

Abstract
The transfer of phosphate groups is an essential function of many intracellular biological enzymes. The transfer is, in many cases, facilitated by a protein scaffold involving two closely spaced magnesium “ions”. It has long been a mystery as to how these “ions” can retain their closely spaced positions throughout enzymatic phosphate transfer: Coulomb’s law would dictate large repulsive forces between these ions at observed distances. Here we show, however, that the electron density can be borrowed from nearby electron rich oxygens to populate a bonding molecular orbital, largely localized between the magnesium “ions”. The result is that the Mg-Mg core of these phosphate transfer enzymes is surprisingly similar to metastable [Mg2]2+ ion in the gas phase, an ion which has been identified experimentally and studied with high level quantum mechanical calculations. This similarity is confirmed with comparative computations of the electron density for [Mg2]2+ in the gas phase and for the Mg-Mg core in the structures derived from QM/MM studies on high resolution x-ray crystal structures. That there is a level of covalent bonding between the two Mg “ions” at the core of these enzymes is a novel concept which enables an improved vision of how these enzymes function at the molecular level. The concept is broader than magnesium—other biologically relevant metals (e.g., Mn and Zn) can also form similar stabilizing covalent Me-Me bonds in both organometallic and inorganic crystals.
TOC Image
I. INTRODUCTION
Bimetallic magnesium catalytic active sites involving phosphate group transfer are widespread in nature. They are essential in DNA and RNA polymerases,1 reverse transcriptases,2 DNA and RNA endo- and exo-nucleases,3 terminases,4 and certain kinases (CDK2).5 It has been common to think that these active site metals function only as charged ions in stabilizing other active site components. We present an alternate view—a view that arises from examining the quantum mechanical nature of the “hidden” covalent metal-metal bond in the bimetallic active site and the relationship to homologous gaseous bimetallic di-cations. This alternate view is found to be remarkably consistent with earlier studies6 of bimetallic magnesium-containing compounds, for which each Mg has an additional electron, and a formal covalent bond between the di-cations is evident.
Observations we wish to understand in the present work are: (1) Why do relatively small divalent-metal cations arrange so close to each other in the active site (for instance, the Mg-Mg distance in these systems has been found to be as short as 2.42 Å)4? (2) Why is it that Mg ions in bimetallic di-cationic interactions observed in crystallographic structures of enzyme active sites do not occupy pseudo-symmetry-related octahedral coordination positions about the individual metals? Instead, metal ions in numerous structures obtained in the last twenty five years interact through the faces of opposing coordination octahedrals as shown in Figure 1. (3) What role does the meta-stable covalent bond between the metal ions (established below) play in enzyme catalysis?
Figure 1. The DNA polymerase β active site.
(pdb ID = 2FMS)7. Each metal ion has octahedral coordination of oxygen (red) ligands. The Mg-Mg bond is indicated by a dotted line. The catalytic Mg (Mg(c)) is magenta and the nucleotide binding Mg (Mg(n)) is orange. Note that the opposing metal ions do not occupy coordination positions, but instead interact along the dotted line.
The central hypothesis is that the bimetallic magnesium core in the active site of many enzymes can be understood in terms of the quantum mechanical properties of gas phase bimetallic di-cations [Me2]2+, that have a rich history in experimental and theoretical chemical physics.8,9 In the simple quantum mechanical description (bond orders, relative degree of stability, bond distances, magnetic properties) of the homonuclear diatomic molecules of the light elements (H2 through Ne2), the electron configuration of the diatom is displayed as an energy diagram from which the atomic orbitals of two adjacent metal atoms interact to form bonding (or antibonding) molecular orbitals (MO). When the existing count of electrons in the diatom is fed into the MO energy diagram starting at the bottom, the result is a MO description for the diatom. For instance,10 H2 : (σ1s)2, He2: (σ1s)2(σ1s*)2, . By this procedure, one finds that He2, Be2 and Ne2 are predicted to not naturally exist, O2 and B2 are predicted to have triplet ground states, and the bond orders for H2, C2 and N2 are predicted to be in the order of 1:2:3. All of these features are experimentally realized. We can also apply this approach to the next period of the Periodic Table (Na2, Mg2,..Ar2). When the same concepts are employed, one finds that Mg2 (same column of Periodic Table as Be2) is predicted to not exist, whereas [Mg2]2+ is predicted to exist (gas phase) with a simple MO bond description of ([Ne2](σ3s)2)2+ and a covalent bond order of 1 (Figure 2).
Figure 2. Standard molecular orbital electronic configurations of the outer electrons of relevant divalent metal di-cations for the first four rows of the period table.
Energy levels and their spacing adjusted for readability. The “*” superscript of the orbital label designates antibonding. He2, Be2, Mg2 and Ca2 (Mn2) have a net formal bond order of zero, whereas their di-cations have a net formal bond order of 1.
The purpose of this work is to seek answers to the questions raised in the early part of the Introduction that investigates the involvement of divalent bimetallic cations in the enzyme catalysis. We have primarily selected the active site of the DNA polymerase β that contains two magnesium ions as our test system since it has been the subject of a large number of structural and theoretical evaluations. In the present study, we have used QM and QM/MM calculations alone with QM charge and electron density evaluations to characterize the behavior of the closely spaced metal ions residing in a heavily ionic environment that may be fostering a pseudo covalent bond between the metal ions.
II. METHODS
Quantum mechanical calculations on various systems
The Gaussian09-version D01 program11 was employed to generate the total energy curves shown in Fig 3. Density functional calculations were performed using the B3LYP exchange–correlation functional and 6-31G* basis set.12
Figure 3. Energy versus metal ion separation distance (R) for various [Mea-Meb]2+ systems.
The curves were adjusted to converge at large R. Calculation details are given in Methods. Multiplicity of Mn complexes are given in parenthesis.
Computation of pre- and post-catalytic geometry representations for DNA polymerase β and the charges of the atoms in the active site
By employing crystallographic structures of the gapped DNA, dCTP, and DNA polymerase β (pdb ID = 2FMS)7 ternary complexes as the starting points, molecular dynamics simulations were carried out in a completely solvated aqueous environment. Valence-filling hydrogens were added, and the systems were neutralized with counter ions while preserving the positions of all the crystal water molecules. The initial triphosphate charge was set to −3e with only one oxygen protonated in the γ-phosphate for the equilibration with the classical force field. The Amber99SB force field was used with the PMEMD module of the Amber12 program for the trajectory calculations.13,14 Water molecules were represented by the TIP3P model.15 Long-range interactions were treated with the particle mesh Ewald method.16,17 Added water molecules were subjected to a constant pressure simulation at low temperatures (<10 K) so that the starting system was at a density near 1 g/cm3. Except for the added waters and counter ions, all heavy atoms in the system were constrained to the crystal positions using a harmonic restraining force with a decreasing force constant from 50 to 0.5 kcal/mol/nm during 10 ns simulations at 300 K at constant volume. This procedure ensures that the crystallographic coordinates, representing a configuration resembling the pre-catalytic state, are not undergoing dramatic perturbation by the molecular dynamics simulation. The final configuration was energy minimized without constraints before initiating the QM/MM steps.
The QM/MM systems were prepared in the following way: the quantum region (or the QM sub-system) includes parts of Asp190, Asp192, Asp256, parts of the primer terminal nucleotide including the phosphate group (excluding the base), and dNTP (excluding the base), two Mg ions, and 8 water molecules of which two coordinate the Mg ions (one on each). One of the magnesium ions is denoted as the catalytic magnesium, Mg(c). In the initial state, Mg(c) coordinates the reactive primer sugar O3′ atom, which in the chain extension reaction, forms the new bond with the Pα atom of the dNTP. Mg(c) also coordinates a non-bridging oxygen on Pα (pro-Rp). The other magnesium ion, the nucleotide binding magnesium, Mg(n), coordinates with non-bridging oxygens of all three phosphates of the incoming dNTP. Atoms within 10 Å of the quantum atoms were treated using a classical MM force field and allowed to move. The remainder of the protein, DNA, and counter-ions were held stationary. Solvent water within 15 Å, but farther than 10 Å from the QM atoms, were included and fixed during the calculation. There were 10158 atoms included in the QM/MM calculations, and 99 atoms in the QM sub-system. The charge for the QM region was −3e, and the MM region was +3e, while the total system remained neutral.
The hybrid QM/MM potential in the ONIOM (MO:MM) method implemented in the program Gaussian09-D01 was used to investigate bond formation during the reaction scheme.18,11 The quantum region was treated using the B3LYP exchange–correlation functional and 6-31g* basis set.12 The classical region was handled using the Amber ff99SB force field within Gaussian09. Calculations were performed with the electronic embedding option to accommodate the polarization of the QM region by the partial atomic charges in the MM region and to provide for the response of the QM region, for which Merz–Kollman (MK) charge fitting was employed to generate partial charges at quantum atoms for classical interactions.19
The coordinates of the initial and final states were used to generate Figure S1. The final, or post-catalytic, state is not a target; it is the natural consequence of systematically stepwise closure of the O3′-Pα distance. The atomic charges for the two metals along the reaction path were computed using the CM520 option (for condensed systems) in the Gaussian program. The charges for both ions stay within the range 0.84–0.89 over the entire reaction path (Figure S2). When preparing a system for QM/MM calculation, one approach is to first carefully equilibrate the protein crystallographic structure using molecular dynamics simulations, after adding missing atoms. Constraints to the crystal structure are initially added and then these constraints are systematically reduced during dynamics. Once the system is relaxed sufficiently, the equilibration is terminated, and the QM/MM boundary is established. From that point on, the enzymatic reaction atoms are treated with QM, adjusting for the charges of the classical atoms outside the QM region, as we step along the reaction path (closing the O3′-Pα distance). It was found in this process that if we equilibrated the system free-of-constraints for too long with classical dynamics, prior to the first QM step, the Mg-Mg distance increased undesirably. But, when we turned on the QM calculation in the early stage of free equilibration, the calculated QM/MM geometry of the reaction scaffold represented the crystallographic structure much better. The rationale, thus, is that when conducting classical dynamics only, the metal ions see only Coulomb repulsion. However, in combination with QM, the covalent bond interaction counters the observed repulsion.
Computation of the pre-catalytic geometry for arginase I
The crystallographic structure of arginase I, with two manganese ions at the active site (pdb ID: 1RLA)21 was used as the starting point for MD simulations. Similar protocols to pol β were followed in running MD for arginase, and the energy optimized final conformation was used to construct the QM/MM system. The QM sub-system contains parts of His101, Asp124, His126, Asp128, Asp232, Asp234, manganese ions and the water molecule that bridges the two metal ions; the MM sub-system contained the rest of the protein and water molecules within 15 Å of the metal ions. Calculation of electron density was similar to that adapted for pol β.
Computation of relative bond orders for several systems (H2, Na2, [Mg2]2+)
The bond character and strength of the metal–metal interactions were investigated with a natural bond orbital (NBO) analysis22,23 were carried out on optimized geometries with NBO 3.1 program implemented in Gaussian.11 It is well known that atomic charges and bond orders between atoms in molecules are not measurable properties and, therefore there is no quantum mechanical operator for them. Many schemes have been originated to attempt to provide chemical insight for atom charges and bond orders in molecules, but all suffer to variable extent due to the “not measurable” label.
Electron density distribution calculations
The B3LYP/6-31G* method/basis (as described above) were used to compute the electron density contours of [Mg2]q+ at distances of 3.63 for (a) q=4 and (b) q=2. System (a) is repulsive at all distances; system (b) is metastable in the approximate range 2.3 to 4.0 . Electron densities were calculated using the cubegen module of Gaussian09.11
A model molecule was constructed for the active site of the initial state by placing hydrogen atoms at the QM/MM link atom positions. The resulting complex has a charge of −3e. The electron density was computed from the wave function of this complex using Gaussian09-D0111 and displayed with the auxiliary program GaussView.24
III. RESULTS AND DISCUSSION
Bimetallic di-cationic species have been observed experimentally25 and the electronic energy dependence on the Mg-Mg distance predicted theoretically with high-level quantum mechanical calculations.26 Here, we have computed the quantum mechanical energy-metal ion separation distance curves of a number of pertinent systems having outer shell 3s-3s σ, 4s-3s σ and 4s-4s σ bonding designations (see Methods) (Figure 3).
Now, intuitively, when we look (e.g., Figure 1 or Figure S1) at an atom/bond diagram of the crystallographic structure of the active site of a DNA polymerase, for instance, we observe two divalent metal ions and generally think of these as being +2 ions. But, in reality, these two ions are surrounded by negatively charged oxygens, and it is not difficult to imagine that these formal +2 charges might be reduced to something of the order of +1, rather than +2. In fact, one observes something close to such a charge distribution for the magnesium ions in QM/MM calculations centered on the active site of this enzyme (Figure S2 and Methods), using a charge computation method appropriate for condensed systems.20
Accordingly, the bimetallic core can be re-pictured as the interaction of two Mg ions, each with ~+1 charge, or as ~[Mg-Mg]+2 or ~[Mg2]2+, which we know has a predicted bond order of 1 (Figure 2). Bond order predictions from quantum mechanical calculations for H2, Na2, and [Mg2]2+ are 1, 1, and 1 (Methods). That is, all of these systems are predicted to have one covalent bond. The region of predicted stability for our quantum mechanical calculations for the gas phase [Mg2]2+ is computed to be at an equilibrium bond distance of ~2.92 Å; this bonding region is separated by a barrier of ~10 kcal/mol occurring at ~4 Å from the much lower reference energy found at infinite separation in Figure 3. Note that the barrier seen in Figure 3 is due to the balance between the Coulomb repulsion between two Mg1+ ions and the stabilizing covalent bond. Since the Coulomb repulsion between two +1 ions is about 110 kcal/mol at a separation of ~3 Å, the net covalent bond strength must exceed ~110 kcal/mol in order to have a net attractive interaction of ~10 kcal/mol. It is interesting that high-level calculations26 gave almost identical ΔE/R results. In the active site of enzymes that exhibit the Mg-Mg core in various pre-and post-catalytic structures, coordinating ligands about each metal ion exhibit an octahedral arrangement. For DNA polymerase β (Figure S1), one apex of each of the octahedra has the same atom, an oxygen atom (proRp) of the α-phosphate of the incoming dNTP (Figure 1). A total of 6 formally negative charges can be found surrounding the two metal ions.
What is the electronic disposition of the Mg-Mg unit buried in the core of the active site of enzymes? The concept of bond order, which works for systems with well-defined bonds, is problematic for our current application to understand the buried Mg-Mg unit. The electron density, on the other hand, is a measurable and computable quantum mechanical variable that allows a view of electron distributions in molecules.
In Figure 4 (panels a and b), the electron density contours are displayed for the simple gas phase bimetal cations with either +4 or +2 charge (illustrating bonding interaction), respectively. In Figure 4c, the calculated electron density for the active site of the initial pre-chemistry state of the reaction of DNA polymerase β is shown. This system was prepared from crystallographic data (Methods). In Figure 4a, one observes the contours defining [Mg2]4+ at the same Mg-Mg distance as Figure 4c. This system is repulsive at all distances with no hint of a bond in the electron density contours. For [Mg2]2+ (Figure 4b), however, one observes the shared contours of electron density that clearly indicate bonding. In Figure 4c, which is centered on the scaffold plane defined by the two magnesium ions and a non-bridging oxygen on Pα of the incoming nucleotide and has the same orientation as Figure 1, one can observe how the electron density of the magnesium ion ligands contribute electron density to the scaffold region, thereby effectively reducing the formal charge of the magnesium atoms and providing electron density for the Mg-Mg bonding interaction in the complex.
Figure 4. Electron density.
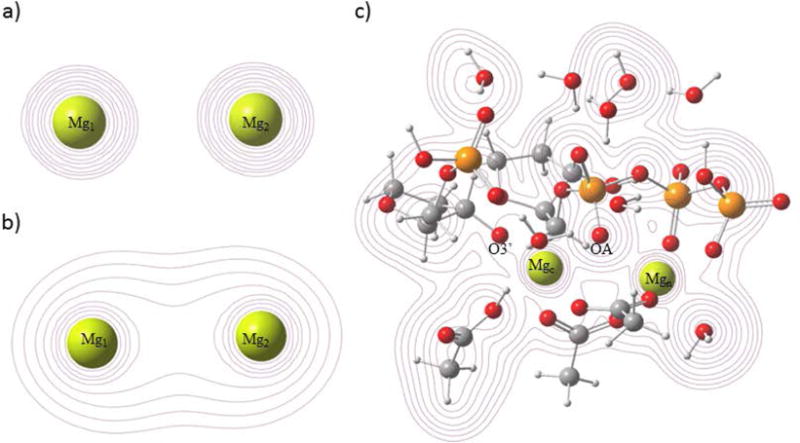
a) A contour map of the charge density for [Mg2]4+ at the Mg-Mg separation of 3.63 . This distance is chosen for display because it is the Mg-Mg distance in the initial state of an ongoing QM/MM model study based on pdb ID 2FMS. b) A contour map of the charge density for [Mg2]2+ at the same Mg-Mg separation of 3.63 . (c) A contour map of the charge density of the QM sub-system (Methods) of pol β from the QM/MM calculations. As seen in Figure 2, the system is likely metastable at this distance. The scaffold resulting from the interaction between OA and the two metals is evident. The initial formal charge on each metal ion is +2 for the quantum calculations. d) The charge density (in ) calculated along the line connecting the two metal centers for the three systems shown above. Metal ion positions are shown with arrows.
In DNA polymerase β, a bridging Pα oxygen atom (OA in Figure 4c) of the incoming nucleotide, along with the two magnesium ions, constitute the reaction scaffold on which a substituted phosphate is transferred from the incoming dNTP to O3′ of the deoxyribose terminus of the primer DNA strand. It is reasonable to imagine that prior to the 3 atom scaffold formation, the dNTP with a charge neutralizing magnesium ion, Mg(n), bound to an oxygen on Pα of the dNTP, approaches the DNA/polymerase complex and binds into a pre-evolved site. By this process, DNA/polymerase/dNTP complex creates a binding site for the Mg(c) ion. Mg(c) coordinates not only with the primer sugar O3′ atom, but also with the same oxygen of Pα to which Mg(n) is bound. At the juncture, two magnesium ions are forced to be geometrically near each other. This situation defines a condition for the Mg(c)-Mg(n) bond formation, the formation of which is enabled by the geometric nearness of the negative oxygen ligands of the magnesium ions to the midpoint of the Mg(c)-Mg(n) unit. In the nucleotide insertion process, the primer strand is elongated, by forming a bond between O3′ and the α-phosphate, with concomitant bond breaking between the α- and β-phosphates generating pyrophosphate (see Figure S1). The finding that both pre- and post-catalytic structures can be obtained crystallographically is due to the stability of the three-atom scaffold upon which the reaction takes place. Pre-catalytic structures were measured by employing nucleotides with a non-hydrolysable bond between the α- and β-phosphates, whereas post-catalytic structures were determined by soaking natural dNTPs with crystals of binary DNA complexes permitting nucleotide insertion in crystallo. For both pre- and post-catalytic structures the three-atom reaction scaffold remains, but the charge state of at least one of the coordinating ligands changes during the reaction. This change, along with the structural forces brought about by bond forming and breaking will modify the three-atom reaction scaffold. We find, for instance, that during QM/MM reaction path calculations, as the reaction advances from its initial state to the transition state, the Mg-Mg distance contracts (Figure S3). This is consistent with the fact that the minimum energy distance of the free-standing bimagnesium di-cation occurs at a somewhat shorter distance than the Mg-Mg distance of the initial state of the enzyme system. It is interesting to envision that the barrier to dissociation of the Mg-Mg interaction is overcome as the reaction proceeds to products, so that the metals enter an electrostatically repulsive region. The catalytic metal becomes destabilized in the Mg-Mg unit as the nucleotide metal bound to product pyrophosphate moves away from the active site. That is, the repulsive part of the divalent di-cation interaction assists in preparing the active site for a subsequent reaction. Thus, in this view of the insertion event of DNA polymerases, the creation and stability of the three-atom reaction scaffold is assisted by formation of the Me-Me covalent bond of considerable plasticity between the metal ions; the release of the products is aided by the repulsion between the metal ions when they become sufficiently separated.
Additional evidence for significant Mg-Mg bonding is provided by the three panels in Figure 4d: the left-most panel shows the electron density along a line that includes two +2 magnesium ions at a separation of 3.63 , the center panel shows the electron density along a line including [Mg2]2+ (at the separation of 3.63 ) and the right-most panel depicts the electron density along a line that includes the two magnesium ions in Figure 4c (separation 3.63 ). Clearly the electron density for the Mg-Mg interaction of the reactive complex and for the [Mg2]2+ ion are quite similar at 3.63 . This comparison offers considerable evidence that the bonding between the magnesium ions in the complex is essentially equivalent to that in the gas phase bimetal di-cation, for which the magnesium ions have a formal single covalent bond.
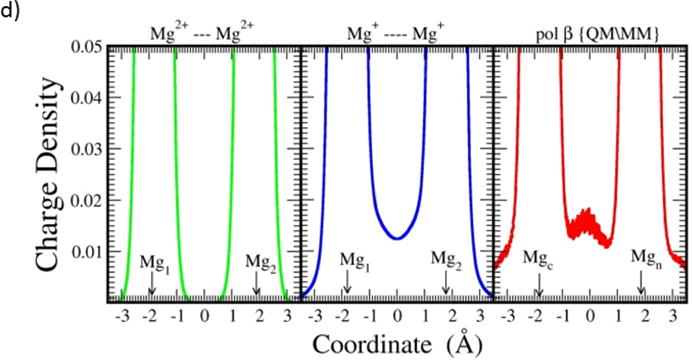
It is clear from the above discussion that the stability of the 3 atom reaction scaffold (Mg(c)..OA..Mg(n)) in the biological systems depends critically on the existence of significant bonding between the two magnesium ions. It is also clear from the pre- and post-catalytic structures (Figure S1), taken together with QM/MM calculations that permit estimation of the transition state between the endpoints, that the insertion reaction takes place as a straight-forward phosphorous inversion with the O3′, Pα and Oαβ atoms approximately inline. Is it possible, then, to relate the scaffold atoms, that depend on the quantum nature of the Mg-Mg interaction, to the linearity requirement of O3′, Pα and Oαβ? Strikingly, this is possible: Figure 5 provides a view of the six atom set (three from the scaffold and three from the reacting atoms). The six atoms are approximately in a plane at all three stages of reaction. It appears thus, that the role of the induced covalent bond is to provide essential structural stability to the core atoms of the active site.
Figure 5. Spatial orientation of the six atoms (O3′, Pα, Oαβ; Mg(c), OA, Mg(n)) in the active site of DNA polymerase β during the insertion reaction.
(Red (initial state), green (transition state), blue (final state); coordinates are from QM/MM study (see Methods) using the frozen outer layer of the system atoms for fitting). The O3′ and Oαβ remain essentially stationary, whereas Pα changes position significantly along with OA. Mg(n) is relatively stationary, while Mg(c) changes position.
Manganese ions can avidly replace magnesium in the three-atom reaction scaffold of some DNA polymerases, with a significant change in catalytic rate and fidelity.27 As seen in Figure 3, the relative positions of the Mg-Mn-6 and Mn-Mn-11 minima are similar to that for Mg-Mg. Calcium can also bind to some DNA polymerase active sites,27 but the insertion reaction does not occur. The energy minima for the Mg-Ca and Ca-Ca interactions in Figure 3 are shifted to larger distances than those for Mg-Mg, Mg-Mn-6 or Mn-Mn-11. The smaller Mg and Mn ions form octahedra preferentially (Figure 1), whereas calcium ions attain higher coordination that leads to distortion of the required alignment of the forming and breaking bonds.
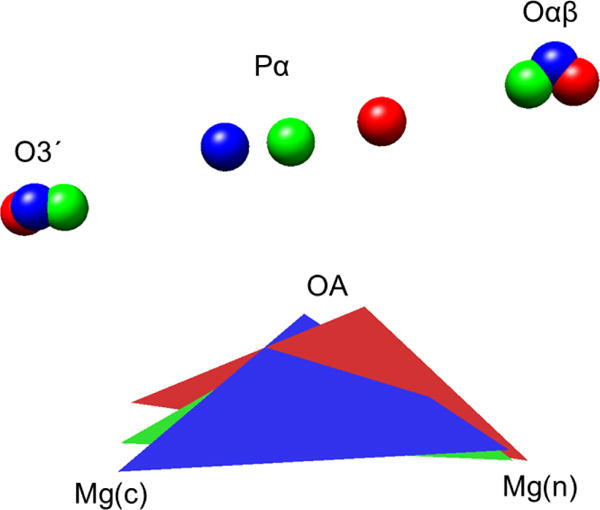
It is evident that [Me-Me]2+ covalent bonds may be of importance in providing structural stability or reactivity to other biological systems, not involving phosphoryl ester reactivity. A system that we have examined under that context of “other than phosphoryl ester reactivity” within the present study is arginase I, an enzyme predominantly found in liver and responsible for removing L-arginine in the form of L-ornithine and urea.28 In the bottom of a deep active site cavity, two manganese ions reside in a close contact to help create a hydroxyl ion nucleophile required for the conversion reaction. These particular metal ions are recognized to be essential for the stabilization and orientation of the nucleophile, a metal bridging hydroxide ion. Removal or substitution of the any of the two manganese ions results in a 20,000-fold reduction in the catalytic efficiency. In the crystal structure of the arginase I system (pdb ID: 1RAL)21, the two metal ions are closely spaced (3.3 ) in the pre-chemistry state where one manganese ion is hexa-coordinated (Mn2, Figure 6) while the other has five coordinating ligands (Mn1, Figure 6).
Figure 6. Electron density of the arginase system.
(a) A contour map of the electron density of the QM sub-system of arginase (described in Methods) used in the QM/MM calculation. (b) The charge density (in ) of [Mn2]2+ (black), [Mn2]4+ (red), and the QM sub-system of arginase (green) along the line connecting the two metal ions. The distance between the two manganese ions is 3.3 in all three systems.
In the contour map in Figure 6a, the electron density is displayed in the plane containing the two metal ions and the oxygen of the water that becomes the nucleophile during the enzymatic reaction. As with the case for pol β, we observe shared contours of electron density between the two metal ions, indicating bonding. In the charge density vs coordination distance plot (Figure 6b), the bimetallic dication at the separation equal to that of the arginase system shows appreciable charge density between the metals (black line). In contrast to [Mg2]4+ that has no computed charge density between the two metal ions, [Mn2]4+ shows a rather small but non-zero electron density in the metal bonding region, probably due to the presence of 10 unpaired electrons in the outmost orbitals of the system. Note that we also evaluated the electron density at this separation (3.3 ) for [Mg2]4+, but found no electron density (results not shown). Also, it is worth noting that there are only 4 negatively charged residues located in the coordination shell for sharing electrons with the manganese ions in arginase, whereas there are 6 negatively charged residues (or groups) available for the magnesium ions in pol β. Since the original charge on each metal ion is +2 and the electron density is borrowed from the coordinating negatively charged oxygens to enhance the metal covalent bond character, effectively, available reduced electron density for the metals may contribute to a slightly reduced electron density between the manganese ions (Figure 6; green curve) when compared with the electron density between the metal ions in the pol β case (Figure 4). However, the positive electron density between the divalent manganese ions in arginase signaling covalent bonding between the metal ions indicates that this metal covalent bonding phenomena is not confined to one type of metal ion or enzyme.
Another example abounds in the Gla domains of Vitamin K-dependent proteins,29 for which up to 5 calcium “ions” are bound to post-translationally modified carboxylates in a torturous chain. The Ca-Ca distances found in the chain are approximately 3.8–4.0 Å, well inside the attractive minima computed for diatomic [Ca-Ca]2+ shown in Figure 3. Protein domains are thought to have relatively small net folding free energies. The structural stability provided by di-cation Me-Me bonding interactions may be critical for proper folding of certain protein domains.
In the realm of inorganic chemistry, also, the magnesium tetrametaphosphate (Mg2P4O12) system,30 for example, has a Mg-Mg distance of 3.23 Å; each Mg is surrounded by an octahedron of oxygen atoms, an environment resembling that of DNA polymerases. Thus, the structure and stability of inorganic crystals that have Mg ions nearer than 3.9 Å will be at least partially dependent on the partial Mg-Mg covalent bond.
CONCLUSIONS
Two Mg2+ ions in a vacuum have a large electrostatic repulsive energy at normal chemical distances. If, however, when these ions are surrounded by electron rich atoms, such as oxygens, electron density from the oxygens can populate the region around the magnesium ions, reducing their positive charge and leading to covalent bond formation between the ions. This bond can provide enough attraction to stabilize the two magnesium ions in the 3–4 Å realm, much as is seen for the ideal case of the [Mg2]2+ di-cation in Figure 3. This quantum mechanical phenomenon provides a novel manner in which to view the Mg-Mg interaction in biological systems. As more structural data, along with theoretical advances for describing chemistry, is gained for enzymes with the bimetallic metal core, we will then be able to appreciate more fully the role of the heretofore “hidden” metal-metal bonding in enzymes.
Supplementary Material
Figure S1; The active site geometry of DNA polymerase β. Figure S2; The metal ion charges as the O3′-Pα distance narrows during the nucleotide insertion path for a DNA polymerase β. Figure S3; The Mg-Mg distance as the nucleotide insertion reaction proceeds with DNA polymerase β. S4: A brief description of terms and techniques.
SYNOPSYS.
Enzyme active sites that feature two magnesium ions in close proximity are common. The nature of the interaction of these metal ions is analyzed quantum mechanically. By analogy to the covalent bonding observed for [Mg2]2+ in the gas phase and in magnesium complexes, it is argued that a covalent bond is generally present in the two-Mg2+−ion active site, as well as in systems (organic and inorganic) with similar electronic structure.
Acknowledgments
This research was supported by Research Projects Z01-ES043010 (to L.P.) and Z01-ES050158 and Z01-ES050159 (to S.H.W.) in the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences and in association with the National Institutes of Health Grant U19CA105010.
Footnotes
Author Contributions
L.P. and L.G.P. developed the research based on discussions with W.A.B. and S.H.W.; L.P. and L.G.P. performed calculations and analyzed the data; All authors wrote the manuscript and provided revisions. All authors contributed to discussions of the results and the manuscript. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci U S A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 3.Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao H, Lin Z, Lynn AY, Varnado B, Beutler JA, Murelli RP, Le Grice SF, Tang L. Two distinct modes of metal ion binding in the nuclease active site of a viral DNA-packaging terminase: insight into the two-metal-ion catalytic mechanism. Nucleic Acids Res. 2015;43:11003–11016. doi: 10.1093/nar/gkv1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobsen DM, Bao ZQ, O’Brien P, Brooks CL, 3rd, Young MA. Price to be paid for two-metal catalysis: magnesium ions that accelerate chemistry unavoidably limit product release from a protein kinase. J Am Chem Soc. 2012;134:15357–15370. doi: 10.1021/ja304419t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green SP, Jones C, Stasch A. Stable magnesium(I) compounds with Mg-Mg bonds. Science. 2007;318:1754–1757. doi: 10.1126/science.1150856. [DOI] [PubMed] [Google Scholar]
- 7.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabzyan H, Keshavarz E, Noorisafa Z. Diatomic dications and dianions. Journal of Inanian Chemical Society. 2014;11:871–945. [Google Scholar]
- 9.Stasch A, Jones C. Stable dimeric magnesium(I) compounds: from chemical landmarks to versatile reagents. Dalton Trans. 2011;40:5659–5672. doi: 10.1039/c0dt01831g. [DOI] [PubMed] [Google Scholar]
- 10.Levine IN. Quantum Chemistry. 6. Prentice-Hall; New Jersey: 2009. [Google Scholar]
- 11.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT, USA: 2009. [Google Scholar]
- 12.Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B. 1988;37:785–797. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 13.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts BP, Hayik S, Roitberg AE, Seabra G, Swails JM, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. Amber.12. University of California; San Francisco: 2012. [Google Scholar]
- 14.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 16.Darden T, York D, Pedersen L. Particle mesh Ewald: an N ln(N) method for Ewald sums in large systems. Journal of Chemical Physics. 1993:10089–10092. [Google Scholar]
- 17.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. Journal of Chemical Physics. 1995;103:8577–8593. [Google Scholar]
- 18.Dapprich S, Komaromi I, Byun KS, Morokuma K, Frisch MJ. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. Journal of Molecular Structure : Theochem. 1999;461:1–21. [Google Scholar]
- 19.Besler BH, Merz KM, Kollman PA. Atomic charges derived from semi-empirical methods. Journal of Computational Chemistry. 1990;11:431–439. [Google Scholar]
- 20.Marenich AV, Jerome SV, Cramer CJ, Truhlar DG. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. Journal of Chemical Theory and Computation. 2012;8:527–541. doi: 10.1021/ct200866d. [DOI] [PubMed] [Google Scholar]
- 21.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 22.Foster JP, Weinhold F. Natural hybrid orbitals. J Am Chem Soc. 1980;102:7211–7218. [Google Scholar]
- 23.Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chemical Reviews. 1988;88:899–926. [Google Scholar]
- 24.Dennington TK, Millam J. Vesion. 5. Semichem Inc.; Shawnee Mission, KS: 2009. Gauss View. [Google Scholar]
- 25.Saito Y, Ishida T, Noda T. Cluster ions ejected from an Li-Mg alloy liquid metal ion source: Observation of MG 2 (2+) and MG 3 (2+) J Am Soc Mass Spectrom. 1991;2:76–80. doi: 10.1016/1044-0305(91)80063-D. [DOI] [PubMed] [Google Scholar]
- 26.Hogreve H. [Mg2]2+: a long-lived metastable dication. Chem Phys letts. 2004;394:32–36. [Google Scholar]
- 27.Freudenthal BD, Beard WA, Shock DD, Wilson SH. Observing a DNA Polymerase Choose Right from Wrong. Cell. 2013;154:157–168. doi: 10.1016/j.cell.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christianson DW. Arginase: Structure, mechanism, and physiological role in male and female sexual arousal. Accounts Chem Res. 2005;38:191–201. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]
- 29.Soriano-Garcia M, Padmanabhan K, de Vos AM, Tulinsky A. The Ca2+ ion and membrane binding structure of the Gla domain of Ca-prothrombin fragment 1. Biochemistry. 1992;31:2554–2566. doi: 10.1021/bi00124a016. [DOI] [PubMed] [Google Scholar]
- 30.Nord AG, Lindberg KB. Crystal-Structure of Magnesium Tetrametaphosphate, Mg2P4O12. Acta Chem Scand. 1975;A29:1–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1; The active site geometry of DNA polymerase β. Figure S2; The metal ion charges as the O3′-Pα distance narrows during the nucleotide insertion path for a DNA polymerase β. Figure S3; The Mg-Mg distance as the nucleotide insertion reaction proceeds with DNA polymerase β. S4: A brief description of terms and techniques.