

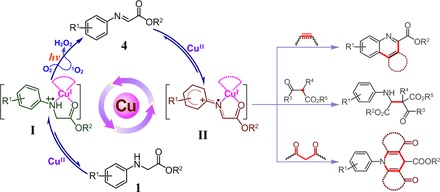
Cu(II) salts can activate C–H bonds of aromatic amines or imines to construct C–C bonds in air without external photosensitizer.
Abstract
Copper compounds involved in photocatalysis have recently spurred considerable interest for their novel transformations. However, mechanistic investigations are still in infancy. We find a new type of reaction, that is, Cu(II) salt–catalyzed C–H functionalization of aromatic amines triggered by visible light irradiation. An array of mechanistic observations, including high-resolution mass spectrometry, ultraviolet-visible absorption spectrum, electron spin resonance, x-ray absorption near-edge structure, and density functional theory calculation, have identified the key intermediates generated in situ in the transformation. Integration of single-electron transfer, singlet oxygen (1O2), and new absorption species, intermediate I and intermediate II formed in situ from Cu(II) salts and substrate amines or imines, respectively, is responsible for the N–H and C–H bond activation of secondary amines to couple with nucleophiles in air, thereby leading to the formation of quinoline, indolo[3,2-c]quinoline, β-amino acid, and 1,4-dihydropyridine derivatives in moderate to good yields under visible light irradiation at room temperature.
INTRODUCTION
Copper salts, which are inexpensive and easily accessible to different oxidation states, have been widely used to activate a variety of C–H bonds for the construction of complex compounds (1–4). Rapid growth of photocatalysis in recent years has introduced new strategies to make chemical reactions occur efficiently under mild conditions (5–10). The use of copper complexes as photoredox catalysts has shown promise for C–C or C–X (X = N, O, or Br) bond formation (11–26). In particular, Ullmann C–N coupling of carbazole with aryl halides could be accomplished at room temperature under ultraviolet (UV) illumination (27). In contrast to the well-established photocatalytic systems, the reaction underwent smoothly without external photosensitizers, and Cu(I) ions, instead of copper complexes, directly initiated the photocatalytic reaction. It was supposed that Cu(I) ion interacted with carbazole substrate to form a Cu(I)-N intermediate in situ that was responsive to UV light for further transformation. As a photochemical key step, the electron transfer from the Cu(I)-N intermediate to the halide was proposed. Furthermore, the combination of CuI and light was also effective for the coupling of alkyl and aryl halides with nitrogen, sulfur, and oxygen nucleophiles (27–36). These stimulating results have revealed the versatile reactivity of copper salts and broad tolerance of functional groups. At the same time, the rapid advances have attracted much interest in monitoring the intermediates generated in situ under illumination.
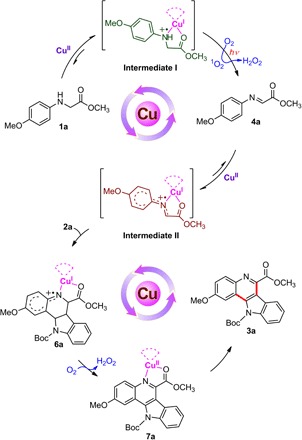
In this contribution, we report a new type of reaction, where Cu(II) salts, instead of copper complexes or Cu(I) ions, are capable of activating C–H bonds of secondary amines 1 to directly construct a range of C–C bonds in air, leading to the formation of quinoline, indolo[3,2-c]quinoline, β-amino acid, and 1,4-dihydropyridine scaffolds in good chemical yields under visible light irradiation. The key intermediates generated in situ in the cascade reaction have been tracked and identified. Studies using high-resolution mass spectrometry (HR-MS), UV-visible (UV-vis) absorption spectrum, electron spin resonance (ESR), x-ray absorption near-edge structure (XANES), and density functional theory (DFT) calculation reveal that simple Cu(II) salts associate with starting material of secondary amines 1 not only to activate the N–H bond and subsequently C–H bond but also to form new absorption species in the visible light region that is essential to ensure that the whole reaction proceeds in the absence of any external photosensitizer. Detailed mechanistic investigations suggest that the initial key step is a single-electron transfer from amine 1 to a Cu(II) ion, forming intermediate I, [Cu(I)-NH+●], that is, NH+● of amine 1 and Cu(I). With visible light irradiation, the intermediate I reacts with oxygen (O2) to generate singlet oxygen (1O2), which can oxidize the NH+● of amine 1 into imine 4. The imine 4 further coordinates with a Cu(II) ion to start the second stage of reaction, that is, a single-electron transfer from imine 4 to a Cu(II) ion, forming intermediate II, [Cu(I)-N+●], that is, N+● of the imine 4 and Cu(I). As a result, the electrophilicity of imine 4 in intermediate II is increased for the subsequent nucleophilic addition (Scheme 1). In addition, molecule O2 is beneficial for releasing the desired product and regenerating the Cu(II) ion to complete the catalytic cycle.
Scheme 1. Cu(II) salt–catalyzed C–H activation of secondary aromatic amines under visible light irradiation.
RESULTS AND DISCUSSION
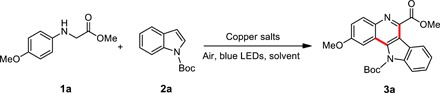
Our initial study focused on the reaction of methyl 2-(4-methoxyphenylamino)acetate 1a and indole 2a. Typically, substrate 1a and tert-butyloxycarbonyl (Boc)–protected 2a were dissolved in MeCN and followed by addition of Cu(OTf)2. The reaction mixture was then irradiated by blue light-emitting diodes (LEDs) at room temperature for 24 hours in air. Surprisingly, 50% yield of the cyclized product 3a was obtained without any external photosensitizer (37, 38). Because substrates 1a and 2a and Cu(II) salts show no absorption in the range of visible light, we believed that some new species formed in situ, which are responsible for this visible light–driven reaction. When Cu(II) salts were added, the solution of 1a in MeCN turned from pale to red immediately, and new absorption peaks around 500 nm appeared (Fig. 1).
Fig. 1. UV-vis spectra of Cu(II) salts, 1a, and 1a with Cu(II) ion in MeCN.
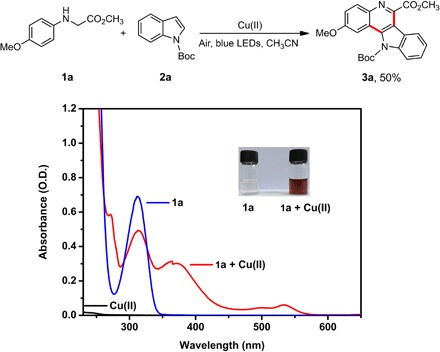
The concentration of Cu(II) ions is 9.0 × 10−5 M, and the concentration of 1a is 3.0 × 10−4 M. The Cu(II) ion used here refers to Cu(OTf)2. O.D., optical density.
The interesting transformation prompted us to identify the new species formed in situ. Fortunately, HR-MS investigation provided direct evidence on the species. The peaks at 258.0182 and 451.0918 corresponding to [1a + Cu]2+ and [1a + Cu + 1a − 2H]2+, respectively, were always detected regardless of the ratio of 1a and Cu(II) salts (fig. S1). To monitor the oxidation states of copper in these species, we performed ESR and XANES experiments. As shown in Fig. 2, a typical peak for Cu(II) was detected when Cu(II) ions were dissolved in MeCN (Fig. 2A, red line) (27, 39). However, this typical peak disappeared immediately upon addition of 1a into the solution, suggesting that Cu(II) ions may interact with 1a intimately to cause a change of oxidation state of copper. This assumption was further verified by the absorption edge of XANES. A typical feature was observed at lower energy (8.979 keV) corresponding to the 1s-3d of the Cu(II) complex (40, 41), whereas a pre-edge at 8.984 keV originated from 1s-4p of Cu(I) indicated the occurrence of the single-electron transfer process upon addition of 1a into the solution of Cu(II) ions in MeCN (Fig. 2B).
Fig. 2. Spectroscopic experiments.
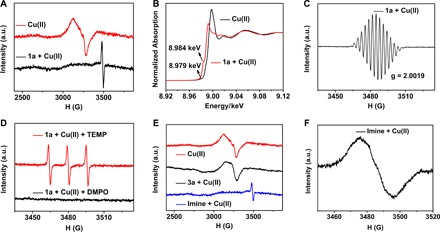
(A) ESR spectra of Cu(II) salts and the mixture of Cu(II) ion with 1a in CH3CN. (B) Normalized Cu XANES spectra of Cu(II) salts and 1a with Cu(II) ion in CH3CN. (C) ESR spectra of the mixture of Cu(II) ion with 1a in CH3CN (the enlarged spectra corresponding to the black line in a). (D) ESR spectra of an air-saturated CH3CN solution of TEMP, Cu(II) ion, and 1a or DMPO, Cu(II) ion, and 1a upon irradiation for 30 s; both NH+● signals of 1a had been deducted. (E) ESR spectra of a solution of Cu(II) salts, 3a with Cu(II) ion, and imine with Cu(II) ion in CH3CN. (F) ESR spectra of the mixture of Cu(II) ion with imine in CH3CN. The Cu(II) ion used here refers to Cu(OTf)2.
After adding 1a into the solution of Cu(II) ions, new peaks that are assigned to NH+● by computational simulation [aNH(N) = 6.16 G; aNH(H) = 6.70 G; aCH2(H) = 6.06 G; aring 2,2′(H) = 3.13 G; aring 3,3′(H) = 3.03 G; fig. S3] (42) were simultaneously detected by ESR (Fig. 2A, black line, corresponding to the enlarged spectra shown in Fig. 2C). This signal increased in the first 2 min, remained unchanged for 1 min, and finally decreased gradually (fig. S4). From these results, we inferred that the simple Cu(II) ions associated with the starting material of secondary amine 1a, leading to the occurrence of the single-electron transfer from 1a to Cu(II) ion directly to afford intermediate I in the dark.
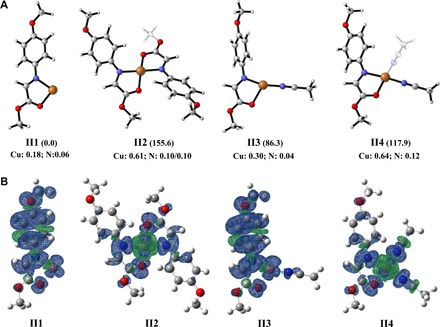
The identity of intermediate I was further confirmed by DFT calculation. As shown in Fig. 3A, four possible intermediates, that is, I1 [1a-Cu]2+, I2 [1a-Cu-1a]2+, I3 [1a-Cu-1MeCN]2+, and I4 [1a-Cu-2MeCN]2+, were involved. The spin density distributions in I1, I3, and I4 were mainly delocalized on 1a (Fig. 3B), with negligible population on the center of Cu, although the spin density of Cu (0.52) in I2 indicated that the single electron mainly populated on the center of Cu. The possible intermediates I1, I3, and I4 involved single-electron transfer from 1a to Cu(II) ion, which was in accordance with the observations supported by XANES and ESR experiments. Among the three possible intermediates, I4 has the most favorable binding energy. Therefore, we speculated that Cu(I)-amine radical cation I4 was the most possible intermediate between 1a and Cu(II) ion.
Fig. 3. Calculation studies of intermediate I.
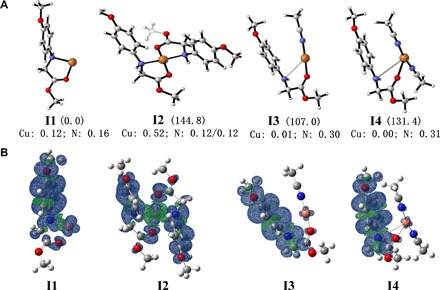
(A) Optimized geometrical structures of intermediate I. (B) Spin density distributions of I1 [1a-Cu]2+, I2 [1a-Cu-1a]2+, I3 [1a-Cu-1MeCN]2+, and I4 [1a-Cu-2MeCN]2+ at the B3LYP+GD3+CPCM/6-311+G** level. The related binding energy (in kilojoules per mole) and spin density of the key atoms (Cu and N of 1a) are indicated in parentheses and at the bottom of the correspondence, respectively. [1a-Cu]2+ was taken as a unit and the energy reference point.
However, the transformation reported in the current study hardly proceeded in the absence of light or air (Table 1, entries 8 and 9). This suggested that another oxygen active species contributed to the unique Cu(II) salt–catalyzed reaction under visible light irradiation. For the active species of oxygen, 2,2,6,6-tetramethyl piperidine (TEMP) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) were used as probes to trap singlet oxygen (1O2) and superoxide radical anion (O2−•), respectively, which could be easily detected by ESR spectroscopy (43, 44). As discussed above, intermediate I formed in situ from 1a and Cu(II) ion has the responsive ability to visible light. Upon irradiation of the solution of TEMP or DMPO, Cu(II) ions, and 1a in air-saturated MeCN solution by visible light (λ = 450 nm; blue LEDs), a single radical was trapped, the spectrum and hyperfine coupling constants of which are consistent with the reported values of nitroxide radical 2,2,6,6-tetramethyl piperidinooxy (TEMPO) from 1O2 and TEMP (Fig. 2D, red line), whereas the adduct of O2−• with DMPO was hardly detected (Fig. 2D, black line). Evidently, singlet oxygen 1O2 was considered as the active species formed under irradiation of 1a and Cu(II) ions. Singlet oxygen 1O2 could oxidize amines into the corresponding imines (45). With visible light irradiation of 1a and Cu(II) ions in air-saturated MeCN solution, 50% yield of imine 4a was isolated (Scheme 2A), whereas no reaction occurred with the addition of specific singlet oxygen (1O2) quencher, that is, DABCO (1,4-diazabicyclo[2.2.2]octane) (46). The isolated imine 4a could further react with nucleophile 2a with the help of Cu(II) salts in the dark to yield 64% of indolo[3,2-c]quinoline product 3a (Scheme 2B). Because no target product could be obtained in the absence of Cu(II) salts (Scheme 2C), Cu(II) salts should contribute to the outcome of this reaction.
Table 1. Impact of reaction parameters for the synthesis of indolo[3,2-c]quinoline 3a.
Unless otherwise specified, the reaction was carried out with substrate 1 (0.25 mmol), 2 (0.1 mmol), and copper salts (0.03 mmol) in CH2Cl2 (2.5 ml) under irradiation with blue LEDs for 24 hours at room temperature.
| Entry | Solvent | Copper salts | Yield (%)* |
| 1 | MeCN | Cu(OTf)2 | 50 |
| 2 | MeOH | Cu(OTf)2 | 0 |
| 3 | THF | Cu(OTf)2 | 0 |
| 4 | DMF | Cu(OTf)2 | 0 |
| 5 | CHCl3 | Cu(OTf)2 | 32 |
| 6 | CF3-Ph | Cu(OTf)2 | 20 |
| 7 | CH2Cl2 | Cu(OTf)2 | 29 |
| 8† | MeCN | Cu(OTf)2 | 5 |
| 9‡ | MeCN | Cu(OTf)2 | 0 |
| 10 | MeCN | — | 0 |
| 11 | CH2Cl2 | CuI | 0 |
| 12 | CH2Cl2 | CuBr | 0 |
| 13 | CH2Cl2 | CuCl2 | 37 |
| 14 | MeCN | CuI | 0 |
| 15 | MeCN | CuBr | 0 |
| 16§ | MeCN | Cu(OTf)2 | 60 |
| 17¶ | MeCN | Cu(OTf)2 | 71 |
*Isolated yields after purification by column chromatography.
†Carried out in the dark.
‡Under argon atmosphere.
§Fifteen milligrams of 4 Å sieves was added.
¶2a was dissolved in MeCN and then injected into the solution for 10 min based on entry 14.
Scheme 2. Control experiments.
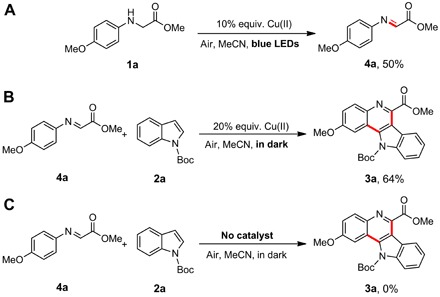
(A) Reaction of 1a in the absence of 2a during the reaction. (B) The reaction of 4a and 2a was carried out in the dark. (C) The reaction of 4a and 2a was carried out without any catalyst.
Similar to that observed in the first stage for the formation of intermediate I, the association of imine 4a with Cu(II) salts was detected. The peaks at 297.0299 and 449.0768 corresponding to [4a + Cu + MeCN]2+ and [4a + Cu + 4a]2+, respectively, were observed in HR-MS spectrometry (fig. S2). With the addition of Cu(II) salts into the MeCN solution of imine 4a, the solution color changed from faint yellow to red, and the signal of Cu(II) ion in ESR spectra disappeared, accompanied by generation of a new one, probably derived from N+● of imine (Fig. 2, E and F). Similar to intermediate I, the single-electron transfer from imine 4a to Cu(II) ion occurred in intermediate II, that is, II1 [4a-Cu]2+, II2 [4a-Cu-4a]2+, II3 [4a-Cu-1MeCN]2+, and II4 [4a-Cu-2MeCN]2+ (Fig. 4). Because the spin density of Cu in II2 (0.61) or in II4 (0.64) mainly populates on the Cu center, we speculated that Cu(I)-imine radical cation II3 is the most possible intermediate. Given the higher conjugated effect of intermediate II3 than intermediate I4, the spin density of N atom in II3 markedly decreased.
Fig. 4. Calculation studies of intermediate II.
(A) Optimized geometrical structures of intermediate II. (B) Spin density distributions of II1 [4a-Cu]2+, II2 [4a-Cu-4a]2+, II3 [4a-Cu-1MeCN]2+, and II4 [4a-Cu-2MeCN]2+ at the B3LYP+GD3+CPCM/6-311+G** level. The related binding energy (in kilojoules per mole) and spin density of the key atoms (Cu and N of 4a) are indicated in parentheses and at the bottom of the correspondence, respectively. [4a-Cu]2+ was taken as a unit and the energy reference point.
On the basis of these results, we proposed that for the cascade photocatalytic process described herein, it is not only intermediate I of Cu(II) ion with 1a and singlet oxygen 1O2 oxidation for the formation of imine (47) but also intermediate II of Cu(II) ion with imine 4a for the key bond-forming step of the product indolo[3,2-c]quinoline 3a as an example shown in Scheme 3. The fact that (i) the oxidation state of Cu(II) remained unchanged in the presence of product 3a (Fig. 2E, black line) and (ii) the signal of Cu(II) disappeared immediately when excess amount of either amine 1a or imine 4a was presented in the solution of 3a (Fig. 2E, blue line) indicated that the interaction of Cu(II) ion with 3a was much weaker than that of its corresponding amine 1a and imine 4a in intermediate I and II, respectively. H2O2 was successfully detected as a by-product in the reaction mixture (see Materials and Methods) (48), suggesting that molecular O2 could regenerate Cu(I) to Cu(II) in the following transformation of 6a to 7a, and Cu(II) ion released from 7a into the solution would coordinate to another imine to complete the reaction cycle.
Scheme 3. Proposed catalytic cycle for the synthesis of indolo[3,2-c]quinoline 3a under visible light irradiation.
By considering that indolo[3,2-c]quinoline and quinoline are important structures of multitudinous natural products and biomolecules (49, 50), we optimized reaction conditions and expanded the scope of this transformation. Screening reaction condition revealed that MeCN was the best choice (Table 1, entries 1 to 7), and Cu(OTf)2 showed better performance than the other Cu(I) and Cu(II) salts for this transformation (Table 1, entries 10 to 15). Addition of 4 Å sieves into the reaction mixture resulted in enhanced reaction efficiency, suggesting that water influenced the transformation to some extent (Table 1, entry 16). Progressive addition of 2a into the solution significantly improved the yield of product 3 to 71% (Table 1, entry 17). Either electron-donating (Me) or electron-withdrawing groups (F, Cl, or Br) at the para-position of aryl amines 1 were tolerated (Table 2, entries 2 to 5). Notably, crystal structure of product 3b confirmed the formation of indolo[3,2-c]quinolines other than indolo[2,3-c]quinolines (fig. S5). Compared to para-substituents, meta-substituted aryl amine gave lower efficiency (Table 2, entry 6). It is of significance that the intramolecular cyclization underwent selectively at the para-position of bromo group rather than at its ortho-position. In addition to methyl ester of the amine, all ethyl-, tert-butyl–, and benzyl-substituted secondary amines 1 were compatible with this reaction (Table 2, entries 7 to 9). In particular, products 3 containing chloro and bromo functionalities can serve as potential intermediates for further transformation. Moreover, a wide range of Boc-protected indoles were able to react with amines 1. Overall, electron-donating groups showed better performance than electron-withdrawing groups regardless of 5- or 6-substituted indole derivatives (Table 2, entries 10 to 14). Next, we evaluated the cascade reaction of amines 1 with other nucleophiles such as arynes or aromatic olefins 8 (51–53). Satisfactorily, all of them could proceed with moderate yields. Increasing the electron density of aromatic rings would be beneficial for the transformation (Table 3).
Table 2. Reaction scope for the synthesis of indolo[3,2-c]quinoline derivatives.
Unless otherwise noted, reactions were run with substrate 1 (0.25 mmol), Cu(OTf)2 (0.03 mmol), and 15 mg of 4 Å sieves in 2.5 ml of MeCN, and 2 (0.1 mmol) in 0.5 ml of MeCN was injected into the solution for 10 min under irradiation with blue LEDs at room temperature.
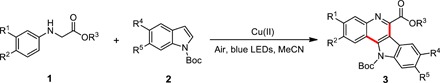
| Entry | R1 | R2 | R3 | R4 | R5 | Product | |
| 3 | Yield (%)* | ||||||
| 1 | H | OMe | Me | H | H | 3a | 71 |
| 2 | H | Me | Me | H | H | 3b | 78 |
| 3 | H | F | Me | H | H | 3c | 52 |
| 4 | H | Cl | Me | H | H | 3d | 80 |
| 5 | H | Br | Me | H | H | 3e | 71 |
| 6 | Br | H | Me | H | H | 3f | 34 |
| 7 | H | OMe | Et | H | H | 3g | 73 |
| 8 | H | OMe | t-Bu | H | H | 3h | 68 |
| 9 | H | OMe | Benzyl | H | H | 3i | 65 |
| 10 | H | Me | Me | H | Me | 3j | 76 |
| 11 | H | Me | Me | H | Cl | 3k | 53 |
| 12 | H | Me | Me | Me | H | 3l | 71 |
| 13 | H | Me | Me | OMe | H | 3m | 76 |
| 14 | H | Me | Me | Br | H | 3n | 34 |
*Isolated yields after purification by column chromatography.
Table 3. Reaction scope for the synthesis of quinoline derivatives under visible light irradiation.
Unless otherwise noted, reaction was carried out with 1a (0.25 mmol), arynes or aromatic olefins 8 (0.1 mmol), and Cu(OTf)2 (0.01 mmol) in 3 ml of MeCN under irradiation with blue LEDs at room temperature.
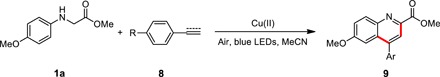
| Entry | R | Product | ||
| 9 | Ar | Yield (%)* | ||
| 1† | H | 9a | Ph | 45 |
| 2† | OMe | 9b | 4-MeOPh | 52 |
| 3† | F | 9c | 4-FPh | 36 |
| 4‡ | H | 9a | Ph | 40 |
| 5‡ | OMe | 9b | 4-MeOPh | 50 |
| 6‡ | F | 9c | 4-FPh | 32 |
*Isolated yields after purification by column chromatography.
†Arynes.
‡Aromatic olefins.
As a further extension, a series of N-substituted glycine esters bearing electron-donating or electron-withdrawing groups in the para-position of phenyl ring coupled with 2-oxocyclopentanecarboxylate 10 were studied (54–56). Most of them could be successfully transformed into desired product 11 in good to excellent yields (Table 4, entries 1 to 6). β-Ketoesters 10 have also a wide range of applicability under the standard conditions. Substituted methyl, ethyl, sterically bulky isopropyl, and tert-butyl as well as benzyl groups are compatible with this protocol, affording excellent yields of products (Table 4, entries 7 to 15). Even the less reactive acyclic β-ketoester could undergo direct alkylation, albeit with a moderate yield (Table 4, entry 16).
Table 4. Reaction scope for the synthesis of β-amino acid derivatives under visible light irradiation.
Unless otherwise noted, reactions were carried out with substrate 1 (0.2 mmol), 10 (0.1 mmol), and Cu(OTf)2 (0.01 mmol) in 3 ml of MeCN under irradiation with blue LEDs at room temperature. d.r., diastereomeric ratio.
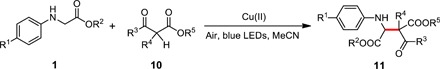
| Entry | R1 | R2 | R3 | R4 | R5 | Product | ||
| 11 | d.r. | Yield (%)* | ||||||
| 1 | OMe | Me | (CH2)3 | Et | 11a | 1.8:1 | 90 | |
| 2 | OMe | Et | (CH2)3 | Et | 11b | 2:1 | 87 | |
| 3 | OMe | Benzyl | (CH2)3 | Et | 11c | 3:1 | 70 | |
| 4 | Cl | Et | (CH2)3 | Et | 11d | 2.3:1 | 64 | |
| 5 | Br | Et | (CH2)3 | Et | 11e | 2.4:1 | 67 | |
| 6 | Me | Et | (CH2)3 | Et | 11f | 1.1:1 | 84 | |
| 7 | OMe | Me | (CH2)3 | Me | 11g | 1.2:1 | 83 | |
| 8 | OMe | Et | (CH2)3 | Me | 11h | 1.5:1 | 93 | |
| 9 | OMe | Me | (CH2)3 | i-Pr | 11i | 3:1 | 91 | |
| 10 | OMe | Et | (CH2)3 | i-Pr | 11j | 2.3:1 | 94 | |
| 11 | OMe | Et | (CH2)3 | t-Bu | 11k | 1.9:1 | 83 | |
| 12 | OMe | Me | (CH2)3 | t-Bu | 11l | 1.8:1 | 84 | |
| 13 | OMe | Et | (CH2)3 | Benzyl | 11m | 2:1 | 72 | |
| 14 | OMe | Me | Ph | H | Et | 11n | 1.2:1 | 81 |
| 15 | OMe | Et | Ph | H | Et | 11o | 1.2:1 | 85 |
| 16 | OMe | Et | Ph | Me | Et | 11p | 1:1 | 55 |
*Isolated yields after purification by column chromatography.
Unexpectedly, when 2,4-pentanedione 12a was used as the nucleophile, trace amount of product could be obtained within 1 hour of irradiation. However, when the reaction time was extended to 12 hours, 1,4-dihydropyridine 13a was isolated as yellow solids at room temperature (scheme S1A). The reaction parameters were optimized for the synthesis of 1,4-dihydropyridine 13a shown in Table 5. With the use of β-ketoester 10a to replace β-diketone 12a, the same situation did not appear. This is different from the case observed in the cascade reaction between secondary amines and β-ketoesters catalyzed by tris(4-bromophenyl)aminium hexachloroantimonate (57). Reaction of imine 4a with 2,4-pentanedione 12a afforded the corresponding 1,4-dihydropyridine 13a, further demonstrating the significance of imine 4a formed in the first stage of the Cu(II) salt–catalyzed cascade reaction (scheme S1B). Monitoring 1H nuclear magnetic resonance (NMR) spectra revealed that the produced imine 4a decomposed slowly into its corresponding aromatic amine 14a and aldehyde 15a. The former (14a) could condense with one molecule of β-diketone to yield enamine 17a, and the latter (15a) was quickly transformed into α,β-unsaturated ketoester 16a. Finally, intermolecular Michael addition of 16a and 17a as well as intramolecular condensation released the product of 1,4-dihydropyridine 13a (Scheme 4) (58, 59).
Table 5. Impact of reaction parameters for the synthesis of 1,4-dihydropyridine 13a.
Unless otherwise specified, the reaction was carried out with substrate 1a (0.1 mmol), 12a, and Cu(OTf)2 in MeCN under irradiation with blue LEDs at room temperature.
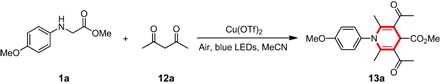
| Entry | 12a (mmol) | Cu(OTf)2 (mmol) | MeCN (ml) | Yield (%)* |
| 1 | 0.2 | 0.02 | 2 | 21 |
| 2 | 0.3 | 0.02 | 2 | 40 |
| 3 | 0.4 | 0.02 | 2 | 47 |
| 4 | 0.5 | 0.02 | 2 | 50 |
| 5 | 0.75 | 0.02 | 2 | 51 |
| 6 | 1 | 0.02 | 2 | 50 |
| 7 | 0.5 | 0.02 | 6 | 35 |
| 8 | 0.5 | 0.02 | 8 | 29 |
| 9 | 0.5 | 0.02 | 1 | 55 |
| 10 | 0.5 | 0.02 | 0.5 | 48 |
| 11 | 0.5 | 0.01 | 1 | 35 |
| 12 | 0.5 | 0.03 | 1 | 52 |
| 13† | 0.5 | 0.02 | 1 | 67 |
*Isolated yields after purification by column chromatography.
†After the reaction was finished, 1 equiv. of NaBH3CN was added and stirred for 1 hour.
Scheme 4. Proposed catalytic cycle for the synthesis of 1,4-dihydropyridine 13a under visible light irradiation.
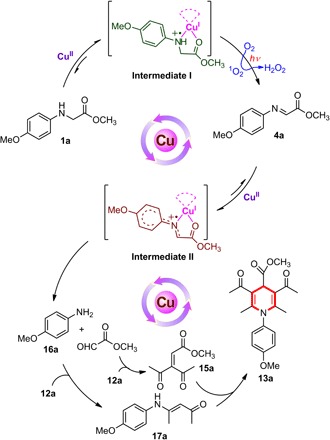
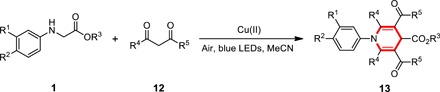
As illustrated in Table 6, a variety of secondary amines with different esters could produce 1,4-dihydroyridines 13a to 13d in good yields. In addition to methoxy-substituted aromatic ring, methyl, fluoro, and bromo groups also functioned well to produce 13e to 13h in moderate yields. Replacing acetylacetone with unsymmetric β-diketones, such as 2,4-hexanedione and 1-phenyl-1,3-butanedione as the coupling partners, yielded 1,4-dihydropyridine 13i and 13j in 60 and 72% yields, respectively. In addition, cyclic β-diketones were well tolerated to produce fused ring compounds 13k and 13l in moderate yields.
Table 6. Reaction scope for the synthesis of 1,4-dihydropyridine derivatives.
Unless otherwise noted, reactions were run with 1 (0.1 mmol), 12 (0.5 mmol), and Cu(OTf)2 (0.02 mmol) in 1 ml of MeCN under irradiation with blue LEDs at room temperature.
| Entry | R1 | R2 | R3 | R4 | R5 | Product | |
| 13 | Yield (%)* | ||||||
| 1 | H | OMe | Me | Me | Me | 13a | 67 |
| 2 | H | OMe | Et | Me | Me | 13b | 68 |
| 3 | H | OMe | Benzyl | Me | Me | 13c | 61 |
| 4 | H | OMe | t-Bu | Me | Me | 13d | 59 |
| 5 | H | Me | Me | Me | Me | 13e | 51 |
| 6 | Me | H | Me | Me | Me | 13f | 35 |
| 7 | H | F | Me | Me | Me | 13g | 51 |
| 8 | H | Br | Me | Me | Me | 13h | 32 |
| 9 | H | OMe | Me | Me | Et | 13i | 60 |
| 10 | H | OMe | Me | Me | Ph | 13j | 72 |
| 11 | H | OMe | Me | (CH2)3 | 13k | 54 | |
| 12 | H | OMe | Me | [CH2C (CH3)2CH2] |
13l | 57 | |
*Isolated yields after purification by column chromatography.
CONCLUSION
In summary, we have demonstrated that Cu(II) salts enable C–H functionalization of aromatic amines to directly form a variety of C–C bonds in air without any external photosensitizer under visible light irradiation at room temperature. A range of quinoline, indolo[3,2-c]quinoline, β-amino acid, and 1,4-dihydropyridine derivatives have been achieved in moderate to good chemical yields. An array of mechanistic studies reveals that the simple Cu(II) ion associates with secondary amine by single-electron transfer to form visible light absorption intermediate I [Cu(I)-NH+●]. Immediately, the N–H and C–H bonds of the amine are activated and further transformed into the corresponding imine by 1O2, generated by irradiation of intermediate I [Cu(I)-NH+●] with O2 under visible light. Next, intermediate II [Cu(I)-N+●] is produced by the single-electron transfer from the imine to Cu(II) ion in the second stage, which increases electrophilicity of the imine for the following addition with nucleophiles. Identifying these key intermediates in the cascade aerobic reaction will facilitate the exploration and exploitation of potential application of Cu(II) salts in synthetic chemistry, thereby leading to lower-cost, milder, and greener processes for catalytic transformations.
MATERIALS AND METHODS
1H NMR spectra were recorded using a Bruker Avance DPX 400 MHz instrument with tetramethylsilane as an internal standard. 13C NMR spectra were obtained at 100 MHz and referenced to the internal solvent signals. HR-MS (ESI) spectra were recorded on a Fourier transform ion cyclotron resonance mass spectrometer at the Analytical Instrumentation Center, Peking University. Steady-state emission spectra were recorded using a PerkinElmer LS50B spectrofluorometer. ESR spectra were recorded at room temperature using a Bruker ESP-300E spectrometer at 9.8 GHz, X-band, with 100-Hz field modulation. X-ray absorption spectroscopic (XANES measurements) data were collected at beamlines 1W1B and 1W2B of the Beijing Synchrotron Radiation Facility (BSRF). All reactions were carried out under air. MeCN was dried by anhydrous MgSO4 before use. Irradiation was carried out with blue LED (3 W). Sieves (4 Å) were activated in a muffle furnace at 250°C for 2 hours. Commercially available reagents were used without further purification.
General procedure for the reaction of aryl amine and Boc-protected indole derivatives
A 10-ml Pyrex tube equipped with a magnetic stir bar was charged with aryl amine (0.25 mmol), Cu(OTf)2 (0.03 mmol), MeCN (2.5 ml), and 15 mg of 4 Å sieves. Boc-protected indole derivative (0.1 mmol) in 0.5 ml of MeCN was slowly injected for 10 min under irradiation with blue LEDs at room temperature. Then, the solution was stirred for another 30 hours under irradiation with blue LEDs at room temperature. Finally, the mixture was evaporated under reduced pressure to remove the solvents, and the residue was purified by flash chromatography on silica gel to afford the desired product.
General procedure for the reaction of aryl amines and alkene or alkyne derivatives
A 10-ml Pyrex tube equipped with a magnetic stir bar was charged with aryl amines (0.2 mmol), alkene or alkyne derivatives (0.1 mmol), and Cu(OTf)2 (0.02 mmol) in MeCN (3 ml) under irradiation with blue LEDs for 6 hours at room temperature. Then, the reaction mixture was evaporated under reduced pressure to remove the solvents, and the residue was purified by flash chromatography on silica gel to afford the desired product.
General procedure for the reaction of aryl amine and β-ketoester derivatives
A 10-ml Pyrex tube equipped with a magnetic stir bar was charged with aryl amine (0.2 mmol), β-ketoester (0.1 mmol), and Cu(OTf)2 (0.01 mmol) in MeCN (3 ml) under irradiation with blue LEDs for 1 hour at room temperature. Then, the mixture was evaporated under reduced pressure to remove the solvents, and the residue was purified by flash chromatography on silica gel to afford the desired product.
General procedure for the reaction of aryl amine and β-diketone derivatives
A 10-ml Pyrex tube equipped with a magnetic stir bar was charged with aryl amine (0.2 mmol), β-diketone (0.5 mmol), and Cu(OTf)2 (0.04 mmol) in MeCN (2 ml) under irradiation with blue LEDs at room temperature. After completion monitored by thin-layer chromatography analysis, the mixture was evaporated under reduced pressure to remove the solvents, and the residue was purified by flash chromatography on silica gel to afford the desired product.
Procedures for ESR experiments
The sample of Cu(OTf)2 (1.6 × 10−2 M), 1a (6.4 × 10−2 M), and imine (6.4 × 10−2 M) in MeCN was prepared. For detection of Cu(II) ions, the solution of Cu(OTf)2 was diluted to 8.0 × 10−3 M with MeCN, and then an aliquot of the solution was transferred into an ESR tube. ESR spectrum was recorded at room temperature using 1500-Hz field modulation. For detection of N radical cation, an equivalent amount of Cu(OTf)2 (1.6 × 10−2 M) solution was mixed with 1a (6.4 × 10−2 M) or imine (6.4 × 10−2 M), and then an aliquot of the solution was transferred into an ESR tube. ESR spectrum was recorded at room temperature using 100-Hz field modulation. For the detection of superoxide radical anion (O2−•), an aliquot of the solution sample containing DMPO (2.0 × 10−2 M), Cu(OTf)2 (1.25 × 10−4 M), 1a (1.25 × 10−3 M), or imine (1.25 × 10−3 M) in air-saturated MeCN was transferred into an ESR tube. Upon irradiation for 30 s, ESR spectrum was recorded at room temperature using 100-Hz field modulation. For detection of singlet oxygen (1O2), TEMP (0.12 M) was used to replace DMPO. NH+● signal of 1a was simulated by Win-ESR software (see below) to yield the hyperfine splitting constants aNH(N) = 6.16 G, aNH(H) = 6.70 G, aCH2(H) = 6.06 G, aring 2,2′(H) = 3.13 G, and aring 3,3′(H) = 3.03 G.
X-ray absorption spectroscopy experiments
X-ray absorption measurements (XANES) were performed at beamlines 1W1B and 1W2B of the BSRF. The data were collected in transmission and fluorescence quick scan mode. The ionization chambers were optimized for the maximal current with linear response (1010 photons detected per second) with 10% absorption (N2) in the incident ion chamber and 70% absorption (60% N2 and 40% Ar) in the incident, transmission, and fluorescent x-ray detector. The samples of Cu(OTf)2 (1.5 × 10−2 M) and 1a (0.1 M) with Cu(OTf)2 (1.5 × 10−2 M) in MeCN were prepared.
DFT calculations
The DFT (60, 61) method B3LYP hybrid functional (62, 63) in combination with the 6-311+G** basis set was used to optimize two series of possible intermediates. One involves 1a, Cu(II) ion, and CH3CN; the other includes 4a, Cu(II) ion, and MeCN. During optimization, the solvent effect was considered via a self-consistent reaction field method based on the conductor-like polarizable continuum model (CPCM) (64, 65) for MeCN, and the dispersion effect was considered with the Grimme’s D3 version (GD3) (66). The frequency analysis validated all optimized structures as the real minima. All calculations were performed with the Gaussian 09 software package (67).
On the basis of the optimized structures, we calculated the related binding energy and spin density. Here, the Gibbs free energy at 298.15 K and 1.0 atm was used to calculate the binding energy. In 1a with Cu(II) ion in MeCN, [1a-Cu]2+ was referred as a unit, and the binding energy of [1a-Cu-1a]2+ ([1a-Cu-1MeCN]2+) was defined as the Gibbs free energy difference between [1a-Cu-1a]2+ ([1a-Cu-1MeCN]2+) and [1a-Cu]2+ with 1a; the binding second MeCN energy for [1a-Cu-1MeCN]2+ was the Gibbs free energy difference between [1a-Cu-2MeCN]2+ and [1a-Cu-1MeCN]2+ with MeCN. The total binding energy of [1a-Cu-2MeCN]2+ was the sum of [1a-Cu]2+ binding two MeCN by two steps. Similar to 1a with Cu(II) ion in MeCN, we calculated the binding energies of possible intermediates in 4a with Cu(II) in MeCN. In combination with the experimental ESR and XANES studies, we proposed the two most possible intermediate I (I4) and intermediate II (II3) at the different reaction stage.
Detection of H2O2
After the reaction was completed, 5 ml of CH2Cl2 was added, the resulting solution was filtered, and 2 ml of saturated aqueous solution of sodium carbonate was introduced into the solution to extract H2O2. Then, 35 ml of isopropanol solution containing 2.5 ml of glacial acetic acid and 2 g of sodium iodide was injected into the extracted H2O2 aqueous solution and heated to reflux for 5 min. In the course of the heating process, the solution color became yellow, featuring H2O2 formation (48).
Acknowledgments
Funding: This work was supported by the Ministry of Science and Technology of China (2013CB834804, 2013CB834505, and 2014CB239402), the National Natural Science Foundation of China (21390404, 91427303, 21473227, and 21402217), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17030400), and the Chinese Academy of Sciences. Author contributions: L.-Z.W. and Q.-Y.M. devised the initial concept of the work and designed the experiments. Q.-Y.M. and X.-W.G. contributed to the synthesis of products and mechanism investigation. T.L. and Z.L. performed the synthesis of starting materials and characterized the products. F.Z., Z.-J.L., and Y.T. performed x-ray absorption experiments. H.X. performed theoretical calculation. L.-Z.W. and Q.-Y.M. wrote the manuscript. J.-J.Z., K.F., B.C., and C.-H.T. helped with the discussion and analysis of the data. Competing interests: L.-Z.W., Q.-Y.M., T.L., X.-W.G., J.-J.Z., B.C., and C.-H.T. are authors on a patent related to this paper (Chinese Patent 201310001383.X, 2013) (68). M. Xiang is also an author on this patent, but is not an author of this paper. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from L.-Z.W. (lzwu@mail.ipc.ac.cn).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700666/DC1
Characterization data for all compounds
1H and 13C NMR
Cartesian coordinates (Å) and Gibbs free energies (Hartree) of all optimized structures
fig. S1. ESI HR-MS for the mixture of 1a and Cu(II) ions.
fig. S2. ESI HR-MS for the mixture of imine and Cu(II) ions.
fig. S3. ESR spectrum of Cu(OTf)2 and 1a in MeCN.
fig. S4. ESR experiments for variation of radical NH+● with the change of time.
fig. S5. X-ray crystal structure of 3b.
scheme S1. Cu(II) salt–catalyzed reactions for the synthesis of 1,4-dihydropyridine 13a.
REFERENCES AND NOTES
- 1.Wendlandt A. E., Suess A. M., Stahl S. S., Copper-catalyzed aerobic oxidative C–H functionalizations: Trends and mechanistic insights. Angew. Chem. Int. Ed. 50, 11062–11087 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Zhang C., Tang C., Jiao N., Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chem. Soc. Rev. 41, 3464–3484 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Girard S. A., Knauber T., Li C.-J., The cross-dehydrogenative coupling of Csp3–H bonds: A versatile strategy for C–C bond formations. Angew. Chem. Int. Ed. 53, 74–100 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Guo X.-X., Gu D.-W., Wu Z., Zhang W., Copper-catalyzed C–H functionalization reactions: Efficient synthesis of heterocycles. Chem. Rev. 115, 1622–1651 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Xuan J., Xiao W.-J., Visible-light photoredox catalysis. Angew. Chem. Int. Ed. 51, 6828–6838 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Nicewicz D. A., Nguyen T. M., Recent applications of organic dyes as photoredox catalysts in organic synthesis. ACS Catal. 4, 355–360 (2013). [Google Scholar]
- 7.Hari D. P., König B., The photocatalyzed Meerwein arylation: Classic reaction of aryl diazonium salts in a new light. Angew. Chem. Int. Ed. 52, 4734–4743 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Prier C. K., Rankic D. A., MacMillan D. W. C., Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kärkäs M. D., Porco J. A., Stephenson C. R. J., Photochemical approaches to complex chemotypes: Applications in natural product synthesis. Chem. Rev. 116, 9683–9747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skubi K. L., Blum T. R., Yoon T. P., Dual catalysis strategies in photochemical synthesis. Chem. Rev. 116, 10035–10074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirtsch M., Paria S., Matsuno T., Isobe H., Reiser O., [Cu(dap)2Cl] as an efficient visible-light-driven photoredox catalyst in carbon–carbon bond-forming reactions. Chem. Eur. J. 18, 7336–7340 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Hernandez-Perez A. C., Vlassova A., Collins S. K., Toward a visible light mediated photocyclization: Cu-based sensitizers for the synthesis of [5]helicene. Org. Lett. 14, 2988–2991 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Paria S., Pirtsch M., Kais V., Reiser O., Visible-light-induced intermolecular atom-transfer radical addition of benzyl halides to olefins: Facile synthesis of tetrahydroquinolines. Synthesis 45, 2689–2698 (2013). [Google Scholar]
- 14.Baralle A., Fensterbank L., Goddard J.-P., Ollivier C., Aryl radical formation by copper(I) photocatalyzed reduction of diaryliodonium salts: NMR evidence for a CuII/CuI mechanism. Chem. Eur. J. 19, 10809–10813 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Hernandez-Perez A. C., Collins S. K., A visible-light-mediated synthesis of carbazoles. Angew. Chem. Int. Ed. 52, 12696–12700 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Paria S., Reiser O., Copper in photocatalysis. ChemCatChem 6, 2477–2483 (2014). [Google Scholar]
- 17.Zhang Z., Tang X., Thomoson C. S., Dolbier W. R. Jr, Photoredox-catalyzed intramolecular aminodifluoromethylation of unactivated alkenes. Org. Lett. 17, 3528–3531 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Tang X.-J., Dolbier W. R. Jr, Efficient Cu-catalyzed atom transfer radical addition reactions of fluoroalkylsulfonyl chlorides with electron-deficient alkenes induced by visible light. Angew. Chem. Int. Ed. 54, 4246–4249 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Knorn M., Rawner T., Czerwieniec R., Reiser O., [Copper(phenanthroline) (bisisonitrile)]+-complexes for the visible-light-mediated atom transfer radical addition and alkylation reactions. ACS Catal. 5, 5186–5193 (2015). [Google Scholar]
- 20.Bagal D. B., Kachkovskyi G., Knorn M., Rawner T., Bhanage B. M., Reiser O., Trifluoromethylchlorosulfonylation of alkenes: Evidence for an inner-sphere mechanism by a copper phenanthroline photoredox catalyst. Angew. Chem. Int. Ed. 54, 6999–7002 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Fumagalli G., Rabet P. T. G., Boyd S., Greaney M. F., Three-component azidation of styrene-type double bonds: Light-switchable behavior of a copper photoredox catalyst. Angew. Chem. Int. Ed. 54, 11481–11484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernandez-Perez A. C., Caron A., Collins S. K., Photochemical synthesis of complex carbazoles: Evaluation of electronic effects in both UV- and visible-light methods in continuous flow. Chem. Eur. J. 21, 16673–16678 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Nicholls T. P., Constable G. E., Robertson J. C., Gardiner M. G., Bissember A. C., Brønsted acid cocatalysis in copper(I)-photocatalyzed α-amino C–H bond functionalization. ACS Catal. 6, 451–457 (2016). [Google Scholar]
- 24.Rabet P. T. G., Fumagalli G., Boyd S., Greaney M. F., Benzylic C–H azidation using the Zhdankin reagent and a copper photoredox catalyst. Org. Lett. 18, 1646–1649 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Rawner T., Knorn M., Lutsker E., Hossain A., Reiser O., Synthesis of trifluoromethylated sultones from alkenols using a copper photoredox catalyst. J. Org. Chem. 81, 7139–7147 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Reiser O., Shining light on copper: Unique opportunities for visible-light-catalyzed atom transfer radical addition reactions and related processes. Acc. Chem. Res. 49, 1990–1996 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Creutz S. E., Lotito K. J., Fu G. C., Peters J. C., Photoinduced Ullmann C–N coupling: Demonstrating the viability of a radical pathway. Science 338, 647–651 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Bissember A. C., Lundgren R. J., Creutz S. E., Peters J. C., Fu G. C., Transition-metal-catalyzed alkylations of amines with alkyl halides: Photoinduced, copper-catalyzed couplings of carbazoles. Angew. Chem. Int. Ed. 52, 5129–5133 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Uyeda C., Tan Y., Fu G. C., Peters J. C., A new family of nucleophiles for photoinduced, copper-catalyzed cross-couplings via single-electron transfer: Reactions of thiols with aryl halides under mild conditions (0°C). J. Am. Chem. Soc. 135, 9548–9552 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Ziegler D. T., Choi J., Muñoz-Molina J. M., Bissember A. C., Peters J. C., Fu G. C., A versatile approach to Ullmann C–N couplings at room temperature: New families of nucleophiles and electrophiles for photoinduced, copper-catalyzed processes. J. Am. Chem. Soc. 135, 13107–13112 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Do H.-Q., Bachman S., Bissember A. C., Peters J. C., Fu G. C., Photoinduced, copper-catalyzed alkylation of amides with unactivated secondary alkyl halides at room temperature. J. Am. Chem. Soc. 136, 2162–2167 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Tan Y., Muñoz-Molina J. M., Fu G. C., Peters J. C., Oxygen nucleophiles as reaction partners in photoinduced, copper-catalyzed cross-couplings: O-arylations of phenols at room temperature. Chem. Sci. 5, 2831–2835 (2014). [Google Scholar]
- 33.Ratani T. S., Bachman S., Fu G. C., Peters J. C., Photoinduced, copper-catalyzed carbon–carbon bond formation with alkyl electrophiles: Cyanation of unactivated secondary alkyl chlorides at room temperature. J. Am. Chem. Soc. 137, 13902–13907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sagadevan A., Ragupathi A., Lin C.-C., Hwu J. R., Hwang K. C., Visible-light initiated copper(I)-catalysed oxidative C–N coupling of anilines with terminal alkynes: One-step synthesis of α-ketoamides. Green Chem. 17, 1113–1119 (2015). [Google Scholar]
- 35.Yoo W.-J., Tsukamoto T., Kobayashi S., Visible light-mediated Ullmann-type C–N coupling reactions of carbazole derivatives and aryl iodides. Org. Lett. 17, 3640–3642 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Kainz Q. M., Matier C. D., Bartoszewicz A., Zultanski S. L., Peters J. C., Fu G. C., Asymmetric copper-catalyzed C-N cross-couplings induced by visible light. Science 351, 681–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z.-Q., Hu M., Huang X.-C., Gong L.-B., Xie Y.-X., Li J.-H., Direct α-arylation of α-amino carbonyl compounds with indoles using visible light photoredox catalysis. J. Org. Chem. 77, 8705–8711 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Zhu S., Rueping M., Merging visible-light photoredox and Lewis acid catalysis for the functionalization and arylation of glycine derivatives and peptides. Chem. Commun. 48, 11960–11962 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Solomon E. I., Spectroscopic methods in bioinorganic chemistry: Blue to green to red copper sites. Inorg. Chem. 45, 8012–8025 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Kau L. S., Spira-Solomon D. J., Penner-Hahn J. E., Hodgson K. O., Solomon E. I., X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc. 109, 6433–6442 (1987). [Google Scholar]
- 41.He C., Zhang G., Ke J., Zhang H., Miller J. T., Kropf A. J., Lei A., Labile Cu(I) catalyst/spectator Cu(II) species in copper-catalyzed C–C coupling reaction: Operando IR, in situ XANES/EXAFS evidence and kinetic investigations. J. Am. Chem. Soc. 135, 488–493 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Josephy P. D., Eling T., Mason R. P., The horseradish peroxidase-catalyzed oxidation of 3,5,3′,5′-tetramethylbenzidine. Free radical and charge-transfer complex intermediates. J. Biol. Chem. 257, 3669–3675 (1982). [PubMed] [Google Scholar]
- 43.Ma J., Zhao J., Jiang L., Effect of structural modification on photodynamic activity of hypocrellins. Photochem. Photobiol. 74, 143–148 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Villamena F. A., Liu Y., Zweier J. L., Superoxide radical anion adduct of 5,5-dimethyl-1-pyrroline N-oxide. 4. Conformational effects on the EPR hyperfine splitting constants. J. Phys. Chem. A 112, 12607–12615 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang G., Chen J., Huang J.-S., Che C.-M., Highly efficient oxidation of amines to imines by singlet oxygen and its application in Ugi-type reactions. Org. Lett. 11, 4568–4571 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Baciocchi E., Del Giacco T., Elisei F., Gerini M. F., Guerra M., Lapi A., Liberali P., Electron transfer and singlet oxygen mechanisms in the photooxygenation of dibutyl sulfide and thioanisole in MeCN sensitized by N-methylquinolinium tetrafluoborate and 9,10-dicyanoanthracene. The probable involvement of a thiadioxirane intermediate in electron transfer photooxygenations. J. Am. Chem. Soc. 125, 16444–16454 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Daimon T., Nosaka Y., Formation and behavior of singlet molecular oxygen in TiO2 photocatalysis studied by detection of near-infrared phosphorescence. J. Phys. Chem. C 111, 4420–4424 (2007). [Google Scholar]
- 48.Mair R. D., Graupner A. J., Determination of organic peroxides by iodine liberation procedures. Anal. Chem. 36, 194–204 (1964). [Google Scholar]
- 49.Paulo A., Gomes E. T., Houghton P. J., New alkaloids from Cryptolepis sanguinolenta. J. Nat. Prod. 58, 1485–1491 (1995). [Google Scholar]
- 50.Van Miert S., Hostyn S., Maes B. U. W., Cimanga K., Brun R., Kaiser M., Mátyus P., Dommisse R., Lemière G., Vlietinck A., Pieters L., Isoneocryptolepine, a synthetic indoloquinoline alkaloid, as an antiplasmodial lead compound. J. Nat. Prod. 68, 674–677 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Richter H., García Mancheño O., TEMPO oxoammonium salt-mediated dehydrogenative Povarov/oxidation tandem reaction of N-alkyl anilines. Org. Lett. 13, 6066–6069 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Huo C., Yuan Y., Wu M., Jia X., Wang X., Chen F., Tang J., Auto-oxidative coupling of glycine derivatives. Angew. Chem. Int. Ed. 53, 13544–13547 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Huo C., Xie H., Wu M., Jia X., Wang X., Chen F., Tang J., CBr4-mediated cross-dehydrogenative coupling reaction of amines. Chem. Eur. J. 21, 5723–5726 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Zhang G., Zhang Y., Wang R., Catalytic asymmetric activation of a –H bond adjacent to a nitrogen atom: A versatile approach to optically active α-alkyl α-amino acids and C1-alkylated tetrahydroisoquinoline derivatives. Angew. Chem. Int. Ed. 50, 10429–10432 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Gao X.-W., Meng Q.-Y., Xiang M., Chen B., Feng K., Tung C.-H., Wu L.-Z., Combining visible light catalysis and transition metal catalysis for the alkylation of secondary amines. Adv. Synth. Catal. 355, 2158–2164 (2013). [Google Scholar]
- 56.Gao X.-W., Meng Q.-Y., Li J.-X., Zhong J.-J., Lei T., Li X.-B., Tung C.-H., Wu L.-Z., Visible light catalysis assisted site-specific functionalization of amino acid derivatives by C–H bond activation without oxidant: Cross-coupling hydrogen evolution reaction. ACS Catal. 5, 2391–2396 (2015). [Google Scholar]
- 57.Jia X., Wang Y., Peng F., Huo C., Yu L., Liu J., Wang X., Catalytic oxidation of C(sp3)–H bonds induced by a radical cation salt: Construction of 1,4-dihydropyridines using a fragment-reassembly strategy. Adv. Synth. Catal. 356, 1210–1216 (2014). [Google Scholar]
- 58.Shen W., Wang L.-M., Tian H., Tang J., Yu J.-j., Brønsted acidic imidazolium salts containing perfluoroalkyl tails catalyzed one-pot synthesis of 1,8-dioxo-decahydroacridines in water. J. Fluorine Chem. 130, 522–527 (2009). [Google Scholar]
- 59.Liu Y.-p., Liu J.-m., Wang X., Cheng T.-m., Li R.-t., Multicomponent reactions leading to symmetric and asymmetric multi-substituted 1,4-dihydropyridines on montmorillonite. Tetrahedron 69, 5242–5247 (2013). [Google Scholar]
- 60.R. G. Parr, W. Yang, Density-Functional Theory of Atoms and Molecules (Oxford Univ. Press, 1989). [Google Scholar]
- 61.Cramer C. J., Truhlar D. G., Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 11, 10757–10816 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Becke A. D., Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- 63.Lee C., Yang W., Parr R. G., Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988). [DOI] [PubMed] [Google Scholar]
- 64.Barone V., Cossi M., Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 (1998). [Google Scholar]
- 65.Cossi M., Rega N., Scalmani G., Barone V., Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 67.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision E.01 (Gaussian Inc., 2013).
- 68.L.-Z. Wu, Q.-Y. Meng, T. Lei, X.-W. Gao, J.-J. Zhong, M. Xiang, B. Chen, C.-H. Tung, Chinese Patent 201310001383.X (2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700666/DC1
Characterization data for all compounds
1H and 13C NMR
Cartesian coordinates (Å) and Gibbs free energies (Hartree) of all optimized structures
fig. S1. ESI HR-MS for the mixture of 1a and Cu(II) ions.
fig. S2. ESI HR-MS for the mixture of imine and Cu(II) ions.
fig. S3. ESR spectrum of Cu(OTf)2 and 1a in MeCN.
fig. S4. ESR experiments for variation of radical NH+● with the change of time.
fig. S5. X-ray crystal structure of 3b.
scheme S1. Cu(II) salt–catalyzed reactions for the synthesis of 1,4-dihydropyridine 13a.