its interaction with partners, such as α-syn or VMAT2, may affect its availability for phosphorylation. In addition, phosphorylation at Ser-31 stabilizes the enzyme, and, in addition, the decreased phosphorylation at Ser-40 would allow more binding of inhibitory catecholamines (16), which would further stabilize THSer(P)-31 (25, 64). This inhibition seems to be congruent with TH being in a stable, and probably in a non-catalytic state during its transport from the cell soma to the terminals (65). Last, further maturation of the vesicles might be accompanied by TH reactivation and increased l-DOPA synthesis upon its proper localization, probably through dephosphorylation, release from the vesicles, or interaction within different protein–protein complexes than during transport.
Synaptic vesicles are transported along the cytoskeleton to the neurites, and disruption of the cytoskeleton network is thus expected to cause the accumulation of the recombinant TH in the cells' soma, as observed here (Fig. 5B). Our results also explain previous observations showing that mice bearing a Cdk5 knock-out mutation show decreased striatal THSer(P)-31 (25). Furthermore, it has been described that the highest stoichiometry of THSer(P)-31 was found in the terminals of the nigrostriatal and mesoaccumbens pathways (23) and that Ser-31 phosphorylation was decreased in conjunction with TH loss following the 6-hydroxydopamine lesion of the nigrostriatal pathway in rats (27), also in agreement with our observation that overexpression of the PD mutant A53T α-syn decreased the amount of the V5-TH1-S31E protein in the distal regions of the neurites (Fig. 6E). Increasingly, Cdk5 and ERK1/2 have been recognized as crucial for many processes in neurodegeneration (66, 67). We therefore cannot rule out effects of these kinases on other important players in axonal transport, which could be expected to have consequences for specific transport of TH beyond Ser-31 phosphorylation.
In conclusion, our results identify a novel role for TH phosphorylation at Ser-31, controlling TH co-distribution with synaptic vesicles through association with VMAT2 and α-syn. Our results also point to the role of Ser-31 phosphorylation of TH on the transport of this enzyme from the cell soma to the terminals using the microtubule network. In addition, the distribution of THSer(P)-31 is affected when the PD mutant α-syn A53T is present. Thus, TH spatial control by Ser-31 phosphorylation implies that TH localization and DA synthesis would be directly affected by disturbances such as the loss of vesicle integrity, the defective vesicle trafficking, and the GC fragmentation described in PD (8, 68–70). Indeed, axonal transport defects correlate with decreased putamen TH levels in early PD patients, whereas in late-stage patients, TH is decreased in both putamen and substantia nigra (7, 71). Our work also points toward the potential of therapeutic avenues aimed at reverting neuronal mislocalization and transport alterations associated with PD (7, 65, 71–73).
Experimental procedures
All reagents were supplied by Sigma except when indicated.
Constructs for transient expression in mammalian cell culture
WT human TH1 coding sequence was inserted into the pcDNA6.2/nTC-Tag-DEST vector. Mutations V5-TH1-S19A, V5-TH1-S19E, V5-TH1-S31A, V5-TH1-S31E, V5-TH1-S40A, and V5-TH1-S40E were introduced using QuikChange Mutagenesis II (Stratagene) and primers specified in Table 1. Mutations were verified by sequencing. Soluble enhanced green fluorescent protein (GFP) was purchased from Clontech. pHM6-α-synuclein-A53T mutant was a gift from David Rubinsztein (Addgene plasmids 40824 and 40825) and described previously (74).
Table 1.
Sequence of primers used for site-directed mutagenesis
| Primer | Forward strand sequence 5′–3′ |
|---|---|
| TH1-S19A | CTTCCGCAGGGCCGTGGCGGAGCTGGACGCCAAGC |
| TH1-S19E | CGCAGGGCCGTGGAGGAGCTGGACGCCAAG |
| TH1-S31A | GGCCATCATGGCCCCGCGGTTC |
| TH1-S31E | CAGAGGCCATCATGGAGCCGCGGTTCATTG |
| TH1-S40A | CATTGGGCGCAGGCAGGCGCTCATCGAGGACGCCCG |
| TH1-S40E | GGGCGCAGGCAGGAACTCATCGAGGAC |
Cell culture
Rat pheochromocytoma PC12Adh cells (ATCC-CRL-1721.1) (<8 passages) were grown in RPMI 1640 medium with 10% horse serum (PAA Laboratories GmbH), 5% fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Adherent PC12 derived from Ref. 35 underwent short-term repeat profiling and presented genetic drift, so it has been denoted PC12* to distinguish it the from PC12Adh strain provided by ATCC (PC12Adh). Culture conditions for both strains were identical. When specified, cells were treated for 48 h with 50 ng/ml 2.5S NGF (Life Technologies, Inc.) in OptiMEM I (Life Technologies), leading to well-developed neurites in PC12*; however, PC12Adh cells do not develop significant projections upon NGF stimulation according to ATCC (strain specifications). Human neuroblastoma SH-SY5Y and HEK293 cells were grown in DMEM with 10% fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were profiled by short tandem repeat. Commercial midbrain dopaminergic neurons generated from human induced pluripotent stem iCell DopaNeuron cells (Cellular Dynamics) were grown following the manufacturer's indications and were analyzed 7–10 days postseeding. For immunofluorescence, PC12* or SH-SY5Y cells were grown for 48 h on poly-l-lysine or PureCol (Inamed Biomaterials) and laminin/collagen–coated coverslips, respectively. iCell DopaNeurons were grown on coverslips coated with polyornithine and laminin. For the coating of the coverslips, a final concentration of 0.1 mg/ml poly-l-lysine, 1.5 mg/ml laminin plus 1.5 mg/ml collagen or a 10 μg/ml polyornithine plus 20 μg/ml laminin solution was placed on the coverslips and incubated for 30 min at 37 °C and washed with PBS before seeding the cells.
Transient transfection of neuroblastoma SH-SY5Y for TH overexpression
For Western blotting, SH-SY5Y or HEK293 cells were seeded on (1 × 106 cells/p60 plate) and grown overnight before transfecting with 5 μg of DNA and Lipofectamine LTX with Plus reagent (Life Technologies), following the manufacturer's instructions. Cells were collected 48 h post-transfection. For imaging experiments, 20,000 SH-SY5Y cells seeded on coverslips were transfected with 0.5 μg of DNA and Lipofectamine LTX with Plus reagent. 48 h post-transfection, cells were subjected to the indicated treatments and fixed with 4% paraformaldehyde. In the case of co-transfections, a 1:1 ratio of both co-transfected plasmids were used except for PLA assays where the ratio of interest/control plasmid was 3:1.
Inhibition of THSer(P)-31 phosphorylation
Cells were incubated 7 h with roscovitine (50 μm) and SL327 (50 μm) before being collected and frozen or subjected to drug washout by incubating with fresh medium. In the case of transfected cells treated with the inhibitors, and to ensure equal expression of the constructs in treated and untreated samples, 24 h post-transfection, cells were split into two p35 plates and were left to grow an additional day before the treatment.
GC and microtubule disassembly
GC was disrupted by incubating PC12* cells for 30 min with 5 μg/ml brefeldin A (Epicenter Technologies). Microtubule disassembly was achieved by a 1-min 4 °C cold shock followed by 30-min 33 μm nocodazole incubation at 37 °C. Cells were fixed in cold methanol. In all cases, drug washout was performed by replacement of drug-containing medium with fresh media for 30 min.
Immunostaining and proximity ligation assays
Samples were fixed with 4% paraformaldehyde for 30 min at room temperature, unless otherwise indicated, and permeabilized and blocked with 0.3% saponin and 5% FBS in PBS for 30 min at room temperature. When stated, a 5-min 0.1% Triton X-100 permeabilization step was performed before blocking. Samples were incubated with specified primary and secondary antibodies in Tables 2 and 3 and, when indicated, stained with 1:200 Oregon Green or tetramethylrhodamine-labeled wheat germ agglutinin (Life Technologies) before being mounted using ProLong Gold with DAPI (Molecular Probes). PLAs were performed using Duolink® in situ according to the manufacturer's instructions and the primary antibody conditions stated above.
Table 2.
Primary antibodies used in Western blotting and immunofluorescence
| Target protein | Host | Supplier | Western blot dilution | Immunofluorescence dilution |
|---|---|---|---|---|
| TH (total) | Rabbit | Thermo Scientific | 1:1000 | 1:100 |
| THSer(P)-19 | Rabbit | Phosphosolutions | 1:1000 | 1:50 |
| THSer(P)-31 | Rabbit | Phosphosolutions | 1:1000 | 1:50 |
| THSer(P)-31 | Rabbit | Ref. 26 | 1:300 | 1:50 |
| THSer(P)-40 | Rabbit | Phosphosolutions | 1:1000 | 1:50 |
| GM130 | Mouse | BD Transduction Laboratories | 1:1000 | 1:100 |
| His | Mouse | GenScript | NAa | 1:100 |
| Synaptotagmin I | Mouse | Abcam | 1:1000 | 1:100 |
| GAPDH | Rabbit | Abcam | 1:1000 | NA |
| V5 | Rabbit | Sigma | 1:2000 | 1:100 |
| V5 | Mouse | Life Technologies | 1:5000 | 1:100 |
| VMAT2 | Rabbit | Millipore | 1:1000 | 1:100 |
| VMAT2 | Goat | Santa Cruz Biotechnology | 1:1000 | 1:100 |
| α-Synuclein 3H9 | Mouse | Abcam | 1:1000 | 1:100 |
| Tubulin | Mouse | Sigma | NA | 1:1000 |
| Clathrin | Mouse | Thermo Scientific | 1:1000 | NA |
| Transferrin receptor | Mouse | Invitrogen | 1:1000 | NA |
| SPC25 | Rabbit | Gift from Stephen High | 1:1000 | NA |
| DOPA-β-hydroxylase | Sheep | Abcam | 1:1000 | NA |
| Chromogranin A | Rabbit | Novus Biologicals | 1:1000 | NA |
a NA, not applicable.
Table 3.
Secondary antibodies used in Western blotting and immunofluorescence
| Target protein | Host | Supplier | Conjugation | Western blot dilution | Immunofluorescence dilution |
|---|---|---|---|---|---|
| IgG Rabbit (H+L) | Goat | Invitrogen | Alexa Fluor 488 | NAa | 1:200 |
| IgG Mouse (H+L) | Goat | Invitrogen | Alexa Fluor 555 | NA | 1:200 |
| IgG Rabbit (H+L) | Goat | Invitrogen | Alexa Fluor 594 | NA | 1:200 |
| IgG Rabbit (H+L) | Goat | Invitrogen | Alexa Fluor 647 | NA | 1:200 |
| IgG Mouse | Goat | Santa Cruz Biotechnology | HRP | 1:1000 | NA |
| IgG Goat | Donkey | Santa Cruz Biotechnology | HRP | 1:1000 | NA |
| IgG Rabbit | Goat | Santa Cruz Biotechnology | HRP | 1:1000 | NA |
| IgG Sheep | Donkey | Santa Cruz Biotechnology | HRP | 1:1000 | NA |
a NA, not applicable.
Confocal laser-scanning microscopy imaging
Confocal imaging was performed on a Leica microscope TCS SP5 in the resonant scanner mode (Leica Microsystems GmbH) using a pinhole airy 1 and a ×63, 1.4 numeric aperture oil immersion objective. For each sample, a stack of images encompassing the complete height of the cell was taken, with a 130-nm step size and using the LasAF software from Leica. Each confocal plane was 512 × 512 pixels with a line average of 20. Stack images were processed in batch using FIJI freeware (75) and/or Photoshop Adobe with minimum adjustments of brightness and background. Single-plane surface plots and signal quantification were prepared using FIJI (75). 3D rendering of Z-stacks was performed using Imaris (Bitplane Inc.), building a co-localization channel between the green and red channels and representing all three using the surface tool. Imaris was employed to quantify the PLA signals by building a co-localization channel between the green (GFP) and red (PLA) signals to eliminate nonspecific PLA signals and set the sphere a radius of 300 nm/signal. The surpass function “spot” tool used to detect the PLA signals, and the same segmentation threshold was used for all images. The numbers of spots obtained were compared using a t test analysis.
For the image analysis of DopaNeuron cells, maximal projections of Z-stacks of the whole cells' height were obtained. Randomly selected cell somas and neurite sections (50–90 μm) located >135 μm away from their soma were manually traced on the tubulin channel using LasAF Lite software (Leica), and the mean intensity of the traced area was recorded for the THSer(P)-31 channel. Average and S.D. values were obtained for each sample set, and p values were calculated using a t test analysis. For the image analysis of the neuroblastoma neurites transfected with V5-TH1 and His-α-syn, maximal projections of Z-stacks of the first micron of the sample were obtained, neurites of a length of approximately 65–95 and 1–2 μm were selected and manually traced on the WGA channel with a 4-pixel-wide line using FIJI, and the intensity profile was recorded for the V5 channel. Using Excel, neurite signal intensity was normalized to the cell soma intensity to minimize variations due to expression levels. The area below the curve corresponding to the most distal 20 μm of the neurite was integrated, average and S.D. values were obtained for each co-transfection set, and p values were calculated using a t test analysis.
Cellular fractionations and chromaffin vesicle membranes
Subcellular fractionation was performed according to Ref. 76 with the only modification that for SH-SY5Y the last centrifugation step to sediment the microsome fraction was carried out for 90 min. Chromaffin vesicle membranes (CVMs) were purified from bovine adrenal medulla as described (46). The CVMs were treated with trypsin (bovine pancreas; Sigma-Aldrich) (1 mg of CVM protein, 1.5 mg of trypsin) for 2 h at 30 °C. The reaction was stopped by adding soybean trypsin inhibitor at a 1:2 ratio (mg of trypsin/mg of inhibitor).
Western blot analysis
Proteins were separated on SDS-PAGE 10% TGXTM gels (Bio-Rad) and transferred onto nitrocellulose or PVDF membranes using the TransBlot Turbo system (Bio-Rad). Membranes were incubated with the primary and secondary antibodies specified in Tables 2 and 3, developed by chemiluminescence, and visualized with a ChemiDoc instrument, and band intensities were quantified by Image Lab software (Bio-Rad). For data analysis, the intensity of target proteins was standardized with the loading control. In the case of each recombinant TH1, the microsomal fraction was normalized against its corresponding whole lysate to minimize the effect of stability and transfection efficiency differences. Treated samples were referenced to untreated, which were given the arbitrary value of 1. The sample size in all cases was n = 3, and Microsoft Excel was used for statistical calculations. Two-way comparison was performed using the t test. Statistical significance was set at p < 0.05.
Preparation of mouse brain lysates and immunoprecipitation
Mouse whole brain was homogenized with a Tissue Lyser II (Qiagen) in IP buffer (20 mm Hepes, pH 7.4, 125 mm NaCl, 1 mm EDTA, 2 mm PMSF), containing protease and phosphatase inhibitors (Roche Applied Science). Extract was clarified by centrifugation at 16,000 × g for 20 min at 4 °C. The supernatant was collected, and Triton X-100 was added to a final concentration of 1%. This sample was centrifuged at 4 °C at 20,000 × g for 15 min after rotation for 1 h at 4 °C. The soluble extract was incubated with antibodies against THSer(P)-31 (rabbit; described previously (26), VMAT2 (C-20) (goat; Millipore), α-synuclein (3H9) (mouse; Abcam), control IgGs (Millipore Merck), or no antibody (only beads) with rotation overnight at 4 °C. Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology, Inc.) were added to samples and rotated for 1 h at 4 °C before samples were pelleted, washed, and incubated at 37 °C for 30 min in 40 μl of sample buffer. Samples were analyzed by SDS-PAGE and immunoblotting with anti-THSer(P)-31, VMAT2 (rabbit; Millipore), and anti-TH (rabbit; Thermo Scientific) antibodies as primary antibodies and anti-rabbit IgG light chain (HRP) (Abcam) antibodies as secondary antibodies, respectively.
TH purification and phosphorylation
Human TH1 was expressed in Escherichia coli (BL21 Codon Plus (DE3), Stratagene) as a His-ZZ-TH1 fusion protein (15) and purified using Talon resin (New England Biolabs) according to the manufacturer's recommendations. The fusion tag was removed by proteolytic cleavage using tobacco etch virus (1:25 (mg) tobacco etch virus/TH) in 15 mm Hepes, pH 7.4, 150 mm NaCl (HBS) for 4 h on ice before centrifugation (13,000 × g, 10 min) and gel filtration (Superdex 200 HR10/30, GE Healthcare). TH1 (50 μm) was phosphorylated for 45 min at 25 °C in HBS buffer using 500 μm ATP, 5 mm MgCl2, and 12.5 units/ml active p35/CDK5 (Millipore; 14-477) to a stoichiometry of 0.5 mol of phosphate/mol of TH subunits as determined by incorporation of 32P using [γ-32P]ATP.
Surface plasmon resonance
The Biacore 3000 system was used with L1 sensor chips and HBS-N buffer (GE Healthcare; BR-1003-69). The L1 surface was loaded with CVM (150 μg of protein/ml, 4–6 min, 3–10 μl/min) according to the manufacturer's recommendation by ensuring surface saturation and minimal binding of BSA. 20 mm CHAPS was used to regenerate the surface. Binding of TH was monitored using different flow rates (5–30 μl/min) and different concentrations of TH (0.1–25 μm).
Author contributions
A. J.-F. performed all cell culture experiments, immnunofluorescence assays, PLAs, and Western blots; designed experiments; interpreted the data; and wrote the paper. R. K. performed SPR experiments, purified and phosphorylated the protein, designed experiments, interpreted the data, and edited the paper. K. J. K. C. performed Western blotting and interpreted the data. M. Y. performed the IP experiments, interpreted the data, and edited the paper. M. F. S. prepared the THSer(P)-31 antibody and edited the paper. M. M., I. R.-M., and J. S. discussed experiments and edited the paper. A. M. coordinated the project, designed experiments, interpreted the data, and wrote the paper.
Acknowledgments
Prof. Clive Bramham kindly provided the SH-SY5Y cells. All imaging was performed at the Molecular Imaging Center, Department of Biomedicine, University of Bergen, Norway.
This work was supported by Research Council of Norway Grant FRIMEDBIO 214012 and the K. G. Jebsen Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
- PD
- Parkinson's disease
- l-DOPA
- l-3,4-dihydroxyphenylalanine
- GC
- Golgi complex
- TH
- tyrosine hydroxylase
- DA
- dopamine
- VMAT2
- vesicular monoamine transporter 2
- NGF
- neuronal growth factor
- α-syn
- α-synuclein
- R/S
- roscovitine and SL327
- sytI
- synaptotagmin I
- PLA
- proximity ligation assay
- WGA
- wheat germ agglutinin
- CVM
- chromaffin vesicle membrane.
References
- 1. Pifl C., Rajput A., Reither H., Blesa J., Cavada C., Obeso J. A., Rajput A. H., and Hornykiewicz O. (2014) Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 34, 8210–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trillo L., Das D., Hsieh W., Medina B., Moghadam S., Lin B., Dang V., Sanchez M. M., De Miguel Z., Ashford J. W., and Salehi A. (2013) Ascending monoaminergic systems alterations in Alzheimer's disease: translating basic science into clinical care. Neurosci. Biobehav. Rev. 37, 1363–1379 [DOI] [PubMed] [Google Scholar]
- 3. Willemsen M. A., Verbeek M. M., Kamsteeg E. J., de Rijk-van Andel J. F., Aeby A., Blau N., Burlina A., Donati M. A., Geurtz B., Grattan-Smith P. J., Haeussler M., Hoffmann G. F., Jung H., de Klerk J. B., van der Knaap M. S., et al. (2010) Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis. Brain 133, 1810–1822 [DOI] [PubMed] [Google Scholar]
- 4. Wijemanne S., and Jankovic J. (2015) Dopa-responsive dystonia–clinical and genetic heterogeneity. Nat. Rev. Neurol. 11, 414–424 [DOI] [PubMed] [Google Scholar]
- 5. Gondré-Lewis M. C., Park J. J., and Loh Y. P. (2012) Cellular mechanisms for the biogenesis and transport of synaptic and dense-core vesicles. Int. Rev. Cell Mol. Biol. 299, 27–115 [DOI] [PubMed] [Google Scholar]
- 6. Alerte T. N., Akinfolarin A. A., Friedrich E. E., Mader S. A., Hong C. S., and Perez R. G. (2008) α-Synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci. Lett. 435, 24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chu Y., Morfini G. A., Langhamer L. B., He Y., Brady S. T., and Kordower J. H. (2012) Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain 135, 2058–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakagomi S., Barsoum M. J., Bossy-Wetzel E., Sütterlin C., Malhotra V., and Lipton S. A. (2008) A Golgi fragmentation pathway in neurodegeneration. Neurobiol. Dis. 29, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cartier E. A., Parra L. A., Baust T. B., Quiroz M., Salazar G., Faundez V., Egaña L., and Torres G. E. (2010) A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J. Biol. Chem. 285, 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haycock J. W. (2002) Species differences in the expression of multiple tyrosine hydroxylase protein isoforms. J. Neurochem. 81, 947–953 [DOI] [PubMed] [Google Scholar]
- 11. Ichikawa S., Ichinose H., and Nagatsu T. (1990) Multiple mRNAs of monkey tyrosine hydroxylase. Biochem. Biophys. Res. 173, 1331–1336 [DOI] [PubMed] [Google Scholar]
- 12. Haycock J. W. (1990) Phosphorylation of tyrosine hydroxylase in situ at serine 8, 19, 31, and 40. J. Biol. Chem. 265, 11682–11691 [PubMed] [Google Scholar]
- 13. Dunkley P. R., Bobrovskaya L., Graham M. E., von Nagy-Felsobuki E. I., and Dickson P. W. (2004) Tyrosine hydroxylase phosphorylation: regulation and consequences. J. Neurochem. 91, 1025–1043 [DOI] [PubMed] [Google Scholar]
- 14. Itagaki C., Isobe T., Taoka M., Natsume T., Nomura N., Horigome T., Omata S., Ichinose H., Nagatsu T., Greene L. A., and Ichimura T. (1999) Stimulus-coupled interaction of tyrosine hydroxylase with 14-3-3 proteins. Biochemistry 38, 15673–15680 [DOI] [PubMed] [Google Scholar]
- 15. Kleppe R., Rosati S., Jorge-Finnigan A., Alvira S., Ghorbani S., Haavik J., Valpuesta J. M., Heck A. J., and Martinez A. (2014) Phosphorylation dependence and stoichiometry of the complex formed by tyrosine hydroxylase and 14-3-3γ. Mol. Cell. Proteomics 13, 2017–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harada K., Wu J., Haycock J. W., and Goldstein M. (1996) Regulation of l-DOPA biosynthesis by site-specific phosphorylation of tyrosine hydroxylase in AtT-20 cells expressing wild-type and serine 40-substituted enzyme. J. Neurochem. 67, 629–635 [DOI] [PubMed] [Google Scholar]
- 17. Gordon S. L., Bobrovskaya L., Dunkley P. R., and Dickson P. W. (2009) Differential regulation of human tyrosine hydroxylase isoforms 1 and 2 in situ: isoform 2 is not phosphorylated at Ser35. Biochim. Biophys. Acta 1793, 1860–1867 [DOI] [PubMed] [Google Scholar]
- 18. Haycock J. W., Ahn N. G., Cobb M. H., and Krebs E. G. (1992) ERK1 and ERK2, two microtubule-associated protein 2 kinases, mediate the phosphorylation of tyrosine hydroxylase at serine-31 in situ. Proc. Natl. Acad. Sci. U.S.A. 89, 2365–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kansy J. W., Daubner S. C., Nishi A., Sotogaku N., Lloyd M. D., Nguyen C., Lu L., Haycock J. W., Hope B. T., Fitzpatrick P. F., and Bibb J. A. (2004) Identification of tyrosine hydroxylase as a physiological substrate for Cdk5. J. Neurochem. 91, 374–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sutherland C., Alterio J., Campbell D. G., Le Bourdellès B., Mallet J., Haavik J., and Cohen P. (1993) Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase and MAP-kinase-activated kinases 1 and 2. Eur. J. Biochem. 217, 715–722 [DOI] [PubMed] [Google Scholar]
- 21. Salvatore M. F., Waymire J. C., and Haycock J. W. (2001) Depolarization-stimulated catecholamine biosynthesis: involvement of protein kinases and tyrosine hydroxylase phosphorylation sites in situ. J. Neurochem. 79, 349–360 [DOI] [PubMed] [Google Scholar]
- 22. Lehmann I. T., Bobrovskaya L., Gordon S. L., Dunkley P. R., and Dickson P. W. (2006) Differential regulation of the human tyrosine hydroxylase isoforms via hierarchical phosphorylation. J. Biol. Chem. 281, 17644–17651 [DOI] [PubMed] [Google Scholar]
- 23. Salvatore M. F., and Pruett B. S. (2012) Dichotomy of tyrosine hydroxylase and dopamine regulation between somatodendritic and terminal field areas of nigrostriatal and mesoaccumbens pathways. PLoS One 7, e29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mitchell J. P., Hardie D. G., and Vulliet P. R. (1990) Site-specific phosphorylation of tyrosine hydroxylase after KCl depolarization and nerve growth factor treatment of PC12 cells. J. Biol. Chem. 265, 22358–22364 [PubMed] [Google Scholar]
- 25. Moy L. Y., and Tsai L. H. (2004) Cyclin-dependent kinase 5 phosphorylates serine 31 of tyrosine hydroxylase and regulates its stability. J. Biol. Chem. 279, 54487–54493 [DOI] [PubMed] [Google Scholar]
- 26. Salvatore M. F., Pruett B. S., Spann S. L., and Dempsey C. (2009) Aging reveals a role for nigral tyrosine hydroxylase Ser31 phosphorylation in locomotor activity generation. PLoS One 4, e8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salvatore M. F. (2014) Ser31 tyrosine hydroxylase phosphorylation parallels differences in dopamine recovery in nigrostriatal pathway following 6-OHDA lesion. J. Neurochem. 129, 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuczenski R. T., and Mandell A. J. (1972) Regulatory properties of soluble and particulate rat brain tyrosine hydroxylase. J. Biol. Chem. 247, 3114–3122 [PubMed] [Google Scholar]
- 29. Parra L. A., Baust T. B., Smith A. D., Jaumotte J. D., Zigmond M. J., Torres S., Leak R. K., Pino J. A., and Torres G. E. (2016) The molecular chaperone Hsc70 interacts with tyrosine hydroxylase to regulate enzyme activity and synaptic vesicle localization. J. Biol. Chem. 291, 17510–17522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 31. Perez R. G., Waymire J. C., Lin E., Liu J. J., Guo F., and Zigmond M. J. (2002) A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Obeso J. A., Rodriguez-Oroz M. C., Goetz C. G., Marin C., Kordower J. H., Rodriguez M., Hirsch E. C., Farrer M., Schapira A. H., and Halliday G. (2010) Missing pieces in the Parkinson's disease puzzle. Nat. Med. 16, 653–661 [DOI] [PubMed] [Google Scholar]
- 33. Jedynak J. P., Ali S. F., Haycock J. W., and Hope B. T. (2002) Acute administration of cocaine regulates the phosphorylation of serine-19, -31 and -40 in tyrosine hydroxylase. J. Neurochem. 82, 382–388 [DOI] [PubMed] [Google Scholar]
- 34. Nakashima A., Mori K., Kaneko Y. S., Hayashi N., Nagatsu T., and Ota A. (2011) Phosphorylation of the N-terminal portion of tyrosine hydroxylase triggers proteasomal digestion of the enzyme. Biochem. Biophys. Res. Commun. 407, 343–347 [DOI] [PubMed] [Google Scholar]
- 35. Sannerud R., Marie M., Hansen B. B., and Saraste J. (2008) Use of polarized PC12 cells to monitor protein localization in the early biosynthetic pathway. Methods Mol. Biol. 457, 253–265 [DOI] [PubMed] [Google Scholar]
- 36. Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H. G., and Døskeland S. O. (2003) cAMP analog mapping of Epac1 and cAMP kinase: discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 278, 35394–35402 [DOI] [PubMed] [Google Scholar]
- 37. Miranda-Barrientos J., Nieto-Mendoza E., and Hernandez-Echeagaray E. (2014) The Cdk5 inhibitor Roscovitine increases LTP induction in corticostriatal synapses. ASN Neuro 10.1042/AN20140006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Groblewski P. A., Franken F. H., and Cunningham C. L. (2011) Inhibition of extracellular signal-regulated kinase (ERK) activity with SL327 does not prevent acquisition, expression, and extinction of ethanol-seeking behavior in mice. Behav. Brain Res. 217, 399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Koike T., and Takashima A. (1984) Clonal variability of Pc12-pheochromocytoma cells with respect to catecholamine biosynthesis. J. Neurochem. 42, 1472–1475 [DOI] [PubMed] [Google Scholar]
- 40. Clementi E., Racchetti G., Zacchetti D., Panzeri M. C., and Meldolesi J. (1992) Differential expression of markers and activities in a group of PC12 nerve cell clones. Eur. J. Neurosci. 4, 944–953 [DOI] [PubMed] [Google Scholar]
- 41. Göttle M., Burhenne H., Sutcliffe D., and Jinnah H. A. (2013) Purine metabolism during neuronal differentiation: the relevance of purine synthesis and recycling. J. Neurochem. 127, 805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alvarez D., Callejo M., Shoucri R., Boyer L., Price G. B., and Zannis-Hadjopoulos M. (2003) Analysis of the cruciform binding activity of recombinant 14-3-3ζ-MBP fusion protein, its heterodimerization profile with endogenous 14-3-3 isoforms, and effect on mammalian DNA replication in vitro. Biochemistry 42, 7205–7215 [DOI] [PubMed] [Google Scholar]
- 43. van Rijn R. M., Chazot P. L., Shenton F. C., Sansuk K., Bakker R. A., and Leurs R. (2006) Oligomerization of recombinant and endogenously expressed human histamine H(4) receptors. Mol. Pharmacol. 70, 604–615 [DOI] [PubMed] [Google Scholar]
- 44. Halskau Ø. Jr., Ying M., Baumann A., Kleppe R., Rodriguez-Larrea D., Almås B., Haavik J., and Martinez A. (2009) Three-way interaction between 14-3-3 proteins, the N-terminal region of tyrosine hydroxylase, and negatively charged membranes. J. Biol. Chem. 284, 32758–32769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nilsson I., Bahram F., Li X., Gualandi L., Koch S., Jarvius M., Söderberg O., Anisimov A., Kholová I., Pytowski B., Baldwin M., Ylä-Herttuala S., Alitalo K., Kreuger J., and Claesson-Welsh L. (2010) VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 29, 1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Terland O., and Flatmark T. (1980) Oxidoreductase activities of chromaffin granule ghosts isolated from the bovine adrenal medulla. Biochim. Biophys. Acta 597, 318–330 [DOI] [PubMed] [Google Scholar]
- 47. Lee H. J., Khoshaghideh F., Lee S., and Lee S. J. (2006) Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur. J. Neurosci. 24, 3153–3162 [DOI] [PubMed] [Google Scholar]
- 48. Koch J. C., Bitow F., Haack J., d'Hedouville Z., Zhang J. N., Tönges L., Michel U., Oliveira L. M., Jovin T. M., Liman J., Tatenhorst L., Bähr M., and Lingor P. (2015) α-Synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis. 6, e1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zatloukal B., Kufferath I., Thueringer A., Landegren U., Zatloukal K., and Haybaeck J. (2014) Sensitivity and specificity of in situ proximity ligation for protein interaction analysis in a model of steatohepatitis with Mallory-Denk bodies. PLoS One 9, e96690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peng X., Tehranian R., Dietrich P., Stefanis L., and Perez R. G. (2005) α-Synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 118, 3523–3530 [DOI] [PubMed] [Google Scholar]
- 51. Guo J. T., Chen A. Q., Kong Q., Zhu H., Ma C. M., and Qin C. (2008) Inhibition of vesicular monoamine transporter-2 activity in α-synuclein stably transfected SH-SY5Y cells. Cell Mol. Neurobiol. 28, 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prado V. F., Roy A., Kolisnyk B., Gros R., and Prado M. A. (2013) Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem. J. 450, 265–274 [DOI] [PubMed] [Google Scholar]
- 53. Buddhala C., Hsu C. C., and Wu J. Y. (2009) A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem. Int. 55, 9–12 [DOI] [PubMed] [Google Scholar]
- 54. Dobransky T., and Rylett R. J. (2005) A model for dynamic regulation of choline acetyltransferase by phosphorylation. J. Neurochem. 95, 305–313 [DOI] [PubMed] [Google Scholar]
- 55. Lou H., Montoya S. E., Alerte T. N., Wang J., Wu J., Peng X., Hong C. S., Friedrich E. E., Mader S. A., Pedersen C. J., Marcus B. S., McCormack A. L., Di Monte D. A., Daubner S. C., and Perez R. G. (2010) Serine 129 phosphorylation reduces the ability of α-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 285, 17648–17661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Requena D. F., Parra L. A., Baust T. B., Quiroz M., Leak R. K., Garcia-Olivares J., and Torres G. E. (2009) The molecular chaperone Hsc70 interacts with the vesicular monoamine transporter-2. J. Neurochem. 110, 581–594 [DOI] [PubMed] [Google Scholar]
- 57. Nirenberg M. J., Chan J., Liu Y., Edwards R. H., and Pickel V. M. (1996) Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J. Neurosci. 16, 4135–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bellucci A., Zaltieri M., Navarria L., Grigoletto J., Missale C., and Spano P. (2012) From α-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson's disease. Brain Res. 1476, 183–202 [DOI] [PubMed] [Google Scholar]
- 59. Cooper A. A., Gitler A. D., Cashikar A., Haynes C. M., Hill K. J., Bhullar B., Liu K., Xu K., Strathearn K. E., Liu F., Cao S., Caldwell K. A., Caldwell G. A., Marsischky G., Kolodner R. D., et al. (2006) α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brimijoin S., and Wiermaa M. J. (1977) Rapid axonal transport of tyrosine hydroxylase in rabbit sciatic nerves. Brain Res. 121, 77–96 [DOI] [PubMed] [Google Scholar]
- 61. Gervasi N. M., Scott S. S., Aschrafi A., Gale J., Vohra S. N., MacGibeny M. A., Kar A. N., Gioio A. E., and Kaplan B. B. (2016) The local expression and trafficking of tyrosine hydroxylase mRNA in the axons of sympathetic neurons. RNA 22, 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wooten G. F., and Coyle J. T. (1973) Axonal transport of catecholamine synthesizing and metabolizing enzymes. J. Neurochem. 20, 1361–1371 [DOI] [PubMed] [Google Scholar]
- 63. Korner G., Noain D., Ying M., Hole M., Flydal M. I., Scherer T., Allegri G., Rassi A., Fingerhut R., Becu-Villalobos D., Pillai S., Wueest S., Konrad D., Lauber-Biason A., Baumann C. R., et al. (2015) Brain catecholamine depletion and motor impairment in a Th knock-in mouse with type B tyrosine hydroxylase deficiency. Brain 138, 2948–2963 [DOI] [PubMed] [Google Scholar]
- 64. Royo M., Fitzpatrick P. F., and Daubner S. C. (2005) Mutation of regulatory serines of rat tyrosine hydroxylase to glutamate: effects on enzyme stability and activity. Arch. Biochem. Biophys. 434, 266–274 [DOI] [PubMed] [Google Scholar]
- 65. Encalada S. E., and Goldstein L. S. (2014) Biophysical challenges to axonal transport: motor-cargo deficiencies and neurodegeneration. Annu. Rev. Biophys. 43, 141–169 [DOI] [PubMed] [Google Scholar]
- 66. Shah K., and Lahiri D. K. (2014) Cdk5 activity in the brain: multiple paths of regulation. J. Cell Sci. 127, 2391–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Subramaniam S., and Unsicker K. (2010) ERK and cell death: ERK1/2 in neuronal death. FEBS J. 277, 22–29 [DOI] [PubMed] [Google Scholar]
- 68. Alter S. P., Lenzi G. M., Bernstein A. I., and Miller G. W. (2013) Vesicular integrity in Parkinson's disease. Curr. Neurol. Neurosci. Rep. 13, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kurian M. A., Gissen P., Smith M., Heales S. Jr, and Clayton P. T. (2011) The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol. 10, 721–733 [DOI] [PubMed] [Google Scholar]
- 70. Sun K. H., de Pablo Y., Vincent F., Johnson E. O., Chavers A. K., and Shah K. (2008) Novel genetic tools reveal Cdk5's major role in Golgi fragmentation in Alzheimer's disease. Mol. Biol. Cell 19, 3052–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kordower J. H., Chu Y., Hauser R. A., Freeman T. B., and Olanow C. W. (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506 [DOI] [PubMed] [Google Scholar]
- 72. Lohr K. M., Bernstein A. I., Stout K. A., Dunn A. R., Lazo C. R., Alter S. P., Wang M., Li Y., Fan X., Hess E. J., Yi H., Vecchio L. M., Goldstein D. S., Guillot T. S., Salahpour A., and Miller G. W. (2014) Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, 9977–9982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Millecamps S., and Julien J. P. (2013) Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neuroscience 14, 161–176 [DOI] [PubMed] [Google Scholar]
- 74. Furlong R. A., Narain Y., Rankin J., Wyttenbach A., and Rubinsztein D. C. (2000) α-Synuclein overexpression promotes aggregation of mutant huntingtin. Biochem. J. 346, 577–581 [PMC free article] [PubMed] [Google Scholar]
- 75. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ying M., Flatmark T., and Saraste J. (2000) The p58-positive pre-Golgi intermediates consist of distinct subpopulations of particles that show differential binding of COPI and COPII coats and contain vacuolar H(+)-ATPase. J. Cell Sci. 113, 3623–3638 [DOI] [PubMed] [Google Scholar]