

Abstract
Phenotypic whole-cell screening in erythrocytic co-cultures of Plasmodium falciparum identified a series of dihydroisoquinolones that possessed potent anti-malarial activity against multiple resistant strains of P. falciparum in vitro and show no cytotoxicity to mammalian cells. Systematic structure-activity studies revealed relationships between potency and modifications at N-2, C-3 and C-4. Careful structure-property relationship studies, coupled with studies of metabolism, addressed the poor aqueous solubility and metabolic vulnerability, as well as potential toxicological effects, inherent in the more potent primary screening hits such as 10b. Analogues 13h and 13i, with structural modifications at each site, were shown to possess excellent anti-malarial activity in vivo. The (+)-(3S,4S) enantiomer of 13i and similar analogues were identified as the more potent. Based on these studies, we have selected (+)-13i for further study as a preclinical candidate.
Graphical Abstract

INTRODUCTION
Despite significant efforts over the last decade to identify and bring to market new agents, malaria remains a significant health issue, with close to 50% of the world’s population currently at risk of infection.1 Of the five species of plasmodium that cause malaria in man, Plasmodium falciparum accounts for the majority of deaths associated with the disease. In most cases, treatment of uncomplicated P. falciparum malaria results in low mortality (ca 0.1%). However, in the absence of effective treatment the disease rapidly progresses and, especially in children, mortality rises to approximately 20%. Drug resistant strains of P. falciparum have emerged for every currently utilized anti-malarial agent.2 Therefore, there is a need for a sustained effort to identify new antimalarial agents that are efficacious against existing resistant strains. Although multiple avenues for addressing these issues are being followed, the use of high-throughput phenotypic screening to identify compounds that kill P. falciparum in human red blood cells has proven to be a particularly effective approach.3, 4 Phenotypic screens offer the potential to identify novel antimalarial chemotypes that fall outside of the known anti-malarial agents and, as a consequence, increase the potential to identify compounds with previously unrecognized or unexploited molecular targets.5, 6
The high-throughput phenotypic screening of over 300,000 compounds against cultures of the 3D7 strain of P. falciparum in human red blood cells resulted in the identification of three series of validated hits with chemical structures that are distinct from previously reported agents possessing anti-malarial activity.7 Subsequent re-synthesis and testing of selected examples from each hit series focused our attention on a series of 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxanilides of general structure 3 (Scheme 1) as our highest priority for hit-to-lead and lead optimization studies. The data available at the initiation of hit-to-lead indicated the compounds were generally potent against all tested resistant strains of malaria and had significant effects on gametocyte development but not on liver stages.8 We have previously described the clinical candidate (+)-SJ733 (13i), which arose from this series.8 In this report, we describe the primary structure activity relationship (SAR) within the series of analogues 3 that resulted in the identification of lead compounds and selection of a preclinical candidate.
Scheme 1.

Synthesis of 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxamides 8
RESULTS AND DISCUSSION
Various 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxylic acid analogues 6 were prepared by the well-documented condensation of an aldimine (3) with homophthalic anhydride (HPA, 4), which, in a single step, provides 5/6 (Scheme 1).9–11 Depending on the reaction conditions and the nature of the aldimine, the reaction affords predominantly the 3,4-cis product, predominantly the 3,4-trans product, or a mixture of both. However, despite the potential for both possible stereoisomers, the cis product (5) can be efficiently converted to the thermodynamically favored trans isomer (6) by heating at reflux in acetic acid solution.12 Thus, the reaction sequence shown in Scheme 1 affords racemic 3,4-trans carboxylic acids 6 with differing substituents at N-2 and C-3, from which various series of carboxamide analogues 8 can be prepared by amide coupling reactions.
The general methods in Scheme 1 served to prepare the compounds discussed below. Formation of the requisite aldimine (1) was generally accomplished by mixing amine 2 and aldehyde 1 in methanol solution with a catalytic amount of acetic acid, followed by stirring at ambient temperature for 4–8 hours and then concentration (Method A). Although this method did allow for the preparation of aldimines derived from many amines, an alternate method was developed for 2,2,2-trifluoroethylamine, given the eventual focus on this N-2 substituent (see below). In this case, a mixture of the amine hydrochloride salt and the requisite aldehyde in toluene was treated with one equivalent of 50% aqueous sodium hydroxide (Method B). The mixture was then allowed to stir for several hours to give, after simple workup, the desired aldimine in high yield. As prepared by either of these conditions, the aldimines are of sufficient purity to be used directly. The preparation of carboxylic acid 6 was accomplished by the addition of HPA to a solution of aldimine 3 in acetic acid solution at room temperature followed by heating at reflux for 16 hours. Concentration of the reaction mixture and either (1) trituration of the resulting residue with a mixture of ethyl acetate / water followed by collection of the crude product by filtration, or (2) extractive work up, generally gave the desired 3,4-trans-1,2,3,4-tetrahydro-1-isoquinolone-4-carboxylates (6) in moderate but reproducible yields (30–60%). Crude trans acids 6 were normally used without further purification, although purification by crystallization from isopropyl alcohol or ethyl acetate proved useful in some cases. This procedure was used for preparation of all trans-acids used for subsequent synthesis. Confirmation of the 3,4-trans configuration relied on analysis of proton NMR spectra, wherein the J3–4 coupling constants are on the order of 1–2 Hz, consistent with literature values.11, 13, 14 For example, carboxylic acid 6 (X = i-Bu and Y = thien-2-yl), exhibits J3–4 = 1.5 Hz, consistent with the assignment as the trans diastereomer. In order to confirm the stereochemistry was as expected, alternate conditions were used to produce the corresponding cis carboxylic acid (5, X = i-Bu and Y = thiophen-2-yl) by conducting the reaction between the aldimine and HPA in dichloromethane solution at –30 °C for several hours followed by additional stirring at ambient temperature. Under these conditions, the cis acid precipitated and was obtained in high purity and acceptable yield by simple filtration of the reaction mixture. The observed J3–4 (5.4 Hz) indicates the cis stereochemistry. These observations support the assumption that, unless otherwise indicated, the compounds evaluated in this series are the 3,4-trans carboxamides (8), consistent with the known thermodynamic preference and the general reaction conditions employed to prepare analogues.
Treatment of a solution of a carboxylic acid (6, Scheme 1), 1.2 equivalents of the requisite amine (7, Z-NH2, generally an aniline), and 2 equivalents of triethylamine with 2.5 equivalents of propanephosphonic acid anhydride (T3P) afforded, after chromatographic purification, 20–70% yields of the desired corresponding carboxamide (8, Method A). Under these conditions, small amounts of cis-amide (cis-8) are sometimes observed, possibly as the result of competitive ketene formation: addition of the amine to an intermediate C-4 ketene can give after protonation some portion of cis-amide. Additionally, small amounts of a product arising from acylation of the product carboxamide were observed in some cases. However, treatment of the crude reaction mixture with methanolic potassium or sodium carbonate equilibrated the mixture to the thermodynamically more stable 3,4-trans carboxamide 8, and also cleaved any bisacylated adduct, and afforded pure 8 after purification by column chromatography.
Unless otherwise noted, Method A served to prepare all of the compounds described below. However, in certain cases superior results were obtained by treating a mixture of carboxylic acid 6 and 1.5–2.0 equiv. of the desired amine (7) with 1.1 equiv. of Scheme 1. Synthesis of 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxamides 8 phosphorous oxychloride in acetonitrile solution at reflux (Method B).15 After extractive work-up, the desired amides (8) were obtained in 60–95% yield. The use of the higher temperature assured that any 3,4-cis diastereomer was converted to the thermodynamically favored 3,4-trans carboxamide, 8.
The initial high throughput screening campaign identified over 40 analogues of 8 covering more than a 100-fold range in potency. Early hit validation studies confirmed the anti-malarial activity, lack of significant mammalian cytotoxicity, acceptable pharmacokinetic properties, and preliminary indication of anti-malarial activity in vivo.7 A major driver for lead optimization of this series was the broad spectrum anti-malarial activity against drug resistant strains of malaria representing a range of resistance mechanisms that was exhibited by the initial hits (< 5-fold range across all genotypes) and generally demonstrated by analogues prepared in this study (generally less than 3-fold; for occasional analogs larger, see Table S-I in Supporting Information).
Preliminary SAR trends were apparent from analysis of the re-synthesized analogues from the screening set. First, from the compounds in the screening set, those bearing a para-methoxy substituent on the aromatic ring of the C-4 carboxamide substituent (Scheme 1, 8, Z = m-MeOPh) were generally the most potent, although there were highly potent compounds possessing alternate substitution patterns. Therefore, careful exploration of the substitution of this ring was included in early plans (see Tables 1 and 2 below). Second, of the compounds represented in the initial screening set, virtually all compounds with EC50 < 500 nM possessed a 2-thienyl substituent at C-3 of the 1,2,3,4-tetrahydro-1-isoquinolone ring (Scheme 1, 8, Y = thiophene). Given the physiochemical and metabolic liabilities potentially embedded in the thiophene, replacing this ring became an initial focus of work (see Table 3 below). Finally, all of the screening hits with submicromolar potency possessed an isobutyl group at N-2 (Scheme 1, 8, X = CH(CH3)CH2CH3), with analogues having smaller or larger substituents showing reduced potency. Thus, based on the confirmation of structure and potency after the re-synthesis of multiple analogues, extensive structure-activity relationship (SAR) studies were undertaken with the series of 3,4-trans-1,2,3,4-tetrahydro-1-isoquinolone-4-carboxamides, 8 to further explore these trends.
Table 1.
C-4 Carboxamide modifications
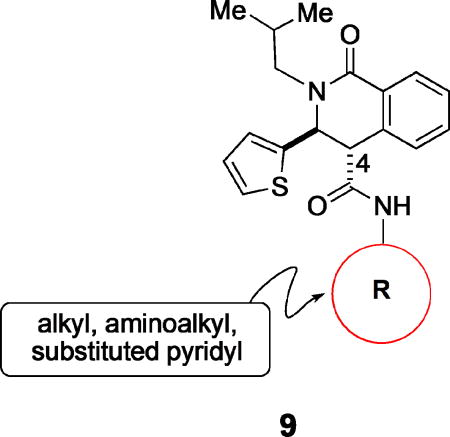
| |||||
|---|---|---|---|---|---|
| Cmpd. | R | EC50(nM)a | Cmpd. | R | EC50(nM)a |
| 9a |

|
>5000 | 9g |
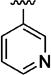
|
179 |
| 9b |
|
>5000 | 9h |
|
3526 |
| 9c |
|
>5000 | 9i |

|
117 |
| 9d |
|
>5000 | 9j |
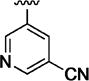
|
140 |
| 9e |
|
>5000 | 9k |
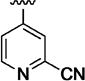
|
48 |
| 9f |
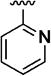
|
454 | 9l |
|
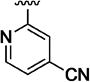
4105 |
Concentration causing 50% reduction in growth of P falciparum 3D7 co-cultured with human erythrocytes; values shown are from two independent experiments done in triplicate.
Table 2.
Carboxanilide substitution effects at C-4

| |||||
|---|---|---|---|---|---|
| Cmpd. | X | ED50(nM)a | Cmpd. | X | EC50(nM)a |
| 10a | H | 97 | 10o | 2'Cl | 1300 |
| 10b | 3'-Cl, 4'-OMe | 53 | 10p | 3'-Cl | 162 |
| 10c (N-Me) | 3'-Cl, 4'-OMe | >5000 | 10q | 4'-C | 497 |
| 10d | 3'-CN | 50 | 10r | 3'-OCF3 | 57 |
| 10e (cis) | 3'-CN | 207 | 10s | 4'-OCF3 | 1022 |
| 10f (N-Me) | 3'-CN | 4115 | 10t | 3'-CF3 | 136 |
| 10g | 2'-CN | 1426 | 10u | 3'-CN, 4'-OMe | 29 |
| 10h | 4'-CN | 1134 | 10v | 3'-CN, 4'-F | 19 |
| 10i | 4'-OMe | 65 | 10w (cis) | 3'-CN, 4'-F | 146 |
| 10j | 3'-OMe | 190 | 10x | 3'-CN, 4'-Cl | 157 |
| 10k | 2'-OMe | 943 | 10y | 3'-CN, 5'-F | 58 |
| 10l | 2'-F | 475 | 10z | 3'-CN, 5'-Cl | 25 |
| 10m | 3'-F | 89 | 10aa | 3'-CN, 5'-CN | 1104 |
| 10n | 4'-F | 56 | |||
Concentration causing 50% reduction in growth of P falciparum 3D7 co-cultured with human erythrocytes; values shown are from two independent experiments done in triplicate.
Table 3.
Modifications at C-3.
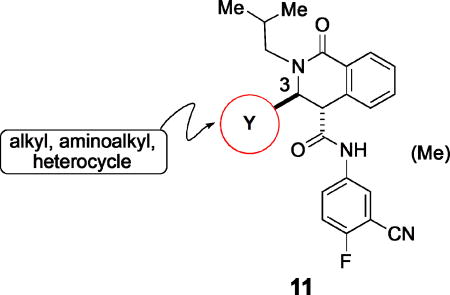
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd. | Y | EC50 (nM)a | Solubilityb | Cmpd. | Y | EC50 (nM)a | Solubilityb |
| 11a |
|
474 | 13 ± 1 | 11n |
|
52 | ND |
| 11b |
|
>5000 | <1 | 11o |
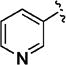
|
25 | 55 ± 2 |
| 11c |

|
>5000 | 54 ± 2 | 11p |
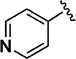
|
87 | 55 ± 2 |
| 11d |
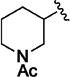
|
>5000 | 69 ± 4 | 11q |
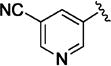
|
164 | 60 ± 2 |
| 11e |
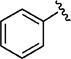
|
68 | ND | 11r |

|
152 | 28 ± 1 |
| 11f |

|
40 | <1 | 11s |
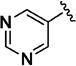
|
486 | 54 ± 7 |
| 11g |
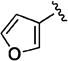
|
73 | 15 ± 2 | 11t |

|
10 | 57 ± 5 |
| 11h |
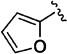
|
150 | 13 ± 1 | 11u |
|
12 | 36 ± 2 |
| 11i |
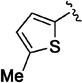
|
44 | <1 | 11v |
|
58 | 63 ± 1 |
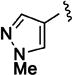
| 11j |
|
126 | ND | 11w |
|
52 | 20 ± 2 |
| 11k |
|
127 | ND | 11x |
|
88 | 53 ± 1 |
| 11l |
|
8 | 4.9 ± 0.5 | 11y |
|
55 | 5 ± 1 |
| 11m |
|
1163 | 19 ± 1 | ||||
As noted above, compounds with a 4-methoxy substituent on the carboxanilide ring were among the most potent screening hits. At the outset, this functionality presented a clear metabolic liability, namely, the potential to generate toxic metabolites resulting from O-demethylation followed by oxidation to an N-acyl-iminoquinone, a behavior precedented by the metabolism of acetaminophen.16 Furthermore, all of the most potent hits feature a carboxanilide at C-4, which presents the potential for metabolic amide hydrolysis and the release of the aniline. Based on these considerations, a thorough investigation of the C-4 carboxamide functionality became an early focus of the program, with the objective of either completely replacing the aniline, or identifying anilines lacking metabolic liabilities and mutagenic properties.
We carried out metabolism and oxidative stability studies using microsomes from rat, dog, and human, as well as hepatocytes from these species (data not shown). The metabolism patterns were consistent across all species, with mouse having the fastest rate. For this reason, and because the mouse is the species used for primary efficacy testing in vivo, we report only the mouse microsomal metabolism data. These studies confirmed that the methoxy substituent on the C-4 carboxanilide aromatic ring of 10b was readily demethylated. Likewise, the 3-cyano carboxanilide ring of 10d was hydroxylated, although the specific site of oxidation was not determined. In both compounds, significant oxidative metabolism of the N-2 isobutyl group was observed. Thus, with respect to maximizing the potential for compounds in this series to express efficacy in vivo, modifications of both the C-4 carboxamide and N-2 substituent would likely be required.
We initially investigated changes to the carboxamide at C-4 (Tables 1 and 2). Substitution of the aniline portion for a benzyl amine, as with N-(4-methoxybenzyl) carboxamide (9a), resulted in more than a 100-fold drop in potency compared to the 4-methoxy carboxanilide (10i). Likewise, both simple alkyl (9b, 9c) or heteroalkyl (9d, 9e) analogues were weakly potent (Table 1). Heteroaromatic surrogates for the C-4 carboxanilide (9f – 9i) were explored with a view toward improving the poor aqueous solubility of the initial screening hits (ca 1–2 µM at pH 7.4). Relative to the parent, unsubstituted C-4 anilide 10a, the pyridin-2-yl amide (9f) was less potent, the pyridin-3-yl amide 9g was equipotent, and the pyridin-4-yl carboxamide 9h was significantly less potent. Incorporation of a cyano group meta to the amide on the pyridyl ring was also explored. Compounds 9i and 9j nitrogen were modestly more potent than the respective unsubstituted pyridyl analogues, 9f and 9g. On the other hand, the N-(4-cyano-2-pyridyl)-carboxamide (9l) was significantly less potent than either 9i or 9f suggesting possibly opposing drivers for binding to the area of the target accommodating this ring. Surprisingly, the 6-cyanopyrid-4-yl derivative (9k) was substantially (73-fold) more potent than the corresponding unsubstituted pyridin-4-yl analogue (9h), and equipotent with the 3-cyano carboxanilide (10d). Thus, with the exception of the N-(2-cyanopyrid-4-yl) analogue (9k), none of the pyridyl analogues were deemed sufficiently potent to merit further investigation. Although 9k possessed potency sufficient to make it attractive for further development, the absence of clear lack of genotoxicity data in the literature for the parent aniline and the tightness of the SAR kept it from frontrunner status. Generally, these analogs maintained broad spectrum activity, although occasionally (10d, 10i) had activity ranges as high as 15-fold, with the V1/S strain consistently being the outlier. Furthermore, the impact on solubility with this series was modest giving less than 10-fold improvements. Overall, these observations, coupled with the data from additional C-4 amides not described here, focused efforts on exploring aromatic substituent effects in a series of carboxanilides. In other words, despite the examination of many possible surrogates, few if any analogues lacking the C-4 carboxanilide functionality maintain acceptable anti-malarial potency in vitro.
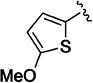
Studies with a series of mono-substituted carboxanilides indicated that electron donating substituents para (4' in Table 2) to the amide nitrogen were favored, whereas electron-withdrawing substituents appeared to be preferred in the meta (3' in Table 2) position relative to the carboxanilide nitrogen. In general, an ortho (2' in Table 2) substituent caused diminished potency relative to the unsubstituted carboxanilide (10a). Among mono-methoxylated analogues, para-substituted analogue 10i was somewhat more potent than its meta-substituted isomer 10j, and both were significantly more potent than 10k, which features an ortho-methoxy carboxanilide. With an electron withdrawing cyano substituent, meta-substituted analogue 10d was 20-fold more potent than either the ortho-(10g) or para-substituted (10h) compounds. Similar effects were seen among a series of ortho-, meta-, and para-substituted fluoro (10l, 10m) and chloro (10o, 10p, and 10q) analogues. Of note was 10n, a clear outlier in this trend, in which the fluoro substituent appears to mimic the effects of the para-methoxy group of 10i. Similarly, comparing the potency of meta-trifluoromethoxy derivative 10r with that of its para-substituted analogue (10s) revealed a 20-fold difference in potency. The meta-trifluoromethyl analogue, 10t, was equipotent with 10p (m-Cl), and slightly less potent than meta-cyano analogue 10d. Thus, within this limited set of mono-substituted C-4 carboxanilides, the most potent compounds possessed either an electron-donating group such as methoxy para to the anilide nitrogen (10i), or an electron-withdrawing group such as cyano (see 10d) meta to the carboxanilide nitrogen.
While this data set did not completely define the nature of the observed substituent effects on compound potency, the potency-enhancing effects of certain para and meta-substituents were maintained in disubstituted analogues. Thus, 10u (EC50 = 29 nM), with meta-cyano and para-methoxy carboxanilide substituents, was of similar potency to mono-substituted analogues 10i (EC50 = 65 nM) and 10d (EC50 = 50 nM). More encouraging, 10v (EC50 = 19 nM), with meta-cyano and para-fluoro aromatic substituents, was somewhat more potent than 10b (EC50 = 53 nM) or 10n (EC50 = 56 nM), and represented a carboxanilide analogue with good potency without the requirement for the para-methoxy substituent. Exchanging the para-fluoro (10v) for the chloro substituent (10x) resulted in a 10-fold reduction in potency, consistent with the trend seen with mono-substituted analogues. Moving the fluoro substituent to a meta-position (10y) led to a modest reduction in potency compared to 10v (EC50 = 58 nM vs. EC50 = 19 nM respectively), although exchanging the meta-fluoro of 10y for the chloro of 10z gave an analogue with similar potency (EC50 = 25 nM). The bis-meta-cyano substitution in 10aa caused a 100-fold drop in potency relative to 10v. Many additional disubstituted carboxanilides were explored, although none proved to be as potent as either 10v or 10z. In this series of compounds, the 3,4-trans-configuration provides the highest potency with the cis isomers 10e and 10w being approximately 10-fold less potent than their respective trans isomers 10d and 10v (see Table 2). Of these two substitution patterns, the meta-cyano-para-fluoro carboxanilide was chosen for subsequent investigations of the effects of modification at N-2 and C-3 of the 1,2,3,4-tetrahydro-1-isoquinolone system. Important to this decision was the observation that the parent 3-cyano-4-fluoroaniline was negative in the Ames' assay.
The importance of the C-4 carboxanilide N-H was probed by preparing two N-methyl analogues. Based on the significant drop in respective potency of 10f and 10c relative to 10d and 10b, the amide N-H appears to be important to potency in this series. Furthermore, the ester analogue of 10d (i. e., “O” replaces “NH”) was also prepared from the corresponding carboxylic acid (see Supporting Information), and was devoid of measurable activity in vitro (EC50 > 11 µM).
Overall, the systematic study of the C4 carboxamide functionality supported a preference for the carboxanilide as indicated by the significant loss of potency observed for the various alkyl, heteroalkyl or heteroaryl amide alternatives. Substituent effects on the carboxanilide appeared to correlate with the inductive effects of the substituent such that electron-donating para-substituents and electron-withdrawing meta-substituents are preferred. These substituent effects could be moderately additive, as evidenced by the potency of 10v compared with that of 10n and 10d; or 10u with 10i and 10d. The loss of potency with the ester analogue of 10d as well as the reduction in potency associated with N-methyl analogues and the reduction in relative potency observed with ortho-substituted carboxanilides indicate the important role of the amide functionality in activity As noted above, the majority of these analogs maintained broad spectrum activity, although occasionally (10d, 10i) had moderately broader activity ranges. A more complete understanding of the substituent effects and the role of the amide group in receptor binding must await structural studies with the target protein.
While exploration of the carboxanilide proved successful with respect to identifying analogues possessing surrogates for the methoxy substituent on carboxanilide aromatic ring and provided analogues with moderately improved potency, no analogues with improved aqueous solubility were found. The majority of these compounds possessed modest solubility (< 5 µM) and some were considerably worse. Consequently, we next focused on modifications at C-3 in order to identify structural modifications improving aqueous solubility (Table 3).
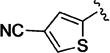
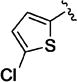
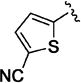
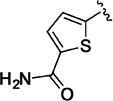
Small heteroaromatic rings appear to be preferred at C-3. Several alkyl substituents were examined, which uniformly caused a large drop in potency as exemplified by 11a and 11b and the piperidin-3-yl analogues (11c and 11d). Replacing the C3 thien-2-yl substituent with a phenyl in 11e caused a 7-fold drop in potency. While the thien-3-yl compound 11f was similar in potency to the thien-2-yl analogue 10v (EC50 = 40 nM and EC50 = 19 nM respectively), there was a small reduction in potency for fur-3-yl analogue 11g and a much more pronounced loss of relative potency with the fur-2-yl analogues (11h). For a series of 5-substituted thien-2-yl analogues, there is either no, or a small, reduction in potency (5-Me-, 11i; 5-OMe-, 11j; 5-Cl – 11k), with the exception of the 5-cyano analogue (11l), where an apparent increase in potency was observed. An attempt to mimic this effect by incorporating the 5-carboxamido functionality (11m) resulted in a marked reduction in potency. The increase in potency was also lost with the 4-cyano-thien-2-yl C-3 substituent in 11n. Overall, this subseries of analogs maintained broad spectrum activity with ranges of potency less than 3-fold.
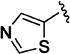
Despite the apparent small improvement in potency associated with analogues such as 11l, solubility in the series remained an issue. In this regard, the pyridin-3-yl analogue (11o) both retained good potency and demonstrated a marked improvement in aqueous solubility (Table 3). For example, 10v, with a C-3 thien-2-yl substituent, was poorly soluble in water (2 µM at pH 7.4), whereas 11o exhibits an aqueous solubility of 59 uM at pH 7.4. While maintaining improved aqueous solubility, the pyridin-4-yl analogue 11p was somewhat less potent, indicating fairly strict structural requirements in this region of the molecule. The addition of a cyano group to the 5-position of the pyridine ring (11q), which is the same relative positioning in the 5-cyano-thien-2-yl analogue (11l), reduced potency relative to 11o. Exchanging the C-3 pyridinyl group for a pyridazin-4-yl group (11r) resulted in a 6-fold reduction in potency compared to 11o, a change that is perhaps related to the difference in potency between the 3-yl and 4-yl C-3 pyridine analogues. The pyrimidin-4-yl C-3 substituent (11s) reduced potency by more than 10-fold relative to 11o (EC50 = 486 nM vs EC50 = 25 nM, respectively) but maintained enhanced aqueous solubility, reflecting the sensitivity to structural changes in this region of the molecule.
There was also an improvement in solubility with a series of C-3 pyrazolyl analogues (Table 3). Given that pyrazole ring nitrogen (pKa of conjugate acid ~ 2.5) is not significantly protonated at physiological pH (compare the pyridine ring, pKa ~ 5.2), the effects on aqueous solubility appear to be related to factors other than simple protonation, such as hydration in this region of the molecule. Pyrazol-4-yl analogue 11t also maintained excellent potency, and was equipotent with the 5-cyano-thien-2-yl analogue (11l), yet much more soluble at pH 7.4 (3 µM vs. 57 µM respectively). N-Methylation of the pyrazole group (11u) had no impact on potency or aqueous solubility.
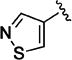
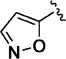
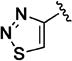
Additional 5-membered nitrogen-containing heterocycles surveyed at C-3 included thiazo-5-yl (11v), isothiazo-4-yl (11w), and isoxazo-5-yl (11x), all of which displayed potency similar to that of pyridin-3-yl analogue 11o and 5–8 fold less than C3 pyrazo-4-yl analogue 11t. With the exception of the thiadiazol-4-yl analogue 11y, each of these analogues maintained enhanced aqueous solubility.
In summary, modification of the C-3 substituent identified analogues such as 11o and 11t, with improved aqueous solubility and good or even slightly improved potency in vitro. The data also indicate a strict structural requirement for the C-3 substituent, with 5- and 6-membered heterocyclic ring systems demonstrating moderate to good potency in vitro. Within this set of analogues, modifications in the position of heteroatoms may have an impact on potency.
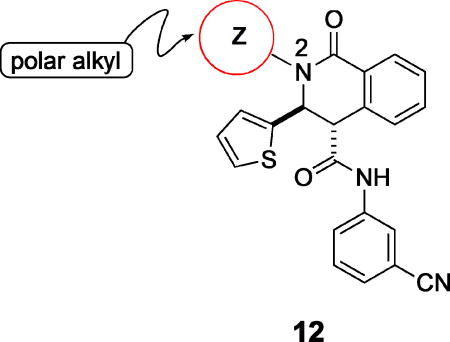
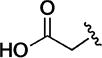
The objectives for investigating modifications of the N-2 substituent were two-fold: improving aqueous solubility of the series, and reducing the metabolic liability associated with oxidation on the N-2 alkyl functionality. Examples of early attempts to address the solubility issue, shown in Table 4, demonstrated that polar functionality in the N-2 alkyl group leads to a marked reduction in potency. Thus, compared to N-2 isobutyl analogue 10d (EC50 = 50 nM), hydroxyalkyl analogues 12a and 12b were significantly less potent, as was 12c, the methyl ether of 12b. Consistent with these data, N-2 substituents such as dimethylaminoethyl (12d), carboxymethyl (12e), and the methyl ester (12f), resulted in compounds devoid of measurable activity. The loss of potency with the N-2 methyl analogue 12g relative to 10d (N-isobutyl) indicates an apparent requirement for a larger, more lipophilic N-2 substituent. In fact, there are multiple examples of N-2 methyl compounds in the original hit set,7 all of which show very low potency relative to their N-2 isobutyl analogues.
Table 4.
Polar alkyl substituents at N-2.
| ||
|---|---|---|
| Cmpd. | Z | EC50 (nM)a |
| 12a |
|
944 |
| 12b |
|
1485 |
| 12c |
|
2841 |
| 12d |
|
>5000 |
| 12e |
|
>5000 |
| 12f |
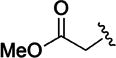
|
>5000 |
| 12g |
|
>5000 |
Concentration causing 50% reduction in growth of P falciparum 3D7 co-cultured with human erythrocytes. Values are from two independent experiments done in triplicate.
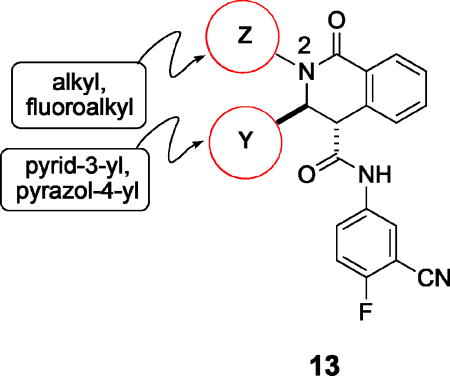
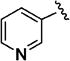
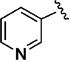
Further studies demonstrated fairly strict structural requirements for the lipophilic N2 substituent. As shown in Table 5, the N-2 n-butyl analogue 13a was more than 20-fold less potent than the corresponding isobutyl derivative, 11n. Similarly, cyclopropylmethyl derivatives 13b and 13c were significantly less potent compared to the corresponding compounds with isobutyl N-2 substituents, 11t and 11n, respectively. Nevertheless, 3-pentyl analogues 13d (EC50 = 22 nM) and 13e (EC50 = 11 nM) were equipotent with their respective isobutyl analogues. There was a slight drop in potency with the cyclopentyl modification (a “cyclized 3-pentyl”) in 13f (EC50 = 51 nM). Finally, replacement of the isobutyl with phenyl, as in 13g, resulted in a significant drop in potency. These data indicate that both the relative lipophilicity and the shape of the N-2 substituent are important to potency in this tetrahydro-1-isoquinolone series.
Table 5.
Modified alkyl substituents at N-2.
| |||
|---|---|---|---|
| Cmpd. | Y | Z | EC50 (nM)a |
| 13a |
|
|
651 |
| 13b |
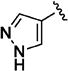
|
|
72 |
| 13c |
|
|
351 |
| 13d |
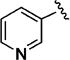
|

|
22 |
| 13e |
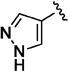
|

|
11 |
| 13f |
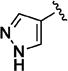
|
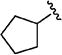
|
51 |
| 13g |

|
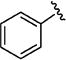
|
648 |
| 13h |
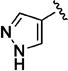
|
|
45 |
| 13i |
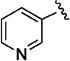
|
|
58 |
| 13j |
|
|
1336 |
| 13k |
|
|
3338 |
Concentration causing 50% reduction in growth of P falciparum 3D7 co-cultured with human erythrocytes; values shown are from two independent experiments done in triplicate.
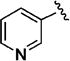
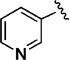
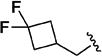
Early studies had shown that the metabolic oxidation of the N-2 iso-butyl group occurred predominately adjacent to nitrogen, which is consistent with the well-documented N-dealkylation of amides.17 Incorporation of an electron-poor N-2 substituent could suppress the observed oxidative process; consequently, the effects of a 2,2,2-trifluoroethyl substituent at N-2 were examined. In general, only a small reduction in potency was observed relative to the corresponding isobutyl. Thus, among the C-3 pyrazol-4-yl compounds, 13h, which possesses a trifluoroethyl substituent, was only 4-fold less potent than iso-butyl derivative 11t (EC50 = 45 nM vs EC50 = 10 nM respectively). For the corresponding C-3 pyridin-3-yl compounds, the trifluoroethyl substituent had little impact on potency (EC50 = 25 nM for 11o vs. EC50 = 58 nM for 13i). Again, relatively minor structural modifications appeared to have a significant impact on potency. For example, extending the length of the alkyl chain in the N-2 substituent, as in 13j, resulted in a 20-fold drop in potency in vitro; an even more pronounced effect on potency (>50-fold) was observed for the difluorocyclobutylmethyl group of 13k. Subsequent metabolism studies in the mouse demonstrated the anticipated metabolic stability of the N-2,2,2-trifluoroethyl substituent was realized, which is clearly reflected in the relative anti-malarial potency in vivo for these analogues, as described below. Overall, this subseries of analogs maintained broad spectrum activity, with potency range less than 4-fold across all genotypes.
The data in Table 6 demonstrate that modification of the C4 carboxanilide aromatic ring substituents and incorporation of the 2,2,2-trifluoroethyl N-2 substituent results in improved metabolic stability in mouse liver microsomes. The T1/2 for 10b, which bears the 4'-methoxy C4 carboxanilide substituent, is lower than the initial 30-minute time point in the microsomal assay. Comparison of the kinetic parameters of 10d and 11t implicates metabolism of the N-2 iso-butyl group as a significant process, given that 11t possesses the 3-cyano-4-fluoro-substituted carboxanilide aromatic ring and the pyrazol-4-yl substituent at C-3 that are stable under these conditions as evidenced by the data with 13h. Nevertheless, the C-3 5-cyano-thiophen-2-yl substituent of 11l does appear to result in improved metabolic stability. Further improvement in stability is realized with analogue 13i, with the N-(2,2,2-trifluoroethyl) substituent, and 13h, which appears to be stable to metabolism in vitro. As noted above, comparing the T1/2 of 11t to that of 13h, which is completely stable in this assay, clearly indicates the improved metabolic stability afforded by the N-2 trifluoroethyl substituent. The difference in metabolic stability between 13i and 13h is associated with oxidative metabolism of the C-3 pyridine ring of 13i. Overall, modification of both the N-2 substituent and the C-4 carboxamide aromatic ring substituents results in improved metabolic stability in the mouse.
Table 6.
Metabolic Stability in Mouse Liver Microsomes
| Cmpd. | T1/2 (hr) | CLint' (ml/min/kg) |
|---|---|---|
| 10d | 0.28 +/− 0.01 | 195.32 +/− 8.91 |
| 11t | 0.33 +/− 0.02 | 162 +/− 10.83 |
| 11l | 1.21 +/− 0.11 | 44.73 +/− 4.25 |
| 13i | 1.63 +/− 0.10 | 33.09 +/− 2.06 |
| 13h | >4 | <13.5 |
In summary, the systematic modification of substituents at N-2, C-3, and C-4 in the 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxanilide series afforded analogues with improved aqueous solubility and addressed the anticipated metabolic and toxicological liabilities, while maintaining acceptable potency in vitro. Although highly polar and/or solubilizing functionality in the substituents at C-4 or N-2 attenuates potency, incorporation of certain small nitrogen-containing heterocycles at C-3 provides analogues with improved aqueous solubility. The data reveal an apparent requirement for a liphophilic substituent at N-2, but seemingly minor changes in structure result in a marked impact on analogue potency. Incorporation of the 2,2,2-trifluoroethyl group at N-2, in place of the metabolically labile iso-butyl group, resulted in significantly increased microsomal stability while causing only a small drop in potency and no decrease in compound solubility. Examination of the anti-malarial activity in vivo of several analogues in this series confirmed that the modifications noted above afford compounds with improved physiochemical and pharmacological properties.
Efficacy testing was performed in vivo using the standard 4-day Peter's test18 in Swiss Webster mice infected with Plasmodium berghei NK65 ANKA strain (Figure 2). The studies were carried out in 2 phases: 1) a screening assay at a fixed dose of 50 mg/kg/d for all compounds tested, and 2) a dose response up to 200 mg/kg/d as well as a single fixed dose of 200 mg/kg for select compounds of interest. These studies demonstrated that the combination of improved solubility and metabolic stability resulted in an increase in efficacy at the doses tested. In this assay, 10d failed to demonstrate significant activity at either a 100 or 300 mg/kg oral dose. Comparing dose-response data for 11l (EC50 = 8 nM) at 50–200 mg/kg oral doses with that of 13i (EC50 = 58 nM) indicated that either the improved solubility of 13i or the increased metabolic stability of the N-2 trifluoroethyl group (compared with the iso-butyl group of 11l) caused in improved potency. In contrast, 11l demonstrated only a small increase in potency above the 100 mg/kg oral dose, with an oral dose of 200 mg/kg being essentially equivalent to 13i at 50 mg/kg. For either compound, a single 200-mg/kg dose showed reductions in parasitemia similar to that observed with 50 mg/kg×4-day dosing schedule. The effects of limiting metabolism at N-2 on efficacy in vivo, as shown in Table 6, were clearly demonstrated by comparing 11t (EC50 = 10 nM) with 13h (EC50 = 45 nM). At 50 mg/kg oral doses, 13h demonstrated a 99% reduction in parasitemia, whereas 11t caused only a 50% reduction despite being 4-fold more potent in vitro. For comparison, amodiaquine caused a 99% reduction in parasitemia at a 5 mg/kg oral dose in this assay. These data indicate that the improvement in analogue solubility and/or modifications that mitigate metabolism of the N-2 substituent result in improved antimalarial activity in vivo upon oral dose. It is important to note that no overt sign of toxicity was observed in any of the efficacy studies that have been performed with this series of compounds.
Figure 2.

Anti-malarial activity of trans-1,2,3,4-tetrahydro-1-isoquinolone-4-carboxanilides in the mouse. q.d. X 4 days, S.D. = single dose
The compounds described above were prepared and tested as racemates. In order to define enantiomeric preference in the series, selected carboxamides, or the esters of acids 6, were separated by chiral SFC or HPLC to obtain individual enantiomers (See Supporting Information). Evaluation of the resolved enantiomers shows the (+)-isomers to be the more potent for the compounds shown in Table 7, although the corresponding (-)-isomers retain measurable potency. Assignment of absolute configuration at C-3 and C-4 was accomplished through resolution of racemic 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxylate 6 (X = 2,2,2-trifluoroethyl, Y = pyridin-3-yl) by chiral SFC to afford the individual enantiomers. Subsequent crystallographic analysis of the crystalline salt formed between the (–)-enantiomer ([α]D = –58; 98% ee by chiral HPLC analysis) and R-(–)-2-amino-1-phenylethanol demonstrates the (3R,4R) absolute configuration for this carboxylic acid (Figure 3).19 Liberation of (–)-6 (X = 2,2,2-trifluoroethyl, Y = pyridin-3-yl) from this salt and subsequent amide formation gave (–)-13i with anti-malarial potency characteristic of the less potent enantiomer (EC50 = 587 nM). The more potent (+)-enantiomers in this series must therefore possess the (3S,4S) configuration.
Table 7.
Comparative potencies of 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxanilide enantiomers.
| Cmpd. | [α]D25 c = 1 | EC50 (nM)a |
|---|---|---|
| 13i | −131 | 587 |
| 13i | +147 | 36 |
| 13h | −154 | 189 |
| 13h | +152 | 21 |
| 11o | −175 | 477 |
| 11o | +159 | 15 |
Concentration causing 50% reduction in growth of P falciparum 3D7 co-cultured with human erythrocytes; values shown are from two independent experiments done in triplicate.
Figure 3.

ORTEP representation of (−)-6 (X = 2,2,2-trifluoroethyl, Y = pyridin-3-yl) as the R-(−)-2-amino-1-phenylethanol salt.
From among the compounds described above, 13i and 13h were selected for consideration as candidates for preclinical development based on their physical properties and potency in vivo. Although the two compounds compare favorably in most respects, especially with respect to potency against P. falciparum in a humanized mouse model,20 the decision was made to pursue 13i for preclinical development.8 This choice is based primarily on the observation that, while 13i demonstrated excellent dose related exposure in the rat, 13h did not (data not shown).
CONCLUSION
In the continuing effort to identify new anti-malarial agents, high-throughput phenotypic screening has proven useful in identifying novel chemotypes with the added potential for identifying agents with novel mechanisms of action. This undertaking resulted in the identification of a series of 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxanilide hits from a high-throughput phenotypic screen against P. falciparum in human erythrocytes. Hit validation studies confirm that the series possesses potent antimalarial activity without measurable cytotoxic activity, and also exhibits potency against a broad range of resistant parasite strains. In preliminary investigations, the compounds were shown to possess very low aqueous solubility, and to be extensively metabolized with the possibility of generating potentially toxic metabolites. SAR studies to address these liabilities focused on the systematic modification of functionality appended to N-2 and C-3 of the ring system, and addressed the nature of the C-4 carboxamide. Studies indicated that a C-4 carboxanilide in which electron-donating groups are para to the amide nitrogen and electron-withdrawing groups are meta to the amide nitrogen appears to provide the more potent analogues. Attempts to improve aqueous solubility by modification in this region of the molecule failed. It was found, however, that incorporation of certain nitrogen-containing rings at C-3, such as pyridin-3-yl or pyrazol-4-yl, improved solubility while maintaining good potency in vitro. Attempts to incorporate polar functionality into the N-2 substituent to improve aqueous solubility resulted in a significant loss of potency. Additionally, the data indicated a rather strict structural requirement for the lipophilic N-2 substituent. It was also observed that alkyl groups at N-2 were subject to significant metabolic oxidation. Ultimately, a compromise was reached with the incorporation of an N-(2,2,2-trifluoroethyl) substituent, which mitigated metabolism without affecting aqueous solubility and has only a minor impact on potency in vitro. The data show that both enantiomers in this series retain measurable activity, with the (+)-enantiomers being significantly more potent than the (-)-isomers of several analogues. Associated crystallographic studies demonstrate that the more potent (+)-enantiomers possess the (3S,4S) absolute configuration. Finally, 13i and 13h demonstrate improved anti-malarial potency in vivo compared to analogues without either the N-(2,2,2-trifluoroethyl) group or enhanced solubility characteristics. Taken together, these preliminary studies identified 13h and 13i as potential lead compounds, with (+)-13i ultimately being selected for preclinical development.
EXPERIMENTAL SECTION
Animal Studies Statement
All animal studies carried out to support this work were executed under approved protocols coverned by the respective Institutional Animal Care and Use Committees (IACUC).
General Procedure for the Preparation of Aldimines (3)
A solution of the 1 equiv of the aldehyde (1) and 1 equiv of the amine (2) in methanol (5 mL per mmol of aldehyde) was treated with a few drops of glacial acid acid, and the resulting mixture was stirred at 25 °C for 4 h. The solution was concentrated, and the crude aldimine (3), containing no more than a trace of aldehyde according to 1H NMR analysis, was used for the next step without further purification. An alternate method was employed to prepare aldimine 3 from 2,2,2-trifluoroethyl amine. Generally, a slurry of aldehyde 1 and 2,2,2-trifluoroethyl amine hydrochloride (1.5 equiv) in toluene (2 mmole aldehyde per mL toluene) was cooled in an ice-bath and treated with 1.5 equiv of 50% aqueous sodium hydroxide. The mixture was then allowed to warm to ambiant temperature and stirred for 12–16 hours. After separation of the phases, the aqueous phase was extracted several time with small amounts of toluene and the combined organic phases were dried over sodium sulfate and concentrated to yield the desired aldimine 3 (X = 2,2,2-trifluoroethyl) in near quantative yield of sufficient purity to be used without further purification.
General Procedure for the Reaction of Homophthalic Anhydride with Imines
A solution of 1 equiv of aldimine 3 and 1 equiv of homophthalic anhydride (4) in glacial acetic acid (2 mL per mmol of aldimine) was heated at reflux for 16 h. The solution was concentrated, and the residue was (usually) crystallized from ethyl acetate to provide the trans-1,2,3,4-tetrahydro-1-isoquinolone-4-carboxylic acid product, 6. In some cases, the product required chromatographic purification on silica with ethyl acetate / methanol as the eluant.
General Procedures for Amide Coupling
Method A
A solution of 1 equiv of the 1,2,3,4-tetrahydro-1-isoquinolone-4-carboxylic acid (6), 1.2 equiv of the amine (7), and 2 equiv of triethylamine in dichloromethane (5 mL per mmol of carboxylic acid) was stirred at 0 °C. Propane-1-phosphonic acid anhydride (T3P, 2.5 equiv) was added, and the reaction mixture was allowed to warm to room temperature and stir for 16 h. The reaction mixture was partitioned between dichloromethane and saturated aqueous sodium bicarbonate, and the organic layer was washed sequentially with water and brine, and then was dried over anhydrous sodium sulfate. In cases where 1H NMR analysis indicated the presence of some cis amide formed during the reaction, the crude product mixture was treated for 15 min with 0.5 mL of 1 N potassium carbonate and 2 mL of methanol per mmol of starting carboxylic acid. This mixture was concentrated, diluted with water, and then extracted with dichloromethane. Concentration and then chromatography on silica with ethyl acetate/petroleum ether as the eluant afforded the trans amide, 8. LC analysis of the amide products indicated >95% purity in each case.
Method B
Phosphoryl chloride (1.1 equiv) was added to a solution of carboxylic acid 6 and 1.5 equiv of amine 7. The resulting mixture was heated to reflux for 3 hr, cooled to room temperature and quenched with ice-water. Standard extractive work-up followed by chromatographic purification as described in Method A afforded 60–95% yields of the desired trans-amides 8.
(3S*,4S*)-2-iso-butyl-1-oxo-3-phenyl-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (6, X = i-Bu, Y = phenyl)
The known aldimine,21 (E)-N-iso-butyl-1-phenylmethanimine, was prepared by the general procedure. A solution of the aldimine (1.3 g, 8.0 mmol) and homophthalic anhydride (1.3 g, 8.0 mmol, 1 equiv) in acetic acid (19 mL) was heated at reflux for 16 h, and worked up according to the general procedure. The crude product was crystallized from ethyl acetate to afford 1.5 g (58%) of the trans-tetrahydroquinolone-4-carboxylic acid (6, X = i-Bu, Y = phenyl): 1HNMR (300 MHz, CD3OD): δ 8.02 – 8.05 (m, 1 H), 7.36 – 7.38 (m, 2 H), 7.12 – 7.22 (m, 4 H), 7.06 (d, 2 H, J = 7 Hz), 5.40 (br s, 1 H), 4.06 (br s, 1 H), 3.96 (dd, 1 H, J = 13 and 9.5 Hz), 2.56 (dd, 1 H, J = 13 and 5.5 Hz), 2.06 – 2.15 (m, 1 H), 0.92 (app d, 6 H, J = 7 Hz); 13C NMR (125 MHz, CD3OD) δ 172.9, 165.6, 139.0, 133.6, 132.2, 129.6, 129.2, 128.6, 128.0, 127.7, 127.2, 126.1, 62.1, 53.6, 51.5, 27.4, 19.7, 19.6; HR – ESI – MS [M+H]+ calcd for C20H22NO3, 324.1599; found, 324.1596.
(3S*,4R*)-N-(3-Cyanophenyl)-2-iso-butyl-1-oxo-3-(thiophen-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (10e)
A stirred solution of aldimine 3 (X = i-Bu, Y = thien-2-yl) (1.0 g, 5.18 mmol, 1 equiv) in 15 mL of dichloromethane was cooled to −30 °C (external temperature, dry ice bath), and then treated with homophthalic anhydride (4, 0.84 g, 5.18 mmol, 1 equiv) in one portion. The reaction mixture was stirred at −30 °C for 2.5 h, then was allowed to warm to room temperature to stir for an additional 16 h. Filtration gave the cis carboxylic acid, 5 (X=i-Bu, Y=thien-3yl), as a white solid (0.98 g, 57% yield), mp 199.1–200.6 °C: 1HNMR (300 MHz, CD3OD) δ 8.07 (d, J = 7.8 Hz, 1 H), 7.82 (d, J = 7.8 Hz, 1 H), 7.58 (t, J = 7.5 Hz, 1 H), 7.48 (td, J = 7.6 and 0.9 Hz, 1 H), 7.17 (d, J = 5.1 Hz, 1 H), 6.92 (d, J = 3.3 Hz, 1 H), 6.84 (td, J = 5.1 and 0.9 Hz, 1 H), 5.42 (d, J = 5.4 Hz, 1 H), 4.77 (d, J = 5.4 Hz, 1 H), 3.88 (dd, J = 13.0 and 7.8 Hz, 1 H), 2.76 (dd, J = 13.0 and 7.8 Hz, 1 H), 2.08 – 2.22 (m, 1 H), 1.03 (d, J = 6.6 Hz, 3 H), 0.98 (d, J = 6.6 Hz, 3 H); 13C NMR (75 MHz, DMSO-d6) δ 170.5, 163.2, 140.3, 134.1, 132.3, 129.6, 128.5, 128.4, 127.9 (2 C), 126.7, 126.3, 58.4, 53.0, 48.3, 27.4, 20.6, 20.5; HPLC: 97.8 % purity; HR – ESI – MS [M+H]+ calcd for C18H20NO3S, 330.1164; found, 330.1166.
Following Method A, a solution of cis carboxylic acid 5 (X = i-Bu, Y = thien-2-yl, 200 mg, 0.607 mmol), 3-aminobenzonitrile (86 mg, 0.73 mmol, 1.2 equiv), and triethylamine (0.34 mL, 2.4 mmol, 4 equiv) in 10 mL of dichloromethane was stirred at 0 °C. Propane-1-phosphonic acid anhydride (T3P, 50% w/v solution in ethyl acetate, 483 mg, 1.5 mmol, 2.5 equiv) was added by drops. The reaction mixture was allowed to warm to room temperature and was stirred for 16 h. Concentration and chromatography on silica by using 20–100% ethyl acetate / hexane mixtures as the eluant afforded 180 mg (69%) of cis amide 10e as a white solid, mp 229–230 °C: 1H NMR (500 MHz, CD3OD) δ 8.01 (d, 1 H, J = 7.5 Hz), 7.97 (s, 1 H), 7.75 (d, 1 H, J = 8 Hz), 7.57 (t, 1 H, J = 7.5 Hz), 7.42 – 7.52 (m, 3 H), 7.40 (d, 1 H, J = 7.5 Hz), 7.20 (d, 1 H, J = 5.5 Hz), 6.85 – 6.90 (m, 1 H), 6.80 – 6.85 (m, 1 H), 5.39 (d, 1 H, J = 6.0 Hz), 4.79 (d, 1 H, J = 6.0 Hz), 3.85 (dd, 1 H, J = 8.5 and 14 Hz), 2.76 (dd, 1 H, J = 8.0 and 14 Hz), 2.10 – 2.22 (m, 1 H), 0.90 – 1.00 (m, 6 H); 13C NMR (125 MHz, CD3OD) δ 168.0, 164.7, 139.5, 139.3, 134.1, 132.5, 130.0, 129.0, 128.1, 128.0, 127.9, 127.8, 127.6, 126.1, 125.8, 124.4, 123.1, 118.2, 112.7, 59.3, 52.9, 50.8, 27.3, 19.5; HR – ESI – MS [M+H]+ calcd for C25H23N3O2S, 448.1495; found, 448.1492.
(3S*,4S*)-N-(3-Cyanophenyl)-2-iso-butyl-N-methyl-1-oxo-3-(thiophen-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (10f)
Following Method B, a solution of carboxylic acid 6 (X = i-Bu, Y = thien-2-yl, 498.5 mg, 1.51 mmol, 1 equiv), 3-(N-methylamino)-benzonitrile (262 mg, 2.27 mmol, 1.5 equiv), and phosphoryl chloride (0.156 mL, 1.66 mmol, 1.1 equiv) in 20 mL of acetonitrile was heated at reflux for 3 h. The reaction mixture was quenched with ice water and extracted with ethyl acetate. The combined organic layer was washed with aqueous sodium carbonate, dried, concentrated, and then chromatographed on silica by using 20–100% ethyl acetate / hexane mixtures as the eluant to afford 543 mg (88%) of the trans carboxanilide 10f as an off white foam: 1HNMR (500 MHz, CDCl3) δ 8.14 (dd, 1 H, J = 7.0, 2.0 Hz), 7.71 (d, 1 H, J = 7.5 Hz), 7.64 (t, 1 H, J = 8.0 Hz), 7.53 (d, 1 H, J = 8.5 Hz), 7.49 (s, 1 H), 7.33 – 7.39 (m, 2 H), 7.04 (d, 1 H, J = 4.5 Hz), 6.84 (d, 1 H, J = 6.0 Hz), 6.74 (t, 1 H, J = 5.0 Hz), 6.47 (s, 1 H), 5.06 (d, 1 H, J = 2.5 Hz), 4.10 (d, 1 H, J = 2.5 Hz), 4.00 (dd, 1 H, J = 13.7, 8.0 Hz), 3.29 (s, 3 H), 2.60 (dd, 1 H, J = 14.0, 6.5 Hz), 1.95 – 2.01 (m, 1 H), 0.95 (d, 3 H, J = 6.5 Hz), 0.93 (d, 3 H, J = 6.5 Hz); 13C NMR (125 MHz, CDCl3) δ 169.6, 164.0, 144.7, 142.8, 133.2, 132.4, 132.2, 132.1, 131.8, 131.3, 130.3, 128.8, 128.6, 127.5, 126.9, 125.7, 125.5, 117.6, 114.9, 59.1, 53.4, 49.5, 38.4, 28.0, 20.7, 20.5; HPLC: 100 % purity; HR – ESI – MS [M+H]+ calcd for C26H25N3O2S, 444.1745; found, 444.1740.
(3S*,4S*)-N-(3-Cyano-4-fluorophenyl)-2-iso-butyl-1-oxo-3-(pyridin-3-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (11o)
Following Method A, a solution of carboxylic acid 6 (X = isobutyl, Y = pyrid-3-yl, 0.25 g, 0.77 mmol, 1 equiv), 3-cyano-4-fluoroaniline (0.13 g, 0.92 mmol, 1.2 equiv), and triethylamine (0.21 mL, 1.54 mmol, 2 equiv) in 5 mL of dichloromethane reaction mixture was stirred at 0 °C. Propane-1-phosphonic acid anhydride (T3P, 50% w/v solution in ethyl acetate, 1.22 g, 1.93 mmol, 2.5 equiv) was added by drops. The reaction mixture was allowed to warm to room temperature and to stir for 16 h. The reaction mixture was worked up according to the general procedures, concentrated, and then dissolved in 0.5 mL of 1 N aqueous potassium carbonate and 2 mL of methanol. Stirring for 15 min converted any cis isomer formed during the amidation reaction to the trans isomer. The reaction mixture was concentrated and partitioned between water and dichloromethane (2 × 10 mL). The organic layer was dried, concentrated, and then chromatographed on silica by using 50%–60% ethyl acetate / petroleum ether mixtures as the eluant to afford 0.10g (30%) of pure carboxanilide 11o: 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1 H), 8.50 (d, 1 H, J = 2.1 Hz), 8.42 (dd, 1 H, J = 4.6 and 1.4 Hz), 8.06 (dd, 1 H, J = 5.7 and 2.7 Hz), 7.97 – 7.99 (m, 1 H), 7.86 (ddd, 1 H, J = 9.6, 4.8 and 2.7 Hz), 7.54 (t, 1 H, J = 9.1 Hz), 7.40 – 7.45 (m, 3 H), 7.29 (dd, 1 H, J = 7.8 and 4.6 Hz), 7.21 – 7.23 (m, 1 H), 5.33 (s, 1 H), 4.24 (s, 1 H), 3.83 (dd, 1 H, J = 13.4 and 8.5 Hz), 2.49 – 2.50 (m, 1 H), 1.82 – 1.87 (m, 1 H), 0.80 (d, 3 H, J = 6.7 Hz), 0.79 (d, 3 H, J = 6.7 Hz); 13C NMR (125 MHz, DMSO-d6) δ 169.6, 163.9, 159.8, 157.7, 149.3, 148.5, 136.4, 136.4, 135.6, 134.2, 133.2, 132.5, 130.5, 128.9, 128.7, 127.6, 127.2, 127.2, 124.0, 123.6, 117.9, 117.7, 114.3, 100.5, 100.4, 60.1, 53.3, 51.8, 27.5, 20.6, 20.5; HR – ESI – MS [M+Na]+ calcd for C26H23FN4NaO2+, 465.1698; found, 465.1697.
(3S*,4S*)-N-(3-Cyano-4-fluorophenyl)-1-oxo-3-(1H-pyrazol-4-yl)-2-(2,2,2-trifluoroethyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (13h)
The carboxylic acid, 6 (X = 2,2,2-trifluoroethyl, Y = 4-pyrazol-yl), was made by the general procedures. Following Method B for the amide formation, a solution of 6 (448 mg, 1.32 mmol) and 5-amino-2-fluorobenzonitrile (269 mg, 1.98 mmol, 1.5 equiv) in 20 mL of acetonitrile was treated with phosphoryl chloride (0.136 mL, 1.45 mmol, 1.1 equiv), and the solution was heated at reflux for 3 h. The reaction mixture was quenched by pouring into ice water, and then extracted with ethyl acetate. The organic solution washed with aqueous sodium carbonate, dried, and then concentrated. The crude product was purified by chromatography on silica by using 20–100% ethyl acetate / hexane mixtures as the eluant to afford 420 mg (70%) of carboxanilide 13h as an off-white foam: 1H NMR (300 MHz, CD3OD) δ 8.11 (dd, 1 H, J = 7.6 and 1.4 Hz), 7.98 (dd, 1 H, J = 5.6 and 2.7 Hz), 7.86 (ddd, 1 H, J = 9.2, 4.7, and 2.7 Hz), 7.58 (td, 1 H, J = 7.6 and 1.2 Hz), 7.52 (td, 1 H, J = 7.6 and 1.2 Hz), 7.37 br s, 1 H), 7.37 (t, 1 H, J = 9.2 Hz), 7.33 (d, 1 H, J = 8.2), 7.26 (br s, 1 H), 5.41 (d, 1 H, J = 1.8 Hz), 4.74 (dq, 1 H, J = 15.2 and 9.2 Hz), 4.21 (d, 1 H, J = 2.0 Hz), 3.80 (dq, 1 H, J = 15.2 and 9.2 Hz); 13C NMR (125 MHz, CD3OD) δ 169.4, 165.3, 159.5 (d, J = 253 Hz), 135.7 (d, J = 3 Hz), 134.6, 133.1, 129.3, 128.8, 128.6, 128.0, 127.1 (d, 2 C’s, J = 8 Hz), 124.9 (q, J = 278 Hz), 124.3 (2 C’s), 119.5, 116.8, 116.7 (d, J = 21 Hz), 113.3, 100.9 (d, J = 16 Hz), 56.9, 52.5, 46.2 (q, J = 34 Hz); HR – ESI – MS [M+H]+ calcd for C22H16F4N5O2, 458.1240; found, 458.1241.
(3S*,4S*)-N-(3-Cyano-4-fluorophenyl)-1-oxo-3-(pyridin-3-yl)-2-(2,2,2-trifluoroethyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (13i)
The carboxylic acid 6 (X = 2,2,2-trifluoroethyl, Y = pyrid-3-yl) was made by the general procedure. Following Method B, a solution of 6 (462 mg, 1.32 mmol, 1 equiv), 5-amino-2-fluorobenzonitrile (269 mg, 1.98 mmol, 1.5 equiv), and phosphoryl chloride (0.136 mL, 1.45 mmol, 1.1 equiv), in 20 mL of acetonitrile was heated at reflux for 3 h. The reaction mixture was quenched with ice water and extracted with ethyl acetate. The combined organic extract was washed with aqueous sodium carbonate, dried, concentrated, and then chromatographed on silica by using 20–100% ethyl acetate / hexane mixtures as the eluant to afford 543 mg (88%) of carboxanilide 13i as an off-white foam: 1H NMR (400 MH, CD3OD) δ 8.48 (d, 1 H, J = 1.8 Hz), 8.42 (d, 1 H, J = 3.9 Hz), 8.12 –8.14 (m, 1 H), 8.01 (dd, 1 H, J = 5.6 and 2.7 Hz), 7.84 (ddd, 1 H, J = 9.4, 4.8, and 2.8 Hz), 7.56 (br d, 1 H, J = 8.0 Hz), 7.51 (app pent, 2 H, J = 4.2 Hz), 7.35 (t, 1 H, J = 9.2 Hz), 7.33 (d, 1 H, J = 8.0 Hz), 7.22 – 7.24 (m, 1 H), 5.56 (s, 1 H), 4.58 (dq, 1 H, J = 15.2 and 9.2 Hz), 4.20 (d, 1 H, J = 1.2 Hz), 4.09 (dq, 1 H, J = 15.2 and 9.2 Hz); 13C NMR (125 MHz, CD3OD) δ 169.0, 165.6, 159.5 (d, J = 253 Hz), 148.6, 147.4, 135.6 (d, J = 2.8 Hz), 135.1, 134.8, 133.3, 133.2, 129.3, 128.9, 128.5, 128.1 (d, J = 4.6 Hz), 127.2 (d, J = 8 Hz), 124.8 (q, J = 278 Hz), 124.4, 124.0, 116.8 (d, J = 21 Hz), 113.4, 100.9 (d, J = 17 Hz), 61.9, 52.4, 47.1 (q, J = 34 Hz); HR – ESI – MS [M+H]+ calcd for C24H17F4N4O2, 469.1288; found, 469.1284.
(3S,4S)-N-(3-Cyano-4-fluorophenyl)-1-oxo-3-(pyridin-3-yl)-2-(3,3,3-trifluoropropyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (13j)
The carboxylic acid 6 (X = 3,3,3-trifluoropropyl, Y = pyrid-3-yl) was made by the general procedure (0.92 g, 100% crude yield): 1HNMR (300 MHz, CD3OD) δ 8.42 (dd, J = 4.6 and 1.5 Hz, 1 H), 8.39 (d, J = 2.1 Hz, 1 H), 8.05 (d, J = 6.9 and 1.8 Hz, 1 H), 7.43 – 7.58 (m, 3 H), 7.27 – 7.35 (m, 2 H), 5.60 (s, 1 H), 4.34 – 4.44 (m, 1 H), 4.16 (s, 1 H), 3.08 – 3.18 (m, 1 H), 2.53 – 2.78 (m, 2 H); 13C NMR (75 MHz, DMSO-d6) δ 172.1, 163.4, 149.2, 148.3, 134.9, 134.2, 133.9, 132.7, 130.3, 128.9, 128.8, 128.5, 127.4, 125.1, 123.9, 58.8, 50.4, 31.5 (q, J = 27 Hz).
Carboxanilide 13j (114 mg, 86%) was prepared from 51 mg of the acid 6 by Method A for amide coupling and then chromatography with 0% – 20% ethyl acetate / hexanes as the eluant: 1H NMR (400 MHz, CD3OD) δ 8.50 – 8.60 (m, 2 H), 8.12 (dd, 1 H, J = 7.2 and 1.7 Hz), 8.01 (dd, 1 H, J = 5.6 and 2.8 Hz), 7.85 (ddd, 1 H, J = 9.2, 4.7, and 2.7 Hz), 7.73 (dd, 1 H, J = 6.4 and 1.2 Hz), 7.45 – 7.60 (m, 3 H), 7.25 – 7.40 (m, 2 H), 5.56 (s, 1 H), 4.33 (ddd, 1 H, J = 15.6, 9.6, and 6.0 Hz), 4.14 (d, 1 H, J = 1.3 Hz), 3.14 (ddd, 1 H, J = 15.2, 10.0, and 5.6 Hz), 2.61 – 2.75 (m, 2 H). 13C NMR (75 MHz, CD3OD) δ 169.3, 164.6, 159.3 (d, J = 253 Hz), 148.6, 147.3, 135.4, 134.7, 132.8, 132.6, 129.1, 128.6, 128.1, 127.5, 126.9 (d, J = 8 Hz), 124.4, 124.0 (d, J = 10 Hz), 122.2 (q, J = 270 Hz), 116.5 (d, J = 21 Hz), 113.1, 100.7 (d, J = 16 Hz), 59.7, 51.8, 31.1 (q, J = 29 Hz). HR – ESI – MS [M+H]+ calcd for C25H19F4N4O2, 483.1444; found, 483.1446.
Separation of the Enantiomers of 13i, 13h and 11o
Enantiomers of 13i
Racemic acid 6 (X=2,2,2-trifluoroethyl, Y=pyridin-3-yl) was converted to the ethyl ester by treatment with ethanolic sulfuric acid. A methanol solution of the racemic ester was loaded on a 250 × 30 mm Chiralpak AD-H column, and eluted with 30% methanol in carbon dioxide at a flow rate of 100 mL/min and 100 bar of back pressure at a rate of 125 mg of sample per 1 mL of solution, giving clean separation of the enantiomers. Isomer 1: retention time 7.6 min, [α]D – 55.6; isomer 2: retention time 11.5 min, [α]D 59.2). The ester enantiomers were then hydrolyzed (lithium hydroxide in aqueous methanol solution) to the respective single enantiomers of 6, which were analyzed by chiral HPLC on a Chiralcel OJH column with 8:2 hexane / ethanol containing 0.1% DEA as the eluant. Isomer 1: retention time 15.6 min, [α]D – 58.0, ee = 98.9%; isomer 2: retention time 17.5 min, [α]D 59.2, ee = 99.9%). Amide formation by using phosphoryl chloride as described for the racemate gave the respective single enantiomers of 13i. Isomer 1: retention time 11.4 min, [α]D −131.0, ee = 99.9%; isomer 2: retention time 16.6 min, [α]D 147, ee = 99.9%).
Enantiomers of 13h
Racemic acid 6 (X=2,2,2-trifluoroethyl, Y= pyrazo-4-yl) was converted to the racemic ethyl ester, and the enantiomers were separated as described above. Isomer 1: retention time 7.2 min, [α]D –112; isomer 2: retention time 8.67 min, [α]D 113. Hydrolysis to the corresponding acids and separation as described above gave the two enantiomers, although the relative retention times of the acid enantiomers are reversed. Isomer 1 (from isomer 1 of the acid): retention time 20.1 min, [α]D –111, ee = 99.9%; isomer 2 (from isomer 2 of the acid): retention time 14.5 min, [α]D 103, ee = 99%). Conversion to the respective amides 13h as described for the racemate afforded the enantiomers of 13h. Isomer 1: retention time 19.0 min, [α]D –154, ee = 99.9%; isomer 2: retention time 25.9 min, [α]D 152, ee = 99%.
Enantiomers of 11o
The enantiomers of racemic amide 11o were purified by chiral HPLC on a Chiralcel OJH column, eluting with 4:1 hexane / ethanol containing 0.1% DEA. Isomer 1: retention time 7.2 min, [α]D –175, ee = 99.9%); isomer 2: retention time 11.3 min, [α]D +159, ee = 98%).
Determination of EC50 against P. falciparum
Two P. falciparum strains, the chloroquine sensitive strain 3D7 and the chloroquine resistant strain K1, were provided by the MR4 Unit of the American Type Culture Collection (ATCC, Manassas, VA). Asynchronous parasites were maintained in culture based on the method of Trager.22 Parasites were grown in the presence of fresh group O-positive erythrocytes (Key Biologics, LLC, Memphis, TN) in Petri dishes at a hematocrite of 4–6% in RPMI based media (RPMI 1640 supplemented with 0.5% AlbuMAX II, 25 mM HEPES, 25 mM NaHCO3 (pH 7.3), 100μg/mL hypoxanthine, and 5 μg/mL gentamycin). Cultures were incubated at 37 °C in a gas mixture of 90% N2, 5% O2, and 5% CO2. For EC50 determinations, 20 μl of RPMI 1640 with 5 μg/ml gentamycin was dispensed per well in an assay plate (384-well microtiter plate, clear bottom, tissue culture treated). An amount of 60 nL of test compound, previously serial-diluted in a separate 384-well white polypropylene plate, was dispensed to the assay plate by hydrodynamic pin transfer, and then an amount of 20 μL of a synchronized culture suspension (1% rings, 4% hematocrite) was added per well, making a final hematocrite and parasitemia of 2% and 1%, respectively. The resulting theoretical compound dilution range was 15 uM to 700 pM (ten 1/3 dilution steps). To increase accuracy, each compound’s actual stock concentration was used for data calculations. Each compound was tested in triplicate by using three different assay plates. DMSO solution was used as the negative control, and 15 uM mefloquine was used as the positive control. Assay plates were incubated for 72 h, and the parasitemia was determined by a method previously described.4 Briefly, an amount of 10 uL of the following solution in PBS (10X Sybr Green I, 0.5% v/v triton, 0.5 mg/ml saponin) was added per well. Assay plates were shaken for 1 min, incubated in the dark for 90 min, and then read with the Envision spectrophotometer at Ex/Em of 485 nm / 535 nm. The EC50 value was calculated with the robust investigation of screening experiments (RISE) with four-parameter logistic equation. Two independent experiments were conducted on different days, resulting in six data points per compound concentration (2 × 3 assay plates).
Curve Fitting
Dose-response curves were calculated from percent activity values and log10-transformed concentrations by using the proprietary Robust Interpretation of Screening Experiments (RISE) application written in Pipeline Pilot (Accelrys, v. 8.5) and the R program.23 Briefly, non-linear regression was performed by using the R drc package with the four-parameter log-logistic function (LL2.4).24 The median value from replicates for each compound was fit three separate times by varying the parameters that were fixed during regression: (1) all parameters free, (2) high response fixed to 100, (3) low response fixed to 0. The best fit from these three nested models was selected by using the anova.drc function, producing 95% confidence intervals.
Solubility Assay
Solubility assays were carried out on a Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) using μSOL Evolution software (pION Inc., Woburn, MA). Compound stock (10 mM in DMSO, 10 μL) was added to 1-propanol (190 μL) to make a reference stock plate. Reference stock solution (5 μL) was mixed with 1-propanol (70 μL) and citrate phosphate buffered saline (pH 7, 75 μL) to make the reference plate which was then measured with UV detection. Test compound stock (10 mM, 6 μL) was added to buffer (594 μL) in a 96-well storage plate and mixed. The storage plate was sealed and incubated at room temperature for 18 hours. The suspension was then filtered through a 96-well filter plate (pION Inc., Woburn, MA). Filtrate (75 μL) was mixed with 1-propanol (75 μL) to make the sample plate, and the UV spectrum (250 nm – 500 nm) of the sample plate was read. Calculation was carried out by μSOL Evolution software based on the AUC (area under curve) of UV spectrum of the sample plate and the reference plate. All compounds were tested in triplicate.
Mouse Liver Microsome Assay
Liver microsomal solutions were prepared by using pooled female CD-1 mouse liver cells (20 mg/ml protein, Fisher Scientific, #NC9567486). Liver microsomes (0.83 mL) were mixed with EDTA solution (0.07 mL, 0.5 M in water) and potassium phosphate buffer (25.35 mL, 0.1M, pH 7.4) to make 26.25 mL of liver microsome solution (20 mg/mL liver microsome protein). 10 mM stocks of compound in DMSO solution were diluted with DMSO and acetonitrile to make an intermediate concentration (0.08 mM) in DMSO:acetonitrile (1:4 v:v). 10 mM stocks of controls in DMSO solution (diphenhydramine HCl and verapamil HCl) were diluted to 0.4 mM concentration in DMSO:acetonitrile (1:4 v:v). Each diluted compound stock (37.83 μL) was added to an aliquot of the liver microsomal solution (3 mL) and vortexed. The resulting solution was added to each of three wells of a master assay plate (pION Inc., MA, #110323). Each plate holds triplicate samples of two controls (0.4 mM) and the compounds (0.08 mM) in mouse microsomes. Aliquots of each well of the plate (175 μL of each well) were transferred from the master plate into five assay plates. For 0-hour time point, pre-cooled (4 °C) internal standard (437.5 μL, 2 μM caffeine in methanol) was added to the first plate before the start of reaction. NADPH regenerating system solution A (1.51 ml, Fisher Scientific, #NC9255727) was combined with NADPH regenerating system solution B (0.30 ml, Fisher Scientific, #NC9016235) in potassium phosphate buffer (3.96 ml, 0.1 M, pH 7.4, 37°C). The resulting NADPH solution (43.75 μL) was added to each well of all the 96-well assay plates and mixed with pipette briefly, making the final protein and compound concentrations in the control and compound wells respectively: medium (4 uM), and low (0.08 uM). The plates were sealed, and all plates except the 0-hr plate were incubated at 37 °C and shaken at a speed of 100 rpm. A single assay plate was tested at each time point: 0.5 h, 1 h, 2 h, and 4 h. At each time point, 437.5 μL of pre-cooled internal standard was added to each well of the plate to quench the reaction. The quenched plate was then centrifuged (model 5810R, Eppendorf, Westbury, NY) at 4000 rpm for 30 min. 100 μL of supernatant was transferred to a 96-well plate and analyzed by UPLC-MS (Waters Inc., Milford, MA). The compounds and internal standard were detected by selected ion recording (SIR).
Calculations
The amount of material was measured as a ratio of peak area to the internal standard and graphed. By using the slope from the most linear portion of this curve, the degradation rate constant was calculated. The rate constant was then used to calculate the half-life in plasma. Intrinsic clearance was calculated as CLint’ = (0.693 / (t1/2)) * (1/microsomal concentration in the reaction solution) * (45 mg microsome/gram liver) * (gram liver/kg by weight), where microsomal concentration in the reaction solution is 0.5 mg/mL, and gram liver/kg by weight of mice is 52. Intrinsic clearance was also calculated as CLint’ (uL/min/mg protein) = (1000)*(0.693/(t1/2*60))/0.5.
| [1] |
| [2] |
Supplementary Material
Figure 1.

Metabolism studies of trans-1,2,3,4-tetrahydro-1-isoquinolone-4-carboxanilides in mouse microsomes.
Acknowledgments
We are grateful to the NIH (AI090662), the Medicines for Malaria Venture (MMV) and the American Lebanese Syrian Associated Charities (ALSAC) for financial support, and to Jayan Joseph and Yuvaraj Sambandan for experimental assistance. The Ames study was conducted by Ricerca Bioscience.
ABBREVIATIONS USED
- Clint
Intrinsic clearance
- HPA
homophthalic anhydride
- IACUC
Institutional Animal Care and Use Committee
- Mmol
millimole
- µM
micromolar
- nM
nanomolar
- P
Plasmodium
- PfATP4
Plasmodium Falciparum ATPase 4
- S.D.
single dose
- T3P
propanephosphonic acid anhydride
Footnotes
This paper is respectfully dedicated to the memory of our friend and colleague Phil Stein.
ANCILLARY INFORMATION: The relative potencies against several resistant strains of P. falciparum (Table S-1), cytotoxicity data, and high resolution mass, 1H, and 13C NMR spectra of all new compounds (Table S-2) are found in the ancillary information, which is available free of charge on the ACS Publications website at DOI:10.1021/acs.jmedchem.xxxxxxx.
References
- 1.World Health Organization. World Malaria Report. World Health Organization; Geneva, Switzerland: p volumes. [Google Scholar]
- 2.Cui L, Mharakurwa S, Ndiaye D, Rathod PK, Rosenthal PJ. Antimalarial Drug Resistance: Literature Review and Activities and Findings of the Icemr Network. Am. J. Trop. Med. Hyg. 2015;93:57–68. doi: 10.4269/ajtmh.15-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria Biology and Disease Pathogenesis: Insights for New Treatments. Nat. Med. 2013;19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nzila A, Rottmann M, Chitnumsub P, Kiara SM, Kamchonwongpaisan S, Maneeruttanarungroj C, Taweechai S, Yeung BK, Goh A, Lakshminarayana SB, Zou B, Wong J, Ma NL, Weaver M, Keller TH, Dartois V, Wittlin S, Brun R, Yuthavong Y, Diagana TT. Preclinical Evaluation of the Antifolate Qn254, 5-Chloro-N'6'-(2,5-Dimethoxy-Benzyl)-Quinazoline-2,4,6-Triamine, as an Antimalarial Drug Candidate. Antimicrob. Agents Chemother. 2010;54:2603–2610. doi: 10.1128/AAC.01526-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Spiroindolones, a Potent Compound Class for the Treatment of Malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. Chemical Genetics of Plasmodium Falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jimenez-Diaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre ME, O'Loughlin KG, Shackleford DM, Justino de Almeida M, Carrillo AK, Clark JA, Dennis AS, Diep J, Deng X, Duffy S, Endsley AN, Fedewa G, Guiguemde WA, Gomez MG, Holbrook G, Horst J, Kim CC, Liu J, Lee MC, Matheny A, Martinez MS, Miller G, Rodriguez-Alejandre A, Sanz L, Sigal M, Spillman NJ, Stein PD, Wang Z, Zhu F, Waterson D, Knapp S, Shelat A, Avery VM, Fidock DA, Gamo FJ, Charman SA, Mirsalis JC, Ma H, Ferrer S, Kirk K, Angulo-Barturen I, Kyle DE, DeRisi JL, Floyd DM, Guy RK. (+)-Sj733, a Clinical Candidate for Malaria That Acts through Atp4 to Induce Rapid Host-Mediated Clearance of Plasmodium. Proc. Natl. Acad. Sci. U S A. 2014;111:E5455–5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cushman M, Gentry J, Dekow FW. Condensation of Imines with Homophthalic Anhydrides. A Convergent Synthesis of Cis- and Trans-13-Methyltetrahydroprotoberberines. J. Org. Chem. 1977;42:1111–1116. doi: 10.1021/jo00427a001. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Lopez M, Shaw JT. Cyclic Anhydrides in Formal Cycloadditions and Multicomponent Reactions. Chem. Rev. 2009;109:164–189. doi: 10.1021/cr8002714. [DOI] [PubMed] [Google Scholar]
- 11.Haimova MA, Mollov NM, Ivanova SC, Dimitrova AI, Ognyanov VI. A Highly Stereoselective Synthesis of 3,4-Dihydro-1(2h)-Isoquinolinones and 8-Oxoberbines from Homophthalic Anhydrides and Azomethines. Tetrahedron. 1977;33:331–336. [Google Scholar]
- 12.Humphries PS, Galang G, Bechle BM, Conn Y, Dirico KJ, Hui Y, Oliver RM, Southers JA, Yang X. Chemically-Enabled Synthesis of 1,2,3,4-Tetrahydroisoquinolin-1-Ones. Tetrahedron Lett. 2009;50:2140–2143. [Google Scholar]
- 13.Cushman M, Choong T-C, Valko JT, Koleck MP. Total Synthesis of (+/−)-Chelidonine. J. Org. Chem. 1980;45:5067–5073. [Google Scholar]
- 14.Vara Y, Bello T, Aldaba E, Arrieta A, Pizarro JL, Arriortua MI, Lopez X, Cossio FP. Trans-Stereoselectivity in the Reaction between Homophthalic Anhydride and Imines. Org. Lett. 2008;10:4759–4762. doi: 10.1021/ol801757r. [DOI] [PubMed] [Google Scholar]
- 15.Rikers DTS, Hemker HC, Nefkens GHL, Tesser GI. The Use of Phosphorous Oxychloride in the Synthesis of P-Nitroanilides. Recl. Trav. Chim. Pays-Bas. 1991;110:347–348. [Google Scholar]
- 16.Hinson JA, Pumford NR, Roberts DW. Mechanisms of Acetaminophen Toxicity: Immunochemical Detection of Drug-Protein Adducts. Drug Metab. Rev. 1995;27:73–92. doi: 10.3109/03602539509029816. [DOI] [PubMed] [Google Scholar]
- 17.Meunier B, de Visser SP, Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 18.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial Drug Discovery: Efficacy Models for Compound Screening. Nat. Rev. Drug Discov. 2004;3:509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Wang Z, Levin A, Emge TJ, Rablen PR, Floyd DM, Knapp S. N-Methylimidazole Promotes the Reaction of Homophthalic Anhydride with Imines. J. Org. Chem. 2014;79:7593–7599. doi: 10.1021/jo501316m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimenez-Diaz MB, Mulet T, Viera S, Gomez V, Garuti H, Ibanez J, Alvarez-Doval A, Shultz LD, Martinez A, Gargallo-Viola D, Angulo-Barturen I. Improved Murine Model of Malaria Using Plasmodium Falciparum Competent Strains and Non-Myelodepleted Nod-Scid Il2rgammanull Mice Engrafted with Human Erythrocytes. Antimicrob. Agents Chemother. 2009;53:4533–4536. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerestif JM, Perrocheau J, Tonnard F, Bazureau JP, Hamelin J. 1,3-Dipolar Cycloaddition of Imidate Ylides on Imino-Alcohols: Synthesis of New Imidazolones Using Solvent Free Conditions. Tetrahedron. 1995;51:6757–6774. [Google Scholar]
- 22.Trager W, Jensen JB. Human Malaria Parasites in Continuous Culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 23.Team RDCR. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2011. [Google Scholar]
- 24.Ritz C, Baty F, Streibig JC, Gerhard D. Dose-Response Analysis Using R. PLoS One. 2015;10:e0146021. doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.