

Abstract
Treatment of (diacetoxyiodo)arenes (1a–1u) with cyclotron-produced [18F]fluoride ion rapidly affords no-carrier-added [18F]fluoroarenes (2a–2u) in useful yields, and constitutes a new method for converting substituted iodoarenes into substituted [18F]fluoroarenes in just two steps.
Graphical Abstract
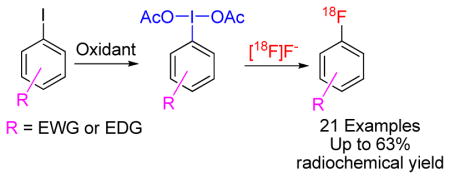
Useful methods for the rapid preparation of [18F]fluoroarenes at high specific activity from cyclotron-produced no-carrier-added (NCA) [18F]fluoride ion (t1/2 = 109.7 min) are in high demand because of increasing interest in the use of 18F-labeled tracers in imaging studies with positron emission tomography (PET).1 In the context of the need for rapidly preparing PET radiotracers for injection into animal or human subjects with minimal separation challenge, metal-free methods are particularly desirable. In this regard, the use of hypervalent diaryliodonium salts2–4 or iodonium ylides5,6 as precursors for preparing [18F]fluoroarenes has already come to the fore in expanding substrate scope beyond those of traditional methods, such as classical aromatic nucleophilic substitution or the earlier Balz Schiemann and Wallach reactions.7,8 Here we report on the reactivity of a further class of hypervalent precursor that also shows value for preparing [18F]fluoroarenes, namely the previously unexplored (diacetoxyiodo)arenes.
(Diacetoxyiodo)arenes are often intermediates in the synthesis of diaryliodonium salts9 and iodonium ylides6,10, and are useful reagents for oxidation reactions.11 They may be prepared from iodoarenes,12–17 do not form extended oligomeric structures, and are readily soluble in many solvents, including DMF. For these encouraging reasons, we were interested to explore whether (diacetoxyiodo)arenes were themselves amenable to radiofluorination to produce NCA [18F]fluoroarenes.
Upon treatment of a λ3-iodane (ArIL2), such as a (diacetoxyiodo)arene, with an external nucleophile (Nu:), ligand exchange may take place to form an ArI(Nu)L adduct.17 Thus, it has been postulated that an ArI(F)OAc adduct forms when a (diacetoxyiodo)arene is treated with HF as a source of F− ion.18 In the presence of excess HF, (diacetoxyiodo)arenes are also known to give the (difluoroiodo)arenes,18 presumably via the postulated adducts. Moreover, treatment of (diacetoxyiodo)benzene with Bu4NF has also been found to constitute a convenient synthesis for the well-known compound,(difluoroiodo)benzene.19 These prior observations reinforced our interest in testing whether treatment of (diacetoxyiodo)arenes with NCA [18F]fluoride ion would generate ArI(18F)OAc adducts and whether they may be converted into NCA [18F]fluoroarenes.
To explore the radiofluorination of (diacetoxyiodo)arenes, several substituted (diacetoxyiodo)arenes (1a–1r) were readily prepared in one step and in moderate to high yields from the corresponding iodoarenes, generally by treatment with peracetic acid (see Experimental).4,11,13,15,20 Exceptionally, oxidation of 1-iodo-4-(trifluoromethyl)benzene by peracetic acid gave not the (diacetoxyiodo)arene but the bridged compound μ-oxa-bis[(acetoxyiodo)-4-trifluoromethylbenzene] (1s; structure shown in Supporting Information), as characterized by NMR and MS data (see Experimental). Such bridged compounds may arise from hydrolysis of the corresponding (diacetoxyiodo)arenes when they bear a strong electron-withdrawing group. Iodoarenes containing a strongly electron-withdrawing nitro substituent in position 4 or a cyano substituent in either positions 3 or 4 were best converted into the corresponding (diacetoxyiodo)arenes by initial oxone oxidation in trifluoroacetic acid followed by ligand exchange in acetic acid (Figure 1).21
Figure 1.

The syntheses of (diacetoxyiodo)arenes (1a–1r) from iodoarenes and their conversion into [18F]fluoroarenes.
Radiochemical experiments were performed within a microfluidic apparatus (NanoTek; Advion, Ithaca, NY), which allowed us to control reagent stoichiometry, temperature and residence time in each single experimental run of a consecutive series on any particular (diacetoxyiodo)arene.20,22
Initially, we explored the reaction of 4-(diacetoxyiodo)benzonitrile (1b) with NCA [18F]fluoride ion, with TBA+ as counterion, in DMF. Gratifyingly, [18F]4-fluorobenzonitrile (2b) was produced rapidly (90 s) in 11% radiochemical yield at 150 °C (entry 1, Table 1) and also in much higher radiochemical yield (31%) at 200 °C (entry 2). Replacement of TBA+ with K+-K 2.2.2 (entry 3), which is also commonly used as a solubilizing counterion for NCA [18F]fluoride ion,1 gave the same radiochemical yield. Reactions performed in either DMSO or DMA (entries 4 and 5) gave inferior radiochemical yields to that in DMF. Optimal residence time in the micro-reactor was found to be 90 s from the use of 5 μL/min flow rate for the input of each reagent (compare entry 2 with entries 6 and 7). The radiochemical yield of 2b increased significantly when the precursor concentration was decreased to 5 mM (entry 8). Therefore, the conditions listed in entry 8 were preferred for subsequent radiofluorination experiments on a range of variously substituted (diacetoxyiodo)arenes, and also on (diacetoxyiodo)benzene (1u).
Table 1.
Yields of [18F]4-fluorobenzonitrile (2b) from 1b under various conditions.a
| Entry | 1b Conc. (mM) | Temp.(°C) | Time (s) | Yieldb (%) |
|---|---|---|---|---|
| 1 | 10 | 150 | 90 | 11 |
| 2 | 10 | 200 | 90 | 31 |
| 3c | 10 | 200 | 90 | 31 |
| 4d | 10 | 200 | 90 | 19 |
| 5e | 10 | 200 | 90 | 7 |
| 6 | 10 | 200 | 45 | 21 |
| 7 | 10 | 200 | 180 | 25 |
| 8 | 5 | 200 | 90 | 54 |
Reactions were performed by treating 1b in DMF with [18F]Bu4N+F− under the stated conditions.
Based on non-decay corrected radiochemical conversion of [18F]fluoride ion measured with HPLC.
Reaction performed with K+-K 2.2.2 complex.
Reaction in DMSO.
Reaction in DMA.
(Diacetoxyiodo)benzene (1u) gave a modest yield (20%) of [18F]fluorobenzene (2u) (Figure 2), whereas, 4-(diacetoxyiodo)anisole (1a) gave no desired product. 4-(Diacetoxyiodo)toluene (1f) also gave a modest yield of [18F]4-fluorotoluene (2f; 20%). As already seen for 1b, radiochemical yield improved appreciably with a weak electron-withdrawing group (CO2Et, 1e; 43%) in para position (Figure 2). However, 4-(diacetoxyiodo)nitrobenzene (1c) gave no desired product (Figure 2).
Figure 2.

Para- and ortho- substituted [18F]fluoroarenes prepared from the corresponding (diacetoxyiodo)arenes (except 2s was prepared from bridged compound 1s). Yields represent radiochemical conversion of [18F]fluoride ion, measured with HPLC.
Interestingly, upon subjecting the bridged compound 1s to the same reaction conditions, [18F]1-fluoro-4-(trifluoromethyl)benzene (2s) was obtained in remarkably high radiochemical yield (51%). This indicates that modification of the substituents bonded through oxygen to the hypervalent iodine could be fruitful for future improvement of reaction yields. Preliminary experiments have shown that replacements of the diacetoxy groups with pivaloyl or trifluoroacetyl groups are tolerated (data not shown).
An unusual reactivity pattern was observed for ortho-substituted (diacetoxyiodo)arenes (Figure 2). Those with electron-withdrawing groups, such as 2-(diacetoxyiodo)bromobenzene (1g) and 2-(diacetoxyiodo)trifluoromethylbenzene (1h) gave the desired [18F]fluoroarenes, 2g and 2h, in low radiochemical yields. By contrast, 2-(diacetoxyiodo)toluene (1i) gave a moderately high yield of [18F]2-fluorotoluene (2i; 49%). This suggested that an ‘ortho effect’ may be operating in a manner similar to that which is well known for the radiofluorination of diaryliodonium salts.20 However, addition of a methyl group to the other ortho position, or to both the other ortho and para positions decreased yields (2j and 2t, respectively). Nonetheless, the tolerance of these reactions for weakly electron-donating methyl groups is quite striking.
With regard to meta-substituted (diacetoxyiodo)arenes, some compounds with electron-withdrawing substituents, such as CF3 (1m), PhCO (1p), and CO2Et (1q), gave appreciable yields of the respective substituted [18F]fluoroarenes (2m, 2p and 2q), whereas the compound with a CN substituent (1d) gave a low yield and the compounds with a nitro (1n) or acetyl (1o) substituent gave no desired product (Figure 3). Generally, the introduction of [18F]fluoride ion into an aryl meta-position with respect to an electron-donating group is especially challenging. Remarkably, the compound with a 3-methyl group (1l) gave 3-[18F]fluorotoluene (2l) with the highest radiochemical yield (63%) of all tested compounds, and the 3,5-dimethyl compound (1k) gave a moderately useful yield of compound (2k) (39%) (Figure 3). The 3-methoxy compound (1r) gave very low yield (3%).
Figure 3.

Meta-substituted [18F]fluoroarenes prepared from the corresponding (diacetoxyiodo)arenes. Yields represent radiochemical conversion of [18F]fluoride ion, measured with HPLC.
In order to gain some mechanistic insight on the reactions observed between [18F]fluoride ion and (diacetoxyiodo)arenes, we performed experiments to examine whether 3-(diacetoxyiodo)toluene (1l) undergoes ligand exchange with Bu4NF·hydrate. The latter (0.83 eq.) was added to 1l (1 eq.) in chloroform. Upon exposing this solution to slow ingress of diethyl ether, crystals formed overnight. 1H-NMR spectroscopy revealed that these crystals were Bu4NOAc unaccompanied by fluoride ion, as determined by absence of an 19F-NMR signal. Furthermore, the 19F-NMR spectrum of the remaining filtrate presented a single signal (− 131 ppm relative to the signal from CFCl3) that was significantly different from that of Bu4NF·hydrate (− 122 ppm). This experiment affirmed the ability of fluoride ion to undergo ligand exchange at the iodine center of a (diacetoxyiodo)arene.
Encouraged by these observations, we also performed quantum chemical calculations to assess the energetics for ligand exchange of fluoride ion with (diacetoxyiodo)benzene (1u) and for subsequent conversion of the postulated intermediate adduct into fluorobenzene. These calculations predict that the formation of (acetoxyfluoroiodo)benzene is thermodynamically favored with a low energy barrier of 16.4 kcal mol–1 (Figure 4). Moreover, the conversion of 1u into fluorobenzene was calculated to have a free energy of activation (ΔG‡) of 33.8 kcal mol–1 with respect to the intermediate state (II), with energetically favoured formation of fluorobenzene (relative free energy − 26.6 kcal mol–1). In terms of bond distances, the covalent Ar–I distance shortens slightly from 2.16 Å in the first stage (I) to 2.13 Å in II while the ionic I–F bond (2.43 Å) in I becomes covalent (2.14 Å) in II. At transition state II (TS-II), this covalent Ar–I bond is weakened to 2.60 Å, and the fluorine forms a partial bond (2.03 Å) with the ipso aryl carbon atom.
Figure 4.

Reaction coordinate for the interaction of fluoride ion with 1u. (See Experimental for quantum chemical calculation). TS-I and TS-II represent transition states 1 and 2, respectively. Atoms are colored as follows: white, hydrogen; grey, carbon; yellow, fluorine; purple, iodine; red, oxygen; blue, potassium. Distances are in Å.
We further noted that radiochemical yields of para-substituted [18F]fluoroarene were not linear with substituent Hammett σp constants23 but showed a bell-shaped relationship (Figure 5). This may indicate that the rate determining step of the reaction (and/or the mechanism itself) changes with increasing σp value. For meta substituted [18F]fluoroarenes, no such clear relationship was seen between radiochemical yields and Hammett σm values. However, a weak inverse relationship between radiochemical yield and the F (field) factors23 was observed (Figure 6). This indicates that inductive effects are somewhat important for the radiofluorination of (diacetoxyiodo)arenes with meta substituents. Overall the mechanisms of these reactions appear complex and heterogeneous, and require deeper investigation for full understanding.
Figure 5.

The radiochemical yields of p-[18F]fluoroarenes show a strong bell-shaped relationship to Hammett σp values. The line represents a Gaussian fit of the data and r the correlation coefficient.
Figure 6.

The radiochemical yields of m-[18F]fluoroarenes show a weak linear relationship with F values. The line represents a linear fit of the data and r the correlation coefficient.
Finally, we explored whether radiofluorinations of (diacetoxyiodo)arenes could also be achieved on a conventional automated batch-reactor platform, such as the TRACERlab FXFN module, which is more widely used for PET radiotracer productions than microfluidic platforms. The treatment of 1d (5 mM) with [18F]fluoride ion-TBA+ in DMF for 5 min at 170 °C gave 2d in 20% radiochemical yield on this module. When the bridged compound 1s was subjected to these conditions, 2s was obtained in 63% radiochemical yield. [18F]N-Succinimidyl 4-fluorobenzoate is a useful labeling synthon for biomolecules, that is usually prepared from [18F]ethyl 4-fluorobenzoate (2e) as intermediate.24,25 We found that the treatment of 1e readily gave 2e in a useful 32% radiochemical yield. Recoveries of radioactivity from the TRACERlab FXFN module in these reactions ranged from 60–79%, which are typical for other radiofluorination procedures. Thus, reactions in the more conventional FXFN module also rapidly gave useful practical yields, even at lower temperature than in the microfluidic apparatus.
In conclusion, the treatment of (diacetoxyiodo)arenes with NCA [18F]fluoride ion proved viable for preparing several substituted [18F]fluoroarenes in moderate to good radiochemical yields, especially [18F]fluoroarenes having a range of strong para electron-withdrawing substituents and [18F]fluoroarenes that have methyl groups in meta position. This method is a useful supplement to others for preparing NCA [18F]fluoroarenes to serve as labeling synthons or radiotracers from hypervalent precursors. Especially, this method constitutes a novel two-step procedure for converting a substituted iodoarene into a substituted [18F]fluoroarene.
EXPERIMENTAL SECTION
Materials and methods
Fluoroarenes, 3-iodobenzophenone, (diacetoxyiodo)benzene (1u), (diacetoxyiodo)2,4,6-trimethylbenzene (1t), and other chemicals and solvents were purchased commercially. 1H (400 MHz), 13C (100 MHz) and 19F (376.5 MHz) NMR spectra were recorded on an Avance 400 instrument at rt in deuterated solvents, as later indicated. 1H and 13C NMR signals are reported as δ (ppm) downfield from the signal for tetramethylsilane. 19F-NMR signals are reported as δ (ppm) downfield from the signal for trichlorofluoromethane. ESI-MS spectra could not be obtained for (diacetoxyiodo)arenes in keeping with the absence of such spectra for all such compounds in this study that had been reported previously. However, ESI-MS data for the bridged compound 1s were successfully obtained. Aqueous Bu4NHCO3 solution of approximately 1 M concentration was prepared for radiochemical experiments by passing carbon dioxide gas through an aqueous solution of Bu4NOH (0.5 mL, 1 M). The pH of the reaction mixture was monitored. As the pH dropped to about 8 and remained stable, the flow of carbon dioxide through the solution mixture was ended and the volume was brought back to 0.5 mL. Bu4NHCO3 was stored at 4 °C and discarded after one week of use.
For radiochemical experiments with Kryptofix 2.2.2 (K 2.2.2) as counter ion, a solution composed of K 2.2.2 (101 mg in 1.8 mL MeCN) and K2CO3 (10.5 mg in 0.2 mL H2O) solution was prepared.
Quantum chemical calculations
The X-ray structure of 1u26 was used to build 1u complexed with potassium fluoride. The geometry and energetics of this complex were then obtained by resorting to quantum chemical calculations at the level of B3LYP/DGDZVP. The reaction path in the gaseous phase was constructed by moving a fluoride ion to the ipso aryl carbon atom in increments of 0.1 Å while relaxing the rest of the geometry. At each stationary point, the geometry was then further refined in the reaction field of MeCN with the polarizable continuum model together with the UAKS parameters set as implemented in Gaussian 09 software.27 A single imaginary frequency was obtained for each TS in Figure 4.
Syntheses of (diacetoxyiodo)arenes
All (diacetoxyiodo)arenes were obtained in ≥ 95% purity, before or after recrystallization from hot ethyl acetate and hexane, as required.
4-(Diacetoxyiodo)anisole (1a)
Sodium perborate monohydrate (30 mmol) was added portion-wise to a solution of 4-iodoanisole (3 mmol) in AcOH: Ac2O (1: 1 v/v, 60 mL) over 5 min at rt. The mixture was then heated to 40 °C and left stirring overnight. Reaction progress was monitored with tlc, and when complete the reaction mixture was filtered. The filtrate was extracted with DCM (50 mL) and washed with water (50 mL × 3). The organic solvent was then evaporated off under vacuum. The residual oil was triturated with ether (10 mL). The generated solid was rinsed with diethyl ether (10 mL × 3) and dried under vacuum to provide 1a as a fluffy white solid (0.475 g, 45%). 1H NMR (CDCl3): δ 8.01 (d, 2H, J = 9.0 Hz), 6.97 (d, 2H, J = 9.0 Hz), 3.86 (s, 3H), 2.00 (s, 6H). These data match literature data.16
General Method A
Oxone monopersulfate (0.92 g, 3 mmol) was added to a solution of the iodoarene in TFA: DCM (70: 30 v/v; 20 mL) and the resultant mixture was stirred at rt. The reaction was monitored with tlc and was over in less than 1 h. Solvents were then removed under vacuum. The residual oil was extracted with DCM and washed with water (50 mL × 3). The organic solvent was then evaporated off under vacuum. The residual oil was re-suspended in AcOH (10 mL) for 1 h. Water (100 mL) was then added, giving a precipitate that was washed successively with water (15 mL × 3), diethyl ether (10 mL × 3) and hexanes (10 mL × 3), and then dried under vacuum for 2 h.
Thus, were prepared:
4-(Diacetoxyiodo)benzonitrile (1b)
4-Iodobenzonitrile (0.69 g, 3 mmol) gave 1b as white crystals (0.781 g, 75%). 1H NMR (d6-DMSO): δ 8.43 (d, 2H, J = 8.7 Hz), 8.04 (d, 2H, J = 8.7 Hz), 1.93 (s, 6H). 13C NMR (d6-DMSO): δ 175.76, 136.25, 134.94, 127.36, 118.09, 114.90, 20.63. These data match literature data.28
4-(Diacetoxyiodo)nitrobenzene (1c)
4-Iodonitrobenzene (0.75 g, 3 mmol) gave 1c as pale yellow crystals (0.781 g, 75%). 1H NMR (d6-DMSO): δ 8.52 (d, 2H, J = 8.9 Hz), 8.34 (d, 2H, J = 9.0 Hz), 1.94 (s, 6H). 13C NMR (d6-DMSO): δ 175.2, 149.1, 136.4, 128.3, 125.7, 20.1. These data match literature data.11
3-(Diacetoxyiodo)benzonitrile (1d)
3-Iodobenzonitrile (0.69 g, 3 mmol) gave 1d as white crystals (0.822 g, 76%). 1H NMR (d6-DMSO): δ 8.36 (t, 1H, J = 1.3 Hz), 8.31 (dq, 1H, J = 5.4, 1.1 Hz), 7.88 (dt, 1H, J = 1.1, 5.4 Hz), 7.64 (t, 1H, J = 8.0 Hz), 2.04 (s, 6 H). 13C NMR (d6-DMSO): δ 176.8, 138.9, 138.1, 134.9, 131.4, 120.9, 116.7, 115.1, 20.3. These data match literature data.22
General Method B
Peracetic acid (32 wt %; 10 mL) was added dropwise to a solution of the appropriate iodoarene (3 mmol) in AcOH: Ac2O (1: 1 v/v; 5 mL) at –10 °C. This mixture was slowly warmed to rt and left stirring. Reaction progress was monitored with tlc and when complete, water (100 mL) was added to the mixture. If a precipitate was generated, this was then washed successively with water (15 mL × 3), ether (10 mL × 3) and hexanes (10 mL × 3), and then dried under vacuum for 2 h to give the product (diacetoxyiodo)arene. In cases of no precipitation, the aqueous mixture was extracted with DCM (50 mL × 3), and the combined organic extracts were evaporated under vacuum. The resultant oil was then triturated with ether (10 mL). Any solid was then rinsed with ether (10 mL × 3) and dried under vacuum to give the product (diacetoxyiodo)arene. In cases where trituration gave no solid, hexane (10 mL) was added and the mixture was cooled in a fridge until the product (diacetoxyiodo)arene precipitated out. This precipitate was then washed with hexanes (10 mL × 3) and dried in vacuo for 2 h.
Thus were prepared:
Ethyl 4-(diacetoxyiodo)benzoate (1e)
White solid (0.576 g, 56%). M.p.: 110–113 °C. 1H NMR (CDCl3): δ 8.17–8.12 (m, 4H), 4.42 (q, 2H, J = 7.1 Hz), 2.02 (s, 6H), 1.41 (t, 3H, J = 7.1 Hz). 13C NMR (CDCl3): δ 176.5, 165.0, 134.9, 133.5, 131.8, 125.7, 61.7, 20.3, 14.3.
4-(Diacetoxyiodo)toluene (1f)
Fluffy white solid (0.323 g, 32%). 1H NMR (CDCl3): δ 7.98 (d, 2H, J = 7.3 Hz), 7.29 (d, 2H, J = 8.0 Hz), 2.44, (s, 3H), 2.00 (s, 6H). 13C NMR (CDCl3): δ 176.0, 142.9, 134.9, 131.4, 117.0, 20.2, 19.1. These data match literature data.11
2-(Diacetoxyiodo)bromobenzene (1g)
White solid (0.806 g, 67%). 1H NMR (d6-DMSO): δ 8.48 (d, 1H, 8.0), 8.03 (d, 1H, J = 8.0 Hz), 7.62 (t, 1H, J = 7.3 Hz), 7.53 (t, 1H, J = 7.2 Hz), 1.92 (s, 6H). 13C NMR (d6-DMSO): δ 176.2, 139.5, 134.8, 133.4, 131.0, 129.0, 127.1, 20.6. These data match literature data.20
2-(Diacetoxyiodo)trifluoromethylbenzene (1h)
White solid (0.644 g, 55%). M.p.: 148–150 °C. 1H NMR (CDCl3): δ 8.48 (d, 1H, J = 7.8 Hz), 7.95 (dd, 1H, J = 6.7, 1.1 Hz), 7.79 (t, 1H, J = 7.6 Hz), 7.67 (dt, 1H, J = 7.6, 1.0 Hz), 1.97 (s, 6H). 13C NMR (CDCl3): δ 176.9, 140.1, 134.3, 132.4, 131.4 (q, J = 32.3 Hz), 128.0 (q, J = 5.1 Hz), 122.6 (q, J = 274.4 Hz), 117.8, 20.2. 19F NMR (CDCl3): δ –60.22.
2-(Diacetoxyiodo)toluene (1i)
White solid (0.655 g, 65%). 1H NMR (CDCl3): δ 8.17 (d, 1H, J = 7.6 Hz), 7.53–7.51 (m, 2H), 7.28–7.25 (m, 1H), 2.72 (s, 3H), 1.99 (s, 6H). 13C NMR (CDCl3): δ 176.4, 140.6, 137.2, 132.7, 130.9, 128.4, 127.2, 25.6, 20.3. These data match literature data.20
2,6-Dimethyl(diacetoxyiodo)benzene (1j)
White solid (0.725 g, 69%). 1H NMR (d6-DMSO): δ 7.39 (dd, 1H, J = 6.4, 1.6 Hz), 7.31–7.27 (m, 2H), 2.75 (s, 6H), 1.98 (s, 6H) ppm. 13C NMR (d6-DMSO): δ 176.5, 141.4, 132.9, 132.5, 128.1, 27.0, 20.3. These data match literature data.20
3,5-Dimethyl (diacetoxyiodo)benzene (1k)
White crystals (0.798 g, 76%). 1H NMR (CDCl3): δ 7.74 (s, 2 H), 7.22 (s, 1H), 2.39 (s, 6H), 2.02 (s, 6H). 13C NMR (DMSO): δ 176.4, 141.1, 133.7, 132.5, 121.3, 21.3, 20.4. These data match literature data.15
3-(Diacetoxyiodo)toluene (1l)
White needles (0.837 g, 83%). 1H NMR (DMSO): δ 8.10 (s, 1H), 8.03 (d, 1H, J = 7.6 Hz), 7.50–7.43 (m, 2H), 2.37 (s, 3H), 1.91 (s, 3H). 13C NMR (DMSO): δ 175.8, 141.5, 135.9, 133.1, 132.8, 131.2, 122.3, 21.2, 20.7. These data match literature data.22
3-(Diacetoxyiodo)trifluoromethylbenzene (1m)
White crystals (0.807 g, 69%). 1H NMR (CDCl3): δ 8.33 (s, 1H), 8.28 (d, 1H, J = 8.2 Hz), 7.85 (d, 1H, J = 7.8 Hz), 7.65 (t, 1H, J = 8.0 Hz), 2.03 (s, 6H). 13C NMR (CDCl3): δ 176.7, 138.2, 133.1 (q, J = 33.6 Hz), 131.8 (q, J = 3.9 Hz), 131.3, 128.5 (q, J = 3.5 Hz), 122.8 (q, J = 273.1 Hz), 20.3 ppm. 19F NMR (CDCl3): δ –62.74 ppm. These data match literature data.22
3-(Diacetoxyiodo)nitrobenzene (1n)
White solid (0.793 g, 72%). 1H NMR (CDCl3): δ 8.95 (t, 1H, J = 1.9 Hz), 8.44 (dd, 1H, J = 6.2, 1.0 Hz), 8.40 (dd, 1H, J = 6.4, 1.0 Hz), 7.73 (t, 1H, J = 8.2, Hz), 2.04 (s, 6H). 13C NMR (CDCl3): δ 176.8, 148.7, 140.4, 131.6, 130.1, 126.4, 120.6, 20.3. These data match literature data.22
3-(Diacetoxyiodo)acetoxybenzene (1o)
Pale yellow solid (0.90 g, 79%). 1H NMR (CDCl3): δ 7.95–7.92 (m, 1H), 7.89 (t, 1H, J = 1.9 Hz), 7.52 (t, 1H, J = 8 Hz), 7.36–7.33 (m, 1H), 2.34 (s, 3H), 2.02 (s, 6H). 13C NMR (CDCl3): δ 176.6, 168.6, 151.3, 132.1, 131.4, 128.3, 125.3, 120.5, 21.1, 20.4. These data match literature data.22
3-(Diacetoxyiodo)benzophenone (1p)
White crystals (0.511 g, 40%). M.p.: 175–178 °C. 1H NMR (CDCl3): δ 8.49 (t, 1H, J = 1.6 Hz), 8.28 (dq, 1H, J = 1.1, 5.1 Hz), 8.02 (dt, 1H, J = 1.2, 5.2 Hz), 7.84–7.82 (m, 2H), 7.67–7.62 (m, 2H), 7.55–7.51 (m, 2H), 2.03 (s, 6H). 13C NMR (CDCl3): δ 194.1, 176.5, 139.8, 138.1, 136.4, 136.2, 133.3, 132.8, 130.8, 130.1, 128.6, 121.1, 20.4.
Ethyl 3-(diacetoxyiodo)benzoate (1q)
White crystals (0.792 g, 67%). 1H NMR (CDCl3): δ 8.74 (t, 1H, J = 1.7 Hz), 8.26 (dd, 2H, J = 1.7, 7.9 Hz), 7.59 (t, 1H, J = 7.9 Hz), 4.43 (q, 2H, J = 7.2), 2.02 (s, 6H), 1.43 (t, 3H, J = 7.1 Hz). 13C NMR (DMSO): δ 176.6, 164.5, 138.9, 136.0, 133.1, 132.6, 130.8, 121.1, 61.9, 20.4, 14.3. These data match literature data.16
3-(Diacetoxyiodo)anisole (1r)
Yellow crystals (0.24 g, 23%). 1H NMR (CDCl3): δ 7.67–7.64 (m, 2H), 7.41 (t, 1H, J = 8.4 Hz), 7.10 (dd, 1H, J = 8.4, 2.4 Hz), 3.87 (s, 3H), 2.02 (s, 6H). 13C NMR (CDCl3): δ 176.4, 160.6, 131.5, 127.1, 121.5, 120.5, 118.0, 55.8, 20.4. These data match literature data.22
μ-Oxa-bis[(acetoxyiodo)-4-trifluoromethylbenzene] (1s)
Application of General Method B to 1-iodo-4-trifluoromethylbenzene gave 1s as a white fluffy solid (0.427 g, 42%). M.p.: 176–178 °C. 1H NMR (CDCl3): δ 7.93 (d, 2H, J = 8.3 Hz), 7.54 (d, 2H, J = 8.7 Hz), 1.94 (s, 3H). 19F NMR (CDCl3): δ − 63.24 ppm. ESI-MS: calculated for C18H14F6I2O5 m/z 677.88, found m/z 677.6. A satisfactory13C-NMR spectrum in CDCl3 was not obtained due to decomposition over the course of the scan time.
Radiochemistry
Radiochemistry was performed in a lead-shielded hot-cell to minimize the radiation exposure to personnel. All radioactivity measurements were performed using a calibrated ionization chamber and were corrected for background.
Production of [18F]fluoride ion reagents
No-carrier-added (NCA) [18F]fluoride ion was obtained through the 18O(p,n)18F nuclear reaction by irradiating [18O]water (95 atom %) for 90–120 min with a proton beam (17 MeV; 20 μA) generated from a biomedical cyclotron.
Aq. Bu4NHCO3 solution (50 μL; 1 M) or K 2.2.2 solution (50 μL) was added to the cyclotron-produced no-carrier-added [18F]fluoride ion (50–200 mCi) in [18O]water (200–500 μL), and four cycles of azeotropic addition and evaporation of acetonitrile (0.65 mL for each addition) were performed, three times at 110 °C for 4 min and once more at 90 °C for 4 min in an oven cavity housed within a modified Synthia29 module.
Configuration and operation of microfluidic reactor apparatus
The microfluidic reactor apparatus was maintained and operated as previously described.20
Radiofluorination reactions
In a microfluidic apparatus
Dry [18F]Bu4NF complex (20–50 mCi) was dissolved in reaction solvent (0.5 mL, typically DMF). A solution of (diacetoxyiodo)arene was prepared in a matching solvent. Each of the two solutions (255 μL) was loaded into separate storage loops of the microfluidic apparatus. For reactions, each solution (10–15 μL) was infused simultaneously into the 4-m-length coiled silica glass tube microreactor of the apparatus at a set flow rate in the range 4–100 μL/min and preset temperature. The microreactor output was quenched with MeCN-H2O (0.5 mL, 1: 1 v/v) at rt. Non-decay-corrected radiochemical yields (RCYs) of the [18F]fluoroarenes were measured by reverse phase radio-HPLC on a Luna C18 column (10 μm, 100 Å, 250 × 4.60 mm) eluted at 2 mL/min with a gradient of MeCN: 0.1% (w/v) HCOONH4, starting at 35% MeCN for 1 min, increased to 90% B over 5 min and maintained at 90% MeCN for 10 min.
In a TracerLab FXFN module
The aqueous [18F]Bu4N+F− reagent was dried within the apparatus by two cycles of addition and evaporation of acetonitrile (2.0 mL) under a nitrogen stream and reduced pressure, first at 65 °C, and then at 88 °C. To the dried [18F]fluoride ion complex, was added a solution of the (diacetoxyiodo)arene in anhydrous DMF (5 mM; 1.0 mL). The reaction mixture was heated to 170 °C for 5 min, and then transferred into water (10 mL) at rt. The mixture was then analyzed by HPLC, as described above.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (NIMH, ZIA-MH002793; CIT, ZIA-CT000265-19). The quantum chemical study utilized PC/linux clusters at the Center for Molecular Modeling of the National Institutes of Health (http://cit.nih.gov).
Footnotes
Electronic supplementary information (ESI) available: 1H, 13C and 19F NMR spectra of (diacetoxyiodo)arenes, and selected radio-chromatograms. The Cartesian coordinates of the geometry optimized structures in Figure 4 are also given. This material is available free of charge via the Internet.
References
- 1.Cai L, Lu S, Pike VW. Eur J Org Chem. 2008:2843–2873. [Google Scholar]
- 2.Pike VW, Aigbirhio FI. J Chem Soc, Chem Commun. 1995:2215–2216. [Google Scholar]
- 3.Ross TL, Ermert J, Hocke C, Coenen HH. J Am Chem Soc. 2007;129:8018–8028. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 4.Chun JH, Pike VW. Org Biomol Chem. 2013:6300–6306. doi: 10.1039/c3ob41353e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satyamurthy N, Barrio JR. 2010117495 A2. WO. 2010
- 6.Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Nature Commun. 2014;5:1–7. doi: 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]
- 7.Tredwell M, Gouverneur V. Angew Chem Int Ed. 2012;51:11426–11437. doi: 10.1002/anie.201204687. [DOI] [PubMed] [Google Scholar]
- 8.Yusubov MS, Svitich DY, Larkina MS, Zhdankin VV. ARKIVOC. 2013;(i):364–395. [Google Scholar]
- 9.Merritt EA, Olofsson B. Angew Chem Int Ed. 2009;48:9052–9070. doi: 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]
- 10.Goudreau SR, Marcoux D, Charette AB. J Org Chem. 2009;74:470–473. doi: 10.1021/jo802208q. [DOI] [PubMed] [Google Scholar]
- 11.Togo H, Iinuma M, Moriyama K. Synlett. 2012;23:2663–2666. [Google Scholar]
- 12.Zielinska A, Skulski L. Molecules. 2002;7:806–809. [Google Scholar]
- 13.Kazmierczak P, Skulski L, Kraszkiewicz L. Molecules. 2001;6:881–891. [Google Scholar]
- 14.Hossain MD, Kitamura T. Synthesis. 2005;12:1932–1934. [Google Scholar]
- 15.Yu P, Zhang G, Chen F, Cheng J. Tetrahedron Lett. 2012;53:4588–4590. [Google Scholar]
- 16.Chun JH, Pike VW. J Org Chem. 2012;77:1931–1938. doi: 10.1021/jo202517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhdankin VV. Hypervalent Iodine Chemistry. John Wiley & Sons; West Sussex: 2014. [Google Scholar]
- 18.Abo-Amer A. PhD Thesis. University of Duisburg-Essen; 2005. p. 35. [Google Scholar]
- 19.Sun H, Wang B, DiMagno SG. Org Lett. 2008;10:4413–4416. doi: 10.1021/ol8015429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chun JH, Lu S, Lee YS, Pike VW. J Org Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasumov TM, Brel VK, Grishin YK, Zefirov NS, Stang PJ. Tetrahedron. 1997;53:1145–1150. [Google Scholar]
- 22.Chun J-H, Lu S, Pike VW. Eur J Org Chem. 2011:4439–4447. doi: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansch C, Leo A, Taft RW. Chem Reviews. 1991;91:165–195. [Google Scholar]
- 24.Vaidyanathan G, Zalutsky MR. Nucl Med Biol. 1992;19:275–281. [Google Scholar]
- 25.Marik J, Sutcliffe JL. Appl Radiat Isot. 2007;65:199–203. doi: 10.1016/j.apradiso.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Lee CK, Mak TCW, Li WK, Kimer JF. Acta Crystallogr, Sect B: Struct Crystallogr Cryst Chem. 1977;33:1620. [Google Scholar]
- 27.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Jr, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Revision A02 edn. Gaussian Inc; Wallingford CT: 2009. [Google Scholar]
- 28.Chun J-H, Telu S, Lu S, Pike VW. Org Biomol Chem. 2013:5094–5099. doi: 10.1039/c3ob40742j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjurling P, Reineck R, Westerburg G, Gee AD, Sutcliffe J, Långström B. In: Proc Sixth Workshop on Targetry and Target Chemistry. Link JM, Ruth TJ, editors. TRIUMF; Vancouver: 1995. pp. 282–284. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.