

ABSTRACT
Histidine kinases are key components of regulatory systems that enable bacteria to respond to environmental changes. Two major classes of histidine kinases are recognized on the basis of their modular design: classical (HKI) and chemotaxis specific (HKII). Recently, a new type of histidine kinase that appeared to have features of both HKIs and HKIIs was identified and termed HKIII; however, the details of HKIII's relationship to other two classes of histidine kinases, their function, and evolutionary history remain unknown. Here, we carried out genomic, phylogenetic, and protein sequence analyses that allowed us to reveal the unusual evolutionary history of this protein family, formalize its distinctive features, and propose its putative function. HKIIIs are characterized by the presence of sensory domains and the lack of a dimerization domain, which is typically present in all histidine kinases. In addition to a single-domain response regulator, HKIII signal transduction systems utilize CheX phosphatase and, in many instances, an unorthodox soluble chemoreceptor that are usual components of chemotaxis signal transduction systems. However, many HKIII genes are found in genomes completely lacking chemotaxis genes, thus decoupling their function from chemotaxis. By contrast, all HKIII-containing genomes also contain pilT, a marker gene for bacterial type IV pilus-based motility, whose regulation is proposed as a putative function for HKIII. These signal transduction systems have a narrow phyletic distribution but are present in many emerging and opportunistic pathogens, thus offering an attractive potential target for future antimicrobial drug design.
IMPORTANCE Bacteria adapt to their environment and their hosts by detecting signals and regulating their cellular functions accordingly. Here, we describe a largely unexplored family of signal transduction histidine kinases, called HKIII, that have a unique modular design. While they are currently identified in a relatively short list of bacterial species, this list contains many emerging pathogens. We show that HKIIIs likely control bacterial motility across solid surfaces, which is a key virulence factor in many bacteria, including those causing severe infections. Full understanding of this putative function may help in designing effective drugs against pathogens that will not affect the majority of the beneficial human microbiome.
KEYWORDS: chemotaxis, evolution, genomics, histidine kinase, motility, pathogens, signal transduction, type IV pili
INTRODUCTION
The transmission of molecular signals in prokaryotes is carried out by the specialized cellular machinery. The simplest signal transduction systems consist of a single protein that is capable of both sensing a signal and directly affecting a cellular response, for example, a ligand-binding transcriptional regulator. Such proteins, termed one-component systems (1), typically use two separate domains: input (also called a sensory domain) and output (also called a regulatory domain). A more complex mode of prokaryotic signal transduction involves two functionally dedicated proteins, a sensor and a response regulator, that make up a two-component system (2). The sensor is a histidine kinase, which consists of an input domain and a transmitter domain that communicates with the receiver domain of the response regulator, which in turn activates the response regulator's output domain. One- and two-component systems share a repertoire of input and output domains, but the main difference is that most one-component systems are known or predicted to detect signals in the cytoplasm, whereas most two-component systems are known or predicted to detect extracellular signals (1, 2). Variation in component design can be seen in both one- and two-component systems. For example, in one-component systems, a single-domain protein can be a sensor and a regulator (3), or multiple sensory and regulatory domains can be present in a single protein (4). In two-component systems, multiple sensory and regulatory domains per system can also exist (5), and additional phosphoacceptor and phosphodonor proteins can extend the system into a more complex phosphorelay (6).
The chemotaxis system, which is a special case of two-component regulatory systems, constitutes yet another mode of bacterial signal transduction. Bacteria navigate in chemical gradients by regulating their motility (7). This behavior, known as chemotaxis, is characterized by high sensitivity and precise adaptation, properties attributed to an assortment of interactions within the multiprotein signal transduction system (7, 8). This pathway utilizes principal modules of two-component systems; however, its design is markedly different. The chemotaxis signal transduction system is best understood in Escherichia coli. The histidine kinase of this system, the CheA protein, is sensorless (no input domain), and the cognate response regulator, the CheY protein, lacks an output domain (9, 10). Although the sequence similarity between the CheA-CheY pair and other two-component regulatory systems was noted early (2, 9), the CheA structure revealed such marked deviation from other known histidine kinases that CheA was proposed to constitute a separate class of histidine kinases, class II (HKII) (11). All other histidine kinases were assigned to class I (HKI).
While tracing the evolutionary history of class II histidine kinases, Wuichet and Zhulin described a protein family that exhibits properties of both class I and class II histidine kinases, which they termed class III histidine kinases (HKIIIs) (12). HKIIIs have an N-terminal sensory module typical of HKIs, but they also have a histidine phosphotransfer (HPT) domain that is N terminal to the kinase domain, similar to that in CheA. A sequence analysis also revealed that the N-box region of the HKIII kinase domain is more similar to that of CheA than to that of HKI (12). Only 4% of analyzed genomes contained HKIII genes, and all of these genomes also contained chemotaxis genes. It was noted that HKIIIs are often encoded near CheY-like proteins and CheX, a CheY phosphatase (12). HKIIIs have features of both HKIs and chemotaxis kinases and thus represent functionally intermediate forms, suggesting a gradual progression from classical two-component systems to the chemotaxis system (12). However, these conclusions were drawn using a very limited set of only 21 HKIII gene sequences from 17 genomes. In this study, we performed a much more in-depth analysis of this unusual group of histidine kinases using improved bioinformatics tools and much larger genomic data sets. While confirming some of the features of HKIII proteins revealed in the early analysis, we were able to identify their novel features, suggest a new evolutionary scenario for their emergence, and propose their putative function.
RESULTS
Identification of HKIIIs in genomic data.
In the first report describing HKIIIs, they were identified in a representative set of bacterial genomes using BLAST searches initiated with CheA sequences (12). In this study, we used the new domain model (HK_CA:Che) constructed specifically for CheA (13) to search the RefSeq database for all protein sequences that contain this domain and the histidine phosphotransfer (HPT) domain but lack the CheW domain, which is typical of CheA (10, 11). All retrieved sequences were then submitted to CDvist (14) to identify their complete domain architecture using a combination of specific and sensitive domain searches implemented in this server. Sequences where the CheW domains were identified as the result of these searches were then removed. The resulting set contained 253 protein sequences identified as HKIIIs (see Data Set S1 in the supplemental material).
Structural features of HKIIIs. (i) Sensory domain repertoire.
Domain architectures for all HKIII protein sequences identified in this study are shown in Fig. S1. Representative domain architectures are shown in Fig. 1. Domain combinations and transmembrane region predictions show that some HKIIIs are transmembrane proteins and some are cytoplasmic. While HKIIs are sensorless and HKIs have a wide repertoire of sensory domains (12), the repertoire of sensory domains in HKIIIs is very limited. The intracellular PAS domain (15) was found in 96% of sequences, always located N terminal to the HPT domain (Fig. 1; see also Fig. S1). The most prevalent extracellular sensory domain in HKIIIs is the four-helix bundle (16, 17) exemplified by the 4HB_MCP_1 and PilJ domain models (Fig. 1; see also Fig. S1). Other sensory domains identified in few instances include NIT (18), GAF (19), CBS (20), Cache (21), etc. (Fig. 1; see also Fig. S1). Notably, all sensory domains identified in HKIIIs are the subset of those found in chemoreceptors (that serve as sensors for HKIIs), which in turn are the subset of those found in HKIs (12).
FIG 1.
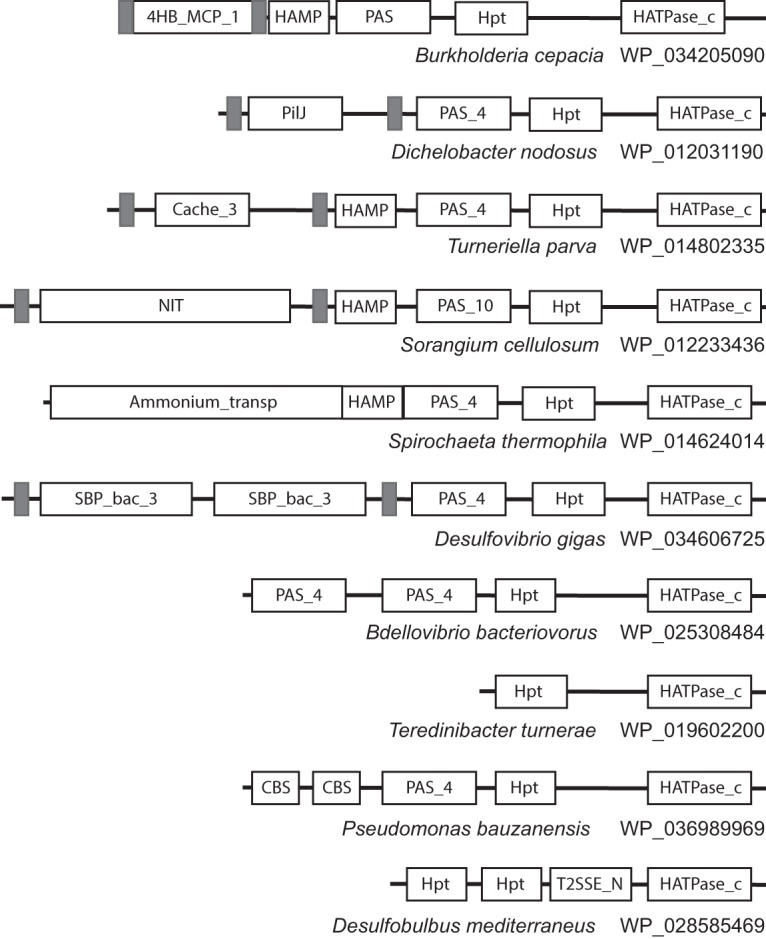
Representative domain architectures of HKIII protein sequences. Domain nomenclature is according to Pfam. Transmembrane regions are shown as gray rectangles. Sequences are identified by species names and NCBI accession numbers.
(ii) HKIIIs lack a dimerization domain.
The key feature differentiating HKIII from both HKI and HKII is the absence of the dimerization domain. In HKIs and HKIIs, the alpha-helical dimerization domains (detectable by Pfam domain model HisKA and related models from the same Pfam clan) are always found N-terminally adjacent to the kinase domains, whereas in HKIIIs, this domain is not found (Fig. 1; see also Fig. S1). HPT and the kinase domain in HKIIIs are connected by a linker, which can be as short as 4 amino acid residues. Even in longer linkers (e.g., 100 amino acid residues), no domains were detected by the most sensitive profile-profile search tool HHsearch (22). Further evidence for the absence of a dimerization domain in HKIIIs was obtained by running exhaustive iterative PSI-BLAST searches with the sequence corresponding to the long putative linker identified in an HKIII protein from Burkholderia cepacia (gi 736061708, residues 513 to 627), which retrieved nearly all HKIII sequences detected in this study, but failed to retrieve any sequences containing the dimerization domain in the region of sequence similarity to the query. In some HKIII sequences, other domains, e.g., T2SSE_N (Fig. 1), were predicted in this region. Finally, we predicted the secondary structure for HKIII sequences that had long putative linkers between HPT and the kinase domain and found that no secondary structure elements were predicted in the largest half of the interdomain region (see Fig. S2). Taken together, the results strongly suggest that HKIII proteins lack a dimerization domain typical of histidine kinases.
HKIII signal transduction systems. (i) Principal components.
Signal transduction systems anchored by histidine kinases contain response regulators and auxiliary components. Both simple two-component (e.g., kinase and regulator only) and much more complex chemosensory signal transduction systems, such as one controlling bacterial chemotaxis (kinase, multiple regulators, multiple sensors, and several auxiliary proteins), are encoded in gene clusters. This appears to be true for more than 90% of cases examined at the genome scale (12, 23). Satisfactorily, we found that more than 93% of HKIII genes have response regulator genes in their gene neighborhoods (see Data Set S2). The majority of response regulators associated with HKIIIs are single-domain proteins similar to the chemotaxis response regulator CheY (24). In addition, two other types of genes were enriched in the HKIII gene neighborhoods: (i) in 58% of cases, it was a gene coding for CheX phosphatase, which is known to dephosphorylate CheY (25), and (ii) in 60% of cases, it was a gene encoding methyl-accepting chemotaxis protein (MCP) (see Fig. S3 and Data Set S2). HKIII-associated MCPs (termed MCPHKIIIs) were much shorter than classical chemoreceptors—just over 200 amino acid residues—and matched the MCPsignal Pfam model depicting a chemoreceptor signaling domain (26). While being similar in length and domain composition to the soluble MCP TM0014 from Thermotoga maritima (27), MCPHKIII sequences formed a monophyletic cluster on a phylogenetic tree, indicating their divergence from TM0014 (see Fig. S4).
(ii) HKIII-associated MCPs retain conserved properties of the kinase-interacting subdomain.
To gain insight into the potential functionality of MCPHKIIIs, we assessed their sequence conservation and built a structural model of a representative protein using the most closely related structural template, the Tsr chemoreceptor from Escherichia coli. We were particularly interested in finding out whether the MCPHKIII model retains the homodimer properties of the template. Therefore, we performed molecular dynamics (MD) simulations of both the model and the template to compare the stability of their dimers (see Materials and Methods for details). The key results can be summarized as follows: (i) MCPHKIII has a conserved subdomain corresponding to a kinase-interacting region in all known MCPs and (ii) as in other MCPs, such as Tsr, this MCPHKIII subdomain forms a stable homodimer, whereas the N- and C-terminal regions of the protein are highly dynamic and do not dimerize.
The MCPHKIII structural model was built as a homodimer, similar to the Tsr template (Fig. 2); however, because its N-terminal region is much longer than the C-terminal region, dimerization occurs only in the bottom half (approximately throughout residues 94 to 236). We assessed potential contacts maintaining intradimer interactions (salt bridges and hydrophobic interactions) and found that, while the numbers of salt bridges are comparable between the model and its template, Tsr has twice as many pairs of interacting hydrophobic residues (see Fig. S5), which likely contributes to the dimer rigidity throughout its length. By contrast, MCPHKIII is much more flexible, although its tip corresponding to the kinase-interacting domain in Tsr is fairly rigid and remains dimerized (Fig. 2). Finally, the MCPHKIII tip retains the highest degree of conservation typical of MCPs (see Fig. S6). Interestingly, not only known intradimer contacts, e.g., a conserved phenylalanine corresponding to F396 in Tsr (28), but also some of the contacts critical for the trimer-of-dimer formation, e.g., positions corresponding to E402 and R404 in Tsr (29), are preserved in MCPHKIII (see Fig. S6).
FIG 2.
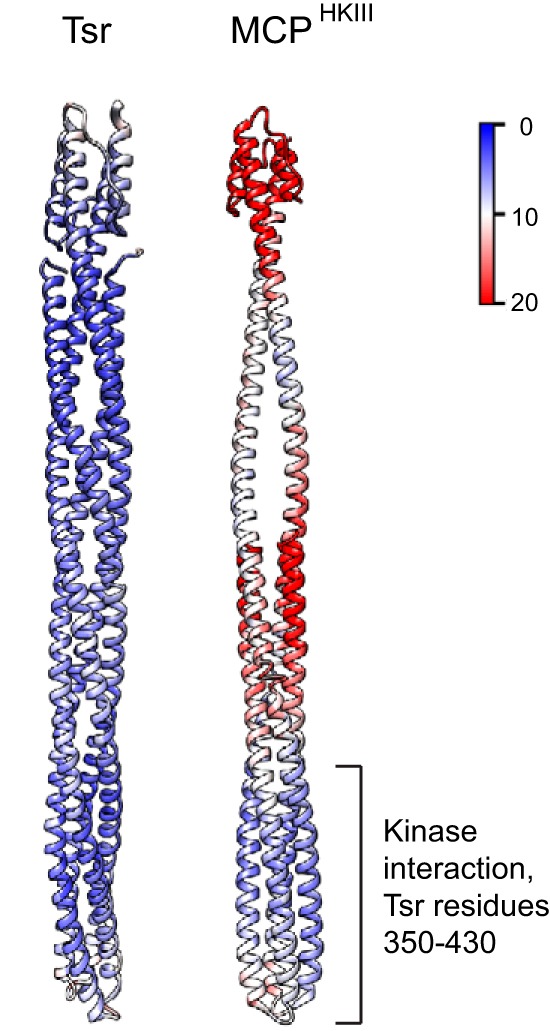
Comparison of dynamic properties of Tsr and MCPHKIII model. The values of root mean square fluctuations (RMSF; a measure of the average atomic mobility) for each residue backbone atom during 10-ns MD simulations are shown on the structure of the Tsr signaling domain and the model. Scale, 0 to 20 Å.
HKIIIs are found predominantly in Betaproteobacteria.
To compare the phyletic distribution of histidine kinases from the three classes, we used the Aquerium tool, which enables the visualization of both the presence and the absence of proteins and protein domains on the taxonomically ranked genome tree (30). The observed distribution of HKIs and HKIIs (Fig. 3) is fully consistent with the previous analyses. HKIs are most widely distributed in prokaryotes and also have representatives in archaea, lower eukaryotes, and plants, where they have been transferred horizontally (26, 31). HKIIs are missing from eukaryotes but are found in all bacterial phyla with a substantial number of sequenced representatives and in archaea, where they also have been transferred horizontally (12, 32). By striking contrast, HKIIIs are limited to very few bacterial phyla. Although we searched comparable numbers of genomes of Firmicutes and Proteobacteria, more than 75% of all sequences were found in representatives of Betaproteobacteria, predominantly in the orders of Burkholderiales and Neisseriales, and less than 2% of HKIII sequences were found in Firmicutes (Fig. 3; see also Data Set S1).
FIG 3.

Phyletic distribution of three classes of histidine kinases. Presences of class I, II, and III histidine kinases (shown in the blue, orange, and red outmost rings, respectively) on the taxonomic tree were revealed using the Aquerium tool (see Materials and Methods for details). Taxonomic ranks are shown in gray. Phyla that contain representatives with HKIIIs are highlighted in pink.
HKIII kinase domain is an intermediate evolutionary step between HKI and HKII.
HKIII relatedness to CheA was initially inferred from H-box and N-box similarity (12). New domain models developed for histidine kinases (13) clearly identify the kinase domain of HKIIIs as more similar to that of HKIIs; both are specifically recognized by the HK_CA:Che model. To further verify these observations, we have compared all histidine kinase domain sequences (matching the Pfam HATPase_c model and the corresponding E. coli CheA sequence WP_024189213, amino acid residues 323 to 506) from HKIIIs to a representative set of those from HKIs and HKIIs by constructing their multiple-sequence alignment and maximum likelihood phylogenetic tree (Fig. 4). The tree suggests that the HKIII domain evolved as an intermediate step in the transition from HKI to HKII. Though HKIIIs are more similar to HKIIs than to HKIs, they form a separate monophyletic group distinct from both HKIs and HKIIs. The sequence logo of HKIIIs shows extensive similarity to the one of HKIIs; however, a few conserved positions that are shared by HKIIIs and HKIs are not observed in HKIIs (see Fig. S7). Moreover, no sequence features that are shared by HKIs and HKIIs are found to be missing from HKIIIs.
FIG 4.
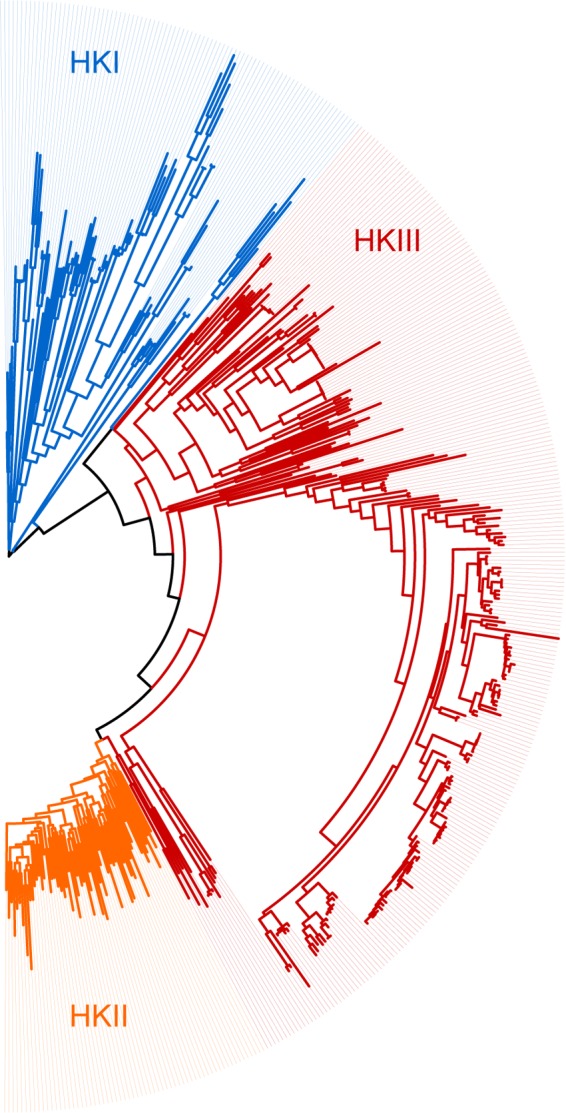
Relationship between HKI, HKII, and HKIII kinase domains. The maximum likelihood phylogenetic tree was built using representative histidine kinase sequences (see Materials and Methods for details).
HKIII genes are found only in genomes containing type IV pilus motility genes.
We have found a single report suggesting the role for the HKIII signal transduction system. Kennan et al. identified a two-component signal transduction system, TwmSR, with features similar to those of both HKIs and HKIIs in Dichelobacter nodosus and showed that its inactivation resulted in a reduced rate of type IV pilus (TFP)-mediated motility due to an overall loss of twitching directionality (33). The earlier report on HKIIIs stated that their genes are found exclusively in genomes that contain chemotaxis genes (12). To determine whether the presence of HKIII genes correlates with the presence of type IV pilus motility and chemotaxis genes, we have searched all genomes that had HKIII genes for the following markers: genes for PilT, a key component of TFP-mediated motility (34), CheA, a chemotaxis (HKII) histidine kinase, which controls flagellar mediated motility, and TFP-type CheA-like (HKII) histidine kinase (12), which mediates TFP motility in many bacterial species, e.g., Pseudomonas aeruginosa, where it is known as ChpA protein (35). We have found that HKIII genes are present in genomes with or without the flagellar CheA gene and with or without the TFP-type CheA gene in various combinations, including, both present, one present, and both absent (Data Set S1). By striking contrast, the PilT gene was found in all HKIII-containing genomes. Although, the opposite was not true; many bacterial genomes that contain the PilT gene lack HKIII genes.
DISCUSSION
Key structural features of HKIIIs.
HKIIIs combine features of both HKIs and HKIIs, resulting in a unique domain architecture. With very few exceptions, HKIIIs contain at least one (usually more than one) sensory domain, as is typical of HKIs; however, their kinase domains are much more similar to those of HKIIs than HKIs. Similar to HKIIs, HKIIIs contain the HPT domain with a conserved phosphodonor histidine; however, HPT in HKIIs is always located at the very N terminus and is separated from the kinase domain by CheY-binding and dimerization domains, whereas in HKIIIs, the HPT domain is located centrally between the sensory domain(s) and the kinase domain. These features can be used routinely for annotating HKIII gene sequences in bacterial genomes. Currently, all these sequences are erroneously annotated as CheA or as hypothetical proteins. The most remarkable molecular characteristic of HKIIIs is the lack of the dimerization domain, which is a key attribute in both HKIs and HKIIs. One unusual way in which transmembrane HKIIIs perhaps can dimerize is by interacting with MCPHKIII (comprised of a shortened signaling domain), which is encoded by a separate gene usually located in the same operon. Homology modeling suggests that a highly conserved tip of MCPHKIII is likely to form a homodimer, similar to that in conventional MCPs (36). Interestingly, in many HKIII kinases, three to four short alpha helices are predicted in the region between HPT and the catalytic domain. Potentially, this region may play the role of an unorthodox dimerization domain. Another potential site for HKIII dimerization is the periplasmic four-helix bundle domain, which is also known to dimerize in conventional MCPs (16). Finally, some cytoplasmic histidine kinases are known to be able to function as monomers (37–39), and this might be the case for HKIIIs.
Evolutionary scenario for HKIII emergence.
Unusual CheA-like proteins that were termed class III histidine kinases were proposed to be intermediate forms in the evolution of HKIs, yielding HKII (12). The rationale behind this argument was based on the observation of the HKIII features that were common to both class II and III histidine kinases. However, at least some of the results obtained in the current study seem to contradict this hypothesis. Several facts support the idea that HKIIIs are a recent innovation in bacterial signal transduction. First, the phyletic distribution of HKIIIs is extremely narrow; they are prevalent in Betaproteobacteria, primarily in the order Burkholderiales, whereas their occurrence in other phyla is limited to a few instances. This distribution favors a recent emergence of HKIIIs in Betaproteobacteria and their horizontal transfer to other species. Second, the sensory domain repertoire in HKIIIs is very limited. In essence, it is a small subset of those in MCPs that are in turn a subset of those found in HKIs. Consequently, because MCPs are evolutionarily younger than HKIs (12), HKIIIs appear to be evolutionarily younger than MCPs based on this feature (the lesser diversity and the younger the system). On the other hand, at least two other lines of evidence support the alternative scenario, where, according to the original proposal (12), HKIIIs predate HKIIs and the associated chemotaxis system. First and most important, the maximum likelihood phylogenetic tree built from HKI, HKII, and HKIII kinase domains (Fig. 4) clearly places HKIII between HKI and HKII. Second, the complexity of the component design in HKIII-based signal transduction systems is intermediate between HKI and HKII, and a gradually increasing level of complexity in signaling systems in evolution is well documented (1, 40). This apparently contradictory view on the evolutionary history of HKIIIs is reconcilable. We propose that the HKIII core—its kinase domain—is indeed an evolutionary intermediate between HKI and HKII. HKIs and HKIIs are extremely successful in what they do, namely, controlling gene expression and motility, correspondingly. HKIII appears to be a “nearly extinct species” that led from HKI to HKII and survived as an evolutionary reservoir for the occasional development of new features. One such new feature was its recent fusion with the MCP four-helix bundle sensory domain and the recruitment of the MCP tip (easily imaginable as an insertion of the HKIII gene into an MCP gene followed by a deletion of half of the MCP gene and its separation). This particular event has likely occurred in the recent ancestor of Burkholderiales, and its success manifests itself in the proliferation of this newly born signal transduction system within this bacterial order. When more bacterial genomes become available, we will most certainly witness other cases of such successful recent accessorizing of the otherwise fairly ancient histidine kinase domain.
Regulation of twitching motility is a likely role for HKIIIs.
The information about a potential function of the HKIII system is limited to a single study, where the HKIII protein in Dichelobacter nodosus was implicated in regulating TFP-mediated twitching motility (33). Our genomic survey shows that HKIII genes are found exclusively in genomes that contain a key component of TFP-based motility, the PilT protein (34). Remarkably, this rule holds for representatives of such distant phyla as Proteobacteria, Spirochaetes, Planctomycetes, Firmicutes, and Fibrobacteres. At the same time, we found no correlation between the presence of HKIII and chemotaxis genes, as was reported in a previous analysis that involved a much smaller number of genomes (12). For example, all representatives of the family Neisseriaceae in Betaproteobacteria and all representatives of Moraxellaceae in Gammaproteobacteria that have HKIIIs lack any components of the chemotaxis machinery. Our survey of HKIIIs is still very limited, and the apparent correlation between HKIII and PilT might as well be by pure chance. However, this hypothesis has a foundation and provides an excellent opportunity for verification, because many of the organisms where it was found are being studied in experimental laboratories around the world.
HKIII in emerging and opportunistic pathogens.
The list of genomes that contain HKIII genes is enriched with plant, animal, and human pathogens, providing additional support for a hypothesis that these signal transduction systems control TFP-mediated motility, a well-known virulence factor (41–46). Many of the species that contain HKIIIs are associated with infections in cystic fibrosis and immunocompromised patients, including Burkholderia cepacia (47), B. cenocepacia (48), B. contaminans (49), B. gladioli (50), B. multivorans (50, 51), B. vietnamiensis (50), Pandoraea apista (50), P. pulmonicola (50), P. sputorum (52), and Ralstonia pickettii (50), among others. Other HKIII-bearing human pathogens include Kingella kingae, which is linked to septic arthritis, osteomyelitis, spondylodiscitis, meningitis, endocarditis, and lower respiratory tract infections (53), as well as Burkholderia ferrarie and B. fungorum, which are implicated in synovial tissue infection and vaginosis (54, 55). Burkholderia ginsengisoli, a plant endosymbiotic bacterium, has recently been reported as a causative agent of bacteremia in a patient with Crohn's disease (56). In a similar fashion, bacteremia caused by the plant-associated bacteria Herbaspirillum huttiense and Acidovorax oryzae were reported in immunocompetent patients (57, 58). Kingella oralis and Neisseria sp. oral taxon 020 are inhabitants of the human oral cavity (59), and Neisseria shayeganii and N. wadsworthii were isolated from clinical specimens (60). HKIII-containing animal pathogens include Dichelobacter nodosus, the causative agent of footrot disease (61) in which HKIII control of TFP-based motility was demonstrated (33), and Moraxella species associated with periodontitis (62) and epistaxis (63). Plant pathogens include Acidovorax avenae and Burkholderia glumae (64), Acidovorax citrulli (65), and Herbaspirillum rubrisubalbicans (66). As the prevalence of antibiotic-resistant strains demands new therapeutic approaches, targeting bacterial functions that inhibit pathogenesis is the future of antivirulence therapies (42). If indeed the key role of HKIII signal transduction systems is to control TFP-mediated motility, they have a potential to become future drug targets.
MATERIALS AND METHODS
Databases and sequence similarity search tools.
The RefSeq database (downloaded on 6 August 2015) of complete nonredundant protein sequences was used in this study. HKs were identified based on the presence of the histidine kinase domain. The histidine kinase domains were classified into HKI and HKII using MiST (13) histidine kinase domain models HK_CA and HK_CA:Che, respectively, and similarity searches with the HMMER v2.4 package (67). Sequences containing the HK_CA:Che domain and HPT domain but lacking CheW domains were considered HKIIIs. Pfam_scan tool (HMMER v3) (68) was used to identify Pfam domains (68). Because domains may not always be sensitively identified by HMMER, the CDvist server (14) was used to perform more sensitive HMM-HMM searches with HHsearch tool (22) against the Pfam 29 database to confirm the absence of CheW domains. Sequences found to contain CheW domains were excluded from the HKIII set.
For the comparative analysis of HK classes, representative sets of HKIs and HKIIs were used against the complete set of HKIII sequences. To select representative HKIs, we retrieved sequences matching only one of the 23 HK subfamily domains retrieved from the MiST database (Agfam1 domains in SeqDepot) (13, 69). Up to five representatives for each subfamily were selected, if available. In the case of HKII domains, representatives were obtained by using CD-Hit (70) at 70% identity to reduce redundancy.
Multiple-sequence alignment and phyletic analyses.
Multiple-sequence alignments were constructed using the MAFFT (v7.154b) E-INSI algorithm (71). The initial HKII phylogenetic tree was built with FastTree (72) with default parameters. A representative set of HKII sequences was chosen from the phylogenetic tree by using a custom script. The final HK tree was built with PhyML (73) with default parameters, and the tree was rooted at the middle point using the Python ETE2 package (74).
The phyletic distribution figure was produced using the Aquerium tool (30), which uses NCBI taxonomic ranks (75) to build the tree. Eukaryotes and Archaea were excluded from the tree, because no HKIIIs were identified in these domains of life.
Gene neighborhoods.
The corresponding nucleotide sequence for each HKIII protein sequence was retrieved from NCBI. Because some protein records became obsolete in the updated NCBI database, the missing sequences were identified by running BLASTp (76) against the nonredundant protein database (December 2015) (see Data Set S2 in the supplemental material). To retrieve gene neighborhoods, six genes upstream and downstream of each HKIII gene were retrieved by parsing the nucleotide files using a custom script.
Structure modeling and MD simulation.
The amino acid sequence of MCPHKIII from Paraburkholderia (formerly, Burkholderia) fungorum (NCBI accession WP_028195998.1) was submitted to I-TASSER (77), which identified the Tsr MCP from Escherichia coli (Protein Data Bank no. 3ZX6, chain A, 303 amino acids) as the best template for modeling (overall sequence identity, >20%; C score = 0.45; TM score = 0.77 ± 0.10). The initial crystal structure of the template protein (homodimer, chains A and B) was obtained from the Protein Data Bank (78). The MCPHKIII homodimer was built by duplicating chain A with chain B and transferring coordinates of both chains obtained by the structural alignment between the model and the template. To rebuild coordinates of missing heavy atoms, the profix module from the Jackal package was used with a “heavy atoms model” option (79). The hydrogen atoms were added to the structures with VMD software v.1.9.1 (topology file from CHARMM27 force field) (80). Then, both structures were subjected to independent structural refinement by NAMD (v.2.9, CHARMM27 force field parameters) (81). For the minimization procedure, we used the generalized born implicit solvent (GBIS) model, implemented in NAMD. The dielectric constant of the implicit solvent was set to 80, and 1 for the protein 20,000-steps conjugate gradient algorithm implemented in NAMD was used to obtain the relaxed configuration with optimized geometric and steric clashes. Both structures were equilibrated for 1 ns followed by the molecular dynamic (MD) simulations for another 10 ns. The temperature of MD simulations was set to 298 K. The snapshots of the protein structures taken every 0.2 ns were used to estimate the probability of proteins to maintain the homodimeric form based on the following parameters: (i) the root mean square fluctuation (RMSF) of residues, (ii) the average number of hydrophobic contacts between monomers estimated by calculating the distance between side chains of hydrophobic residues at a ≤4-Å cutoff, and (iii) the average number of salt bridges between monomers, estimated based on the distance (≤3.2 Å) between oxygen atoms of the COO− group of acidic residues and the nitrogen atoms of the NH3+ group of basic residues.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants GM072285 and DE024463 from the National Institutes of Health (to I.B.Z.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00218-17.
REFERENCES
- 1.Ulrich LE, Koonin EV, Zhulin IB. 2005. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol 13:52–56. doi: 10.1016/j.tim.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu Rev Biochem 69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 3.Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF. 2001. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 A resolution. Nat Struct Biol 8:710–714. doi: 10.1038/90429. [DOI] [PubMed] [Google Scholar]
- 4.Tuckerman JR, Gonzalez G, Sousa EH, Wan X, Saito JA, Alam M, Gilles-Gonzalez MA. 2009. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry 48:9764–9774. doi: 10.1021/bi901409g. [DOI] [PubMed] [Google Scholar]
- 5.Zhulin IB, Nikolskaya AN, Galperin MY. 2003. Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. J Bacteriol 185:285–294. doi: 10.1128/JB.185.1.285-294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Appleby JL, Parkinson JS, Bourret RB. 1996. Signal transduction via the multi-step phosphorelay: not necessarily a road less traveled. Cell 86:845–848. doi: 10.1016/S0092-8674(00)80158-0. [DOI] [PubMed] [Google Scholar]
- 7.Wadhams GH, Armitage JP. 2004. Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol 5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 8.Hazelbauer GL, Falke JJ, Parkinson JS. 2008. Bacterial chemoreceptors: high-performance signaling in networked arrays. Trends Biochem Sci 33:9–19. doi: 10.1016/j.tibs.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kofoid EC, Parkinson JS. 1988. Transmitter and receiver modules in bacterial signaling proteins. Proc Natl Acad Sci U S A 85:4981–4985. doi: 10.1073/pnas.85.14.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wuichet K, Alexander RP, Zhulin IB. 2007. Comparative genomic and protein sequence analyses of a complex system controlling bacterial chemotaxis. Methods Enzymol 422:1–31. doi: 10.1016/S0076-6879(06)22001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bilwes AM, Alex LA, Crane BR, Simon MI. 1999. Structure of CheA, a signal-transducing histidine kinase. Cell 96:131–141. doi: 10.1016/S0092-8674(00)80966-6. [DOI] [PubMed] [Google Scholar]
- 12.Wuichet K, Zhulin IB. 2010. Origins and diversification of a complex signal transduction system in prokaryotes. Sci Signal 3:ra50. doi: 10.1126/scisignal.2000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulrich LE, Zhulin IB. 2010. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res 38:D401–D407. doi: 10.1093/nar/gkp940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adebali O, Ortega DR, Zhulin IB. 2015. CDvist: a webserver for identification and visualization of conserved domains in protein sequences. Bioinformatics 31:1475–1477. doi: 10.1093/bioinformatics/btu836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor BL, Zhulin IB. 1999. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev 63:479–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milburn MV, Prive GG, Milligan DL, Scott WG, Yeh J, Jancarik J, Koshland DE Jr, Kim SH. 1991. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science 254:1342–1347. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- 17.Ulrich LE, Zhulin IB. 2005. Four-helix bundle: a ubiquitous sensory module in prokaryotic signal transduction. Bioinformatics 21 Suppl 3:iii45–iii48. doi: 10.1093/bioinformatics/bti1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shu CJ, Ulrich LE, Zhulin IB. 2003. The NIT domain: a predicted nitrate-responsive module in bacterial sensory receptors. Trends Biochem Sci 28:121–124. doi: 10.1016/S0968-0004(03)00032-X. [DOI] [PubMed] [Google Scholar]
- 19.Aravind L, Ponting CP. 1997. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem Sci 22:458–459. doi: 10.1016/S0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- 20.Bateman A. 1997. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 22:12–13. doi: 10.1016/S0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 21.Upadhyay AA, Fleetwood AD, Adebali O, Finn RD, Zhulin IB. 2016. Cache domains that are homologous to, but different from PAS domains comprise the largest superfamily of extracellular sensors in prokaryotes. PLoS Comput Biol 12:e1004862. doi: 10.1371/journal.pcbi.1004862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams RH, Whitworth DE. 2010. The genetic organisation of prokaryotic two-component system signalling pathways. BMC Genomics 11:720. doi: 10.1186/1471-2164-11-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenal U, Galperin MY. 2009. Single domain response regulators: molecular switches with emerging roles in cell organization and dynamics. Curr Opin Microbiol 12:152–160. doi: 10.1016/j.mib.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park SY, Chao X, Gonzalez-Bonet G, Beel BD, Bilwes AM, Crane BR. 2004. Structure and function of an unusual family of protein phosphatases: the bacterial chemotaxis proteins CheC and CheX. Mol Cell 16:563–574. doi: 10.1016/j.molcel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Wuichet K, Cantwell BJ, Zhulin IB. 2010. Evolution and phyletic distribution of two-component signal transduction systems. Curr Opin Microbiol 13:219–225. doi: 10.1016/j.mib.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard AM, Bilwes AM, Crane BR. 2009. The structure of a soluble chemoreceptor suggests a mechanism for propagating conformational signals. Biochemistry 48:1936–1944. doi: 10.1021/bi801727m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ortega DR, Yang C, Ames P, Baudry J, Parkinson JS, Zhulin IB. 2013. A phenylalanine rotameric switch for signal-state control in bacterial chemoreceptors. Nat Commun 4:2881. doi: 10.1038/ncomms3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai RZ, Gosink KK, Parkinson JS. 2017. Signaling consequences of structural lesions that alter the stability of chemoreceptor trimers of dimers. J Mol Biol 429:823–835. doi: 10.1016/j.jmb.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adebali O, Zhulin IB. 2017. Aquerium: a web application for comparative exploration of domain-based protein occurrences on the taxonomically clustered genome tree. Proteins 85:72–77. doi: 10.1002/prot.25199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koretke KK, Lupas AN, Warren PV, Rosenberg M, Brown JR. 2000. Evolution of two-component signal transduction. Mol Biol Evol 17:1956–1970. doi: 10.1093/oxfordjournals.molbev.a026297. [DOI] [PubMed] [Google Scholar]
- 32.Briegel A, Ortega DR, Huang AN, Oikonomou CM, Gunsalus RP, Jensen GJ. 2015. Structural conservation of chemotaxis machinery across Archaea and Bacteria. Environ Microbiol Rep 7:414–419. doi: 10.1111/1758-2229.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennan RM, Lovitt CJ, Han X, Parker D, Turnbull L, Whitchurch CB, Rood JI. 2015. A two-component regulatory system modulates twitching motility in Dichelobacter nodosus. Vet Microbiol 179:34–41. doi: 10.1016/j.vetmic.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 34.Merz AJ, So M, Sheetz MP. 2000. Pilus retraction powers bacterial twitching motility. Nature 407:98–102. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- 35.Whitchurch CB, Leech AJ, Young MD, Kennedy D, Sargent JL, Bertrand JJ, Semmler AB, Mellick AS, Martin PR, Alm RA, Hobbs M, Beatson SA, Huang B, Nguyen L, Commolli JC, Engel JN, Darzins A, Mattick JS. 2004. Characterization of a complex chemosensory signal transduction system which controls twitching motility in Pseudomonas aeruginosa. Mol Microbiol 52:873–893. doi: 10.1111/j.1365-2958.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- 36.Kim KK, Yokota H, Kim SH. 1999. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature 400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 37.Rivera-Cancel G, Ko WH, Tomchick DR, Correa F, Gardner KH. 2014. Full-length structure of a monomeric histidine kinase reveals basis for sensory regulation. Proc Natl Acad Sci U S A 111:17839–17844. doi: 10.1073/pnas.1413983111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rinaldi J, Arrar M, Sycz G, Cerutti ML, Berguer PM, Paris G, Estrin DA, Marti MA, Klinke S, Goldbaum FA. 2016. Structural insights into the HWE histidine kinase family: the Brucella blue light-activated histidine kinase domain. J Mol Biol 428:1165–1179. doi: 10.1016/j.jmb.2016.01.026. [DOI] [PubMed] [Google Scholar]
- 39.Herrou J, Crosson S, Fiebig A. 2017. Structure and function of HWE/HisKA2-family sensor histidine kinases. Curr Opin Microbiol 36:47–54. doi: 10.1016/j.mib.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anantharaman V, Iyer LM, Aravind L. 2007. Comparative genomics of protists: new insights into the evolution of eukaryotic signal transduction and gene regulation. Annu Rev Microbiol 61:453–475. doi: 10.1146/annurev.micro.61.080706.093309. [DOI] [PubMed] [Google Scholar]
- 41.Persat A, Inclan YF, Engel JN, Stone HA, Gitai Z. 2015. Type IV pili mechanochemically regulate virulence factors in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 112:7563–7568. doi: 10.1073/pnas.1502025112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. 2008. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol 6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coureuil M, Bourdoulous S, Marullo S, Nassif X. 2014. Invasive meningococcal disease: a disease of the endothelial cells. Trends Mol Med 20:571–578. doi: 10.1016/j.molmed.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Lowry R, Balboa S, Parker JL, Shaw JG. 2014. Aeromonas flagella and colonisation mechanisms. Adv Microb Physiol 65:203–256. doi: 10.1016/bs.ampbs.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 45.Ceroni D, Dubois-Ferriere V, Cherkaoui A, Lamah L, Renzi G, Lascombes P, Wilson B, Schrenzel J. 2013. 30 years of study of Kingella kingae: post tenebras, lux. Future Microbiol 8:233–245. doi: 10.2217/fmb.12.144. [DOI] [PubMed] [Google Scholar]
- 46.Mhedbi-Hajri N, Jacques MA, Koebnik R. 2011. Adhesion mechanisms of plant-pathogenic Xanthomonadaceae. Adv Exp Med Biol 715:71–89. doi: 10.1007/978-94-007-0940-9_5. [DOI] [PubMed] [Google Scholar]
- 47.Parkins MD, Floto RA. 2015. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J Cyst Fibros 14:293–304. doi: 10.1016/j.jcf.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Lewis ER, Torres AG. 2016. The art of persistence-the secrets to Burkholderia chronic infections. Pathog Dis 74:ftw070. doi: 10.1093/femspd/ftw070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nunvar J, Kalferstova L, Bloodworth RA, Kolar M, Degrossi J, Lubovich S, Cardona ST, Drevinek P. 2016. Understanding the pathogenicity of Burkholderia contaminans, an emerging pathogen in cystic fibrosis. PLoS One 11:e0160975. doi: 10.1371/journal.pone.0160975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lipuma JJ. 2010. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev 23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zlosnik JE, Zhou G, Brant R, Henry DA, Hird TJ, Mahenthiralingam E, Chilvers MA, Wilcox P, Speert DP. 2015. Burkholderia species infections in patients with cystic fibrosis in British Columbia, Canada. 30 years' experience. Ann Am Thorac Soc 12:70–78. doi: 10.1513/AnnalsATS.201408-395OC. [DOI] [PubMed] [Google Scholar]
- 52.Martinez-Lamas L, Rabade Castedo C, Martin Romero Dominguez M, Barbeito Castineiras G, Palacios Bartolome A, Perez Del Molino Bernal ML. 2011. Pandoraea sputorum colonization in a patient with cystic fibrosis. Arch Bronconeumol 47:571–574. doi: 10.1016/j.arbr.2011.06.014 (In Spanish.) [DOI] [PubMed] [Google Scholar]
- 53.Yagupsky P. 2015. Kingella kingae: carriage, transmission, and disease. Clin Microbiol Rev 28:54–79. doi: 10.1128/CMR.00028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loong SK, Soh YH, Mahfodz NH, Johari J, AbuBakar S. 2016. Synovial tissue infection with Burkholderia fungorum. Emerg Infect Dis 22:1834–1835. doi: 10.3201/eid2210.151114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia Q, Cheng L, Zhang H, Sun S, Liu F, Li H, Yuan J, Liu Z, Diao Y. 2016. Identification of vaginal bacteria diversity and its association with clinically diagnosed bacterial vaginosis by denaturing gradient gel electrophoresis and correspondence analysis. Infect Genet Evol 44:479–486. doi: 10.1016/j.meegid.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Marks LR, Dodd H, Russo TA, Berenson CS. 2016. Burkholderia ginsengisoli bacteraemia: emergence of a novel pathogen. BMJ Case Rep 2016:bcr2015213584. doi: 10.1136/bcr-2015-213584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Regunath H, Kimball J, Smith LP, Salzer W. 2015. Severe community-acquired pneumonia with bacteremia caused by Herbaspirillum aquaticum or Herbaspirillum huttiense in an immune-competent adult. J Clin Microbiol 53:3086–3088. doi: 10.1128/JCM.01324-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orsborne C, Hardy A, Isalska B, Williams SG, Muldoon EG. 2014. Acidovorax oryzae catheter-associated bloodstream infection. J Clin Microbiol 52:4421–4424. doi: 10.1128/JCM.00657-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaddox LM, Huang H, Lin T, Hou W, Harrison PL, Aukhil I, Walker CB, Klepac-Ceraj V, Paster BJ. 2012. Microbiological characterization in children with aggressive periodontitis. J Dent Res 91:927–933. doi: 10.1177/0022034512456039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolfgang WJ, Carpenter AN, Cole JA, Gronow S, Habura A, Jose S, Nazarian EJ, Kohlerschmidt DJ, Limberger R, Schoonmaker-Bopp D, Sproer C, Musser KA. 2011. Neisseria wadsworthii sp. nov. and Neisseria shayeganii sp. nov., isolated from clinical specimens. Int J Syst Evol Microbiol 61:91–98. doi: 10.1099/ijs.0.022426-0. [DOI] [PubMed] [Google Scholar]
- 61.Kennan RM, Gilhuus M, Frosth S, Seemann T, Dhungyel OP, Whittington RJ, Boyce JD, Powell DR, Aspan A, Jorgensen HJ, Bulach DM, Rood JI. 2014. Genomic evidence for a globally distributed, bimodal population in the ovine footrot pathogen Dichelobacter nodosus. mBio 5:e01821-14. doi: 10.1128/mBio.01821-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Riggio MP, Jonsson N, Bennett D. 2013. Culture-independent identification of bacteria associated with ovine ‘broken mouth’ periodontitis. Vet Microbiol 166:664–669. doi: 10.1016/j.vetmic.2013.06.034. [DOI] [PubMed] [Google Scholar]
- 63.Ladner JT, Whitehouse CA, Koroleva GI, Palacios GF. 2013. Genome sequence of Moraxella macacae 0408225, a novel bacterial species isolated from a cynomolgus macaque with epistaxis. Genome Announc 1:e00188-12. doi: 10.1128/genomeA.00188-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang IJ, Kang MH, Noh TH, Shim HK, Shin DB, Heu S. 2016. Simultaneous detection of three bacterial seed-borne diseases in rice using multiplex polymerase chain reaction. Plant Pathol J 32:575–579. doi: 10.5423/PPJ.NT.05.2016.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang T, Guan W, Huang Q, Yang Y, Yan W, Sun B, Zhao T. 2016. Quorum-sensing contributes to virulence, twitching motility, seed attachment and biofilm formation in the wild type strain Aac-5 of Acidovorax citrulli. Microb Pathog 100:133–140. doi: 10.1016/j.micpath.2016.08.039. [DOI] [PubMed] [Google Scholar]
- 66.Monteiro RA, Balsanelli E, Tuleski T, Faoro H, Cruz LM, Wassem R, de Baura VA, Tadra-Sfeir MZ, Weiss V, DaRocha WD, Muller-Santos M, Chubatsu LS, Huergo LF, Pedrosa FO, de Souza EM. 2012. Genomic comparison of the endophyte Herbaspirillum seropedicae SmR1 and the phytopathogen Herbaspirillum rubrisubalbicans M1 by suppressive subtractive hybridization and partial genome sequencing. FEMS Microbiol Ecol 80:441–451. [DOI] [PubMed] [Google Scholar]
- 67.Johnson LS, Eddy SR, Portugaly E. 2010. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41:e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ulrich LE, Zhulin IB. 2014. SeqDepot: streamlined database of biological sequences and precomputed features. Bioinformatics 30:295–297. doi: 10.1093/bioinformatics/btt658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 74.Huerta-Cepas J, Dopazo J, Gabaldon T. 2010. ETE: a python Environment for Tree Exploration. BMC Bioinformatics 11:24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Federhen S. 2012. The NCBI taxonomy database. Nucleic Acids Res 40:D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER suite: protein structure and function prediction. Nat Methods 12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov IN, Bourne PE. 2000. The protein data bank. Nucleic Acids Res 28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiang JZ, Honig B. 2002. JACKAL: a protein structure modeling package. Columbia University, Howard Hughes Medical Institute, New York, NY. [Google Scholar]
- 80.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 81.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.