

Abstract
Background and Purpose
Cardiac glycosides are Na+/K+‐ATPases inhibitors used to treat congestive heart failure and cardiac arrhythmias. Epidemiological studies indicate that patients on digitalis therapy are more protected from cancer. Evidence of a selective cytotoxicity against cancer cells has suggested their potential use as anticancer drugs. The effect on angiogenesis of clinically used cardiac glycosides has not been extensively explored.
Experimental Approach
We studied the effect of digoxin, digitoxin and ouabain on early events of the angiogenic process in HUVECs. We determined HUVEC viability, proliferation, migration and differentiation into capillary tube‐like structures. We also tested drug activity using an in vivo angiogenesis model. Activation of protein tyrosine kinase 2 (FAK) and signalling proteins associated with the Na+/K+‐ATPase signalosome was determined by Western blotting.
Key Results
Digitoxin and ouabain (1–100 nM) inhibited HUVEC migration, concentration‐dependently, without affecting cell viability, while digoxin induced apoptosis at the same concentrations. Digitoxin antagonized growth factor‐induced migration and tubularization at concentrations (1–25 nM) within its plasma therapeutic range. The anti‐angiogenic effect of digitoxin was confirmed also by in vivo studies. Digitoxin induced Src, Akt and ERK1/2 phosphorylation but did not affect FAK autophosphorylation at Tyr397. However, it significantly inhibited growth factor‐induced FAK phosphorylation at Tyr576/577.
Conclusions and Implications
Therapeutic concentrations of digitoxin inhibited angiogenesis and FAK activation by several pro‐angiogenic stimuli. These novel findings suggest a potential repositioning of digitoxin as a broad‐spectrum anti‐angiogenic drug for diseases where pathological angiogenesis is involved.
Abbreviations
- bFGF
basic FGF
- ECGS
endothelial cell growth supplement
- FAK
focal adhesion kinase (protein tyrosine kinase 2)
- MVD
microvessel density
Introduction
The cardiac glycosides, digoxin, digitoxin and ouabain, have been used to treat congestive heart failure and cardiac arrhythmias. Cardiac effects of this class of drugs have been related to the inhibition of Na+/K+‐ATPases (also known as the sodium pump) that leads to an increase of intracellular Na+, which in turn induces higher levels of intracellular Ca2+ (Smith, 1988). Digoxin and digitoxin are still used worldwide, with a predominant use of digoxin because of its faster elimination (t 1/2 36–48 h for digoxin and 7 days for digitoxin in patients with normal renal function), which is advantageous in case of drug intoxication. Cardiac glycosides have, in fact, a narrow therapeutic window: plasma therapeutic concentrations are 0.5–2 ng·mL−1 (0.6–2.5 nM) for digoxin and 10–30 ng·mL−1 (10–40 nM) for digitoxin, while toxic effects have been reported at plasma concentrations of >2 and >39 ng·mL−1 for digoxin and digitoxin respectively (Belz et al., 2001; Ehle et al., 2011). At present, ouabain is mostly used as a research tool, but in the past, it was clinically employed for prevention and treatment of acute heart attacks, left ventricular insufficiencies, angina and digoxin intoxication (Fürstenwerth, 2010).
Besides the well‐known cardiac effects, a large number of studies carried out mainly in cancer cells show that cardiac glycosides affect several cellular processes, including cell survival or death, differentiation, proliferation and migration, with mechanisms that are unrelated to pump inhibition (Schoner and Scheiner‐Bobis, 2007). In fact, it has been demonstrated that binding of cardiac glycosides to a pool of Na+/K+‐ATPases localized in caveolae causes conformational changes to neighbouring proteins, leading to the activation of multiple signalling cascades. This evidence indicates that, besides its pumping function, Na+/K+‐ATPases act as a signal transducer in a protein complex that has been called the Na+/K+‐ATPase signalosome (Pierre and Xie, 2006). Interestingly, many cardiac glycosides in nanomolar concentrations (not leading to calcium overload) show a selective cytotoxic and antiproliferative effect against cancer cells, suggesting their use in anticancer therapy (Newman et al., 2008; Prassas and Diamandis, 2008; Cerella et al., 2013; Trenti et al., 2014). The potential repositioning of these drugs is supported by epidemiological data reporting that patients who have received digitalis therapy are more protected from some types of malignancies such as breast, lymphoma/leukaemia and prostate/urinary cancers (Stenkvist, 1999; Haux et al., 2001; Menger et al., 2013; Shim and Liu, 2014).
Angiogenesis is the process of formation of new blood vessels through vessel sprouting from pre‐existing ones (Potente et al., 2011). It is essential for physiological events, such as organ growth and repair, but its imbalance contributes to numerous malignant disorders including cancer (Carmeliet, 2005). Angiogenesis plays an important role in tumour growth, invasion and metastasis, by providing nutrients, oxygen and growth factors to the growing tumour mass, facilitating the removal of metabolic wastes and allowing the spread of cancer cells through the circulation. For these reasons in the last years, angiogenesis has become a therapeutic target for cancer treatment. Evidence of the effect of clinically used cardiac glycosides on angiogenesis is scarce and mostly focused on the production of pro‐angiogenic factors by cancer cells. In fact, it has been demonstrated that digoxin inhibits hypoxia‐inducible factor 1α (HIF‐1α) transcriptional activity in hypoxic tumour cells or retinal epithelial cells, thus reducing the production of growth factors, in particular VEGF, necessary to stimulate the angiogenic process (Zhang et al., 2008; Yoshida et al., 2010). Accordingly, inhibition of HIF‐1α and blood vessel formation by digoxin has been shown in mice (Gayed et al., 2012). Furthermore, a recent study has shown that ouabain inhibits the HIF‐1α pathway in placental cytotrophoblasts, leading to a decrease of fms‐like tyrosine kinase 1 (sFlt1), a soluble version of the VEGF receptor that is abnormally increased in preeclampsia (Rana et al., 2014). Besides this evidence, ouabain activates signalling pathways in endothelial cells (such as PI3K‐Akt and ERK) that promote cell survival or proliferation (Trevisi et al., 2004; Eva et al., 2006), suggesting a potential pro‐angiogenic activity. On the other hand, bufadienolides (bufalin and arenobufagin), another group of cardiac glycosides contained in toad venom and used in traditional Chinese medicine to treat cancer, have shown a direct anti‐angiogenic action on endothelial cells (Lee et al., 1997; Li et al., 2012). Based on these data, we studied the effect of clinically used cardiac glycosides on human endothelial cells, in terms of their proliferation, migration and differentiation into capillary‐like structures, three key events in the angiogenic process. We also performed studies using an in vivo angiogenesis model. Then we explored the signalling proteins involved, in particular those activated by the Na+/K+‐ATPase signalosome, such as Src kinase, Akt and ERK, and those linked to angiogenesis, such as protein tyrosine kinase 2, previously known as focal adhesion kinase (FAK). Our results show for the first time the inhibition of angiogenesis and FAK activation by digitoxin at concentrations within its plasma therapeutic range.
Methods
Cell culture
HUVEC isolation and culture
HUVEC were isolated in our laboratory as previously described (Vinci et al., 2004) from human umbilical cords. Umbilical cords were collected after delivery, from full‐term normal pregnancies at the Obstetrics and Gynaecological Unit of Padua University Hospital. The donors gave their informed consent, and the collected cords were non‐identifiable. The procedure was approved by the local Ethics Committee (Comitato Etico per la Sperimentazione Clinica della Provincia di Padova). Each HUVEC preparation was derived from at least three donors and were pooled after isolation. Cells were grown at 37°C under 5% CO2 in medium M199 supplemented with 15% FBS, 100 μg·mL−1 endothelial cell growth supplement (ECGS), 100 U·mL−1 heparin, 2 mM l‐glutamine, 100 U·mL−1 penicillin‐G and 100 μg·mL−1 streptomycin. HUVECs were used from passages 2 to 6.
Tumour cell lines
IGROV‐1 cells were purchased from American Type Culture Collection, and OC316 cells were provided by S. Ferrini (IST, Genoa, Italy). Both human ovarian cancer cell lines were grown in RPMI 1640 (Euroclone, Pero, Milan, Italy) supplemented with 10% fetal calf serum (Life Technologies, Carlsbad, CA, USA), 1% HEPES (10 mmol·L−1; Cambrex Bioscience, East Rutherford, NJ, USA), 1% l‐glutamine (2 mmol·L−1), 1% sodium pyruvate (1 mmol·L−1) and 1% antibiotic‐antimycotic mix (Gibco‐BRL, Life Technologies). Cultures were maintained at 37°C in a humidified 5% CO2/95% air atmosphere.
HUVEC proliferation and viability assays
Crystal violet
HUVEC (104 cells per well) were plated in 96‐well culture plate and incubated in complete culture medium. The next day, the medium was replaced with a fresh one containing the tested cardiac glycosides (1–100 nM) and the cells were incubated for 48 h. At the end of the treatment, plates were washed with PBS, stained with 100 μL of 0.1% crystal violet solution in 70% ethanol for 20 min, washed with deionized water and air‐dried. The bound dye was dissolved with 100 μL of 10% acetic acid. Optical density was measured with a multilabel plate counter (VICTOR2–Wallac, Perkin Elmer, Waltham, MA, USA) at 570 nm. Background absorbance from control wells (media without cells) was subtracted. Four replicates for each independent experiment were performed to test the reliability of single values.
ATP detection assay
Cell viability of HUVEC in apoptotic conditions was assessed with ATPlite assay kit (Promega), according to the manufacturer's instructions. Briefly, HUVEC (104 cells per well) were plated in 96‐well culture plate and incubated in complete culture medium. The next day, the medium was replaced with a fresh one containing only 1% FBS with or without digitoxin (0–100 nM) or ouabain (100 nM), and the cells were treated for further 48 h. At the end of the treatment, an equal volume of reconstituted reagent was added to each well, and the plate was mixed at 500 r.p.m. on an orbital shaker for 60 s, shielded from ambient light. After incubation for 10 min at room temperature in the dark, the luminescence of each sample was measured in a plate‐reading luminometer (VICTOR2–Wallac). For each independent experiment, four replicates were performed to test the reliability of single values, and data were normalized to control cells (cells incubated in complete culture medium).
Analysis of annexin V and PI binding
HUVEC (3 × 105 per well in a six‐well plates) were treated with digoxin (1–100 nM) for 24 h. At the end of the treatment, flow cytometry analysis of annexin V‐FITC and propidium iodide (PI) were performed as previously described (Trenti et al., 2014) using FC500 (Beckman Coulter, Brea, CA, USA). Bivariate analysis of FL1 and FL3, as well as forward scatter and side scatter (morphology parameters), were performed to discriminate live cells (annexin V‐FITC‐, PI−) from cells undergoing programmed cell death (annexin V‐FITC+, PI− and annexin V‐FITC+, PI+). For each sample, 10 000 events were collected. Data acquisition and analysis were done using the CXP Analysis software (Beckman Coulter).
Chemotaxis assay
Chemotaxis experiments were performed in a 48‐well microchemotaxis chamber (Neuro Probe, Gaithersburg, MD, USA) using 8 μm polyvinylpyrrolidone‐free polycarbonate filters coated with 10 μg·mL−1 of collagen (rat tail, Roche, Basel, Switzerland). Lower chambers were filled with 28 μL of M199 supplemented with 100 U·mL−1 heparin in presence of 15% FBS plus 100 μg·mL−1 ECGS, 10 ng·mL−1 VEGF, 10 ng·mL−1 EGF or 10 ng·mL−1 basic FGF (bFGF) respectively. Upper chambers were filled with 50 μL HUVEC suspension (1.6 × 105 cells per mL in M199 supplemented with 1% FBS and 100 U·mL−1 heparin). For the evaluation of the basal motility, M199 supplemented with 100 U·mL−1 heparin was used in the lower chamber. The tested cardiac glycosides (1–100 nM) were added both in the upper and lower compartment. After 6 h of incubation at 37°C, the non‐migrated HUVEC on the upper surface of the filter were removed by scraping. The cells that migrated to the lower side of the filter were stained with Diff‐Quick stain (Medion Diagnostics, Duedingen, Switzerland) and were counted at 400× magnification in eight random fields with a phase contrast inverted microscope (Nikon Eclipse Ti, Shinagawa, Tokyo, Japan). Six replicates for each independent experiment were performed to test the reliability of single values.
Wound healing assay
HUVEC (105 cells) were seeded in complete culture medium on 24‐well culture plate and allowed to reach confluence. One scratch was made, and the media was replaced with fresh complete medium containing or not digitoxin (0.1–25 nM). Three images of each well were captured at 40× with a phase contrast inverted microscope (Nikon Eclipse Ti) equipped with a digital camera, immediately after the scratch was made (time 0) and after 16 h of incubation. The wound spaces were measured from nine random fields of view using ImageJ software. Quantitative analysis of cell migration was performed using the average wound space from those random fields of view. The percentage of change in the wound space was calculated using the following formula:
Values were expressed as % change from control cells.
Capillary tube‐like formation assay
HUVEC (4 × 104 cells per well) were plated onto a thin layer of basement membrane matrix (Matrigel™, Corning Corp., Corning, NY, USA) in 24‐well plates. The cells were incubated at 37°C for 8 h in complete cell culture medium with or without digitoxin (1–25 nM). Three images per well were captured at 40× with a phase contrast inverted microscope (Nikon Eclipse Ti) equipped with a digital camera. Images were analysed using Angiogenesis Analyzer, a plugin developed for the ImageJ software (Carpentier et al., 2012). Data on dimensional parameters (total tubule length) and topological parameters (number of junctions and meshes and total mesh area) of the capillary‐like network (Guidolin et al., 2004) were analysed in all the images obtained from control and treated wells. Data are expressed as absolute values.
In vivo angiogenesis
Animals and treatment
All procedures involving animals and their care conformed to institutional guidelines that comply with national and international laws and policies (EEC Council Directive 86/609, OJ L 358, 12 December 1987) and were authorized by the Italian Ministry of Health (Authorization n. 129/2017‐PR). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). During in vivo experiments, animals in all experimental groups were examined daily for a decrease in physical activity and other signs of disease or drug toxicity. Six to eight‐week‐old female NOD/SCID‐γ−/− (NSG) mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed in our specific pathogen‐free animal facility in Allentown IVC cages (floor area 542 cm2) with a maximum of six mice per cage. All mice received water and food ad libitum and were kept under a 12 h light/dark cycle in a well‐ventilated room at an approximate temperature of 22°C. Mice acclimatized for a minimum of 7 days and a maximum of 15 days before being randomly assigned to treatment or vehicle groups.
The Matrigel sponge model of in vivo angiogenesis introduced by Passaniti et al. (1992) and Albini et al. (1994) was used. For angiogenesis to be induced, NSG mice were randomly divided into two groups of five animals each and injected s.c. into both flanks with 400 μL Matrigel supplemented with 500 ng bFGF and IGROV‐1 tumour cells (5 × 105 cells per injection), along with either digitoxin (25 nM) or vehicle (DMSO). Seven days after injection, mice were anaesthetized with isoflurane/oxygen and killed via cervical dislocation, and Matrigel pellets (two pellets per mouse) were collected.
Evaluation of MVD
Four‐μm‐thick frozen sections of Matrigel pellets were processed for immunohistochemistry as previously reported (Nardo et al., 2011). Microvessels were stained by rat anti‐CD31 mAb (1:50 dilution; Becton Dickinson, East Rutherford, NJ, USA); immunostaining was performed using the avidin–biotin–peroxidase complex technique and 3–3′ diaminobenzidine as chromogen (Vector Laboratories, Burlingame, CA, USA), and the sections were then lightly counterstained with Mayer's haematoxylin. Parallel negative controls, obtained by replacing primary Abs with PBS, were run. Microvessel density (MVD) was quantified both by screening the CD31‐stained sections for the areas of highest vascularity and by calculating the area of the sample containing microvessels as percentage of the total sample area. The number of fields analysed varied between 5 and 10 per sample, depending on the sample size. Images were collected at a total magnification of ×200. For each animal, the mean value of replicates was used for statistical analysis; five animals per group were analysed.
Western blotting analysis
Cells (3 × 105 cells per well in six‐well plates) were seeded in complete culture medium. After reaching confluence, the medium was replaced with M199 supplemented with 1% FBS and 100 UI·mL−1 heparin for 16 h. Then the cells were treated with digitoxin as indicated in the Results section and lysed with 100 μL lysis buffer (PBS containing 1% Triton X‐100, Sigma inhibitor cocktail 1X, NaF 1.25 mM, Na4P207 1 mM, Na‐orthovanadate 2 mM). After centrifugation at 10 000× g for 15 min, supernatants were collected for SDS‐PAGE and western blotting. Protein quantification was performed by the BCA assay (Sigma‐Aldrich, St. Louis, MO, USA). Proteins (60 μg) were separated on SDS‐PAGE, and gels were transferred on PVDF membranes (Hybond‐P, Amersham, Little Chalfont, UK). Membranes were blocked and probed using the following primary antibodies: rabbit anti‐phospho‐FAK Tyr397 , rabbit anti‐phospho‐FAK Tyr567/577, rabbit anti‐phospho‐FAK Tyr397, rabbit anti‐phospho‐Src Tyr416, rabbit anti‐phospho‐ERK1/2 and rabbit anti‐phospho‐Akt Ser473 (1:1000 dilution, Cell Signaling, Danvers, MA, USA); mouse anti‐FAK and rabbit anti‐Src (1:1000 dilution, GeneTex, Irvine, CA, USA); rabbit anti‐Akt (1: 1000 dilution), rabbit anti‐ERK (1:1000 dilution) and rabbit anti‐GAPDH (1:5000 dilution) from Santa Cruz Biotechnology (Dallas, TX, USA). After washing, the membranes were incubated with the appropriate secondary antibodies HRP‐conjugated (Santa Cruz Biotechnology) at 1:2500 dilution. Bands were detected by chemiluminescence using the LiteAblot Extend (Euroclone). Images were acquired with VersaDoc™ 4000 Imaging System (Bio Rad, Hercules, CA, USA). Densitometric analysis of the bands was performed with Image J 1,47v (NIH, USA). Densitometric values of phospho‐proteins were normalized to the respective GAPDH levels and expressed as % of untreated cells.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are presented as mean ± SEM. For in vitro studies, collected data were analysed by a blinded investigator; statistical analysis are from at least five independent experiments, performed with different HUVEC preparations. Graphs and statistical analysis were performed using GraphPad Prism version 5.0 for Windows (GraphPad Software Inc, San Diego, CA, USA). The differences between control and experimental groups were analysed by ANOVA followed by post hoc test as detailed in the figure legends; post hoc tests were run only for data where F achieved P < 0.05 and there was no significant variance inhomogeneity. For in vivo studies, collected data were analysed by two different blinded investigators; the differences between control and experimental groups were analysed by Student's t‐test (for normally distributed data: analysis of percentage of vascularized area) or the non‐parametric test Mann–Whitney Wilcoxon test (for non‐normally distributed data: analysis of MVD), as detailed in figure legends, using SigmaPlot (Systat Software Inc, London, UK). Differences were considered statistically significant at P < 0.05.
Materials
Cell culture reagents, FBS and ECGS, for HUVEC culture were from Sigma‐Aldrich (St. Louis, MO, USA). Ouabain, digoxin, digitoxin, crystal violet and PI were also supplied by Sigma‐Aldrich. VEGF‐A, bFGF and EGF were from ImmunoTools (Friesoythe, Germany). Annexin V‐FITC was from Immunotech (Beckman Coulter, Brea, CA, USA). Matrigel™ was purchased from Corning Corp. Digitoxin and digoxin were dissolved in DMSO, whereas ouabain was dissolved in water. Growth factors were dissolved following manufacturer's instructions.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Clinically used cardiac glycosides differently affect HUVEC viability and proliferation
We first examined the effect of digitoxin, ouabain and digoxin on HUVEC proliferation and viability. Drug chemical structures (Figure 1A) show the common molecular motif, a steroid nucleus containing an unsaturated lactone at the C17 position and one (ouabain) or three (digitoxin, digoxin) glycosidic residues at C3. Ouabain is the most hydrophilic compound, while digitoxin is the most lipophilic and differs from digoxin only by the absence of a hydroxyl group at C12. In exponentially growing HUVEC, digitoxin and ouabain (1–100 nM) did not affect cell proliferation and viability, determined by crystal violet assay (Figure 1B). Unexpectedly, at the same concentrations, digoxin decreased dye uptake and induced cell death (observed under the microscope). Flow‐cytometric analysis of annexin V/PI binding in HUVEC confirmed induction of cell death by digoxin in a concentration‐dependent manner (Figure 1C). As previously demonstrated for ouabain (Trevisi et al., 2004), we observed that also digitoxin protects HUVEC from apoptosis induced by growth factor deprivation. Figure 1D shows that the addition of digitoxin (or ouabain) was able to counteract the decrease of HUVEC viability caused by incubation with culture medium containing 1% FBS (without added growth factors) for 48 h. Overall, our results demonstrate that digitoxin and ouabain have effects on HUVEC viability which differ from those of digoxin, although the three drugs have the same pharmacological target, that is, Na+/K+‐ATPase.
Figure 1.
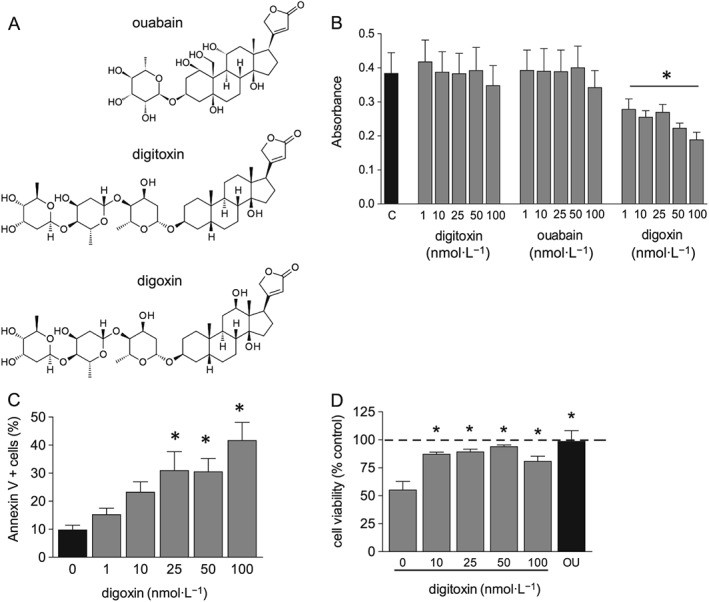
Effect of cardiac glycosides on cell viability and proliferation. (A) Chemical structure of cardiac glycosides tested. (B) HUVEC were plated in 96‐well plates and incubated in complete culture medium with digitoxin, ouabain or digoxin (1–100 nM) for 48 h. C, control; cell number was measured by crystal violet assay. Data are expressed as mean ± SEM, of six independent experiments performed in quadruplicate; statistical analysis: *P < 0.05 for cells treated with digoxin (one‐way ANOVA, post test for linear trend). (C) HUVEC were plated in six‐well plates and incubated in complete culture medium with digoxin (0–100 nM) for 24 h, then flow cytometry analysis of annexin V and PI binding was performed as described in the Methods section. Data on graph indicate % of annexin V positive cells (both PI+ and PI−) and are expressed as mean ± SEM (n = 5). * P < 0.05,significantly different from control cells; one‐way ANOVA, with Dunnett's test. (D) HUVEC were plated in 96‐well plates and incubated in medium containing 1% FBS with digitoxin (0–100 nM) or ouabain (100 nM) 48 h, ATP content was measured and is shown as % ATP content in control cells (cells incubated in complete culture medium). Data are expressed as mean ± SEM, of six independent experiments performed in quadruplicate; *P < 0.05, significantly different from 1% FBS‐treated cells without digitoxin; one‐way ANOVA, with Dunnett's test.
Cardiac glycosides inhibit HUVEC migration
Chemotaxis assays are useful to measure HUVEC migration in response to an attractant gradient, an essential step in tumour angiogenesis. Therefore, we investigated the effect of ouabain and digitoxin on HUVEC migration, through a microchemotaxis chamber, using several different chemoattractant stimuli, added to the cell culture medium: VEGF (10 ng·mL−1), bFGF (10 ng·mL−1), EGF (10 ng·mL−1) and 15% FBS plus 100 μg·mL−1 ECGS (a brain bovine extract containing a mixture of growth factors). We did not test the effect of digoxin on HUVEC chemotaxis due to its cytotoxic effect. Figure 2 shows that digitoxin and ouabain inhibited HUVEC migration in a concentration‐dependent manner. Interestingly, inhibition of migration was observed in response to all the stimuli tested, indicating that cardiac glycosides do not interfere with the binding of a specific growth factor to its receptor.
Figure 2.
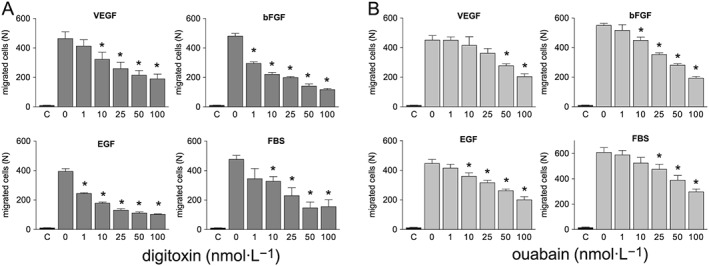
Effect of cardiac glycosides on HUVEC chemotaxis. HUVEC migration towards different chemotactic stimuli (10 ng·mL−1 VEGF, 10 ng·mL−1 bFGF, 10 ng·mL−1 EGF, 15% FBS plus 100 μg·mL−1 ECGS) was measured in a microchemotaxis chamber in the presence or absence of digitoxin (A) or ouabain (B) . C, basal migration (without chemotactic stimulus). Data are expressed as mean ± SEM of five independent experiments performed in sextuplicate. *P < 0.05, significantly different from their respective controls; one‐way ANOVA, with Dunnett's test.
Subsequent to this first screening, we chose digitoxin for further studies, as it was active on HUVEC chemotaxis at concentrations within the therapeutic plasma levels for congestive heart failure, established at 10–40 nM (Belz et al., 2001). We tested the effect of digitoxin on HUVEC collective bi‐dimensional migration (not stimulated by soluble chemoattractants) through the wound healing assay and found a slight reduction of wound closure, after overnight incubation, in digitoxin‐treated respect to control cells (Figure 3). Considering that digitoxin did not affect HUVEC proliferation (as shown in Figure 1B), we assume that the inhibition of wound closure by digitoxin is due to an effect on HUVEC migration.
Figure 3.
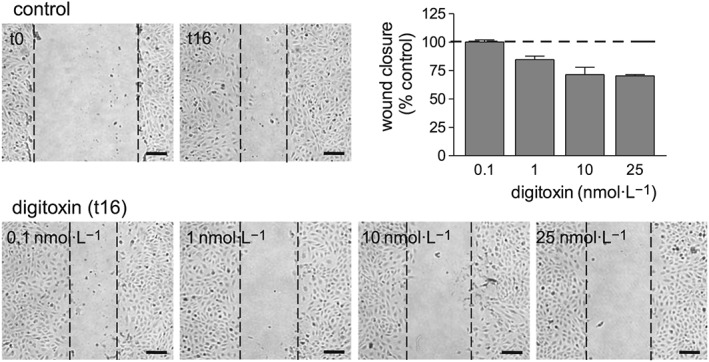
Effect of digitoxin on HUVEC migration determined by the wound healing assay. HUVEC were plated in 24‐well plates; the assay was performed in confluent cells as described in the Methods section. Wound closure was measured after 16 h incubation in complete culture medium in the presence or absence of digitoxin (0.1–25 nM). For each well, three micrograph images were taken at t0 (immediately after wounding) and t16 (after 16 h incubation); wound closure was then calculated. The figure shows representative images of an experiment (scale bar 100 μm). Data on graph are the mean ± SEM of three independent experiments.
Digitoxin inhibits capillary tube‐like formation
We then analysed the effect of digitoxin on tubularization, the process of organization of endothelial cells in capillary tube‐like structures when cultured onto extracellular matrix proteins. As shown in Figure 4, digitoxin (1–25 nM) inhibited tubularization of HUVEC seeded onto Matrigel and incubated in cell culture medium containing pro‐angiogenic factors (FBS plus ECGS). The inhibitory effect was observable already at 1 nM. These results clearly indicate that digitoxin has an in vitro anti‐angiogenic effect at concentrations that are within, and even below, the therapeutic plasma concentration range of digitoxin.
Figure 4.
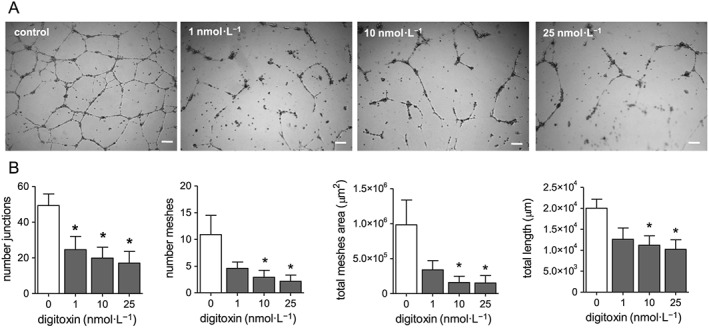
Effect of digitoxin on capillary tube‐like formation. HUVEC were seeded onto 24‐well plates coated with Matrigel, in complete culture medium in the presence and absence of digitoxin (1–25 nM). After 8 h incubation three micrograph images per well were taken. (A) Representative images of an experiment, scale bar: 200 μm. (B) Analysis of some parameters of capillary tube formation determined using Angiogenesis Analyser (ImageJ); data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control; one‐way ANOVA, with Dunnett's test.
Digitoxin inhibits angiogenesis in vivo
To confirm in vitro data, we tested the effect of digitoxin (25 nM) on in vivo angiogenesis using the Matrigel sponge model enriched with IGROV‐1 ovarian cells, which were known from our previous in vivo studies to induce angiogenesis and increase sample consistency at the time of Matrigel sponge recovery (Nardo et al., 2011). IGROV‐1 cells were chosen to avoid misleading results due to potential cytotoxic effects of digitoxin on cancer cells. Indeed, in vitro tests of cell viability, showed a higher resistance of IGROV‐1 to digitoxin compared to OC316 ovarian cancer cells (see Supporting Information).
Assessment of angiogenesis was achieved by scoring regions of Matrigel pellet sections for vascular density, as described in the Methods section. Figure 5 shows that MVD (Figure 5B, upper panel) and fraction of tumour area containing microvessels (Figure 5B, lower panel) were significantly reduced in mice treated with digitoxin, compared with controls.
Figure 5.
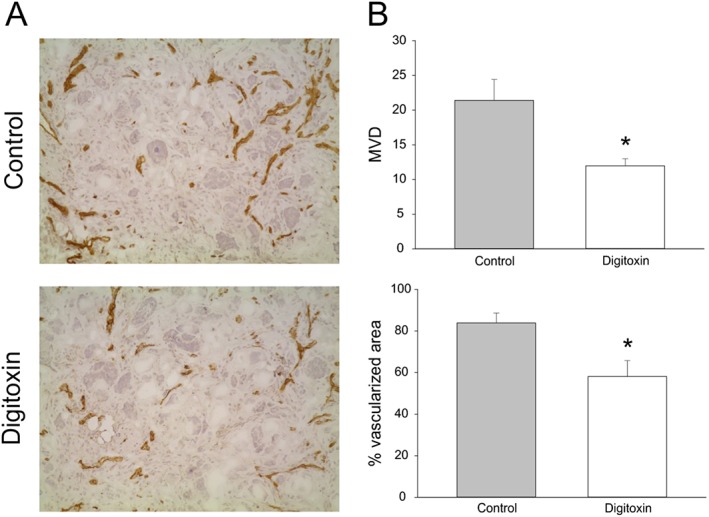
Effect of digitoxin on bFGF‐induced angiogenesis in vivo. Vascularization of Matrigel pellets by staining with anti‐CD31 mAb and calculation of MVD. For angiogenesis to be induced, on day 0 NOD/SCID‐γ−/− (NSG) mice were injected s.c. with Matrigel (400 μL per injection) supplemented with bFGF (500 ng) and IGROV‐1 tumour cells (5 × 105 cells per injection), along with either digitoxin (25 nM) or vehicle (DMSO). Seven days later, animals were killed and Matrigel pellets obtained, frozen and processed for immunohistochemical analysis . (A) Representative microphotographs are shown (original magnification: ×200). (B) Columns show mean ± SEM values of MVD (upper panel) and percentage ± SEM of vascularized area (lower panel); n = 5 animals per group. *P < 0.05, significantly different from control group; MVD: Mann–Whitney Wilcoxon test; % vascularized area: t‐test.
Digitoxin activates the Na+/K+‐ATPase signalosome in HUVEC
In the last years many laboratories have extensively studied the ability of ouabain and other cardiac glycosides to activate signalling pathways that regulate cell proliferation and death. These include activation of the PI3K/Akt pathway and Src‐dependent transactivation of the EGF receptor (EGFR), which may lead to the stimulation of the Ras–Raf–MEK–ERK cascade (Pierre and Xie, 2006; Schoner and Scheiner‐Bobis, 2007; Newman et al., 2008). We previously demonstrated that the anti‐apoptotic effect of ouabain in HUVEC was dependent on PI3K and ERK activation (Trevisi et al., 2004). In agreement with our findings, digitoxin (10 nM) caused a rapid and persistent increase of Src kinase, ERK1/2 and Akt phosphorylation in HUVEC incubated with low serum medium without added growth factors (Figure 6), indicating that digitoxin elicits signalling pathways comparable with that of ouabain, probably by activation of the Na+/K+‐ATPase signalosome.
Figure 6.
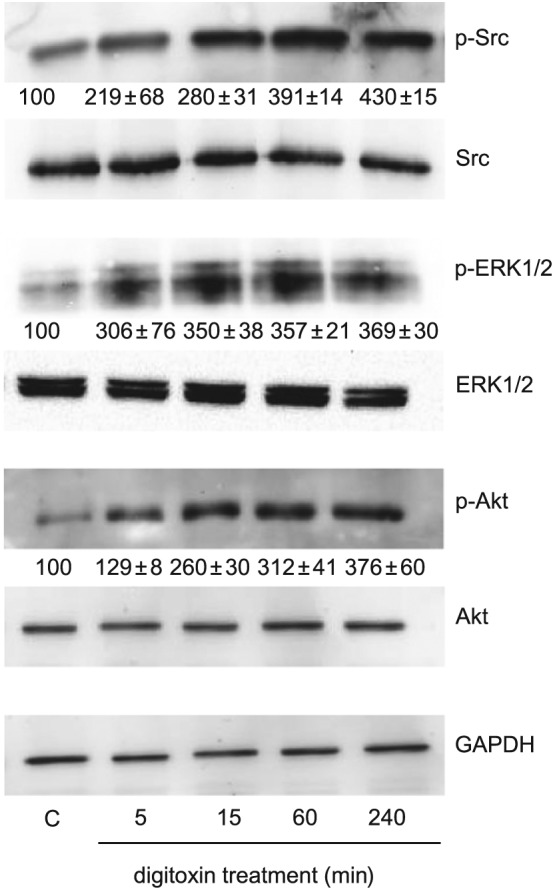
Signalling pathways activated by digitoxin in HUVEC. Cells were grown in six‐well plates and, after reaching confluence, were incubated overnight in culture medium containing 1% FBS. Then cells were stimulated with digitoxin (10 nM) for the time indicated. Cell lysates were analysed by Western blotting. Representative blots of an experiment showing the expression of phosphorylated and total Src, phosphorylated and total ERK1/2, phosphorylated and total Akt; GAPDH expression was used as loading control. Below each blot of phosphorylated proteins is shown the densitometric analysis of the bands, normalized to GAPDH levels, expressed as % of control (mean ± SEM from three independent experiments).
Digitoxin inhibits growth factor‐induced FAK activation
Studies performed in cultured cells and in knockout mouse models showed the critical role of FAK in angiogenesis during embryonic development and cancer progression (Polte et al., 1994; Ilić et al., 1995; Haskell et al., 2003; Tavora et al., 2010). FAK is a non‐receptor tyrosine kinase, which integrates signals from both extracellular matrix and growth factors to promote cell migration (Sieg et al., 2000). In particular, cell adhesion to extracellular matrix induces integrin clustering causing FAK autophosphorylation at Tyr397, which is a binding site for other signalling proteins such as Src family kinases. Src binding results in its conformational activation and subsequent FAK phosphorylation in the kinase domain activation loop at Tyr567 and Tyr577. FAK phosphorylation at Tyr567/577 promotes its maximal catalytic activation (Mitra et al., 2005; Zhao and Guan, 2011).
Considering the significance of Src‐mediated FAK activation in angiogenesis, we explored the effect of digitoxin on Src and FAK phosphorylation in HUVEC using VEGF as pro‐angiogenic stimulus. In Figure 7 is shown that, in our experimental conditions (subconfluent HUVEC cultured on gelatin‐coated dishes) VEGF (10 ng·mL−1) increased phosphorylation of Src as well as FAK at its catalytic domain (Tyr567/577) without affecting FAK phosphorylation at Tyr397. Interestingly, treatment of HUVEC with 10 nM digitoxin inhibited FAK phosphorylation at Tyr567/577 and left unaffected both FAK autophosphorylation at Tyr397 and Src activation (Figure 7). Similarly, digitoxin inhibited FAK phosphorylation at the catalytic domain induced by bFGF and EGF (Figure 8), thus confirming the ability of digitoxin to affect the actions of several different pro‐angiogenic factors.
Figure 7.
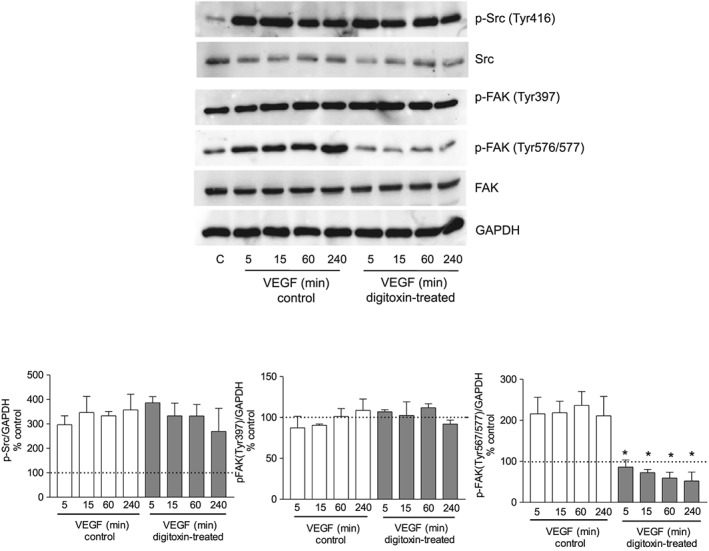
Effect of digitoxin on Src and FAK activation by VEGF. HUVEC were grown in six‐well plates and, after reaching confluence, were incubated overnight in culture medium containing 1% FBS. Cells were then treated in the absence or presence of digitoxin (10 nM) for 60 min and subsequently stimulated with 10 ng·mL−1 VEGF for the time indicated in the presence or absence of digitoxin. Cell lysates were analysed by Western blotting. Representative blots showing the expression of p‐Src (Tyr416), total Src, p‐FAK (Tyr397), p‐FAK (Tyr576/577) and total FAK; GAPDH expression was used as loading control. Below is shown the densitometric analysis of the bands, normalized to GAPDH levels, expressed as % of untreated cells (mean ± SEM; n = 5). *P < 0.05, significantly different from VEGF control at each time point; one‐way ANOVA, with Dunnett's test.
Figure 8.
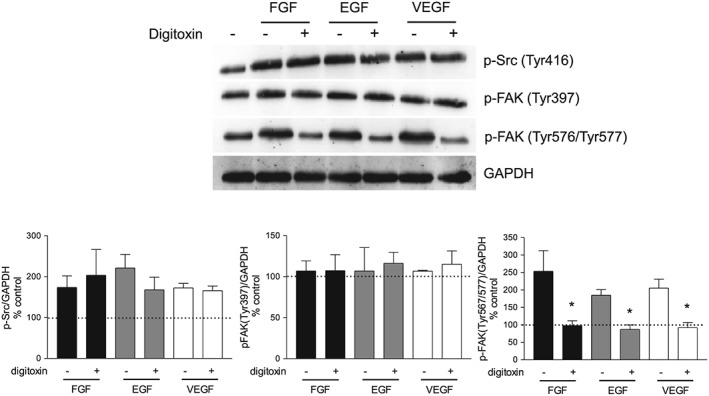
Effect of digitoxin on Src and FAK activation by three different pro‐angiogenic growth factors. HUVEC were grown in six‐well plates and, after reaching confluence, were incubated overnight in culture medium containing 1% FBS. HUVEC were stimulated with 10 ng·mL−1 of bFGF, EGF or VEGF for 30 min in the presence or absence of digitoxin. Cell lysates were analysed by Western blotting . Representative blots showing the expression of p‐Src (Tyr416), p‐FAK (Tyr397) and p‐FAK (Tyr576/577); GAPDH expression was used as loading control. Below is shown the densitometric analysis of the bands, normalized to GAPDH levels, expressed as % of untreated cells (mean ± SEM; n = 5), *P < 0.05, significantly different from GF‐stimulated without digitoxin; one‐way ANOVA, with Dunnett's test.
Discussion and conclusions
Cardiac glycosides have been used for more than 200 years for the treatment of congestive heart failure and cardiac arrhythmias. Digoxin and digitoxin are still used worldwide but, due to the shorter t 1/2, prescription of digoxin is prevalent over digitoxin. More recently, the potential use of cardiac glycosides as anticancer drugs, alone or in combination with traditional chemotherapy, has emerged from preclinical studies (including drug repositioning screenings) and some clinical studies, both retrospective and prospective (Stenkvist et al., 1982; Stenkvist, 1999; Haux et al., 2001; Platz et al., 2011; Menger et al., 2013). The anticancer effect of clinically used cardiac glycosides has been mostly related to their ability to specifically block cancer cell proliferation or induce cell death (López‐Lázaro, 2007; Newman et al., 2008; Prassas and Diamandis, 2008; Cerella et al., 2013). In this context, it is of interest to determine the effect of cardiac glycosides on tumour angiogenesis.
The main finding of our study is that digitoxin has a potent anti‐angiogenic effect, demonstrated in vitro on migration and tubularization of human endothelial cells and which was further supported by an in vivo model of tumour vessel growth. Notably, the anti‐angiogenic effect was observed at concentrations within (and even below) the therapeutic plasma concentration range of digitoxin, which is 10–40 nM (Belz et al., 2001). This is an important issue considering the low therapeutic index of cardiac glycosides. In fact, in almost all in vitro studies, the anticancer effects of the most widely used glycoside, digoxin, have been shown at concentrations 5–50‐times the therapeutic plasma concentrations (about 2 nM), not achievable with standard therapeutic regimens because of the risk of life‐threatening cardiac arrhythmias (Kometiani et al., 2005; Zhang et al., 2008; Platz et al., 2011; Lu et al., 2014; Shim and Liu, 2014). To our knowledge, this is the first evidence of an anti‐angiogenic activity of digitoxin. Moreover, we found that digoxin induced apoptosis in HUVEC at 10–100 nM, concentrations in which both digitoxin and ouabain did not affect cell viability and proliferation. Our observation is in agreement with previous data from Qiu et al. (2008) who demonstrated caspase‐3 activation and apoptosis in HUVEC treated with digoxin at concentrations ≥40 nM. It is interesting that digitoxin and digoxin, which differ only by a hydroxyl group at C12, have opposite effects on HUVEC viability. Understanding the basis of the different effects of digitoxin, digoxin and ouabain on HUVEC viability is beyond the objective of the present study but it is worthy of further investigation. We hypothesize that the ability of the three glycosides to differently affect HUVEC survival and death is not correlated to inhibition of Na+/K+‐ATPase activity. In fact, Paula et al. (2005) determined the relative binding affinities (RBA) and inhibitory potencies (RIP) of 37 cardiac glycosides, relative to those of ouabain, and found that the RBA and RIP of digoxin were 2.13 and 2.06, respectively (lower binding affinity and inhibition than ouabain) while binding affinity and inhibition potency of digitoxin were higher than ouabain (RBA 0.5 and RIP 0.6).
Digitoxin and ouabain blocked HUVEC migration towards different growth factors: VEGF, bFGF, EGF, FBS plus ECGS. Inhibition of cell migration by several cardiac glycosides (ouabain, arenobufagin, bufalin, the cardenolide analogue UNBS1450) has been demonstrated in other cell types, in particular cancer cells, indicating that it might be a common biological effect of this class of drugs (Mijatovic et al., 2007; Li et al., 2012; Chueh et al., 2014; Magpusao et al., 2015). Interfering with cell migration in the tumour context affects several key processes for cancer development and progression such as angiogenesis, recruitment of inflammatory cells, tumour cells invasion and metastasis. Therefore, inhibition of cell migration could explain, at least in part, the anticancer effect of cardiac glycosides.
Previous studies have demonstrated that, besides its pumping function, Na+/K+‐ATPases may act as a signal transducer, affecting the activity of several kinases. A non‐pumping pool of Na+/K+‐ATPase (localized in caveolae) establishes interactions with the cytoskeleton and with intracellular signalling proteins, such as Src, PI3K and phospholipase C, which are stimulated upon binding of cardiac glycosides to Na+/K+‐ATPases (Pierre and Xie, 2006; Schoner and Scheiner‐Bobis, 2007; Silva and Soares‐da‐Silva, 2012). One of the early events, described by Xie and co‐workers, is Src activation. They demonstrated that Na+/K+‐ATPase α1 interacts with the SH2 domain of Src kinase, keeping Src in an inactive state, and binding of ouabain to Na+/K+‐ATPases releases the kinase domain of Src resulting in its activation (Tian et al., 2006). Src then can recruit other kinases and transactivates EGF receptors, leading to the stimulation of the Ras–Raf–MEK‐ERK1/2 cascade. Consequently, this multiprotein caveolar complex, called the Na+/K+‐ATPase signalosome, is able to regulate many cellular processes including cell survival, differentiation, proliferation and migration (Schoner and Scheiner‐Bobis, 2007). In this study, we show that digitoxin increased Src, ERK1/2 and Akt phosphorylation, in line with the hypothesis of the signalosome activation by this glycoside in HUVEC. Compatible with the role of Akt in promoting cell survival, we demonstrated that digitoxin protected HUVEC against growth factor deprivation. Although we did not explore the mechanism of the antiapoptotic effect of digitoxin, this is likely related to the activation of Akt, as previously demonstrated for ouabain (Trevisi et al., 2004).
In addition to stimulating antiapoptotic pathways, digitoxin inhibited the activation of FAK, a key signalling protein involved in cell migration and angiogenesis, which is in agreement with the observed reduction of cell migration and tubularization without affecting HUVEC viability, induced by the glycoside. FAK is a non‐receptor tyrosine kinase, located at sites of integrin clustering (focal adhesions) where it contributes to focal adhesion scaffolding and transmission of adhesion‐dependent and growth factor‐dependent intracellular signals to promote cell migration (Sieg et al., 2000; McLean et al., 2005). The mutual activation of FAK and Src family tyrosine kinases is an essential step in this process (Hanks et al., 2003). Among the Src‐specific sites, phosphorylation of FAK on Tyr567/577 (within the kinase domain activation loop) is necessary for maximal FAK catalytic activity (Zhao and Guan, 2011; Mitra et al., 2005). Src family kinases are also stimulated by several different growth factors and are required for VEGF‐ and bFGF‐mediated angiogenesis (Klint et al., 1999; Eliceiri et al., 2002). Our study shows that digitoxin treatment of HUVEC did not affect either FAK autophosphorylation at Tyr397 (a binding site for Src kinase) or Src phosphorylation by VEGF, bFGF and EGF; suggesting that the glycoside does not interfere with integrin clustering and Src activation by growth factors. However, digitoxin caused a significant reduction of growth factor‐induced phosphorylation of FAK at Tyr567/577, thus preventing FAK maximal catalytic activity, which is compatible with the observed inhibition of cell migration and tubularization. It is intriguing that digitoxin treatment inhibited FAK Tyr567/577 phosphorylation, even though Src activation was not affected. We cannot exclude the possibility that digitoxin affects cell migration and angiogenesis acting on other proteins, such as the Rho family of GTPases, which in turn could influence FAK activity and cytoskeleton remodelling (Lamalice et al., 2007). How the inhibitory signal to FAK is transmitted and whether switching on the Na+/K+‐ATPase signalosome is essential for the anti‐angiogenic effect of digitoxin remain to be elucidated.
It is worth noting that the effect of digitoxin on both FAK activation and cell migration were observed in the presence of several different growth factors, as well as complete culture medium containing FBS plus ECGS. The ability of digitoxin to hinder a signalling protein downstream multiple angiogenic stimuli is particularly relevant as, in the tumour micro‐environment, several pro‐angiogenic factors are produced that may account for cancer resistance to anti‐angiogenic therapy. In fact, in the last years anti‐angiogenic therapy has proceeded from single‐target drugs, such as bevacizumab (the monoclonal antibody against VEGF), to drugs able to inhibit the signalling of several growth factor receptors (receptor tyrosine kinase inhibitors such as sunitinib). Therefore, broad‐spectrum molecules like digitoxin, which interfere with signalling proteins including FAK, might be the best strategy to target tumour angiogenesis (Weis and Cheresh, 2011; Limaverde‐Sousa et al., 2014). Being an important regulator of cell migration and angiogenesis, FAK represents an important target for anticancer therapy. Indeed, FAK inhibitors have been recently developed and evaluated in both preclinical studies and human clinical trials (Zhao and Guan, 2011; Sulzmaier et al., 2014).
In conclusion, our results demonstrate for the first time that digitoxin inhibits in vitro and in vivo angiogenesis as well as FAK activation by several different pro‐angiogenic stimuli, suggesting the potential use of digitoxin as a broad‐spectrum anti‐angiogenic drug. The fact that the anti‐angiogenic activity is observed at concentrations within the plasma therapeutic range, makes digitoxin a good candidate for repositioning as anticancer drug (alone or in combination with traditional chemotherapy) or for the use of digitoxin in other diseases where pathological angiogenesis is involved, such as age‐related macular degeneration and psoriasis.
Author contributions
A.T. designed and performed the research, analysed the data. E.Z. performed in vivo experiments, analysed the data. L.P. performed IHC analysis, analysed the data. S.I. designed the in vivo experiments and revised the paper. C.B. analysed the data, revised critically the paper. L.T. designed the work, analysed the data, wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of digitoxin on cell viability and proliferation of IGROV‐1 and OC316 ovarian cancer cells. Cells (5 × 103 cells/well) were plated in 96‐well plates and incubated in complete culture medium with digitoxin (1–50 nM) for 48 h, C: control; cell viability was measured by MTT assay. Data are expressed as mean ± SEM, of 6 independent experiments performed in sextuplicate. * P < 0.05 for cells treated with digoxin (one‐way ANOVA, with Dunnett's test).
Acknowledgements
We wish to thank Sisto Luciani for valuable comments and Andrea Pagetta for technical assistance. This work was supported by grants of the University of Padova (60A04‐0220/12, 60A04‐0443/13 CPDA121973 to L.T. and 60A04‐9153/15 to C.B.) and Associazione Italiana per la Ricerca sul Cancro (IG 18803 to S.I.).
Trenti, A. , Zulato, E. , Pasqualini, L. , Indraccolo, S. , Bolego, C. , and Trevisi, L. (2017) Therapeutic concentrations of digitoxin inhibit endothelial focal adhesion kinase and angiogenesis induced by different growth factors. British Journal of Pharmacology, 174: 3094–3106. doi: 10.1111/bph.13944.
References
- Albini A, Fontanini G, Masiello L, Tacchetti C, Bigini D, Luzzi P et al. (1994). Angiogenic potential in vivo by Kaposi's sarcoma cell‐free supernatants and HIV‐1 tat product: inhibition of KS‐like lesions by tissue inhibitor of metalloproteinase‐2. AIDS 8: 1237–1244. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belz GG, Breithaupt‐Grögler K, Osowski U (2001). Treatment of congestive heart failure‑current status of use of digitoxin. Eur J Clin Invest 31 (Suppl 2): 10–17. [DOI] [PubMed] [Google Scholar]
- Carmeliet P (2005). Angiogenesis in life, disease and medicine. Nature 438: 932–936. [DOI] [PubMed] [Google Scholar]
- Carpentier G, Martinelli M, Courty J, Cascone I (2012). Angiogenesis Analyzer for ImageJ In: 4th ImageJ User and Developer Conference Proceedings. Mondorf‐les‐Bains: Luxembourg, pp. 198–201. ISBN:2‐919941‐18‐6. [Google Scholar]
- Cerella C, Dicato M, Diederich M (2013). Assembling the puzzle of anti‐cancer mechanisms triggered by cardiac glycosides. Mitochondrion 13: 225–234. [DOI] [PubMed] [Google Scholar]
- Chueh FS, Chen YY, Huang AC, Ho HC, Liao CL, Yang JS et al. (2014). Bufalin‐inhibited migration and invasion in human osteosarcoma U‐2 OS cells is carried out by suppression of the matrix metalloproteinase‐2, ERK, and JNK signaling pathways. Environ Toxicol 29: 21–29. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehle M, Patel C, Giugliano RP (2011). Digoxin: clinical highlights. A review of digoxin and its use in contemporary medicine. Crit Pathw Cardiol 10: 93–98. [DOI] [PubMed] [Google Scholar]
- Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ et al. (2002). Src‐mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J Cell Biol 157: 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eva A, Kirch U, Scheiner‐Bobis G (2006). Signaling pathways involving the sodium pump stimulate NO production in endothelial cells. Biochim Biophys Acta 1758: 1809–1814. [DOI] [PubMed] [Google Scholar]
- Fürstenwerth H (2010). Ouabain – the insulin of the heart. Int J of Clin Pract 64: 1591–1594. [DOI] [PubMed] [Google Scholar]
- Gayed BA, O'Malley KJ, Pilch J, Wang Z (2012). Digoxin inhibits blood vessel density and HIF‐1a expression in castration‐resistant C4‐2 xenograft prostate tumors. Clin Transl Sci 5: 39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidolin D, Vacca A, Nussdorfer GG, Ribatti D (2004). A new image analysis method based on topological and fractal parameters to evaluate the angiostatic activity of docetaxel by using the Matrigel assay in vitro. Microvasc Res 67: 117–124. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Ryzhova L, Shin NY, Brábek J (2003). Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci 8: 982–996. [DOI] [PubMed] [Google Scholar]
- Haskell H, Natarajan M, Hecker TP, Ding Q, Stewart J Jr, Grammer JR et al. (2003). Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin Cancer Res 9: 2157–2165. [PubMed] [Google Scholar]
- Haux J, Klepp O, Spigset O, Tretli S (2001). Digitoxin medication and cancer; case control and internal dose–response studies. BMC Cancer 1: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilić D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N et al. (1995). Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK‐deficient mice. Nature 377: 539–544. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klint P, Kanda S, Kloog Y, Claesson‐Welsh L (1999). Contribution of Src and Ras pathways in FGF‐2 induced endothelial cell differentiation. Oncogene 18: 3354–3364. [DOI] [PubMed] [Google Scholar]
- Kometiani P, Liu L, Askari A (2005). Digitalis‐induced signaling by Na+/K+−ATPase in human breast cancer cells. Mol Pharmacol 67: 929–936. [DOI] [PubMed] [Google Scholar]
- Lamalice L, Le Boeuf F, Huot J (2007). Endothelial cell migration during angiogenesis. Circ Res 100: 782–794. [DOI] [PubMed] [Google Scholar]
- Lee DY, Yasuda M, Yamamoto T, Yoshida T, Kuroiwa Y (1997). Bufalin inhibits endothelial cell proliferation and angiogenesis in vitro. Life Sci 60: 127–134. [DOI] [PubMed] [Google Scholar]
- Li M, Wu S, Liu Z, Zhang W, Xu J, Wang Y et al. (2012). Arenobufagin, a bufadienolide compound from toad venom, inhibits VEGF‐mediated angiogenesis through suppression of VEGFR‐2 signaling pathway. Biochem Pharmacol 83: 1251–1260. [DOI] [PubMed] [Google Scholar]
- Limaverde‐Sousa G, Sternberg C, Ferreira CG (2014). Antiangiogenesis beyond VEGF inhibition: a journey from antiangiogenic single‐target to broad‐spectrum agents. Cancer Treat Rev 40: 548–557. [DOI] [PubMed] [Google Scholar]
- López‐Lázaro M (2007). Digitoxin as an anticancer agent with selectivity for cancer cells: possible mechanisms involved. Expert Opin Ther Targets 11: 1043–1053. [DOI] [PubMed] [Google Scholar]
- Lu GY, Liu ST, Huang SM, Chang YL, Lin WS (2014). Multiple effects of digoxin on subsets of cancer‐associated genes through the alternative splicing pathway. Biochimie 106: 131–139. [DOI] [PubMed] [Google Scholar]
- Magpusao AN, Omolloh G, Johnson J, Gascón J, Peczuh MW, Fenteany G (2015). Cardiac glycoside activities link Na(+)/K(+) ATPase ion‐transport to breast cancer cell migration via correlative SAR. ACS Chem Biol 10: 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC (2005). The role of focal‐adhesion kinase in cancer – a new therapeutic opportunity. Nat Rev Cancer 5: 505–515. [DOI] [PubMed] [Google Scholar]
- Menger L, Vacchelli E, Kepp O, Eggermont A, Tartour E, Zitvogel L et al. (2013). Trial watch: cardiac glycosides and cancer therapy. Oncoimmunology 2: e23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijatovic T, Roland I, Van Quaquebeke E, Nilsson B, Mathieu A, Van Vynckt F et al. (2007). The alpha1 subunit of the sodium pump could represent a novel target to combat non‐small cell lung cancers. J Pathol 212: 170–179. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD (2005). Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6: 56–68. [DOI] [PubMed] [Google Scholar]
- Nardo G, Favaro E, Curtarello M, Moserle L, Zulato E, Persano L et al. (2011). Glycolytic phenotype and AMP kinase modify the pathologic response of tumor xenografts to VEGF neutralization. Cancer Res 71: 4214–4225. [DOI] [PubMed] [Google Scholar]
- Newman RA, Yang P, Pawlus AD, Block KI (2008). Cardiac glycosides as novel cancer therapeutic agents. Mol Interv 8: 36–49. [DOI] [PubMed] [Google Scholar]
- Passaniti A, Taylor RM, Pili R, Guo Y, Long PV, Haney JA et al. (1992). A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab Invest 67: 519–528. [PubMed] [Google Scholar]
- Paula S, Tabet MR, Ball WJ Jr (2005). Interactions between cardiac glycosides and sodium/potassium‐ATPase: three‐dimensional structure–activity relationship models for ligand binding to the E2‐Pi form of the enzyme versus activity inhibition. Biochemistry 44: 498–510. [DOI] [PubMed] [Google Scholar]
- Pierre SV, Xie Z (2006). The Na,K‐ATPase receptor complex: its organization and membership. Cell Biochem Biophys 46: 303–316. [DOI] [PubMed] [Google Scholar]
- Platz EA, Yegnasubramanian S, Liu JO, Chong CR, Shim JS, Kenfield SA et al. (2011). A novel two‐stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov 1: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte TR, Naftilan AJ, Hanks SK (1994). Focal adhesion kinase is abundant in developing blood vessels and elevation of its phosphotyrosine content in vascular smooth muscle cells is a rapid response to angiotensin II. J Cell Biochem 55: 106–119. [DOI] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P (2011). Basic and therapeutic aspects of angiogenesis. Cell 146: 873–887. [DOI] [PubMed] [Google Scholar]
- Prassas I, Diamandis EP (2008). Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov 7: 926–935. [DOI] [PubMed] [Google Scholar]
- Qiu J, Gao HQ, Liang Y, Yu H, Zhou RH (2008). Comparative proteomics analysis reveals role of heat shock protein 60 in digoxin‐induced toxicity in human endothelial cells. Biochim Biophys Acta 1784: 1857–1864. [DOI] [PubMed] [Google Scholar]
- Rana S, Rajakumar A, Geahchan C, Salahuddin S, Cerdeira AS, Burke SD et al. (2014). Ouabain inhibits placental sFlt1 production by repressing HSP27‐dependent HIF‐1α pathway. FASEB J 28: 4324–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoner W, Scheiner‐Bobis G (2007). Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol 293: C509–C536. [DOI] [PubMed] [Google Scholar]
- Shim JS, Liu JO (2014). Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci 10: 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH et al. (2000). FAK integrates growth‐factor and integrin signals to promote cell migration. Nat Cell Biol 2: 249–256. [DOI] [PubMed] [Google Scholar]
- Silva E, Soares‐da‐Silva P (2012). New insights into the regulation of Na+,K+−ATPase by ouabain. Int Rev Cell Mol Biol 294: 99–132. [DOI] [PubMed] [Google Scholar]
- Smith TW (1988). Digitalis. Mechanisms of action and clinical use. N Engl J Med 318: 358–365. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkvist B (1999). Is digitalis a therapy for breast carcinoma? Oncol Rep 6: 493–496. [PubMed] [Google Scholar]
- Stenkvist B, Pengtsson E, Dahlquist B, Eriksson O, Jarkrans T, Nordin B (1982). Cardiac glycosides and breast cancer, revisited. N Engl J Med 306: 484. [PubMed] [Google Scholar]
- Sulzmaier FJ, Jean C, Schlaepfer DD (2014). FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14: 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavora B, Batista S, Reynolds LE, Jadeja S, Robinson S, Kostourou V et al. (2010). Endothelial FAK is required for tumour angiogenesis. EMBO Mol Med 2: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M et al. (2006). Binding of Src to Na+/K+−ATPase forms a functional signaling complex. Mol Biol Cell 17: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenti A, Grumati P, Cusinato F, Orso G, Bonaldo P, Trevisi L (2014). Cardiac glycoside ouabain induces autophagic cell death in non‐small cell lung cancer cells via a JNK‐dependent decrease of Bcl‐2. Biochem Pharmacol 89: 197–209. [DOI] [PubMed] [Google Scholar]
- Trevisi L, Visentin B, Cusinato F, Pighin I, Luciani S (2004). Antiapoptotic effect of ouabain on human umbilical vein endothelial cells. Biochem Biophys Res Commun 321: 716–721. [DOI] [PubMed] [Google Scholar]
- Vinci MC, Visentin B, Cusinato F, Nardelli GB, Trevisi L, Luciani S (2004). Effect of vascular endothelial growth factor and epidermal growth factor on iatrogenic apoptosis in human endothelial cells. Biochem Pharmacol 67: 277–284. [DOI] [PubMed] [Google Scholar]
- Weis SM, Cheresh DA (2011). Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17: 1359–1370. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Zhang H, Iwase T, Shen J, Semenza GL, Campochiaro PA (2010). Digoxin inhibits retinal ischemia‐induced HIF‐1alpha expression and ocular neovascularization. FASEB J 24: 1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR et al. (2008). Digoxin and other cardiac glycosides inhibit HIF‐1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A 105: 19579–19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Guan JL (2011). Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev 63: 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of digitoxin on cell viability and proliferation of IGROV‐1 and OC316 ovarian cancer cells. Cells (5 × 103 cells/well) were plated in 96‐well plates and incubated in complete culture medium with digitoxin (1–50 nM) for 48 h, C: control; cell viability was measured by MTT assay. Data are expressed as mean ± SEM, of 6 independent experiments performed in sextuplicate. * P < 0.05 for cells treated with digoxin (one‐way ANOVA, with Dunnett's test).