

Abstract
Smooth muscle cell (SMC) differentiation is essential for vascular development, and TGF-β signaling plays a critical role in this process. Although long non-coding RNAs (lncRNAs) regulate various cellular events, their functions in SMC differentiation remain largely unknown. Here, we demonstrate that the lncRNA growth arrest-specific 5 (GAS5) suppresses TGF-β/Smad3 signaling in smooth muscle cell differentiation of mesenchymal progenitor cells. We found that forced expression of GAS5 blocked, but knockdown of GAS5 increased, the expression of SMC contractile proteins. Mechanistically, GAS5 competitively bound Smad3 protein via multiple RNA Smad–binding elements (rSBEs), which prevented Smad3 from binding to SBE DNA in TGF-β–responsive SMC gene promoters, resulting in suppression of SMC marker gene transcription and, consequently, in inhibition of TGF-β/Smad3-mediated SMC differentiation. Importantly, other lncRNAs or artificially synthesized RNA molecules that contained rSBEs also effectively inhibited TGF-β/Smad3 signaling, suggesting that lncRNA–rSBE may be a general mechanism used by cells to fine-tune Smad3 activity in both basal and TGF-β–stimulated states. Taken together, our results have uncovered an lncRNA-based mechanism that modulates TGF-β/Smad3 signaling during SMC differentiation.
Keywords: differentiation; long non-coding RNA (long ncRNA, lncRNA); signal transduction; smooth muscle; TGF-β; GAS5; Smad3
Introduction
Smooth muscle cell (SMC)3 differentiation is a pivotal process during vascular development. Defects in this process at embryonic stages are usually lethal (1). Functional impairment in SMC in adults often leads to various cardiovascular diseases, including heart failure, atherosclerosis, hypertension, and stroke (2). SMC can be differentiated from multiple progenitors, such as the neural crest, secondary heart field, somites, mesoangioblasts, proepicardium, splanchnic mesoderm, and mesothelium (3). These differentiation processes are controlled at transcriptional and post-transcriptional levels, including long non-coding RNA (lncRNA) (4–6). However, the mechanisms underlying lncRNA function in SMC differentiation remain largely unknown.
LncRNAs are non-coding RNA molecules longer than 200 nucleotides (7). LncRNAs are produced from various genome contexts, such as antisense transcripts from promoter/enhancer or coding regions, transcripts from intron or intergenic regions (8), or spliced transcripts of pre-mRNAs (9). When transcribed/spliced/processed, most lncRNAs are recruited back to nuclei, where they regulate target gene transcription. Compelling evidence supports that lncRNA regulates gene expression at various levels, including chromatin remodeling (10), transcription (11), translation (12), and protein stability (13). A common machinery controlling lncRNA functions, however, has not been identified. It is generally acknowledged that lncRNAs require protein partners to exert their functions. Indeed, lncRNAs have been shown to directly bind transcription factors to participate in gene transcription (14, 15). However, the general recognition pattern between lncRNA and its protein partners remains unknown.
TGF-β is one of the most potent stimulators for SMC differentiation (16). TGF-β transduces its signal mainly through Smad-dependent pathways (17). The ligands bind and activate TGF-β receptors, leading to Smad phosphorylation and translocation into nuclei, where they activate target gene transcription via binding to Smad-binding elements (SBEs) in gene promoters. It has been proved that lncRNAs are involved in various cardiovascular diseases (5). Although lncRNAs have been shown to regulate TGF-β downstream target genes (18), and TGF-β can activate some of the lncRNAs (19), lncRNAs that directly regulate TGF-β signaling and the related SMC differentiation remain largely unknown.
Growth arrest-specific 5 (GAS5) is a well-known tumor suppressor lncRNA down-regulated in various tumors (20, 21). It functions as a microRNA sponge interacting with miR-21 in breast cancer cells (22). Nuclear GAS5 appears to serve as a protein sponge for glucocorticoid receptors in regulating cell growth arrest (23). GAS5 has also been shown to regulate SMC growth (24, 25) In this study, we identified a previously unknown function of GAS5 in TGF-β signaling. We found that GAS5 negatively regulates Smad3 activity in TGF-β–induced SMC differentiation. Mechanistic studies showed that GAS5 competitively binds to Smad3 via RNA SBE (rSBE), which prevents Smad3 from binding to the SBE in TGF-β target gene promoters, resulting in a negative regulation of Smad3 signaling. Importantly, rSBE modulation of Smad3 activity represents a general mechanism for lncRNA regulation of TGF-β/Smad3 signaling.
Results
GAS5 is physically associated with Smad3 proteins
TGF-β/Smad3 signaling is known to play an important role in SMC differentiation (16). To identify lncRNAs with potential function in TGF-β–induced SMC differentiation, we first screened Smad protein–associated lncRNAs. Thus, RNA immunoprecipitation (RIP) assays were performed to detect Smad-bound lncRNAs in C3H10T1/2 (10T1/2) cells, a mesenchymal cell line widely used as SMC progenitors. As shown in Fig. 1A, among 45 previously annotated lncRNAs, GAS5 showed a high affinity in binding Smad3 and Smad4 but a low affinity in association with Smad2. Most other lncRNAs, including nucleus enriched U6 RNA, showed a low affinity with Smads, whereas a few others, such as B2 short interspersed element (B2 SINE), polymorphic derived intron-containing (Pldi), and Y RNA, showed a better association with Smad2 than Smad3/Smad4. Because Smad3 and Smad4, but not Smad2, directly bind to gene promoter SBE DNA (26), we tested whether there is a physical interaction between GAS5 RNA and Smad3 by performing a biotin-avidin pulldown assay. As shown in Fig. 1B, biotin-labeled GAS5, but not the antisense control, physically associated with endogenous Smad3 in 10T1/2 cells. FISH of GAS5 and immunostaining of Smad3 showed that GAS5 was co-localized with Smad3 in 10T1/2 cells (supplemental Fig. S1), further supporting their physical interaction.
Figure 1.

LncRNA GAS5 physically associates with Smad3 proteins. A, Smad-associated-lncRNA profiling in 10T1/2 cells. RNA–protein complexes were pulled down using anti-Smad2, Smad3, or Smad4 antibody. LncRNAs that were associated with Smad proteins were detected by quantitative RT-PCR. GAS5 strongly associated with Smad3 and Smad4 but not Smad2. **, p < 0.01 compared with the IgG group; n = 3. B, Smad3 co-immunoprecipitated with biotin-labeled GAS5. 10T1/2 cells were transfected with biotin-labeled GAS5, and RNA–protein complexes were pulled down using avidin beads. The presence of Smad3 in the complex was detected by Western blotting. C and D, TGF-β induced GAS5 nuclear export. 10T1/2 cells were treated with 5 ng/ml of TGF-β for 0, 1, 2, 4, 8, and 24 h. Total and nuclear RNA were extracted, and GAS5 was detected by quantitative RT-PCR. GAS5 levels were relative to vehicle-treated cells (0h) after being normalized to GAPDH (for total RNA) or U6 (for nuclear RNA). **, p < 0.01 compared with untreated cells (0h); n = 9. E, TGF-β induced GAS5 nuclear export, as shown by FISH for GAS5. 10T1/2 cells were treated with vehicle (0h) or TGF-β (5 ng/ml) for 2 h (2h). A GAS5 sense probe (sense Ctrl) was used as a control for FISH. Scale bar = 20 μm. F, TGF-β (5 ng/ml) suppressed endogenous Smad3 binding to GAS5 in 10T1/2 cells at 2 h of treatment, as measured by RIP assay. **, p < 0.05 compared with the control (Ctrl/IgG) group; #, p < 0.01 compared with cells without TGF-β induction (Ctrl) but pulled down with Smad3 antibody (Smad3 Ab) (Student's t test, n = 9).
TGF-β induces GAS5 nuclear export
Because Smad3 is translocated into nuclei of cells upon TGF-β induction, we sought to determine whether TGF-β/Smad3 alters GAS5 cellular location. Thus, we treated 10T1/2 cells with 5 ng/ml TGF-β for 0, 1, 2, 4, 8, and 24 h and extracted nuclear RNA using a nucleus extraction kit (EMD Millipore). Total and nuclear GAS5 expression were detected by quantitative RT-PCR. As shown in Fig. 1C, the GAS5 level was not affected during the initial TGF-β treatment (0–4 h) but was decreased at later times (8–24 h, Fig. 1C). Interestingly, a portion of GAS5 was time-dependently exported from nuclei of 10T1/2 cells in the early response to TGF-β treatment (Fig. 1D), when the Smad3 is translocated to the nuclei. At later times, when Smad3 normally shuttles back to the cytoplasm (after 4–8 h of TGF-β induction), GAS5 nuclear accumulation was increased (Fig. 1D). These data indicate that Smad3 may not be involved in GAS5 processing, which was different from microRNAs processed by the Smad3–rSBE interaction (27). GAS5 nuclear exportation was also confirmed by FISH of endogenous GAS5 in 10T1/2 cells treated with 5 ng/ml TGF-β for 2 h (Fig. 1E). Consistently, TGF-β treatment suppressed endogenous GAS5 binding to Smad3 at 2 h (Fig. 1F). Nuclear export of GAS5 or dissociation of GAS5 from Smad3 is likely to be essential for TGF-β signaling transduction.
GAS5 negatively regulates TGF-β-induced smooth muscle cell differentiation
TGF-β induces SMC differentiation of 10T1/2 cells in a Smad3-dependent manner (28). Because GAS5 physically binds Smad3, we sought to determine whether GAS5 affects TGF-β/Smad3-induced SMC differentiation. Forced expression of GAS5 by adenoviral transduction, which increased the total cellular as well as the nuclear GAS5 levels (supplemental Fig. S2, A and B), suppressed both the mRNA and protein expression of SMC-specific genes that were induced by TGF-β (Fig. 2, A–C). Conversely, knockdown of GAS5 by its specific siRNA increased SMC marker gene mRNA and protein expression (Fig. 2, D—F, and supplemental Fig. S2C). Moreover, overexpression of GAS5 diminished TGF-β–induced morphological alteration of 10T1/2 cells (Fig. 2G), further supporting the role of GAS5 in suppressing SMC differentiation.
Figure 2.

GAS5 inhibits TGF-β-induced SMC differentiation. 10T1/2 cells were transduced with an adenovirus expressing GAS5 cDNA (Ad-GAS5) or transfected with GAS5 siRNA (siGAS5) for 24 h and then treated with 5 ng/ml TGF-β for 12 h (for mRNA detection) or 24 h (for protein detection). A and B, forced expression of GAS5 suppressed the mRNA (A) and protein (B) expression of the SMC marker smooth muscle myosin heavy chain (SMMHC), SM22α, and calponin (CNN1). C, quantification of the protein levels shown in B by normalizing to α-tubulin. **, p < 0.01 compared with TGF-β–treated cells with Ad-GFP transduction for each corresponding gene in A and C. D and E, knockdown of GAS5 by siRNA increased SMC marker mRNA (D) and protein (E) expression. F, quantification of the protein levels shown in E by normalizing to α-tubulin. **, p < 0.01 compared with the control siRNA–treated group (siCtrl) with or without TGF-β for each corresponding gene in D and F; n = 9. G, overexpression of GAS5 blocked TGF-β (5 ng/ml)–induced morphological changes in 10T1/2 cells.
To test whether GAS5 suppressed SMC differentiation via inhibition of Smad3 activity, we overexpressed GAS5 along with GFP-tagged Smad3 in 10T1/2 cells. Although GAS5 inhibited SMC marker SM22α expression without affecting Smad3 expression or phosphorylation (Fig. 3, A and B), overexpression of Smad3 restored the SM22α expression suppressed by GAS5 (Fig. 3, A and B). In contrast, knockdown of GAS5 enhanced SM22α expression without altering Smad3 expression or phosphorylation (Fig. 3, C and D). However, blockade of Smad3 activity by its inhibitor SIS3 attenuated GAS5 knockdown-enhanced SM22α expression (Fig. 3, C and D), indicating that GAS5 inhibits SMC differentiation by altering Smad3 activity. Serum response factor (SRF) is known to play a critical role in TGF-β-induced SMC marker gene expression (29). However, GAS5 bound Smad3, but not SRF, in 10T1/2 cells (Fig. 3E), suggesting that GAS5 modulation of SMC differentiation was not related to SRF but mainly a result of blocking Smad3 activity.
Figure 3.

GAS5 suppresses SMC marker expression by inhibiting Smad3 activity. A, GAS5 did not affect Smad3 expression or phosphorylation (pSmad3) but inhibited SM22α expression, which was reversed by Smad3 overexpression. 10T1/2 cells were transfected with pcDNA or pcDNA-Smad3 (79 kDa) and transduced with an adenovirus expressing GFP (Ad-GFP) or GAS5 (Ad-GAS5) for 24 h before treatment with 5 ng/ml of TGF-β. Western blotting was performed to detect SM22α, Smad3, and pSmad3 levels. B, quantification of the protein levels shown in A by normalizing to α-tubulin. **, p < 0.05; n = 9. C, GAS5 knockdown–enhanced SM22α expression was suppressed by the Smad3 inhibitor (SIS3). 10T1/2 cells were transfected with GAS5 siRNA for 24 h and then treated with SIS3 (10 μm) for 24 h. Western blot was performed to detect SM22α, Smad3, and pSmad3 protein levels. D, quantification of the protein levels shown in C by normalizing to α-tubulin. The pSmad3 level was normalized to total Smad3, which was normalized to α-tubulin. **, p < 0.05; n = 9. E, GAS5 associated with Smad3, but not SRF, in 10T1/2 cells, as measured by RIP assay. **, p < 0.01 (Student's t test); n = 9.
GAS5 inhibits Smad3 binding to the SMC gene promoter
Because GAS5 physically interacted with Smad3 and suppressed TGF-β-induced SMC differentiation independent of Smad3 phosphorylation (Figs. 1–3), we sought to determine whether GAS5 is involved in other Smad3 function-related processes. Smad3 is known to translocate into nuclei of cells upon activation or phosphorylation. However, neither overexpression nor knockdown of GAS5 appeared to alter Smad3 nuclear translocation, as observed at 2 h of TGF-β treatment (Fig. 4, A and B). By examining the promoter activity of a Smad3 downstream target gene, SM22α, we found that overexpression of GAS5 dramatically suppressed TGF-β-induced SM22α promoter activity (Fig. 4C). Likewise, knockdown of GAS5 enhanced the promoter activity (Fig. 4D). Because TGF-β–induced SM22α promoter activity is mediated by Smad3 via binding to SBE in the promoter (28), we tested whether GAS5 blocked the promoter activity by preventing Smad3 from binding to the promoter SBE. A ChIP assay showed that GAS5 indeed attenuated Smad3 binding to the SBEs in the SM22α promoter in a chromatin setting (Fig. 4, E and F). Moreover, GAS5 blocked TGF-β/Smad3 activity in activating the SBE-LUX reporter, in which the promoter mainly contains 6×SBEs (Fig. 4G). These results demonstrate that GAS5 is a suppressor of Smad3-mediated gene transcription.
Figure 4.

GAS5 did not impact Smad3 nuclear translocation but inhibited its transcription activity. A and B, forced expression (Ad-GAS5, A) or knockdown of GAS5 (siGAS5, B) did not alter TGF-β-induced Smad3 nuclear translocation. Shown is immunostaining of Smad3 with different treatments as indicated. Scale bars = 20 μm. C and D, forced GAS5 expression (Ad-GAS5) suppressed, but GAS5 knockdown (siGAS5) enhanced, TGF-β–induced SM22α promoter activity. 10T1/2 cells were transfected with an SM22α promoter luciferase reporter, followed by transduction of a control adenovirus (Ad-GFP) or Ad-GAS5 (C) or transfection of control siRNA (siCtrl) or siGAS5 (D) for 24 h prior to TGF-β treatment (5 ng/ml) for 8 h. Luciferase assays were performed. **, p < 0.01 compared with the Ad-GFP/TGF-β or siCtrl/TGF-β group (n = 9). E, forced GAS5 expression suppressed Smad3 binding to the SBE in the SM22α promoter in a chromatin setting, as measured by ChIP assay. IP, immunoprecipitation. F, quantification of Smad3 binding enrichment on the SM22α promoter. Shown are the relative -fold changes. **, p < 0.01 compared with the Ad-GFP/Smad3 group; n = 9. G, forced GAS5 expression suppressed the activity of the promoter containing only SBE elements (SBE-LUX). **, p < 0.01 compared with the Ad-GFP/TGF-β group (Student's t test, n = 9).
GAS5 blocks Smad3 activity via its rSBEs
Because GAS5 bound to Smad3 and competitively blocked Smad3 binding to the SBEs in the SM22α promoter, we analyzed the potential binding elements in GAS5. Surprisingly, 11 tentative rSBE elements were observed in the GAS5 RNA sequence (Fig. 5A). Most of these elements were located in the stem structures (Fig. 5B), as predicted using the RNAalifold Web Server (30). This exciting finding prompted us to hypothesize that GAS5 may competitively bind to Smad3 via rSBEs and thus prevent Smad3 from binding to SBE DNA in TGF-β target gene promoters. To test this hypothesis, we first truncated GAS5 into six different fragments with or without rSBEs and co-transfected each individual fragment with an SM22α promoter luciferase reporter into 10T1/2 cells. As shown in Fig. 5C, most fragments containing rSBEs (F1, F4, and F5) suppressed TGF-β–induced promoter activities. The only exception was fragment F3, which contained an rSBE but failed to suppress the TGF-β signal, probably because of the incompetent secondary structure.
Figure 5.
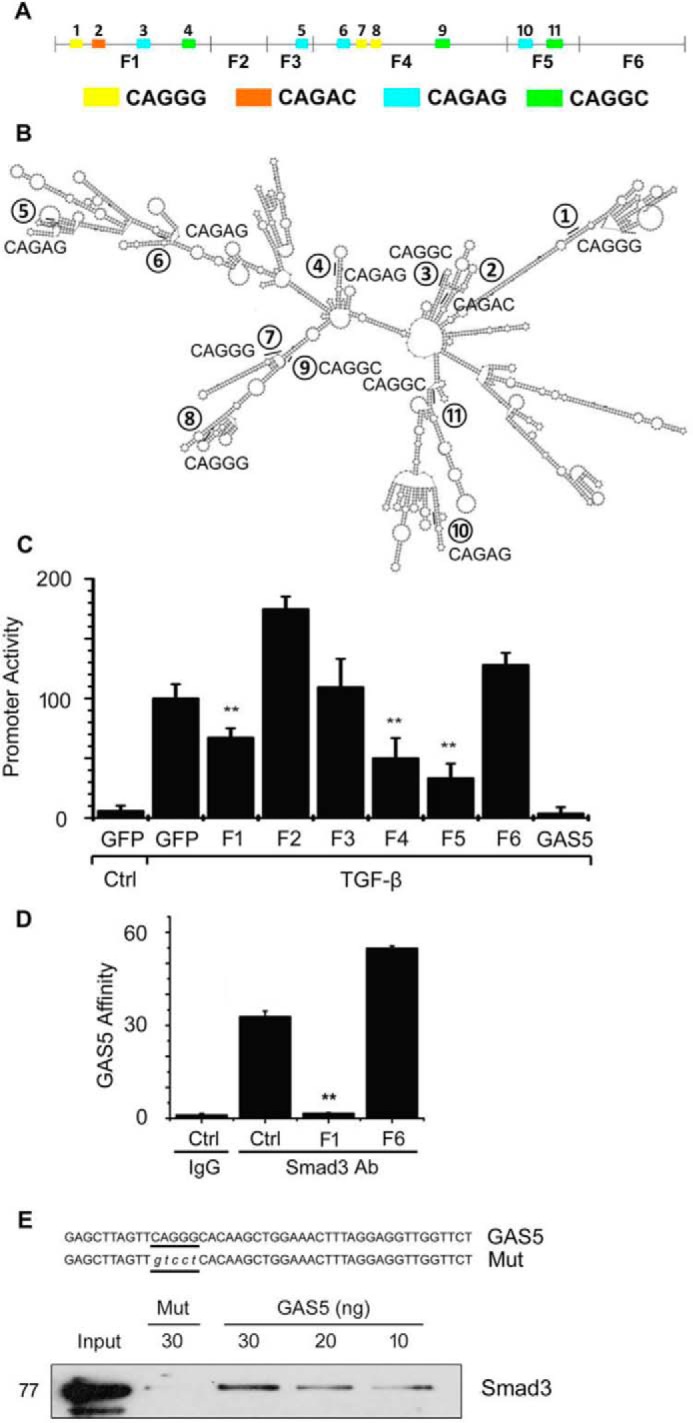
GAS5 attenuates Smad3 activity via its rSBE elements. A, schematic locations of mouse GAS5 fragments. Colors indicate the locations of corresponding rSBE elements. B, predicted secondary structure of mouse GAS5. Numbers label the positions of rSBE elements, with sequences indicated. C, effect of different GAS5 fragments with or without rSBE on SM22α promoter activity. GAS5 fragments with rSBE (F1, F4, and F5) suppressed TGF-β–induced SM22α promoter activity. **, p < 0.01 compared with TGF-β-treated cells without GAS5 fragment (GFP/TGF-β); n = 9. D, rSBE-containing F1, but not the F6 fragment without rSBE, prevented endogenous GAS5–Smad3 binding. 10T1/2 cells were untransfected (Ctrl) or transfected with shuttle vectors expressing F1 or F6 GAS5. 24 h later, the cells were treated with 1% PFA, and the RNA–protein complexes were pulled down by Smad3 antibody. The presence of GAS5 was detected by qPCR. **, p < 0.01 compared with the control group (Student's t test, n = 9). E, recombinant Smad3 bound to GAS5 F1 containing the wild type (GAS5) but not the mutant rSBE (Mut). GAS5 F1 sequences with wild-type or mutated rSBE are shown in the top panel. The mutated rSBE is indicated as lowercase letters. The in vitro Smad3-GAS5 binding assay is described under “Experimental procedures.”
To determine whether Smad3 specifically binds to the rSBE in GAS5, we transfected F1 (with rSBEs) and F6 (without rSBE) separately into 10T1/2 cells and tested whether these fragments affect Smad3 binding to the endogenous GAS5. As shown in Fig. 5D, rSBE-containing F1, but not the rSBE-lacking F6, competitively suppressed endogenous GAS5–Smad3 binding. To further confirm whether GAS5 directly binds to Smad3 via rSBE elements, we performed RIP assays with the purified recombinant Smad3 and biotin-labeled GAS5 F1 containing either wild-type or mutant rSBE. As shown in Fig. 5E, Smad3 bound to the GAS5 fragment with wild-type rSBE in a dose-dependent manner. However, the rSBE mutation diminished the binding, demonstrating that rSBE mediated the GAS5–Smad3 interaction.
LncRNA rSBE suppression of TGF-β/Smad signaling is a general mechanism
To determine whether the effect of lncRNA rSBE on TGF-β/Smad3 signaling is specific to GAS5 or a general phenomenon, we examined other rSBE-containing lncRNAs. The rat GAS5 homologue (rGAS5) exhibits only a 12% similarity to the mouse GAS5 in the primary sequence. However, rGAS5 also contains rSBEs (supplemental Fig. S3A) and was able to inhibit Smad3-dependent promoter activities of SM22α and smooth muscle α-actin (α-SMA) genes (Fig. 6A), similar to mouse GAS5. Consistently, rGAS5 also inhibited TGF-β-induced SMC contractile protein expression in mouse 10T1/2 cells (Fig. 6, B and C), suggesting that the titration of Smad3 activity by lncRNA rSBE is not species- but rather sequence-dependent. Moreover, rSBE-containing lncRNA1257, which exhibited Smad3 binding in the RIP assay (supplemental Fig. S3B and Fig. 1A) also effectively suppressed Smad3-dependent promoter activities (Fig. 6D). We then synthesized an artificial RNA fragment that only contained tandem rSBE repeats (Fig. 6E). The RIP assay showed that the artificial rSBE RNA fragment directly bound to Smad3 and Smad4 (Fig. 6F). Importantly, different artificial rSBE-contained RNA fragments were all able to suppress TGF-β/Smad3-mediated promoter activity (Fig. 6G). These results indicate that lncRNA rSBE suppression of Smad3 activity is a general mechanism limiting TGF-β signaling.
Figure 6.

LncRNA rSBEs suppresses TGF-β/Smad3 signaling as a general mechanism. A, rat GAS5 (Ad-rGAS5) effectively suppressed the promoter activities of the TGF-β/Smad3 target genes SM22α and α-SMA in mouse 10T1/2 cells. **, p < 0.01 compared with TGF-β-treated cells with Ad-GFP transduction (n = 9). B, Ad-rGAS5 also suppressed TGF-β-induced SMC marker expression in 10T1/2 cells. C, quantification of the protein levels shown in B by normalizing to α-tubulin. **, p < 0.01 compared with TGF-β–treated cells with Ad-GFP transduction (n = 9). D, rSBE-contained lncRNA-1257 suppressed TGF-β–induced SBE promoter (SBE-Lux) activities. **, p < 0.01 compared with control adenovirus–transduced cells (GFP) in either the control or TGF-β–treated group (n = 9). E, artificially synthesized RNA molecule containing four tandem rSBE repeats. The location and sequence of rSBEs are indicated. F, the artificial RNA molecule with rSBE repeats physically bound Smad3 and Smad4 proteins, as measured by RIP assay. **, p < 0.01 compared with the IgG group (n = 9). G, artificial RNA with different rSBEs suppressed TGF-β–induced SM22α promoter activities. **, p < 0.01 compared with TGF-β–treated cells without the artificial RNA molecule (Ctrl). n = 9.
Discussion
We have identified GAS5 as a novel lncRNA regulator for Smad3 function in SMC differentiation. GAS5 binds to Smad3 via its rSBEs, which demolishes Smad3 binding to SBE DNA in the promoter of SMC contractile genes. Through this competitive binding, GAS5 is able to negatively regulate TGF-β/Smad3 signaling and suppress TGF-β–induced SMC differentiation.
Smad3 proteins continuously shuttle between the cytoplasm and nuclei of cells, even without TGF-β induction (31). Therefore, in the basal state, a certain number of Smad3 proteins are present in the nuclei of cells. The Smad3 activity in the basal state is likely to be suppressed by GAS5, which may be important for maintaining the homeostasis of the cells. In the quiescent state, GAS5 is located in both the cytoplasm and nuclei of cells. TGF-β induction causes a major portion of GAS5 export from nuclei, which is opposite to the nuclear translocation of Smad3. Although the mechanism governing the nuclear export of GAS5 needs further investigation, this export is likely to be essential for the nuclear activity of Smad3, when cells are under TGF-β induction, because excessive GAS5 in the nuclei blocks Smad3 function, as shown by the decreased promoter activity of TGF-β/Smad3 target genes. In addition, the residual nuclear GAS5 that is not exported by TGF-β may fine-tune Smad3 activity in the nuclei. Interestingly, although GAS5 is abundant in the cytoplasm, it does not affect Smad3 phosphorylation or its nuclear translocation. Whether GAS5 regulates the Smad3 stability or function of other TGF-β signaling–related proteins requires extensive future investigation. Nevertheless, GAS5 appears to serve as a molecular break for Smad3 signaling in quiescent cells while modulating Smad3 activity under TGF-β induction.
rSBE-containing lncRNA binding to Smad3 and suppressing its activity appears to be a general mechanism in regulating TGF-β signaling. This is supported by two observations. First, rat GAS5, although with a very small similarity to mouse GAS5, can block TGF-β/Smad3 signaling in mouse cells. Second, multiple lncRNAs that contain rSBEs can block TGF-β/Smad3 activity. The existence of multiple different rSBE-containing lncRNAs may be essential for blocking excessive Smad3 activity in different cellular contexts or even in different cells under physiological conditions.
rSBE location in the lncRNA fragment appears to be important for its binding to Smad3 because GAS5 F3 fails to bind Smad3, although it contains an rSBE in its primary sequence. Unlike other fragments, the rSBE in F3 is located inside a large internal loop, which prevents it from forming a secondary structure that may be required for the recognition of Smad3 proteins.
In summary, we have demonstrated, for the first time, that lncRNA GAS5 can directly bind Smad3 and suppress TGF-β/Smad3 signaling in SMC differentiation. The lncRNA rSBE appears to be a novel general mechanism inhibiting Smad3 signaling in both quiescent and TGF-β–treated cells.
Experimental procedures
Cells and reagents
C3H10T1/2 (10T1/2) cells were purchased from the ATCC. Cells were maintained at 37 °C in a humidified 5% CO2 incubator in Dulbecco's modified Eagle's medium (Gibco) containing 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin. TGF-β1 was obtained from R&D Systems (Minneapolis, MN). Mouse GAS5 siRNA (n251731) was purchased from Life Technologies. Recombinant Smad3 (SRP5132) and the Smad3 inhibitor SIS3 was purchased from Sigma-Aldrich (St. Louis, MO). Smad2 (5339S), Smad3 (9523S), phospho-Smad3 (9520S), and Smad4 (38454) antibodies were purchased from Cell Signaling Technology (Danvers, MA). SM22α (ab10135) and CNN1 (ab46794) antibodies were purchased from Abcam (Cambridge, MA). Smooth muscle myosin heavy chain (BT-562) antibody was purchased from Biomedical Technologies Inc. (Stoughton, MA). GAPDH (G8795) and α-SMA (A2547) antibodies were purchased from Sigma-Aldrich.
Quantitative RT-PCR (qPCR)
Total RNA was extracted from 10T/2 cells using TRIzol reagent (Life Technologies) and reverse-transcribed to cDNA using the iScriptTM cDNA synthesis kit (Bio-Rad). qPCR was performed using a Stratagene Mx3005 qPCR thermocycler (Agilent Technologies, La Jolla, CA). All reactions, including no-template controls, were run in triplicate. After the reaction, the threshold cycle values were determined using fixed threshold settings. LncRNA expression in total cell lysates or cytoplasm was normalized to cyclophilin; lncRNA expression in cell nuclei was normalized to U6. The primer sequences used in this study are listed in supplemental Table S1.
RIP assay
The RIP assay was performed as described previously (32). Cells at 80–90% confluence on 15-cm culture dishes were fixed with 1% paraformaldehyde (PFA) before being scraped off and lysed in FA lysis buffer (50 mm HEPES, 140 mm NaCl, 1 mm EDTA, 1% (v/v) Triton X-100, and 0.1% (w/v) sodium deoxycholate (pH 7.5)) containing 40 units/ml RNase inhibitor (Sigma-Aldrich) and 1× HaltTM protease inhibitor mixture (Thermo Fisher Scientific, Grand Island, NY). After four to six rounds of 50% power output sonication, 300 μl of whole cell lysates (around 500 μg of total proteins) was incubated with normal rabbit IgG, Smad2, Smad3, or Smad4 antibodies (1 μg) at 4 °C overnight. The next day, the immunoprecipitates were captured with protein A/G-agarose beads (50 μl) (Santa Cruz Biotechnology, Dallas, TX). After washing with FA lysis buffer, the samples were incubated with Proteinase K at 42 °C for 1 h to digest the proteins. Then immunoprecipitated RNA was isolated. Purified RNA was subjected to qRT-PCR analyses to detect the presence of lncRNAs using their respective primers.
GAS5 RNA labeling and biotin–avidin pulldown assay
Mouse GAS5 full-length RNA, F1 containing the wild-type CAGGG-SBE element, or F1 with the mutated rSBE element were synthesized and labeled with biotin-UTP using an in vitro transcription kit (Roche). For the Smad3 pulldown assay, 1 μg of biotin-labeled GAS5 RNAs (sense/antisense) was transfected into 10T1/2 cells in 10-cm culture dishes and incubated at 37 °C overnight. Cells were cross-linked by adding 1% formaldehyde and then lysed in FA lysis buffer containing 40 units/ml RNase inhibitor (Sigma-Aldrich) and 1× HaltTM protease inhibitor mixture (Thermo Fisher Scientific) at 4 °C for 30 min. Cell lysates were treated with DNase I at 37 °C for 20 min, followed by spinning down at maximum speed for 10 min. The supernatant (around 200 μl) was then incubated with 20 μl of avidin-coated agarose beads (Thermo Fisher Scientific) at 25 °C for 1 h. After washing four times with washing/binding buffer (PBS with 0.1% SDS and 1% Nonidet P-40 or 0.5% sodium deoxycholate), the bead—RNA—protein complex was spun down, and the presence of Smad3 was detected by Western blotting. For the in vitro Smad3–GAS5 binding assay, labeled GAS5 F1 fragments (30 ng) were incubated with 0.8 μg of recombinant Smad3 (Sigma-Aldrich) in 50 μl of PBS at 4 °C overnight. The next day, 50 μl of avidin-coated agarose beads were applied to pull down the RNA–protein complex. After washing with washing/binding buffer (PBS with 0.1% SDS and 1% Nonidet P-40 or 0.5% sodium deoxycholate) four times, the presence of Smad3 was detected by Western blotting.
Western blotting
10T1/2 cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, and 0.1% SDS) and incubated at 4 °C with continuous rotation for 10 min, followed by centrifugation at 12,000 × g for 10 min. The supernatant was collected, and the protein concentration was determined by BCA assay (Pierce). Protein extracts (60–100 μg) were dissolved on 10% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk in TBS plus 0.1% Tween 20 (TBST) at room temperature for 1 h, followed by incubation with primary antibodies diluted in TBST at 4 °C overnight. After three 10-min washes with TBST, the blots were incubated with the appropriate secondary antibody conjugated to HRP at room temperature for 1 h. Protein expression was detected with enhanced chemiluminescence reagents.
Cell fractionation
Cell fractionation was performed using a nuclear extraction kit (EMD Millipore, Temecula, CA) following standard procedures. Briefly, 10T1/2 cells on 10-cm culture dishes were grown until 80% confluence. After trypsinization, cells were collected by spinning down at 500 × g for 10 min. Cell pellets were washed with ice-cold PBS, and the cells were resuspended with ice-cold cytoplasmic lysis buffer (provided by the kit) containing 40 units/ml RNase inhibitor. The cells were lysed by passing through a syringe with a 27-gauge needle. The cell lysates were then centrifuged at 8000 × g for 20 min. The remaining pellet contained the nuclear fraction. Nuclear fractions were used for RNA extraction and RT-PCR analysis.
FISH
GAS5 RNA probes (sense/antisense, supplemental Table S1) were synthesized and labeled with Alexa Fluor 594 using the FISH tag RNA multicolor kit (Life Technologies). 10T1/2 cells grown on coverglasses were treated with 20 μg/ml proteinase K at 37 °C for 1 h and washed with 2× SSC solution followed by water at room temperature. The cells were then incubated with predenatured GAS5 probes in a dark and humid environment at 55 °C for 24 h to allow hybridization. The cells were then washed with 50% formamide in 2× SSC four times before mounting. Nuclei were counterstained with DAPI.
Immunofluorescent staining
Cells were seeded on coverglasses placed in a 24-well plate and allowed to adhere overnight. Cells were then treated with TGF-β1 for 1 h (for detecting Smad3) or 24 h (for SM22α), fixed with 1% PFA in PBS, permeabilized with 0.1% Triton X-100/PBS for 5 min, followed by blocking with 10% goat serum and incubation with Smad3 or SM22α antibody at 4 °C overnight. Tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibodies were used to detect target protein expression.
ChIP
1% formaldehyde was directly added to 10T1/2 cell culture medium. The cross-linked cells were collected by centrifugation at 4 °C and washed with PBS containing protease inhibitors before final collection. The cells were then resuspended by rotating in 1% SDS lysis buffer at 4 °C for 20 min, followed by sonication on ice to shear DNA into 500- to 1000-bp fragments. The lysates were immunoprecipitated with 2 μg of IgG (negative control) or Smad3 antibody using coimmunoprecipitation reagent (17-195, Millipore). Semiquantitative and quantitative PCR were performed to amplify the SM22α gene promoter region containing the SBE.
Luciferase reporter assay
250 ng of either empty vector or pShuttle-mGAS5 was co-transfected with 250 ng of firefly luciferase reporter driven by SM22α or α-SMA promoter into 10T1/2 cells in 12-well plates using Lipofectamine LTX (Invitrogen). Cells were treated with 5 ng/ml of TGF-β1 for 8 h, and luciferase activities were measured using a luciferase assay kit (Promega) according to the protocol of the manufacturer. The experiments were repeated three times in triplicate.
Statistical analysis
Experiments were performed in triplicate wells for each condition and repeated three times. All values are presented as means ± S.E. Comparisons of parameters between two groups were made by unpaired Student's t tests. p values from 0.01 to 0.05 and lower than 0.01 were considered significant (*) and very significant (**), respectively. Sample or experiment sizes were determined empirically to achieve sufficient statistical power. In all experiments reported in this study, no data point was excluded. No randomization was used in this study. Data distribution was assumed to be normal, but this was not formally tested. The variance was similar between groups that were being statistically compared.
Author contributions
R. T. and S. Y. C. conceived and coordinated the study. R. T. designed and performed the experiments in Figs. 1–6. G. Z. provided technical assistance and contributed to the experiments in Figs. 1–6. X. M. and Y. C. W. performed the experiments in supplemental Figs. S1 and S2. R. T. analyzed the results. R. T. and S. Y. C. wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by NHLBI, National Institutes of Health Grants HL119053, HL123302, and HL135854. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S3 and Table S1.
- SMC
- smooth muscle cell(s)
- lncRNA
- long non-coding RNA
- SBE
- Smad-binding element
- rSBE
- RNA Smad—binding element
- RIP
- RNA immunoprecipitation
- SRF
- serum response factor
- F1
- fragment 1
- α-SMA
- α-smooth muscle actin
- qPCR
- quantitative RT-PCR
- PFA
- paraformaldehyde
- TRITC
- tetramethylrhodamine isothiocyanate
- Ad
- adenovirus.
References
- 1. Li S., Wang D.-Z., Wang Z., Richardson J. A., and Olson E. N. (2003) The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. U.S.A. 100, 9366–9370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gittenberger-de Groot A. C., DeRuiter M. C., Bergwerff M., and Poelmann R. E. (1999) Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler. Thromb. Vasc. Biol. 19, 1589–1594 [DOI] [PubMed] [Google Scholar]
- 3. Majesky M. W. (2007) Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 27, 1248–1258 [DOI] [PubMed] [Google Scholar]
- 4. Zhao J., Zhang W., Lin M., Wu W., Jiang P., Tou E., Xue M., Richards A., Jourd'heuil D., Asif A., Zheng D., Singer H. A., Miano J. M., and Long X. (2016) MYOSLID is a novel serum response factor-dependent long noncoding RNA that amplifies the vascular smooth muscle differentiation program. Arterioscler. Thromb. Vasc. Biol. 10, 2088–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freedman J. E., Miano J. M., and National Heart, Lung, and Blood Institute Workshop Participants (2017) Challenges and opportunities in linking long noncoding RNAs to cardiovascular, lung, and blood diseases. Arterioscler. Thromb. Vasc. Biol. 37, 21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bell R. D., Long X., Lin M., Bergmann J. H., Nanda V., Cowan S. L., Zhou Q., Han Y., Spector D. L., Zheng D., and Miano J. M. (2014) Identification and initial functional characterization of a human vascular cell–enriched long noncoding RNA. Arterioscler. Thromb. Vasc. Biol. 34, 1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mercer T. R., Dinger M. E., and Mattick J. S. (2009) Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159 [DOI] [PubMed] [Google Scholar]
- 8. Tsai M.-C., Spitale R. C., and Chang H. Y. (2011) Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 71, 3–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sehgal L., Mathur R., Braun F. K., Wise J. F., Berkova Z., Neelapu S., Kwak L. W., and Samaniego F. (2014). FAS-antisense 1 lcRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia 28, 2376–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu Y., Rowley M. J., Böhmdorfer G., and Wierzbicki A. T. (2013) A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol. Cell 49, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang B., Arun G., Mao Y. S., Lazar Z., Hung G., Bhattacharjee G., Xiao X., Booth C. J., Wu J., Zhang C., and Spector D. L. (2012) The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2, 111–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnsson P., Ackley A., Vidarsdottir L., Lui W.-O., Corcoran M., Grandér D., and Morris K. V. (2013) A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 20, 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoon J.-H., Abdelmohsen K., Kim J., Yang X., Martindale J. L., Tominaga-Yamanaka K., White E. J., Orjalo A. V., Rinn J. L., Kreft S. G., Wilson G. M., and Gorospe M. (2013) Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat. Commun. 4, 2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sigova A. A., Abraham B. J., Ji X., Molinie B., Hannett N. M., Guo Y. E., Jangi M., Giallourakis C. C., Sharp P. A., and Young R. A. (2015) Transcription factor trapping by RNA in gene regulatory elements. Science 350, 978–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou L., Sun K., Zhao Y., Zhang S., Wang X., Li Y., Lu L., Chen X., Chen F., Bao X., Zhu X, Wang L., Tang L. Y., Esteban M. A., Wang C. C., et al. (2015) Linc-YY1 promotes myogenic differentiation and muscle regeneration through an interaction with the transcription factor YY1. Nat. Commun. 6, 10026. [DOI] [PubMed] [Google Scholar]
- 16. Hirschi K. K., Rohovsky S. A., and D'Amore P. A. (1998) PDGF, TGF-β, and heterotypic cell–cell interactions mediate endothelial cell–induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 141, 805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi Y., and Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 18. Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A. R., Hoberg E., Moustakas A, Gyllensten U., Jones S. J., Gustafsson C. M., Sims A. H., et al. (2015) MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 6, 7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng W., Si S., Zhang Q., Li C., Zhao F., Wang F., Yu J., and Ma R. (2015) Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate gastric cancer progression. J. Exp. Clin. Cancer Res. 34, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pickard M. R., Mourtada-Maarabouni M., and Williams G. T. (2013) Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim. Biophys. Acta 1832, 1613–1623 [DOI] [PubMed] [Google Scholar]
- 21. Mourtada-Maarabouni M., Pickard M. R., Hedge V. L., Farzaneh F., and Williams G. T. (2009) GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 28, 195–208 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Z., Zhu Z., Watabe K., Zhang X., Bai C., Xu M., Wu F., and Mo Y. (2013) Negative regulation of lncRNA GAS5 by miR-21. Cell Death Differ. 20, 1558–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kino T., Hurt D. E., Ichijo T., Nader N., and Chrousos G. P. (2010) Noncoding RNA Gas5 is a growth arrest and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 3, ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li L., Li X., The E., Wang L.-J., Yuan T.-Y., Wang S.-Y., Feng J., Wang J., Liu Y., Wu Y.-H., Ma X.-E., Ge J., Cui Y. Y., and Jiang X. Y. (2015) Low expression of lncRNA-GAS5 is implicated in human primary varicose great saphenous veins. PLoS ONE 10, e0120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y. N., Shan K., Yao M.-D., Yao J., Wang J.-J., Li X., Liu B., Zhang Y.-Y., Ji Y., Jiang Q., and Yan B. (2016) Long noncoding RNA-GAS5: a novel regulator of hypertension-induced vascular remodeling. Hypertension 68, 736–748 [DOI] [PubMed] [Google Scholar]
- 26. Shi Y., Wang Y.-F., Jayaraman L., Yang H., Massagué J., and Pavletich N. P. (1998) Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell 94, 585–594 [DOI] [PubMed] [Google Scholar]
- 27. Davis B. N., Hilyard A. C., Nguyen P. H., Lagna G., and Hata A. (2010) Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 39, 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen S., Kulik M., and Lechleider R. J. (2003) Smad proteins regulate transcriptional induction of the SM22α gene by TGF-β. Nucleic Acids Res. 31, 1302–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirschi K. K., Lai L., Belaguli N. S., Dean D. A., Schwartz R. J., and Zimmer W. E. (2002) Transforming growth factor-β induction of smooth muscle cell phenotype requires transcriptional and post-transcriptional control of serum response factor. J. Biol. Chem. 277, 6287–6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gruber A. R., Lorenz R., Bernhart S. H., Neuböck R., and Hofacker I. L. (2008) The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao Z., Watson N., Rodriguez C., and Lodish H. F. (2001) Nucleocytoplasmic shuttling of Smad1 conferred by its nuclear localization and nuclear export signals. J. Biol. Chem. 276, 39404–39410 [DOI] [PubMed] [Google Scholar]
- 32. Niranjanakumari S., Lasda E., Brazas R., and Garcia-Blanco M. A. (2002) Reversible cross-linking combined with immunoprecipitation to study RNA–protein interactions in vivo. Methods 26, 182–190 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.