

ABSTRACT
Pseudomonas aeruginosa is an important pathogen of the immunocompromised, causing both acute and chronic infections. In cystic fibrosis (CF) patients, P. aeruginosa causes chronic disease. The impressive sensory network of P. aeruginosa allows the bacterium to sense and respond to a variety of stimuli found in diverse environments. Transcriptional regulators, including alternative sigma factors and response regulators, integrate signals changing gene expression, allowing P. aeruginosa to cause infection. The two-component transcriptional regulator AlgR is important in P. aeruginosa pathogenesis in both acute and chronic infections. In chronic infections, AlgR and the alternative sigma factor AlgU activate the genes responsible for alginate production. Previous work demonstrated that AlgU controls rsmA expression. RsmA is a posttranscriptional regulator that is antagonized by two small RNAs, RsmY and RsmZ. In this work, we demonstrate that AlgR directly activates rsmA expression from the same promoter as AlgU. In addition, phosphorylation was not necessary for AlgR activation of rsmA using algR and algZ mutant strains. AlgU and AlgR appear to affect the antagonizing small RNAs rsmY and rsmZ indirectly. RsmA was active in a mucA22 mutant strain using leader fusions of two RsmA targets, tssA1 and hcnA. AlgU and AlgR were necessary for posttranscriptional regulation of tssA1 and hcnA. Altogether, our work demonstrates that the alginate regulators AlgU and AlgR are important in the control of the RsmA posttranscriptional regulatory system. These findings suggest that RsmA plays an unknown role in mucoid strains due to AlgU and AlgR activities.
IMPORTANCE P. aeruginosa infections are difficult to treat and frequently cause significant mortality in CF patients. Understanding the mechanisms of persistence is important. Our work has demonstrated that the alginate regulatory system also significantly impacts the posttranscriptional regulator system RsmA/Y/Z. We demonstrate that AlgR directly activates rsmA expression, and this impacts the RsmA regulon. This leads to the possibility that the RsmA/Y/Z system plays a role in helping P. aeruginosa persist during chronic infection. In addition, this furthers our understanding of the reach of the alginate regulators AlgU and AlgR.
KEYWORDS: P. aeruginosa, RsmA, AlgR, mucoid, Pseudomonas aeruginosa, cystic fibrosis, mucA, two-component regulatory systems
INTRODUCTION
The opportunistic pathogen Pseudomonas aeruginosa possesses multiple virulence factors for causing disease. This allows P. aeruginosa to cause both acute and chronic infections. Acute infecting strains are characterized by the presence of type IV pili (T4P), flagella, and a type III secretion system (T3SS) (1–3). In contrast, chronic infecting strains diversify (4, 5) and frequently do not express T3SS, T4P, or flagella (6, 7). Chronic infecting strains often form biofilms composed of exopolysaccharides, such as alginate, and signal a decline in lung function in CF patients (8–11). Alginate biosynthesis requires activation of the alternative sigma factor AlgU (12, 13) and the two-component transcriptional regulator AlgR (12, 14, 15).
Two-component signal transduction systems are important in regulating the bacterial response to environmental conditions. AlgR controls both acute and chronic virulence genes (16–18). In the case of acute infections, AlgZ phosphorylates AlgR and activates the fimU operon encoding minor pilins important in pilus biogenesis (19–21). The production of alginate, indicative of chronic infection, does not require AlgR phosphorylation (22). This fact has been demonstrated by deletion of the histidine sensor kinase gene algZ (23) and by the expression of an algR mutant allele encoding an asparagine in place of the conserved aspartate (22).
In chronic P. aeruginosa infections, the bacteria frequently acquire spontaneous mucA mutations that free the alternative sigma factor AlgU (12, 14, 24–26). AlgU and AlgR regulate the alginate biosynthesis genes producing mucoid colonies (12). In addition, AlgR decreases the expression of acute virulence factors, such as the T3SS in chronic infections when mucA mutations occur (7, 27). Therefore, AlgR has multiple roles depending on the type of infection and the phosphorylation state of AlgR: phosphorylated in acute infections activating the fimU operon and unphosphorylated in chronic infections activating alginate biosynthesis.
Previous work implicated the AlgZ/R system in the control of rsmA expression (28), but a mechanism for AlgR activation of rsmA was not investigated. RsmA is considered a global regulator that controls the expression of many P. aeruginosa genes by binding to mRNAs (29). Two noncoding RNAs, RsmY and RsmZ, counteract RsmA (29, 30). In this study, we sought to determine how AlgR activates rsmA and further investigate the role of RsmA in P. aeruginosa strains containing an mucA mutation. We further examined rsmY and rsmZ expression in algR mutant strains.
We demonstrate that AlgR directly activates rsmA expression in a phosphorylation-independent manner and that both AlgR and AlgU are important in activating rsmA expression in mucoid strains. The activity of RsmA is not thought to be significant in chronic infecting strains, such as mucA mutants. However, we also provide evidence that RsmA is active in mucoid P. aeruginosa. Altogether, our work shows that the two-component regulator AlgR can affect gene expression through RsmA, providing another mechanism for how AlgR impacts virulence gene regulation. We postulate that AlgR and RsmA likely function as a rheostat, as opposed to a switch, and suggest that RsmA plays a role in chronic infections.
RESULTS
mucA mutant strains require AlgR for increased rsmA expression.
Previous studies suggested that AlgU and AlgR control rsmA expression (28, 31). However, whether both AlgZ and AlgR were involved in the control of rsmA expression was not tested. We constructed and assayed an rsmA transcriptional fusion (Fig. 1A) that contains both rsmA promoters (rsmATF1-lacZ). The rsmATF1-lacZ fusion was analyzed in the wild-type strain P. aeruginosa PAO1, ΔalgR and algZ mutants, and in the corresponding mucA22 mutants. The algZ mutant has a mutation in the conserved histidine residue, which prevents AlgZ-mediated phosphorylation of AlgR (19, 32) but does not disrupt the internal algR promoter (33). As shown in Fig. 1B, there was a slight increase in rsmATF1-lacZ activity in a ΔalgR mutant. When the rsmATF1-lacZ fusion was assayed in an algZ mutant strain, there was also a slight increase in reporter activity (Fig. 1B). The modest increase in reporter activity suggests that AlgZ and AlgR have a minor role in rsmA regulation in strain PAO1.
FIG 1.

AlgR, but not AlgZ, is necessary for increased rsmA expression in mucoid strains. (A) Schematic of rsmA genomic region. The sequence below the genomic schematic indicates the AlgU-dependent promoter region. Underlined sequences are potential AlgR-binding sites. The bent arrow and bold nucleotide indicate the transcriptional start sites. The rsmA transcriptional fusion rsmATF1-lacZ used in this study is listed below. Arrows indicate the primers used and the numbers indicate the primer locations relative to the translational start site. (B) The transcriptional fusion rsmATF1-lacZ (Fig. 1A) was assayed in the indicated strains after growth for 8 h in LB broth. Significant differences from the wild type were determined using a one-way analysis of variance and Tukey's posttest. Asterisks indicate P values of 0.01 (*) and <0.0001 (***). (C) Western blot analysis of the indicated strains containing an HA-tagged rsmA allele. PAO1 without the rsmAHA allele was run as a negative control. Densitometry analysis was performed using a duplicate gel and staining for total protein using Coomassie blue, and all strains were normalized to PAO1 containing the rsmAHA allele. Western blotting was performed four times, and densitometry analysis is indicated below the Western blot. A representative Western blot is shown above the densitometry. A one-way ANOVA with Tukey's posttest was used to determine statistical significance. **, P < 0.001; ***, P < 0.0001.
As previous studies also indicated a role for AlgU in regulating rsmA expression, the rsmATF1-lacZ transcriptional fusion was assayed in a mucA22 strain, where AlgU is most active. Reporter activity was increased ∼3-fold in a mucA22 strain, as previously described (31) (Fig. 1B). However, in the mucA22 ΔalgR double mutant, there was a drastic decrease (∼3-fold) in rsmATF1-lacZ activity (Fig. 1B). The mucA22 algZ mutant strain had rsmATF1-lacZ activity almost identical to that of the mucA22 mutant strain (Fig. 1B). These data implicate AlgR but not AlgZ in rsmA regulation in a mucA22 mutant strain and suggest that AlgR phosphorylation is not necessary for rsmA activation.
To confirm that AlgR affected rsmA expression, an epitope-tagged rsmA allele was introduced into the wild-type strain (PAO1), mucA22 mutant, and respective algR mutant strains and analyzed by Western blot analysis. As shown in Fig. 1C, a slight decrease in RsmA levels in the ΔalgR mutant strain was detected compared to the wild-type strain PAO1. As reported previously (31), a mucA22 mutant strain had drastically increased RsmA levels (Fig. 1C). The mucA22 ΔalgR mutant strain had significantly decreased RsmA compared with the mucA22 mutant strain (Fig. 1C). The Western blot analysis confirmed the transcriptional fusion analysis and supported a significant role for AlgR activating rsmA in the mucoid mucA22 mutant strain but not in the nonmucoid strain PAO1.
AlgR phosphorylation is not required for activation of rsmA expression.
To further investigate the role of AlgR phosphorylation in the regulation of rsmA, algR site-directed mutants were constructed that mimic either the unphosphorylated (D54N) or phosphorylated form of AlgR (D54E). The algRD54N and algRD54E mutant alleles were also constructed in the mucA22 mutant background to confirm a phosphorylation-independent mode of AlgR activation of rsmA. Both the mucA22 algRD54N and the mucA22 algRD54E mutant strains were mucoid (data not shown), supporting the notion that phosphorylation is not necessary for alginate production and that the mutant AlgR proteins produced in these strains were still functional. The mucA22D54N mutant had increased rsmA reporter activity compared to a mucA22 mutant strain (Fig. 2). The mucA22 algRD54E strain had decreased rsmATF1-lacZ activity compared to the mucA22 mutant (Fig. 2). Because the mucA22 algRD54N and the mucA22 algZ mutant strains had elevated and similar levels, respectively, of rsmATF1-lacZ activity (Fig. 2 and 1B, respectively) compared to the mucA22 mutant strain, this suggests that phosphorylation of AlgR is not required for rsmA activation.
FIG 2.
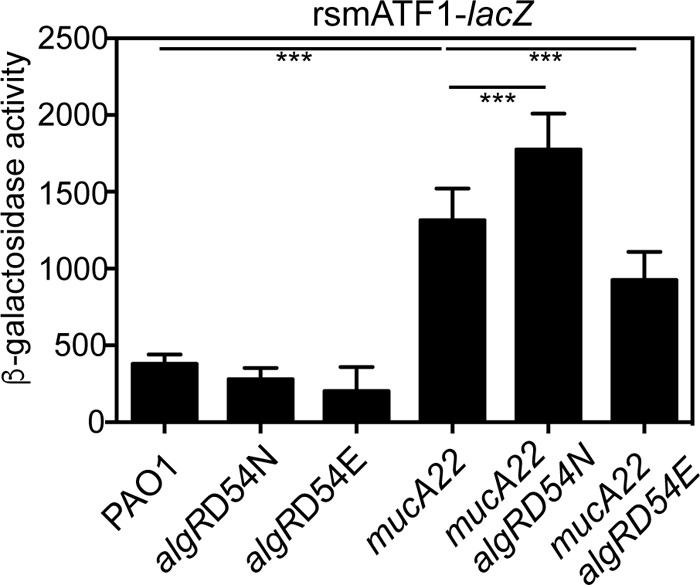
AlgR phosphorylation is not required for rsmA activation. Mutants containing either a mutated aspartate to asparagine (D54N) or aspartate to glutamate (D54E) were constructed in both the wild-type background PAO1 and the mucA22 mutant background. The rsmA transcriptional fusion rsmATF1-lacZ was introduced into the strains indicated, resulting in single-copy chromosomal transcriptional fusions. The indicated strains were grown for 8 h in LB broth and assayed for β-galactosidase activity. Values indicate the actual β-galactosidase activity minus the vector control (−28 Miller units). Differences from the wild-type strain PAO1 or mucA22 mutant were determined using a one-way analysis of variance and Tukey's posttest. Triple asterisks indicate P values of <0.0001.
AlgR regulates the distal rsmA promoter.
Previous work determined that rsmA has two promoters (31). An RNase protection assay was performed using a probe spanning the upstream region of rsmA (Fig. 3A) to determine which rsmA message AlgR affected. As we previously reported (31), two rsmA messages were seen in both the wild-type strain PAO1 and the mucA22 mutant strain, with the longer transcript increased in the mucA22 mutant strain (Fig. 3B). There was little difference in the rsmA transcripts between PAO1 and the ΔalgR mutant (Fig. 3B). In contrast, there was a substantial decrease in the longer rsmA transcript in the mucA22 ΔalgR mutant strain compared to the mucA22 parent strain (Fig. 3B). The specificity of the rsmA probe was confirmed using the probe incubated with yeast tRNA (+RNase) and using a ΔrsmA mutant (Fig. 3B). These results suggest that AlgR is most important for rsmA control in the mucA22 background and that AlgR controls the distal rsmA promoter that is also under control by AlgU.
FIG 3.
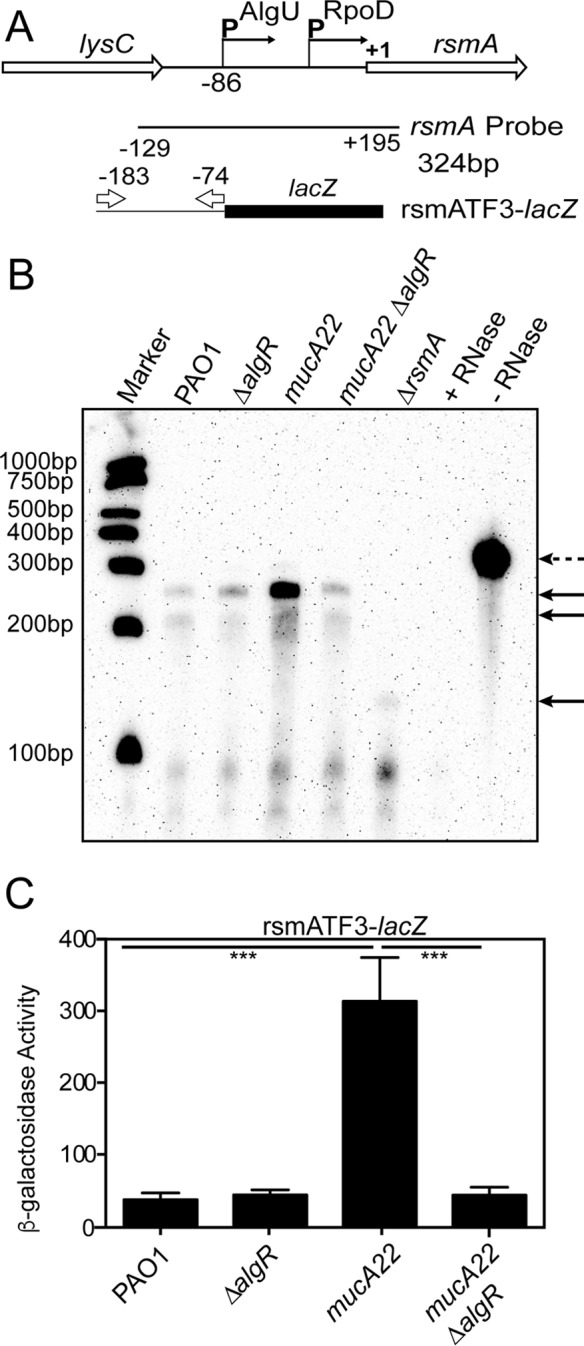
AlgR controls the distal AlgU-dependent rsmA promoter. (A) Schematic of the RNase protection assay probe used. Numbers indicate distances from the rsmA translational start site. The transcriptional fusion rsmATF3-lacZ is indicated at the bottom. Arrows indicate the primers used and their location in reference to the rsmA translational start site. (B) A representative RNase protection assay performed on the indicated strains after 8 h of growth in LB broth. Total RNA was isolated from each strain and hybridized to 800 pg of biotinylated rsmA probe. After treatment with RNase A/RNase T1, protected probe fragments were detected after electrophoresis through a denaturing polyacrylamide gel and transfer to a nylon membrane. Protected probe fragments are indicated by solid arrows. The broken arrow indicates a full-length probe. Biotinylated molecular size markers are indicated to the left. RNase protection assays were performed three times. (C) The rsmA transcriptional fusion rsmATF3-lacZ was introduced into the indicated strains to generate strains containing single-copy chromosomal fusions. The strains were grown for 8 h in LB broth and assayed for β-galactosidase activity minus the vector control. Differences from the wild-type strain PAO1 or mucA22 mutant were determined using a one-way analysis of variance and Tukey's posttest. Triple asterisks indicate P values of <0.0001. Fusion analysis was performed in triplicate three times.
To confirm that AlgR regulates the distal rsmA promoter, transcriptional fusions containing individual rsmA promoters were used (Fig. 3A; see also Fig. S1A in the supplemental material). The transcriptional fusion rsmATF2-lacZ contains only the proximal rsmA promoter (Fig. S1A). The transcriptional fusion rsmATF3-lacZ (Fig. 3A) contains only the distal promoter that has been shown to be controlled by AlgU (31). The deletion of algR had no effect in the rsmATF2-lacZ transcriptional fusion in the wild-type or a mucA22 background (Fig. S1B). The transcriptional fusion rsmATF3-lacZ had increased reporter activity in a mucA22 mutant strain compared to that in PAO1 (Fig. 3C). Compared to PAO1, no change in rsmTF3-lacZ activity was seen in a ΔalgR mutant strain (Fig. 3C). However, algR deletion in a mucA22 mutant strain (mucA22 ΔalgR) resulted in a significant decrease in rsmATF3-lacZ activity (Fig. 3C), supporting the RNase protection assay. Taking these results together, we conclude that AlgR activates the distal rsmA promoter, and this further suggests that AlgU and AlgR both are required to activate rsmA transcription from the distal promoter.
AlgR directly binds the distal rsmA promoter.
We hypothesized that AlgR directly activated the distal rsmA promoter. In support of this idea, two potential AlgR-binding sites are located upstream of rsmA. One of the potential AlgR-binding sites is 2 nucleotides away from the AlgU −35 consensus promoter (Fig. 4A). Purified AlgR was tested in gel shift studies with biotinylated rsmA promoter fragments (Fig. 4A). A biotinylated pscF gene, a component of the T3SS (34, 35), was used as a negative control and did not shift when incubated with purified AlgR (Fig. 4B to D). A PCR fragment representing the entire rsmA upstream region containing both promoters was dramatically shifted using purified AlgR (Fig. 4B), suggesting that AlgR directly binds to at least one of the AlgR-binding sites upstream of rsmA. To further define the region upstream of rsmA that was bound by AlgR, PCR amplicons that corresponded to the transcriptional fusion constructs containing individual rsmA promoters (Fig. 4A) were biotinylated and tested via gel shift analysis. A biotinylated probe containing the proximal promoter (TF2) and upstream sequence to the more distal transcriptional start site did not shift when incubated with purified AlgR (Fig. 4B). This result suggested that AlgR does not bind the proximal rsmA promoter. As shown in Fig. 4B, AlgR bound the distal promoter sequence fragment, TF3, containing the AlgU-dependent promoter and potential AlgR-binding sites. The TF3 fragment containing the AlgU promoter was tested in a competition assay using increasing concentrations of AlgR and unlabeled probe. A 1:1 ratio of labeled to unlabeled probe was capable of decreasing the shift of the TF3 probe (Fig. 4C). Competition with the unlabeled specific probe in a 1:5 or 1:10 ratio was able to compete for AlgR binding, as indicated by severe reduction of the probe shift (Fig. 4C). These data suggest that AlgR specifically binds the AlgU-dependent promoter region and strongly suggests that AlgR binds at least one of the AlgR consensus sequences located in this promoter region.
FIG 4.

AlgR directly binds the AlgU-dependent rsmA promoter and requires both AlgR-binding sites. Purified AlgR was incubated with either biotinylated PCR products or annealed primers. (A). Schematic of the rsmA genomic region. Below are fragments or annealed primers and the approximate location upstream of rsmA. Promoters are indicated above as bent arrows and are denoted by PAlgU or PRpoD. The arrowheads indicate the approximate location of the putative AlgR-binding sites. (B) Analysis of biotinylated PCR fragments in gel shift studies. The PCR fragment TF1 represents the entire rsmA upstream region (see panel A). Fragments TF2 and TF3 correspond to the separated rsmA promoters (see Fig. 1). TF2 corresponds to the proximal rsmA promoter, and TF3 corresponds to the distal AlgU-dependent promoter. A minus sign indicates probe alone. A plus sign indicates AlgR concentration of 2.5 μM. pscF was used as a negative control. Shaded arrowheads indicate shifted fragments. White arrowheads indicate unbound probe. (C) Titration and competition assay using purified AlgR and the TF3 fragment corresponding to the distal rsmA promoter. The pscF fragment was used as a negative control. TF3 was incubated with different concentrations of AlgR (indicated by the graded triangle; 0.05, 0.25, 1.25, and 2.5 μM). TF3 was also used in a competition assay using unlabeled TF3 as a competitive inhibitor. Ratios below indicate the ratio of labeled to unlabeled probe. Shaded arrowheads indicate shifted fragments. White arrowheads indicate unbound probe. A minus sign indicates probe alone. (D) Two AlgR-binding sites are upstream of the distal rsmA promoter. Annealed primers containing either wild-type or two different mutated AlgR-binding sites were tested in gel shift experiments. The pscF fragment was used as a negative control. The rsmA lanes indicate the probe using annealed primers with both AlgR-binding sites intact. M1 indicates the same as rsmA, except that the furthest upstream AlgR-binding site was mutated. M2 indicates the same as rsmA, except that the further downstream AlgR-binding site is mutated. Shaded arrowheads indicate shifted fragments. White arrowheads indicate unbound probe. Minus signs indicate probe alone. Plus signs indicate an AlgR concentration of 2.5 μM.
AlgR has previously been shown to bind an 11-bp consensus sequence, with nucleotides 2 to 10 being the most conserved (36). To determine if one, or both, of the putative AlgR-binding sites were required for AlgR binding, we used annealed primers upstream of the distal promoter in gel shift analyses. Approximately 30-bp primers were annealed and biotinylated that contained the predicted AlgR-binding sites. AlgR was able to shift the annealed biotinylated primers containing the two predicted AlgR-binding sites (Fig. 4D). The first putative AlgR-binding site, CCTTTTGTC, was mutated, and this mutation resulted in a loss of AlgR binding (Fig. 4D), suggesting that the first AlgR-binding site is required for AlgR binding to the rsmA promoter. The second putative AlgR-binding site, CCGTTTGGC, is located 7 bp downstream and directly adjacent to the AlgU −35 consensus sequence. When the second AlgR-binding site was mutated, AlgR was no longer able to bind to the annealed primers (Fig. 4D). These data suggest that AlgR binds both of the consensus sequences in order to activate rsmA expression.
AlgR regulates rsmY expression indirectly.
A previous study indicated that rsmY and rsmZ expression was also increased in a mucA mutant background and required AlgR (28). The transcriptional start site of rsmY was determined in Pseudomonas fluorescens using 5′ rapid amplification of cDNA ends (RACE), and this information was used to deduce the transcriptional start site in P. aeruginosa (30, 37). To confirm the P. aeruginosa rsmY transcriptional start site, we performed primer extension. As indicated in Fig. 5A, a single extension product was obtained after reverse transcription of total RNA that confirmed the rsmY transcriptional start site assigned using P. fluorescens.
FIG 5.

AlgR indirectly increases rsmY expression in a mucA22 mutant strain. (A) Primer extension analysis performed on total RNA from strain PAO1. GATC indicates the sequencing ladder. -C is a control without reverse transcriptase. PAO1 is reverse-transcribed PAO1 mRNA. Below is the sequence of the rsmY promoter region. The asterisk denotes the transcriptional start site identified. (B) Schematic of the rsmY genomic region. The UAS is indicated between the numbers from the transcriptional start site. The bent arrows above indicate the primers used for the transcriptional fusions, and the numbers correspond to the distance from the transcriptional start site. P and the bent arrow indicate the start site of transcription. The arrowheads represent potential AlgR-binding sites. The bent arrow below indicates the primer used in the primer extension experiment. Indicated below is the probe generated by in vitro transcription for Northern analysis. Transcriptional fusions are indicated below. Numbers above the arrows indicate the distance from the transcriptional start site. (C) Transcriptional fusion rsmYTF1-lacZ was introduced into the indicated strains in single copy, grown for 8 h in LB broth, and assayed for β-galactosidase activity. Significant differences from the wild type were determined using a one-way analysis of variance and Tukey's posttest. Asterisks indicate P values of 0.001 (**) and <0.0001 (***). (D) Northern blot analysis of RsmY in the indicated strains after growth for 8 h in LB broth. The ΔrsmY mutant was used as a control. The top half is a membrane probed with the RsmY probe, and the bottom portion is a second independent blot of the loading control probed with a 5S rRNA probe. Northern blotting was performed at least three times. (E) Transcriptional fusion rsmYTF2-lacZ was introduced into the indicated strains in single copy, grown for 8 h in LB broth, and assayed for β-galactosidase activity. Significant differences from the wild type were determined using a one-way analysis of variance and Tukey's posttest. Triple asterisks indicate P values of <0.0001. All transcriptional fusion analyses were performed at least three times.
A transcriptional fusion, rsmYTF1-lacZ (Fig. 5B), was constructed and assayed in algR and algZ mutants in both the PAO1 (wild-type) and the mucA22 background. When a ΔalgR or algZ mutant was tested for rsmY reporter activity, there was no decrease compared to PAO1 (Fig. 5C). There was a >3-fold increase in rsmYTF1-lacZ activity in a mucA22 mutant strain compared to PAO1 (Fig. 5C). When tested in a mucA22 ΔalgR mutant strain, the increased activity in the mucA22 mutant strain was reduced to PAO1 levels (Fig. 5C). In the case of the mucA22 algZ mutant strain, there was a slight but statistically significant decrease in rsmYTF1-lacZ activity (Fig. 5C). Overall, the fusion results suggest that AlgR affects rsmY expression, but only in the mucA22 background.
To confirm that the rsmY levels decreased, Northern blotting was performed on the same strains. Two bands hybridized to the rsmY probe, as has been seen previously (Fig. 5D) (30, 38). Little difference was seen between a ΔalgR mutant strain and the wild-type PAO1 (data not shown). However, an increase in both hybridizing RNAs was seen in a mucA22 mutant strain (Fig. 5D). The mucA22 ΔalgR and mucA22 algZ mutant strains had decreased hybridizing fragments (Fig. 5D). The loading control used (5S rRNA) was consistently uneven upon repeated attempts, but the Northern blotting does confirm the transcriptional fusion analysis. A second probe using proC was also used and gave similar results. A ΔrsmY strain had no hybridizing RNA, confirming the specificity of the rsmY probe. Overall, these results support AlgR control of rsmY expression in the mucA22 strain.
Both rsmY and rsmZ contain upstream activating sequences (UAS) that GacA controls (39). A potential AlgR-binding site is located 23 nucleotides upstream of the UAS for rsmY. An additional AlgR-binding site is present further upstream on the opposite strand (Fig. 5B). Two additional transcriptional fusions, rsmYTF2-lacZ and rsmYTF3-lacZ (Fig. 5B and S2A), were constructed to determine the sequences required for AlgR control. The rsmYTF2-lacZ transcriptional fusion deletes the AlgR-binding sites but retains the UAS sequence (Fig. 5B). When assayed in the algR mutant strains in both PAO1 and mucA22 backgrounds, a difference was only seen in the mucA22 background (Fig. 5E). This result suggests that the potential AlgR-binding sites are not necessary for increased rsmY reporter activity.
An additional rsmY reporter, rsmYTF3-lacZ, was constructed that does not contain the UAS sequence. This fusion had very low activity in both PAO1 and mucA22 (Fig. S2B). This result suggests that the UAS is required for increased rsmY reporter activity in a mucA22 mutant strain as well as PAO1. Overall, the data suggest that AlgR and AlgU indirectly affect rsmY expression.
AlgR regulates rsmZ expression indirectly.
The regulation of rsmZ is more complex than rsmY and includes regulators other than GacA (38, 40, 41). Primer extension analysis confirmed the location of the transcriptional start site in P. aeruginosa that was predicted from P. fluorescens (Fig. 6A) (42, 43). A transcriptional fusion, rsmZTF1-lacZ, was assayed in the algZ and algR mutants (Fig. 6B). There was no difference in the rsmZ reporter in the ΔalgR or algZ mutant compared to PAO1 (Fig. 6C). There was a 5-fold increase in rsmZTF1-lacZ activity in the mucA22 mutant strain compared to PAO1 (Fig. 6C). In a mucA22 ΔalgR mutant strain, there was a significant decrease in rsmZTF1-lacZ activity. In a mucA22 algZ mutant strain, there was a significant but slight decrease in rsmZTF1-lacZ activity compared to the mucA22 mutant (Fig. 6C). In all, these results suggest that AlgR plays a major role and AlgZ plays only a minor role, if any, in terms of rsmZ expression in the mucA mutant background.
FIG 6.

AlgR increases rsmZ expression in a mucA22 mutant strain indirectly. (A) Primer extension analysis using total RNA isolated from PAO1. GATC indicates the sequencing ladder. -C is a control without reverse transcriptase. PAO1 is reverse-transcribed PAO1 mRNA. Below is the sequence of the rsmZ promoter region. The asterisk denotes the transcriptional start site identified. (B) Schematic of the rsmZ genomic region. The bent arrow on top indicates the primer used for transcriptional fusion construction. The bent arrow indicated by a “P” indicates the transcriptional start site. The bent arrow below indicates the primer used in primer extension. (C) Transcriptional fusion ZTF1-lacZ was introduced into the indicated strains in single copy, grown for 8 h in LB broth, and assayed for β-galactosidase activity. Significant differences from the wild type were determined using a one-way analysis of variance and Tukey's posttest. Asterisks indicate P values of 0.001 (**) and <0.0001 (***). Transcriptional fusion analysis was performed in triplicate three times. (D) Northern analysis of the indicated strains grown for 8 h in LB broth and total RNA probed with and rsmZ probe. A ΔrsmZ mutant strain was used to denote the specificity of the probe. A separate blot of the same strains was probed using a 5S rRNA probe (shown below). Northern blotting was performed three times.
To confirm the transcriptional fusion results, we again wanted to monitor the actual RNA levels. Using Northern blot analysis, a trend was seen similar to that in the transcriptional fusions. As shown in Fig. 6D, there were two bands detected, with the larger fragment being the most intense. Previous studies utilizing Northern blotting suggested that the smaller hybridizing fragment represents the small RNA lacking the stem-loop found at the 3′ end of the small RNA (30). Therefore, the smaller hybridizing bands may represent the small RNAs lacking a transcriptional terminator. A ΔrsmZ mutant was used as a negative control. The mucA22 mutant strain had increased RsmZ compared to PAO1 (Fig. 6D). The mucA22 ΔalgR mutant strain had the most drastic decrease in RsmZ levels, and there was a slight decrease in RsmZ levels in the mucA22 algZ mutant (Fig. 6D). Altogether, these results suggest that AlgR plays a role in increasing the RsmZ small RNA in the mucA22 background.
We hypothesized that like rsmY expression, AlgR indirectly affects rsmZ expression. An additional transcriptional fusion, rsmZTF2-lacZ (Fig. S3A), was constructed and assayed in the wild-type strain PAO1 and a mucA22 mutant strain, which lacks the UAS sequence. Both strains had drastically decreased reporter activity (Fig. S3B). Interestingly, the mucA22 mutant strain had significantly decreased activity compared to PAO1 (Fig. S3B). Further support for an indirect mechanism of AlgR activation was obtained using gel shift analysis of a PCR amplicon of the rsmZ upstream region (Fig. S3A). Purified AlgR was not able to shift the rsmZ upstream region tested (Fig. S3C), suggesting AlgR does not directly activate rsmZ expression. These results suggest that the UAS is required for increased rsmZ expression, and we conclude that AlgR affects rsmZ expression indirectly.
AlgR is necessary for RsmA activity in a mucA mutant background.
To ascertain whether AlgR control of rsmA was significant in a biological context, the direct RsmA targets hcnA and tssA1 were used to assess RsmA activity (29). A new integrating vector was constructed that contains the lacUV5 promoter and lacks a ribosome-binding site or a start codon for lacZ. When tested in the wild-type strain PAO1, there was no activity of the vector alone.
The hcnA gene encodes part of the hydrogen cyanide synthase enzyme that is implicated in virulence (44, 45). RsmA was shown to previously negatively affect a leader fusion using the tac promoter and the ribosome-binding site region of hcnA (45). An hcnA leader fusion was constructed by annealing 33 nucleotides, including one of the predicted RsmA-binding sites, the ribosome-binding site, and the translational start codon of hcnA. As shown in Fig. 7A, the lacUV5 hcnA-lacZ fusion had activity in strain PAO1. When assayed in a mucA22 mutant strain, there was an ∼2.5-fold decrease in reporter expression (Fig. 7A), consistent with increased RsmA levels in the mucA22 mutant strain. If rsmA expression requires AlgR and AlgU, mutating algR and algU should result in increased fusion activity in the mucA22 background due to decreased rsmA expression. Both the mucA22 ΔalgR and the mucA22 ΔalgU mutant strains had at least a 3-fold increase in reporter expression compared to the mucA22 strain (Fig. 7A). The mucA22 ΔrsmA mutant strain was assayed and had a 3.6-fold increase in reporter activity compared to the mucA22 mutant (Fig. 7A). Overall, these results are consistent with the conclusion that RsmA is active in the mucA22 strain and that AlgU and AlgR are important for the decreased posttranscriptional regulation of hcnA.
FIG 7.
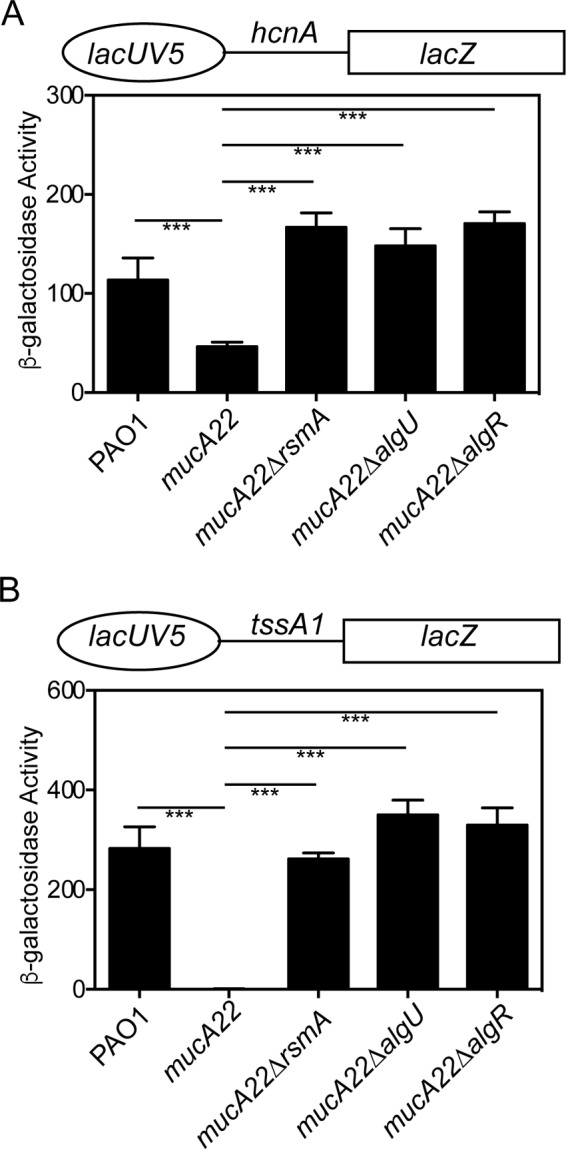
RsmA is active in a mucA22 mutant strain. (A) Leader/translational fusion lacUV5 hcnA-lacZ (above graph) was introduced into the indicated strains in single copy, grown for 8 h in LB broth, and assayed for β-galactosidase activity. Significant differences from the wild type were determined using a one-way analysis of variance and Tukey's posttest. Triple asterisks indicate P values of <0.0001. (B) Leader/translational fusion lacUV5 tssA1-lacZ (above graph) was introduced into the indicated strains in single copy, grown for 8 h in LB broth, and assayed for β-galactosidase activity. Significant differences from the wild type were determined using a one-way analysis of variance and Tukey's posttest. Triple asterisks indicate P values of <0.0001. All leader/translational fusions were assayed in triplicate three times.
Another RsmA target, tssA1, was also analyzed. The tssA1 gene encodes a portion of the T6SS and is directly regulated by RsmA (29, 46). Previous studies have used a lacUV5 tssA1 construct containing 227 bp of tssA1 upstream sequence that had extremely low activity (28, 29, 31). However, this fusion was found to contain additional transcriptional controls that resulted in low activity (data not shown). To more carefully assess RsmA activity using tssA1, we constructed a new leader fusion by annealing 31 nucleotides, including the predicted RsmA-binding site, the ribosome-binding site, and the translational start codon of tssA1. The lacUV5 tssA1-lacZ fusion had approximately 200 times the activity as the previously constructed lacUV5 tssA1-lacZ fusion containing 227 bp of upstream sequence in the wild-type strain PAO1 (Fig. S4). There was no activity when this fusion was assayed in a mucA22 mutant strain (Fig. 7B and S4). The lacUV5 tssA1-lacZ fusion confirmed our previous result (31), demonstrating increased activity in the mucA22 ΔalgU mutant strain, validating the use of this construct. When a mucA22 ΔalgR mutant was tested, the fusion was also increased from the mucA22 mutant strain to levels similar to those of the mucA22 ΔalgU mutant strain (Fig. 7B). A mucA22 ΔrsmA mutant strain also had statistically significant activity compared to that of the mucA22 mutant (Fig. 7B). These results demonstrate that AlgR and AlgU are necessary for posttranscriptional regulation of tssA1. From these data, we conclude that AlgU and AlgR are both required for increased rsmA expression in the mucA mutant background and that this leads to posttranscriptional regulation on two known RsmA targets, most likely through RsmA.
DISCUSSION
AlgR is an important two-component regulator having roles in both acute and chronic infections. In the case of acute infections, AlgR activates the fimU operon, enabling the production of T4P (19, 21, 47). In chronic infections, AlgR, AlgU, and other transcriptional regulators activate the production of alginate (22, 48–50). These and other virulence factors in P. aeruginosa are often considered mutually exclusive, depending on whether there is an acute or chronic infection (51–53). This work demonstrates that another effect of a mucA mutation is that AlgR activity is directed at increasing the posttranscriptional regulator RsmA. AlgR activation of rsmA may help explain how AlgR participates in the mutually exclusive production of some virulence factors depending on acute or chronic infection.
The initial description of AlgR as a regulator of rsmA (28) did not evaluate the contribution of AlgR phosphorylation by AlgZ or a mechanism for AlgR activation of rsmA. Because AlgR is part of a two-component system that is important for both acute and chronic infections (19, 32), it was necessary to address the mechanism for AlgR activation of rsmA in order to create a framework to understand how AlgR regulation of rsmA might impact virulence gene expression. AlgR binds a consensus sequence CCGTTCGTC (21, 48, 49), and phosphorylation is thought to enable AlgR to bind potential binding sites that deviate from this consensus, such as the sites found in the fimU promoter (32, 47). However, the rsmA promoter deviates from the AlgR-binding consensus, and AlgR phosphorylation was not required for rsmA expression. This argues against phosphorylation as the sole mechanism for AlgR binding less well-conserved consensus sequences. The studies using the mucA22 mutant strain, the algZ mutant, and the D54N mutant are consistent with the conclusion that AlgR phosphorylation is not required for rsmA activation. As AlgR phosphorylation is not required for alginate expression (22), our results are consistent with the conclusion that AlgR activates rsmA expression in mucoid strains.
The mechanism for AlgR activation of rsmA is by directly binding upstream of the AlgU-dependent promoter, further supporting our model (Fig. 8). Gel shift studies determined that two AlgR-binding sites are required for in vitro binding. The location of the AlgR-binding sites, the in vitro gel shift analysis, transcriptional fusions, and RNase protection assay all support AlgR binding the distal promoter region of rsmA and are in accord with the activation of the AlgU-dependent promoter.
FIG 8.
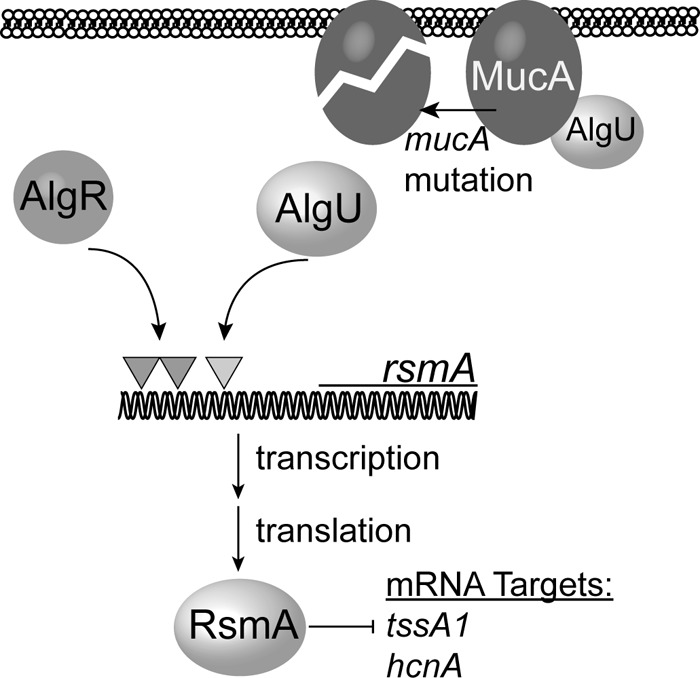
Model for AlgR activation of rsmA and RsmA activity in mucoid strains. In chronic infections, mucA mutants arise, leading to increased AlgU activity. In the case of rsmA, AlgR and AlgU activate the distal rsmA promoter. The dark-shaded triangles indicate the AlgR-binding sites located upstream of rsmA. The light-shaded triangle indicates the AlgU-dependent promoter. The model predicts that increased expression of rsmA leads to increased RsmA activity, as indicated by the negative regulation of the RsmA targets, hcnA and tssA1.
Our data suggest that AlgR plays a more important role in regulating rsmA expression in mucA mutant strains, such as those found in CF patients. If AlgU and AlgR activate rsmA expression, as predicted by our model (Fig. 8), this would explain why AlgR did not appear to play a significant role in regulating rsmA in the wild-type PAO1 strain (Fig. 1B and C, 2, and 3). From this work, we conclude that AlgR and AlgU play a greater role in rsmA activation in mucA mutant strains. However, it is possible that in vivo conditions differ and that nonmucoid wild-type strains also have increased regulation of rsmA under other conditions.
The mucA mutation leads to AlgU and AlgR activation of rsmA, suggesting that RsmA may regulate specific targets in mucA mutant strains. RsmA is a posttranscriptional regulator that binds mRNAs, in many cases, at or near the ribosome-binding site of targets (54). It is likely that RsmA has unknown targets in a mucA mutant background due to transcriptional differences between nonmucoid and mucoid strains (55–59). Therefore, RsmA will bind only to targets that are present at a given time, supporting RsmA working as a rheostat. An alternative explanation is that our conditions of growth in the laboratory environment do not represent the conditions in the CF lung. While this is quite obvious, it is known that the mutation in mucA leads to alginate production in vivo (11, 60, 61). In addition, nonmucoid P. aeruginosa also has active AlgU and AlgR during infection because of alginate secretion by nonmucoid P. aeruginosa from CF patients (62, 63). Therefore, it is highly probable that AlgU and AlgR activity in vivo would increase RsmA levels during infection. What role RsmA plays due to AlgU and AlgR regulation in vivo is not known.
The significance of AlgU and AlgR control of rsmA is demonstrated by the analyses of RsmA targets. The posttranscriptional activity of the RsmA targets, tssA1 and hcnA, was greater in the mucA22 background than in the wild-type PAO1 strain (Fig. 7). We also observed a lack of posttranscriptional activity on RsmA targets when algU or algR was inactivated in a mucA22 mutant strain (Fig. 7), due to the decreased expression of rsmA. Therefore, we conclude that RsmA is active in an mucA22 mutant strain and that RsmA activity requires AlgU and AlgR to increase rsmA gene expression in this background. The correlation between increased AlgR activity and increased RsmA activity in mucA mutants also supports these systems acting as a rheostat to fine-tune gene expression, as opposed to an on/off switch. When AlgU and AlgR increase RsmA levels, the indirect effects of these two regulators may be due to RsmA. Further work is necessary to pursue this exciting discovery that could help explain the exclusive expression of particular virulence genes in a given background.
The role of AlgU and AlgR on the RsmA-antagonizing small RNAs rsmY and rsmZ is likely indirect. While our Northern blot data were not strong, they did support our transcriptional fusion analysis. Our transcriptional fusions mirrored what was seen in a previous study (28), supporting the idea of AlgR indirectly controlling rsmY and rsmZ expression. We hypothesize that AlgU and AlgR activities may coincide with factors that lead to increased GacA phosphorylation, resulting in the increased expression of rsmY and rsmZ. Further work is necessary to establish the mechanism for increased expression of the small RNAs in a mucA background.
Another question raised by our study is how RsmA remains active when the antagonizing small RNAs are increased. One possibility is that RsmA preferentially binds its targets in the mucA mutant strains better than the antagonizing small RNAs. This might also result from other regulators, either protein or other small RNAs that are currently unknown and affect the ability of RsmA to interact with the antagonizing small RNAs RsmY and RsmZ.
Altogether, our work demonstrates that AlgR is required for increased rsmA expression in mucA mutant strains. Phosphorylation of AlgR was not required for rsmA activation. The increased RsmA levels in a mucA22 mutant strain result in increased RsmA activity, even though the antagonizing small RNAs are also increased. What roles RsmA plays and how RsmA functions in chronic infecting strains are not known. To better understand the role of RsmA, we are currently investigating new possible RsmA targets in AlgU-active strains. A further understanding of the RsmA regulon in mucA mutants may provide additional insight into how P. aeruginosa becomes such a successful CF pathogen and has implications for the important role of RsmA in all types of P. aeruginosa infections.
MATERIALS AND METHODS
Strains, plasmids, and media.
The strains used in this study are presented in Table 1. Escherichia coli strains were maintained on LB (Difco) plates or broth without or with antibiotics as appropriate. Pseudomonas aeruginosa strains were grown on Pseudomonas isolation agar (PIA), LB, or Vogel-Bonner minimal medium (64) and supplemented with the appropriate concentration of antibiotics. For E. coli, antibiotics were used at the following concentrations when appropriate: 10 μg/ml tetracycline, 15 μg/ml gentamicin, 100 μg/ml ampicillin, and 35 μg/ml kanamycin. For Pseudomonas strains, antibiotics were used at the following concentrations: 150 μg/ml gentamicin, 50 μg/ml tetracycline, and 300 μg/ml carbenicillin. For allelic exchange, sucrose was supplemented at 10% in YT (1% tryptone and 0.5% yeast extract) medium.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant propertiesa | Reference or source |
|---|---|---|
| E. coli strains | ||
| NEB5α | fhuA2 Δ(argF-lacZ)U169 phoA glnV44 ϕ80Δ(lacZ)M15 gyrA96 | New England BioLabs |
| SM10 | thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu Kmr | 72 |
| pRK2013 | Helper strain | 67 |
| Plasmids | ||
| pEX18Tc | Allelic exchange vector | 66 |
| pEX18Gm | Allelic exchange vector | 66 |
| ΔrsmA pEX18Tc | Allelic exchange for rsmA nonpolar deletion | 31 |
| ΔalgR pEX18Tc | Allelic exchange for algR nonpolar deletion | 47 |
| algZHSDM/pEX18Gm | Allelic exchange for making algZ mutant | This study |
| rsmAHA/pEX18Gm | Allelic exchange for rsmA HA allele | 31 |
| pGEX4T-3 | GST fusion vector | GE Healthcare |
| pGEX4T-3 algR | AlgR purification | This study |
| miniCTXlacZ | Transcriptional fusion vector | 73 |
| rsmATF1-lacZ | Transcriptional fusion | 31 |
| rsmATF3-lacZ | Transcriptional fusion | 31 |
| rsmATF2-lacZ | Transcriptional fusion | 31 |
| rsmYTF1-lacZ | Transcriptional fusion | This study |
| rsmYTF2-lacZ | Transcriptional fusion | This study |
| rsmYTF3-lacZ | Transcriptional fusion | This study |
| rsmZTF1-lacZ | Transcriptional fusion | This study |
| rsmZTF2-lacZ | Transcriptional fusion | This study |
| lacUV5CTXCP | Leader fusion vector | This study |
| lacUV5 tssA-lacZ | tssA1 leader fusion | This study |
| lacUV5 hcnA-lacZ | hcnA leader fusion | This study |
| P. aeruginosa strains | ||
| PAO1 | Wild type | 74 |
| ΔalgR mutant | algR mutant | 75 |
| D54N mutant | algR with asparagine instead of aspartate at residue 54 | This study |
| D54E mutant | algR with glutamate instead of aspartate at residue 54 | This study |
| algZ mutant | algZ mutant | This study |
| mucA22 mutant | mucA22 mutant | 76 |
| mucA22 ΔalgR mutant | mucA22 ΔalgR mutant | This study |
| mucA22D54N mutant | mucA22 mutant strain with algR with asparagine instead of aspartate at residue 54 | This study |
| mucA22D54E mutant | mucA22 mutant strain with algR with glutamate instead of aspartate at residue 54 | This study |
| mucA22 algZ mutant | mucA22 mutant strain, algZ mutation | This study |
| ΔrsmA mutant | rsmA nonpolar deletion | 31 |
| mucA22 ΔrsmA mutant | rsmA nonpolar deletion in mucA22 strain | 31 |
| ΔrsmY mutant | rsmY nonpolar deletion mutant | This study |
| ΔrsmZ mutant | rsmZ nonpolar deletion mutant | This study |
| PAO1 rsmATF1-lacZ | rsmA transcriptional fusion strain | 31 |
| ΔalgR TF1-lacZ mutant | rsmA transcriptional fusion strain | This study |
| D54N rsmATF1-lacZ mutant | rsmA transcriptional fusion strain | This study |
| D54E rsmATF1-lacZ mutant | rsmA transcriptional fusion strain | This study |
| mucA22D54N rsmATF1-lacZ mutant | rsmA transcriptional fusion strain | This study |
| mucA22D54E rsmATF1-lacZ mutant | rsmA transcriptional fusion strain | This study |
| mucA22 algZ mutant | rsmA transcriptional fusion strain | This study |
| mucA22 TF1-lacZ mutant | rsmA transcriptional fusion strain | 31 |
| mucA22 ΔalgR rsmATF1-lacZ mutant | rsmA transcriptional fusion strain | This study |
| PAO1 rsmATF3-lacZ | rsmA transcriptional fusion strain | 31 |
| ΔalgR rsmATF3-lacZ mutant | rsmA transcriptional fusion strain | This study |
| mucA22 TF3-lacZ mutant | rsmA transcriptional fusion strain | 31 |
| mucA22 ΔalgR rsmATF3-lacZ mutant | rsmA transcriptional fusion strain | This study |
| PAO1 rsmATF2-lacZ | rsmA transcriptional fusion strain | 31 |
| ΔalgR rsmATF2-lacZ mutant | rsmA transcriptional fusion strain | This study |
| mucA22 rsmATF2-lacZ mutant | rsmA transcriptional fusion strain | 31 |
| mucA22 ΔalgR rsmATF2-lacZ mutant | rsmA transcriptional fusion strain | This study |
| PAO1 rsmYTF1-lacZ mutant | rsmY transcriptional fusion strain | This study |
| ΔalgR rsmYTF1-lacZ mutant | rsmY transcriptional fusion strain | This study |
| algZ rsmYTF1-lacZ mutant | rsmY transcriptional fusion strain | This study |
| mucA22 rsmYTF1-lacZ mutant | rsmY transcriptional fusion strain | This study |
| mucA22 ΔalgR rsmYTF1-lacZ mutant | rsmY transcriptional fusion strain | This study |
| mucA22 algZ mutant | rsmY transcriptional fusion strain | This study |
| PAO1 rsmYTF2-lacZ | rsmY transcriptional fusion strain | This study |
| ΔalgR rsmYTF2-lacZ mutant | rsmY transcriptional fusion strain | This study |
| mucA22 rsmYTF2-lacZ mutant | rsmY transcriptional fusion strain | This study |
| mucA22 ΔalgR rsmYTF2-lacZ mutant | rsmY transcriptional fusion strain | This study |
| PAO1 rsmYTF3-lacZ | rsmY transcriptional fusion strain | This study |
| mucA22 rsmYTF3-lacZ mutant | rsmY transcriptional fusion strain | This study |
| PAO1 rsmZTF1-lacZ | rsmZ transcriptional fusion strain | This study |
| ΔalgR rsmZTF1-lacZ mutant | rsmZ transcriptional fusion strain | This study |
| algZ rsmZTF1-lacZ mutant | rsmZ transcriptional fusion strain | This study |
| mucA22 mutant | rsmZ transcriptional fusion strain | This study |
| mucA22 ΔalgR rsmZTF1-lacZ mutant | rsmZ transcriptional fusion strain | This study |
| mucA22 algZ rsmZTF1-lacZ mutant | rsmZ transcriptional fusion strain | This study |
| PAO1 rsmZTF2-lacZ | rsmZ transcriptional fusion strain | This study |
| mucA22 rsmZTF2-lacZ mutant | rsmZ transcriptional fusion strain | This study |
| PAO1HA | PAO1 with epitope-tagged RsmA | 31 |
| ΔalgRHA mutant | ΔalgR mutant with epitope-tagged RsmA | This study |
| mucA22HA mutant | mucA22 mutant with epitope-tagged RsmA | 31 |
| mucA22 ΔalgRHA mutant | mucA22 ΔalgR mutant with epitope-tagged RsmA | This study |
| PAO1 lacUV5 hcnA-lacZ mutant | PAO1 with hcnA leader fusion | This study |
| mucA22 lacUV5 hcnA-lacZ | mucA22 mutant with hcnA leader fusion | This study |
| mucA22 ΔrsmA lacUV5 hcnA-lacZ mutant | mucA22 ΔrsmA mutant with hcnA leader fusion | This study |
| mucA22 ΔalgU lacUV5 hcnA-lacZ mutant | mucA22 ΔalgU mutant with hcnA leader fusion | This study |
| mucA22 ΔalgR lacUV5 hcnA-lacZ mutant | mucA22 ΔalgR mutant with hcnA leader fusion | This study |
| PAO1 lacUV5 tssA1-lacZ | PAO1 with tssA1 leader fusion | This study |
| mucA22 lacUV5 tssA1-lacZ mutant | mucA22 mutant with tssA1 leader fusion | This study |
| mucA22 ΔrsmA lacUV5 tssA1-lacZ mutant | mucA22 ΔrsmA mutant with tssA1 leader fusion | This study |
| mucA22 ΔalgU lacUV5 tssA1-lacZ mutant | mucA22 ΔalgU mutant with tssA1 leader fusion | This study |
| mucA22 ΔalgR lacUV5 hcnA-lacZ mutant | mucA22 ΔalgR mutant with tssA1 leader fusion | This study |
Kmr, kanamycin resistance.
Mutant construction.
All PCR products were amplified from P. aeruginosa PAO1, unless otherwise noted, using Q5 polymerase (New England BioLabs). Crossover PCR (SOE'ing) (65) was used to construct deletion mutations and to clone into the suicide vector pEX18Tc or pEX18Gm (66). All cloned constructs were confirmed via sequencing. P. aeruginosa strains were conjugated with E. coli as a donor strain and the pRK2013-containing helper strain (67). Conjugations were performed overnight on LB plates at 30°C, and conjugations were plated for single-crossover mutants on the appropriate selective media. Merodiploids were grown without selection and then screened for sucrose sensitivity on YT–10% sucrose plates. Mutations were confirmed using PCR with primers containing the suffix intF and intR shown in Table 1 and sequencing of the resulting PCR fragment. Hemagglutinin (HA) tagging of proteins was accomplished using primers containing the HA tag at the 3′ end of the gene and introduced as described above using the suicide vector pEX18Gm.
algR mutant construction.
The wild-type algR genomic region was amplified using Q5 (NEB) and primers algRXbaIR and algRHindIIIF and cloned into pEX18Tc. Site-directed mutagenesis was performed using the algRD54XbaIF/algRD54NR primers for the D54N allele or the algRD54EF/algRD54ER primer pair for the D54E mutation. The algZ mutant was constructed using the algZHSDMF/algZHSDMR primers. The primers were phosphorylated and used in site-directed mutagenesis, in accordance with the manufacturer's instructions, using Q5 (NEB). Constructs were analyzed by restriction enzyme analysis and sequencing. Mutant strains were constructed using homologous recombination, as described above, and were checked using PCR and the algRintF/algRintR primer pair and digestion with the appropriate restriction enzyme. Additional PCR amplicons were sequenced to confirm the mutation in each strain using the same primers. Further confirmation of mutants was done using phenotypic assays.
Transcriptional and translational leader fusion analysis.
Upstream DNA fragments containing promoter regions were generated by using primers listed in Table 1 in conjunction with Q5 polymerase (New England BioLabs). PAO1 genomic DNA was used as the template. PCR products were cloned into pMiniT (NEB) and then subcloned into miniCTXlacZ using the restriction enzymes HindIII and BamHI, HindIII and EcoRI, or KpnI and BamHI (NEB). The rsmA transcriptional fusions have been previously described (31). To construct rsmY and rsmZ transcriptional fusions, the rsmYTFFEcoRI/rsmYTFR and rsmZTFF/rsmZTFR primer pairs, respectively, were used. PCR products were purified, cut with restriction enzymes, and inserted into the EcoRI and BamHI sites of miniCTXlacZ using T4 DNA ligase (NEB). The leader fusion vector was constructed by annealing the lacUV5NotI/lacUV5BamHI primer pair together and cloning into the NotI/BamHI site of CTXCP (31). Translational/leader fusions were constructed using the tssA1annealF/R and hcnAannealF/R primer pairs (Table 1) and were cloned into the ScaI/BamHI site of the leader fusion vector. Fusion constructs were confirmed by sequencing and conjugated into P. aeruginosa strains by triparental conjugation. Strains were selected for tetracycline resistance and then conjugated with pFLP2 to remove vector sequences (66). Strains were selected for carbenicillin resistance, grown overnight without selection, and plated on YT medium with 10% sucrose to select for the loss of pFLP2. Individual colonies were patch-plated onto VBMM CB300 and PIA to ensure the loss of pFLP2. To confirm the presence of the fusion constructs, PCR was performed using the forward primer used to construct the fusion and the reverse primer lacZRforTF (Table 1). β-Galactosidase activity was determined by incubating cell extracts with o-nitrophenyl-β-d-galactopyranoside (ONPG) (4 mg/ml), as described by Miller (68). A strain carrying the empty vector miniCTXlacZ was also conjugated into PAO1 and assayed, and this background (28 Miller units) was subtracted from all transcriptional fusions. The translational/leader fusion backbone CTXCPlacUV5 had no background activity. All mucoid strains were confirmed mucoid at the end of each experiment by plating on PIA plates to ensure all colonies were mucoid. Three biological replicates were reproduced for all assays.
AlgR purification.
The algR gene was PCR amplified using Q5 (NEB) and PAO1 chromosomal DNA using oligonucleotides algRSal1F and algRNot1R (Table 1). A 754-bp SalI/NotI fragment was cloned into pGEX-4T-3 (Novagen) to be expressed as a glutathione S-transferase–AlgR (GST-AlgR) fusion protein. The resulting plasmid (pGEX-4TAlgR) was transformed into E. coli BL21(DE3) (NEB) cells and incubated overnight. The colonies from this transformation were collected, inoculated into LB supplemented with 100 μg/ml ampicillin, and grown to an optical density at 600 nm (OD600) of 0.6; 0.2 mM isopropyl-β-d-galactopyranoside (IPTG) was added to induce AlgR expression for 4 h at 15°C. The cells were collected by centrifugation (6,740 × g, 15 min), washed once in 20 ml of phosphate-buffered saline (PBS), and resuspended in 1 ml of PBS containing protease inhibitors (Thermo Fisher). AlgR was purified from this supernatant using the GST spin purification kit (Thermo Fisher). After binding of the fusion protein and washing the column, AlgR was cleaved away from GST with 10 U of thrombin/column overnight at 22°C. Purified protein was dialyzed using a Slide-A-Lyzer (Thermo Fisher) and storage buffer (20% glycerol, 20 mM Tris [pH 7.5], 5 mM MgCl2, and 1 mM dithiothreitol [DTT]) overnight at 22°C. The identity of the purified protein was determined by mass spectrometry. The purity of AlgR was visually determined in a Coomassie-stained 7.5% electrophoresis gel (SDS-PAGE).
EMSAs.
Purified AlgR was used in electrophoretic mobility shift assays (EMSAs) using either PCR amplicons or annealed primers using the LightShift kit, in accordance with the manufacturer's instructions (Thermo Fisher Scientific). For rsmA, the rsmAPE3F/rsmAPE3R, rsmAPE1F/rsmAEcoRIR, or rsmAPE3F/rsmAEcoRIR primer pair was used to produce PCR amplicons using Taq polymerase (NEB). Amplicons were gel extracted and biotinylated using the 3′ biotinylation kit (Thermo Fisher Scientific). Annealed primers were heated to 95°C for 5 min and cooled to room temperature. The annealed primers were biotinylated and incubated with purified AlgR. Purified AlgR was incubated at increasing concentrations to determine the suitable concentrations to be used. The nonspecific competitor poly(dI-dC) at 25 ng/μl was used for all gel shift reactions. DNA and protein were electrophoresed through 5% or 10% native polyacrylamide gels, transferred to a nylon membrane using a semidry apparatus (Hoefer), UV cross-linked, developed using the chemiluminescence detection kit (Thermo Fisher Scientific), and visualized using a charge-coupled-device (CCD) camera (ProteinSimple). The gel shift assays were repeated at least two times.
Western blot analysis.
P. aeruginosa strains were grown in LB at 37°C for 8 h. The bacteria were collected by centrifugation, resuspended in sterile PBS, and lysed by sonication using a Branson sonifier. Total protein concentrations were determined by the Bradford protein assay (Bio-Rad, Carlsbad, CA). Cell extracts (5 to 10 μg) were separated by 15% SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane (Bio-Rad). The membranes were blocked and probed using a 1:2,500 dilution of anti-HA monoclonal antibody in blocking buffer (Thermo Fisher, Pittsburgh, PA), followed by a 1:30,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit antibody (69). Densitometry of Coomassie-stained SDS-PAGE gels was used to standardize the Western blots using total protein analysis, as described previously (70, 71). Detection was performed using the ECL Plus kit (Thermo Fisher) and chemiluminescence detection (ProteinSimple, Santa Clara, CA). The blots and Coomassie-stained SDS-PAGE were analyzed using ImageJ analysis. All Western blotting was repeated at least four times.
Primer extension.
Total RNA (15 μg) from P. aeruginosa was isolated using the PureLink RNA minikit (Life Technologies, Grand Island, NY). Radiolabeled primers rsmYPE1 and rsmZPE1 (see Table 1) were used in a reverse transcription reaction using ThermoScript (Life Technologies). A temperature of 42°C was used for the extension. The same primer used for primer extension was also used in a sequencing reaction with the rsmY or rsmZ upstream sequence in pGEM-T (Promega, Madison, WI) using the Sequenase 7-deaza-deoxy-GTP (7-deaza-dGTP) kit (Affymetrix, Cleveland, OH). Both primer extensions and sequencing reactions were run on a 6% acrylamide-8 M urea gel. The gel was dried and extension products detected using autoradiography.
In vitro transcription and RPA.
The rsmA, rsmY, and rsmZ probes for RNase protection assay (RPA) were synthesized using a PCR product generated with primers that incorporated a T7 promoter sequence at the 5′ end and using the MAXIscript T7 kit (Life Technologies) (Table 1). The reverse primer containing the T7 promoter sequence contained a nonhomologous sequence to discriminate between the full-length probe and protected fragments. The primers rsmASDMAlgRF and rsmARPAR were used to produce the PCR product for rsmA. The rsmYRPAT&R/rsmYRPAF primer pair for rsmY or rsmZRPAT7RPAF/rsmZRPAF primer pair for rsmZ was used to produce probes for the appropriate target. Probes were labeled by biotin-11-UTP (Life Technologies) at a ratio of 4:6 with UTP and were gel purified after in vitro transcription. Five micrograms of total RNA from each P. aeruginosa strain was precipitated with 800 pg of labeled probe and resuspended in hybridization buffer [80% formamide, 400 mM NaCl, 40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.4), 1 mM EDTA], heated 95°C for 5 min, and incubated overnight at 42°C. Negative controls using the probe incubated with 10 μg of yeast RNA were also included. Digestion of unhybridized RNA was performed using RNase A at 25 μg/ml and RNase T1 at 10 U/ml. A biotinylated RNA ladder (Kerafast) was also used to determine the size of protected probe fragments. The reactions were run on a 5% acrylamide-8 M urea gel for rsmA and a 10% acrylamide-8 M urea gel for rsmY and rsmZ. RNA was transferred to a positively charged membrane (Hybond N+; Amersham Biosciences) using a semidry blotting system and 0.5× Tris-borate-EDTA (TBE; Hoefer, Holliston, MA). Nucleic acid was UV cross-linked and probes detected using the chemiluminescent nucleic acid detection module (Thermo Scientific). After washing, the blot was incubated for 5 min and developed using a FluorChem M (ProteinSimple). RNase protection assays were performed at least three times.
Northern blot analysis.
Total RNA was extracted from strains using the PureLink RNA minikit (Thermo Fisher). RNA was quantitated using a NanoDrop (Thermo Fisher). Two to 5 μg of total RNA was electrophoresed through a 10% acrylamide-8 M urea gel using formaldehyde loading buffer. After electrophoresis, RNA was transferred using a semidry blotter (Hoefer, Holliston, MA) at 300 mAmp for 45 min and the membranes cross-linked using UV light (Bio-Rad). Probes produced by in vitro transcription were biotin labeled and hybridized to the membrane using ULTRAhyb buffer (Thermo Fisher) at 65°C overnight. The primers used to make probes for rsmY and rsmZ are listed in Table 2. For rsmY, the rsmYRPAF/rsmYT7RPAR primer pair was used. For rsmZ, the rsmZRPAF/rsmZRPART7 primer pair was used. Membranes were washed with two 5-min low-stringency washes at room temperature and 2 high-stringency washes for 15 min at 65°C. Blots were developed using the chemiluminescence detection kit (Thermo Fisher), in accordance with the manufacturer's instructions. For normalization, 5S RNA probes (Table 2) made by in vitro transcription were used using primers 5SrRNAI and 5SrRNA2T7. Northern blotting was repeated more than three times.
TABLE 2.
Primers used in this study
| Primer name | Sequence | Usea |
|---|---|---|
| algRintF | GCAACTGGACTGGCAGGTGC | Mutant |
| algRintR | CGCGACTGGTCATCGGCAG | Mutant |
| algRBamHIR | GCGCGGATCCGTCAGAGCTGATGCATCAGACG | Mutant |
| algZHindIIIF | GCGCAAGCTTCTCTCGCTGCAACAAGAAACGG | Mutant |
| algZSDMcheckF | CAGCTGGGCGGAGAACTGAC | Mutant |
| algRD54XbaIF | AACATCCGCATGCCCGGTCTAGACGGC | Mutant |
| algRD54NR | CAGCAGGACGATATCGGGCTTGA | Mutant |
| algRD54EF | GTCCTTCTAGAGATCCGCATGC | Mutant |
| algRD54ER | GATATCGGGCTTGAGGCTGTCG | Mutant |
| algZHSDMF | GAATTCCTGTTCAACAGCCTGAACAG | Mutant |
| algZHSDMR | CGGGCGAATCCGCGCCTGCA | Mutant |
| algRSalIF | CCTGTCGACATGAATGTCCTGATTGTCGATGAC | AlgR purification |
| algRNotIR | AGCCGCGGCCGCTCAGAGCTGATGCATCAGACG | AlgR purification |
| rsmARXbaI | GCGCTCTAGAGCACGGTGATCCTGCAGACC | Mutant |
| rsmAFHindIII | GCGCAAGCTTCGGCAACATCACCACGCTGGG | Mutant |
| rsmASDMcheckF | GCCAAGGTTTCCATCGTCGG | PCR check |
| rsmYRPAF | CTTGGACGTCAGGACATTGCGCAGGAAG | Northern blot |
| rsmYT7RPAR | TAATACGACTCACTATAGGGAGACAATAAAAAACCCCGCCTTTTGGG | Northern blot |
| rsmZRPAF | GGGCCCCACTCCTGCGTACAGGGAACAC | Northern blot |
| rsmZRPART7 | TAATACGACTCACTATAGGGAGACAAAAAAAAGGGGCGGGGTATTAC | Northern blot |
| rsmYTFFEcoRI | GCGCGAATTCGTGTTGCCGTCGGTCCGGC | TF |
| rsmYTFRBamHI | GCGCGGATCCTTCCTGCGCAATGTCCTGACG | TF |
| rsmYTFFUAS | GCGCGCGGCCGCCAGCGTGTAAGCCAAGGCTTAC | TF |
| rsmYshortEcoRIF | GCGCGAATTCAAACGGCGGTGGTTTTGGCTG | TF |
| dnrTFRBamHI | GCGCGGATCCCCATGCTGGGAAGGCTCGC | TF |
| rsmZKpnIF | GCGCGGTACCCGTCTGGCGCAGAAGGGCG | Mutant |
| rsmZXbaIR | GCGCTCTAGAGAGCGAAACCGCCAACATCC | Mutant |
| rsmZSOEF | GTTTTCTGGCAGGTTGCGGGATTTGCCTGCCGTTTTACTCGTC | Mutant |
| rsmZSOER | GACGAGTAAAACGGCAGGCAAATCCCGCAACCTGCCAGAAAAC | Mutant |
| rsmZintF | GCGCGCGGCCGCACCGATCCGTGCGAGCTG | Mutant |
| rsmZintR | CATGCAGGAATTCATCGAGCTG | Mutant |
| rsmYKOF | CGCGGAGCTCCTGTTCACTCGAAGCACTCCAG | Mutant |
| rsmYKOR | CGCGTCTAGATTCGCCAACTCCGCTATTTCG | Mutant |
| rsmYSOEF | GCGTAATCTTCAAACCGTCAGGATCCTGCGGCCCGAGGAAAACCGCGTCG | Mutant |
| rsmYSOER | CGACGCGGTTTTCCTCGGGCCGCAGGATCCTGACGGTTTGAAGATTACGC | Mutant |
| rsmYintF | CCTGGAGCTGGACGGGAG | Mutant |
| rsmYintR | GGAATTCAGGAAGGTGTCCC | Mutant |
| lacUV5NotI | GGCCGCTTTACACTTTATGCTTCCGGCTCGTATAATGTGTGGAG | Leader fusion vector |
| lacUV5BamHI | GATCCTCCACACATTATACGAGCCGGAAGCATAAACTGTAAAGC | Leader fusion vector |
| tssA1annealF | GATCCTTCGATGATAGGGAGATCGTCACCGTGCTG | Leader fusion |
| tssA1annealR | CAGCACGGTGACGATCTCCCTATCATCGAAG | Leader fusion |
| hcnAannealF | GATCCACTCTCTCTCACGGATGAAAGGGCAATGCAC | Leader fusion |
| hcnAannealR | GTGCATTGCCCTTTCATCCGTGAGAGAGAGTG | Leader fusion |
| 5SrRNAI | TTGGACAGGATGGGGTTGGA | Northern blot |
| 5SrRNA2T7 | TAATACGACTCACTATAGGGA GACGATTGTGTGTTGTAAGG | Northern blot |
| lacZScaIF | GCTATGACCATGATTAGTACTGATTCACTGGCCGTC | TLF vector |
| lacZRXhoI | CCCCTCGAGCAGACATGGCCTGCCCGGTTATTA | TLF vector |
| lacZRforTF | GATGTGCTGCAAGGCGATTAAG | SEQ |
| rsmASDMAlgRF | GAATTCGGCAGGAACTTTCATTCCGGC | RPA |
| rsmARPAR | TAATACGACTCACTATAGGGAGAGATAAAAATTAATGGTTTGGCTCTTG | RPA |
| rsmARSDMF | CCTTTTGTCGTTTTTGAGAATTCGGCAGGAACTTTCATTC | Gel shift |
| rsmARSDMR | GAATGAAAGTTCCTGCCGAATTCTCAAAAACGACAAAAGG | Gel shift |
| algDBS1F | CGCCTCCTGGCGCTACCGTTCGTCCCTCCGACACCCCTGCTGCGTCGC | Gel shift |
| algDBS1R | GCGACGCAGCAGGGGTGTCGGAGGGACGAACGGTAGCGCCAGGAGGCG | Gel shift |
| rsmARBS1F | GCGCCTTTTGTCGTTTTTGACCGTTTGGCAGGAACTTTCATTCCG | Gel shift |
| rsmARBS1R | CGGAATGAAAGTTCCTGCCAAACGGTCAAAAACGACAAAAGGCGC | Gel shift |
| pscFRBSF | GCTGGCGGAGTGTCGCCGGGAACTGGCCAGAGG | Gel shift |
| pscFRBSR | CCTCTGGCCAGTTCCCGGCGACACTCCGCCAGC | Gel shift |
| rsmAPE1F | GCGCGGTACCAAGGATCGCGCTCTTGATTTCTGCGGATCCGCCGCCATTTCTT | Gel shift |
| rsmAPE3F | GCGCGGTACCGCTGACAGGCGAAAGGCG | Gel shift |
| rsmAPE3R | GCGCGGATCCTCACCCAGTATTGACCAGTCC | Gel shift |
| rsmAEcoRIR | GCGCGAATTCACGAGTCAGAATCAGCATTCCTTTC | Gel shift |
| rsmAR1SDMannealF | CCGCGGAATTCGTCGTTTTTGACCGTTTGGCAGGAACTTTC | Gel shift |
| rsmAR1SDMannealR | GAAAGTTCCTGCCAAACGGTCAAAAACGACGAATTCCGCGG | Gel shift |
| rsmAR2SDMannealF | CCGCGGAATTCGTCGTTTTTGAGAATTCGGCAGGAACTTTC | Gel shift |
| rsmAR2SDMannealR | GAAAGTTCCTGCCGAATTCTCAAAAACGACGAATTCCGCGG | Gel shift |
| rsmAGSWTannealF | CCGCGCCTTTTGTCGTTTTTGACCGTTTGGCAGGAACTTTC | Gel shift |
| rsmAGSWTannealR | GAAAGTTCCTGCCAAACGGTCAAAAACGACAAAAGGCGCGG | Gel shift |
TF, transcriptional fusion; TLF, translational fusion; SEQ, sequencing primer; RPA, RNase protection assay.
Statistical analyses.
Statistical analyses were performed using Prism 6.0 (GraphPad software, La Jolla, CA). Transcriptional and translational fusions and densitometry analysis were compared using a one-way analysis of variance (ANOVA) with Tukey's posttest.
Supplementary Material
ACKNOWLEDGMENTS
We thank Laraine Powers (ETSU) for critical review of the manuscript. We thank Eric Jones for expert technical assistance.
This work was supported by Research and Development Committee (RDC) major grants (RDC12-013M and RDC17-007M) and startup funds to C.L.P.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00048-17.
REFERENCES
- 1.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. 1999. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun 67:3625–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burrows LL. 2012. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol 66:493–520. doi: 10.1146/annurev-micro-092611-150055. [DOI] [PubMed] [Google Scholar]
- 4.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet 47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 6.Garrett ES, Perlegas D, Wozniak DJ. 1999. Negative control of flagellum synthesis in Pseudomonas aeruginosa is modulated by the alternative sigma factor AlgT (AlgU). J Bacteriol 181:7401–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu W, Badrane H, Arora S, Baker HV, Jin S. 2004. MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J Bacteriol 186:7575–7585. doi: 10.1128/JB.186.22.7575-7585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjarnsholt T, Jensen PO, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Hoiby N. 2009. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol 44:547–558. doi: 10.1002/ppul.21011. [DOI] [PubMed] [Google Scholar]
- 9.Hoiby N, Ciofu O, Bjarnsholt T. 2010. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol 5:1663–1674. doi: 10.2217/fmb.10.125. [DOI] [PubMed] [Google Scholar]
- 10.Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, Collins J, Rock MJ, Splaingard ML. 2005. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293:581–588. doi: 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- 11.Pedersen SS. 1992. Lung infection with alginate-producing, mucoid Pseudomonas aeruginosa in cystic fibrosis. APMIS Suppl 28:1–79. [PubMed] [Google Scholar]
- 12.Govan JR, Deretic V. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60:539–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hershberger CD, Ye RW, Parsek MR, Xie Z-D, Chakrabarty AM. 1995. The algT (algU) gene of Pseudomonas aeruginosa, a key regulator involved in alginate biosynthesis, encodes an alternative sigma factor (sigmaE). Proc Natl Acad Sci U S A 92:7941–7945. doi: 10.1073/pnas.92.17.7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wozniak DJ, Ohman DE. 1994. Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J Bacteriol 176:6007–6014. doi: 10.1128/jb.176.19.6007-6014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deretic V, Dikshit R, Konyecsni WM, Chakrabarty AM, Misra TK. 1989. The algR gene, which regulates mucoidy in Pseudomonas aeruginosa, belongs to a class of environmentally responsive genes. J Bacteriol 171:1278–1283. doi: 10.1128/jb.171.3.1278-1283.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deretic V, Schurr MJ, Yu H. 1995. Pseudomonas aeruginosa, mucoidy and chronic infection phenotype in cystic fibrosis. Trends Microbiol 3:351–356. doi: 10.1016/S0966-842X(00)88974-X. [DOI] [PubMed] [Google Scholar]
- 17.Morici LA, Carterson AJ, Wagner VE, Frisk A, Schurr JR, Zu Bentrup KH, Hassett DJ, Iglewski BH, Sauer K, Schurr MJ. 2007. Pseudomonas aeruginosa AlgR repressed the Rhl quorum-sensing system in a biofilm-specific manner. J Bacteriol 189:7752–7764. doi: 10.1128/JB.01797-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lizewski SE, Lundberg DS, Schurr MJ. 2002. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect Immun 70:6083–6093. doi: 10.1128/IAI.70.11.6083-6093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitchurch CB, Alm RA, Mattick JS. 1996. The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 93:9839–9843. doi: 10.1073/pnas.93.18.9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitchurch CB, Erova TE, Emery JA, Sargent JL, Harris JM, Semmler AB, Young MD, Mattick JS, Wozniak DJ. 2002. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J Bacteriol 184:4544–4554. doi: 10.1128/JB.184.16.4544-4554.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belete B, Lu H, Wozniak DJ. 2008. Pseudomonas aeruginosa AlgR regulates type IV pilus biosynthesis by activating transcription of the fimU-pilVWXY1Y2E operon. J Bacteriol 190:2023–2030. doi: 10.1128/JB.01623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma S, Selvaraj U, Ohman DE, Quarless R, Hassett DJ, Wozniak DJ. 1998. Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J Bacteriol 180:956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu H, Mudd M, Boucher JC, Schurr MJ, Deretic V. 1997. Identification of the algZ gene upstream of the response regulator AlgR and its participation in control of alginate production in Pseudomonas aeruginosa. J Bacteriol 179:187–193. doi: 10.1128/jb.179.1.187-193.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathee K, McPherson CJ, Ohman DE. 1997. Posttranslational control of the AlgT (AlgU)-encoded sigma22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (AlgN). J Bacteriol 179:3711–3720. doi: 10.1128/jb.179.11.3711-3720.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boucher JC, Yu H, Mudd MH, Deretic V. 1997. Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect Immun 65:3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schurr MJ, Yu H, Martinez-Salazar JM, Boucher JC, Deretic V. 1996. Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J Bacteriol 178:4997–5004. doi: 10.1128/jb.178.16.4997-5004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones AK, Fulcher NB, Balzer GJ, Urbanowski ML, Pritchett CL, Schurr MJ, Yahr TL, Wolfgang MC. 2010. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J Bacteriol 192:5709–5717. doi: 10.1128/JB.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Intile PJ, Diaz MR, Urbanowski ML, Wolfgang MC, Yahr TL. 2014. The AlgZR two-component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol 196:357–366. doi: 10.1128/JB.01199-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brencic A, Lory S. 2009. Determination of the regulon and identification of novel mRNA targets of Pseudomonas aeruginosa RsmA. Mol Microbiol 72:612–632. doi: 10.1111/j.1365-2958.2009.06670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kay E, Humair B, Denervaud V, Riedel K, Spahr S, Eberl L, Valverde C, Haas D. 2006. Two GacA-dependent small RNAs modulate the quorum-sensing response in Pseudomonas aeruginosa. J Bacteriol 188:6026–6033. doi: 10.1128/JB.00409-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stacey SD, Pritchett CL. 2016. Pseudomonas aeruginosa AlgU contributes to posttranscriptional activity by increasing rsmA expression in a mucA22 strain. J Bacteriol 198:1812–1826. doi: 10.1128/JB.00133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okkotsu Y, Little AS, Schurr MJ. 2014. The Pseudomonas aeruginosa AlgZR two-component system coordinates multiple phenotypes. Front Cell Infect Microbiol 4:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pritchett CL, Little AS, Okkotsu Y, Frisk A, Cody WL, Covey CR, Schurr MJ. 2015. Expression analysis of the Pseudomonas aeruginosa AlgZR two-component regulatory system. J Bacteriol 197:736–748. doi: 10.1128/JB.02290-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pastor A, Chabert J, Louwagie M, Garin J, Attree I. 2005. PscF is a major component of the Pseudomonas aeruginosa type III secretion needle. FEMS Microbiol Lett 253:95–101. doi: 10.1016/j.femsle.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 35.Quinaud M, Chabert J, Faudry E, Neumann E, Lemaire D, Pastor A, Elsen S, Dessen A, Attree I. 2005. The PscE-PscF-PscG complex controls type III secretion needle biogenesis in Pseudomonas aeruginosa. J Biol Chem 280:36293–36300. doi: 10.1074/jbc.M508089200. [DOI] [PubMed] [Google Scholar]
- 36.Kong W, Zhao J, Kang H, Zhu M, Zhou T, Deng X, Liang H. 2015. ChIP-seq reveals the global regulator AlgR mediating cyclic di-GMP synthesis in Pseudomonas aeruginosa. Nucleic Acids Res 43:8268–8282. doi: 10.1093/nar/gkv747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valverde C, Heeb S, Keel C, Haas D. 2003. RsmY, a small regulatory RNA, is required in concert with RsmZ for GacA-dependent expression of biocontrol traits in Pseudomonas fluorescens CHA0. Mol Microbiol 50:1361–1379. doi: 10.1046/j.1365-2958.2003.03774.x. [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Martínez I, Haas D. 2011. Azithromycin inhibits expression of the GacA-dependent small RNAs RsmY and RsmZ in Pseudomonas aeruginosa. Antimicrob Agents Chemother 55:3399–3405. doi: 10.1128/AAC.01801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brencic A, McFarland KA, McManus HR, Castang S, Mogno I, Dove SL, Lory S. 2009. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol 73:434–445. doi: 10.1111/j.1365-2958.2009.06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Ye F, Kumar V, Gao YG, Zhang LH. 2014. BswR controls bacterial motility and biofilm formation in Pseudomonas aeruginosa through modulation of the small RNA rsmZ. Nucleic Acids Res 42:4563–4576. doi: 10.1093/nar/gku106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrova OE, Sauer K. 2010. The novel two-component regulatory system BfiSR regulates biofilm development by controlling the small RNA rsmZ through CafA. J Bacteriol 192:5275–5288. doi: 10.1128/JB.00387-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heurlier K, Williams F, Heeb S, Dormond C, Pessi G, Singer D, Camara M, Williams P, Haas D. 2004. Positive control of swarming, rhamnolipid synthesis, and lipase production by the posttranscriptional RsmA/RsmZ system in Pseudomonas aeruginosa PAO1. J Bacteriol 186:2936–2945. doi: 10.1128/JB.186.10.2936-2945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aarons S, Abbas A, Adams C, Fenton A, O'Gara F. 2000. A regulatory RNA (PrrB RNA) modulates expression of secondary metabolite genes in Pseudomonas fluorescens F113. J Bacteriol 182:3913–3919. doi: 10.1128/JB.182.14.3913-3919.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gallagher LA, Manoil C. 2001. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183:6207–6214. doi: 10.1128/JB.183.21.6207-6214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pessi G, Williams F, Hindle Z, Heurlier K, Holden MT, Camara M, Haas D, Williams P. 2001. The global posttranscriptional regulator RsmA modulates production of virulence determinants and N-acylhomoserine lactones in Pseudomonas aeruginosa. J Bacteriol 183:6676–6683. doi: 10.1128/JB.183.22.6676-6683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marden JN, Diaz MR, Walton WG, Gode CJ, Betts L, Urbanowski ML, Redinbo MR, Yahr TL, Wolfgang MC. 2013. An unusual CsrA family member operates in series with RsmA to amplify posttranscriptional responses in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 110:15055–15060. doi: 10.1073/pnas.1307217110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okkotsu Y, Tieku P, Fitzsimmons LF, Churchill ME, Schurr MJ. 2013. Pseudomonas aeruginosa AlgR phosphorylation modulates rhamnolipid production and motility. J Bacteriol 195:5499–5515. doi: 10.1128/JB.00726-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohr CD, Hibler NS, Deretic V. 1991. AlgR, a response regulator controlling mucoidy in Pseudomonas aeruginosa, binds to the FUS sites of the algD promoter located unusually far upstream from the mRNA start site. J Bacteriol 173:5136–5143. doi: 10.1128/jb.173.16.5136-5143.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohr CD, Leveau JH, Krieg DP, Hibler NS, Deretic V. 1992. AlgR-binding sites within the algD promoter make up a set of inverted repeats separated by a large intervening segment of DNA. J Bacteriol 174:6624–6633. doi: 10.1128/jb.174.20.6624-6633.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramsey DM, Wozniak DJ. 2005. Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol 56:309–322. doi: 10.1111/j.1365-2958.2005.04552.x. [DOI] [PubMed] [Google Scholar]
- 51.Goodman AL, Lory S. 2004. Analysis of regulatory networks in Pseudomonas aeruginosa by genomewide transcriptional profiling. Curr Opin Microbiol 7:39–44. doi: 10.1016/j.mib.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski MA, Kazmierczak BI. 2006. Mutational analysis of RetS, an unusual sensor kinase-response regulator hybrid required for Pseudomonas aeruginosa virulence. Infect Immun 74:4462–4473. doi: 10.1128/IAI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ventre I, Goodman AL, Vallet-Gely I, Vasseur P, Soscia C, Molin S, Bleves S, Lazdunski A, Lory S, Filloux A. 2006. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc Natl Acad Sci U S A 103:171–176. doi: 10.1073/pnas.0507407103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH. 2007. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol 14:807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- 55.Firoved AM, Boucher JC, Deretic V. 2002. Global genomic analysis of AlgU (sigma(E))-dependent promoters (Sigmulon) in Pseudomonas aeruginosa and implications for inflammatory processes in cystic fibrosis. J Bacteriol 184:1057–1064. doi: 10.1128/jb.184.4.1057-1064.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Firoved AM, Deretic V. 2003. Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J Bacteriol 185:1071–1081. doi: 10.1128/JB.185.3.1071-1081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Firoved AM, Ornatowski W, Deretic V. 2004. Microarray analysis reveals induction of lipoprotein genes in mucoid Pseudomonas aeruginosa: implications for inflammation in cystic fibrosis. Infect Immun 72:5012–5018. doi: 10.1128/IAI.72.9.5012-5018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood LF, Ohman DE. 2009. Use of cell wall stress to characterize sigma 22 (AlgT/U) activation by regulated proteolysis and its regulon in Pseudomonas aeruginosa. Mol Microbiol 72:183–201. doi: 10.1111/j.1365-2958.2009.06635.x. [DOI] [PubMed] [Google Scholar]
- 59.Wood LF, Ohman DE. 2015. Cell wall stress activates expression of a novel stress response facilitator (SrfA) under sigma22 (AlgT/U) control in Pseudomonas aeruginosa. Microbiology 161:30–40. doi: 10.1099/mic.0.081182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pier GB, Saunders JM, Ames P, Edwards MS, Auerbach H, Goldfarb J, Speert DP, Hurwitch S. 1987. Opsonophagocytic killing antibody to Pseudomonas aeruginosa mucoid exopolysaccharide in older noncolonized patients with cystic fibrosis. N Engl J Med 317:793–798. doi: 10.1056/NEJM198709243171303. [DOI] [PubMed] [Google Scholar]
- 61.Ames P, DesJardins D, Pier GB. 1985. Opsonophagocytic killing activity of rabbit antibody to Pseudomonas aeruginosa mucoid exopolysaccharide. Infect Immun 49:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bragonzi A, Worlitzsch D, Pier GB, Timpert P, Ulrich M, Hentzer M, Andersen JB, Givskov M, Conese M, Doring G. 2005. Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. J Infect Dis 192:410–419. doi: 10.1086/431516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anastassiou ED, Mintzas AC, Kounavis C, Dimitracopoulos G. 1987. Alginate production by clinical nonmucoid Pseudomonas aeruginosa. J Clin Microbiol 25:656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schweizer HP. 1991. The agmR gene, an environmentally responsive gene, complements defective glpR, which encodes the putative activator for glycerol metabolism in Pseudomonas aeruginosa. J Bacteriol 173:6798–6806. doi: 10.1128/jb.173.21.6798-6806.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Higuchi R. 1989. Using PCR to engineer DNA, p 61–70. In Erlich HA. (ed), PCR technology: principles and applications for DNA amplification. Stockton Press, New York, NY. [Google Scholar]
- 66.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 67.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller JF. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, New York, NY. [Google Scholar]
- 69.Deretic V, Leveau JH, Mohr CD, Hibler NS. 1992. In vitro phosphorylation of AlgR, a regulator of mucoidy in Pseudomonas aeruginosa, by a histidine protein kinase and effects of small phospho-donor molecules. Mol Microbiol 6:2761–2767. doi: 10.1111/j.1365-2958.1992.tb01455.x. [DOI] [PubMed] [Google Scholar]
- 70.Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. 2008. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods 172:250–254. doi: 10.1016/j.jneumeth.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eaton SL, Roche SL, Llavero Hurtado M, Oldknow KJ, Farquharson C, Gillingwater TH, Wishart TM. 2013. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS One 8:e72457. doi: 10.1371/journal.pone.0072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Becher A, Schweizer HP. 2000. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29:948–950, 952. [DOI] [PubMed] [Google Scholar]
- 74.Holloway BW, Krishnapillai V, Morgan AF. 1979. Chromosomal genetics of Pseudomonas. Microbiol Rev 43:73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lizewski SE, Schurr JR, Jackson DW, Frisk A, Carterson AJ, Schurr MJ. 2004. Identification of AlgR-regulated genes in Pseudomonas aeruginosa by use of microarray analysis. J Bacteriol 186:5672–5684. doi: 10.1128/JB.186.17.5672-5684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JI, Jensen P, Johnsen AH, Givskov M, Ohman DE, Molin S, Hoiby N, Kharazmi A. 1999. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145:1349–1357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.