

Abstract
Background and Purpose
Liver fibrosis is a major cause of liver‐related mortality and, so far, no effective antifibrotic drug is available. Galunisertib, a TGF‐β receptor type I kinase inhibitor, is a potential candidate for the treatment of liver fibrosis. Here, we evaluated the potency of galunisertib in a human ex vivo model of liver fibrosis.
Experimental Approach
Antifibrotic potency and associated mechanisms were studied ex vivo, using both healthy and cirrhotic human precision‐cut liver slices. Fibrosis‐related parameters, both transcriptional and translational level, were assessed after treatment with galunisertib.
Key Results
Galunisertib showed a prominent antifibrotic potency. Phosphorylation of SMAD2 was inhibited, while that of SMAD1 remained unchanged. In healthy and cirrhotic human livers, spontaneous transcription of numerous genes encoding collagens, including collagen type I, α 1, collagen maturation, non‐collageneous extracellular matrix (ECM) components, ECM remodelling and selected ECM receptors was significantly decreased. The reduction of fibrosis‐related transcription was paralleled by a significant inhibition of procollagen I C‐peptide released by both healthy and cirrhotic human liver slices. Moreover, galunisertib showed similar antifibrotic potency in human and rat lives.
Conclusions and Implications
Galunisertib is a drug that deserves to be further investigated for the treatment of liver fibrosis. Inhibition of SMAD2 phosphorylation is probably a central mechanism of action. In addition, blocking the production and maturation of collagens and promoting their degradation are related to the antifibrotic action of galunisertib.
Abbreviations
- COL1A1
collagen type I, α 1
- ECM
extracellular matrix
- FN2
fibronectin
- HSCs
hepatic stellate cells
- HSP47
heat shock protein 47
- LDA
low‐density array
- PAI‐1
plasminogen activator inhibitor 1
- PCLS
precision‐cut liver slices
- PICP
procollagen I C‐peptide
- αSMA
α‐smooth muscle actin
Introduction
Advanced liver fibrosis, and its end‐stage condition, cirrhosis, is characterized by aberrant and excessive accumulation of extracellular matrix (ECM) components, with architectural and vascular distortion, and an excess risk of mortality due to functional liver failure, variceal bleeding, infection and emerging liver cancer (Schuppan and Afdhal, 2008; Torok et al., 2015; Trautwein et al., 2015). Liver fibrosis is mainly caused by viral infections, chronic alcohol abuse, non‐alcoholic steatohepatitis and biliary/autoimmune diseases (Schuppan and Afdhal, 2008). To date, liver transplantation remains the sole therapeutic option for advanced cirrhosis. However transplantation is an invasive and high‐risk surgical procedure and limited to only few patients at need (Zarrinpar and Busuttil, 2013). Thus, there is an urgent and unmet need for effective antifibrotic drugs.
Among the spectrum of mediators possessing profibrotic properties, TGF‐β1 has always been recognized as a key profibrogenic cytokine in liver fibrosis (Akhurst and Hata, 2012; Dooley and ten Dijke, 2012; Giannelli et al., 2014). TGF‐β1 affects various liver‐specific cells, including fibrogenic stimulation of hepatic stellate cells (HSCs) and portal fibroblasts towards excessive ECM producing myofibroblasts, the major effector cells of fibrosis (Hellerbrand et al., 1999; Dooley et al., 2000; Dooley and ten Dijke, 2012). In hepatocytes, TGF‐β1 can induce apoptosis or mitogenic pathways leading to various pathological conditions (Dooley and ten Dijke, 2012; Yang et al., 2013). It is well established that TGF‐β1 signalling in liver fibrosis is mediated by the transcription factor SMAD2 via activin receptor‐like kinase 5 (ALK5 or TGF‐β receptor type I) (Schmierer and Hill, 2007; Akhurst and Hata, 2012; Dooley and ten Dijke, 2012). Recently, it was discovered that TGF‐β1 might also activate transcription factor SMAD1 via ALK1 signalling in certain cell types; however, the role of this pathway in liver fibrosis remains unclear (Wiercinska et al., 2006; Munoz‐Felix et al., 2013).
Even though inhibition of TGF‐β1 signalling has been studied as a potential therapeutic target for fibrosis in the past (Akhurst and Hata, 2012; Giannelli et al., 2014), there is currently no specific inhibitor in clinical studies for fibrosis. However, galunisertib, a TGF‐β receptor type I kinase inhibitor, is presently studied for the treatment of different cancers, including hepatocellular carcinoma (Eli‐Lilly, 2010; Eli‐Lilly, 2015; Giannelli et al., 2014; Herbertz et al., 2015; Rodon et al., 2015). Due to its mode of action and attractive pharmacokinetic/pharmacodynamic properties (Herbertz et al., 2015), galunisertib may also qualify as a potential candidate for the treatment of liver fibrosis.
Previously, rodent and human ex vivo model of liver fibrosis, precision‐cut liver slices (PCLS), have been successfully used to test the potency of a variety of putative antifibrotic compounds (Olinga and Schuppan, 2013; Westra et al., 2014; Westra et al., 2016). The main advantages of PCLS pertain to the facts that all cell types are preserved in their original environment and the possibility for studying in human tissues. Therefore, our study aimed to evaluate the antifibrotic potency of galunisertib in both early‐onset and end‐stage of fibrosis in human PCLS, as an immediate translation towards further studies. The use of human PCLS allowed us to assess the potency and possibility of divergent molecular mechanism of action of galunisertib, between rodent and human livers.
Methods
Human livers
The experimental protocols in this study were approved by the Medical Ethical Committee of the University Medical Center Groningen. Early‐onset fibrosis was studied in PCLS prepared from surgical excess material of donor livers which were regarded as clinically healthy (hPCLS). End‐stage fibrosis was studied in PCLS prepared from explanted cirrhotic livers of clinically diagnosed end‐stage liver disease patients undergoing liver transplantation (chPCLS).
Animal livers
All animal care and experimental procedures were approved by the Animal Ethical Committee of the University of Groningen. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Rat precision‐cut liver slices (rPCLS) were prepared from adult male, 12–16 weeks old, Wistar rats, purchased from Charles River (Sulzfeld, Germany).
Precision‐cut liver slices
PCLS were made as previously described (Olinga and Schuppan, 2013). The slices, with an estimated thickness of 250–300 μm, were exposed to galunisertib (Selleckchem, Munich, Germany) or solvent control (DMSO; final concentration < 0.4%) for 48 h (human) and 72 h (rat). The selected incubation periods correspond to the timeframe in which slices remain viable during culture (Westra et al., 2016). Galunisertib was first tested at 0.625, 2.5 and 10 μM in rPCLS and hPCLS to identify the most effective concentration. Preliminary results indicated that 10 μM of galunisertib elicited a maximal effect while not influencing slice viability. Therefore, 10 μM was used in subsequent experiments including chPCLS. For testing the potency of galunisertib in TGF‐β1‐induced fibrosis, human recombinant TGF‐β1 (Roche Diagnostics, Mannheim, Germany) was added in culture medium, containing 1 μg·mL−1 bovine serum albumin, to yield 1 ng·mL−1 TGF‐β1. Every 24 h, PCLS were transferred into new plates containing fresh culture medium.
ATP determination
Viability of PCLS was evaluated by assessing ATP content (de Graaf et al., 2010) using a bioluminescence kit (Roche Diagnostics). ATP values were corrected for the total protein content, estimated by the Lowry assay (Bio‐Rad DC Protein Assay, Hercules, USA), of each PCLS. Values for the treated groups are expressed as relative value as compared to the control group.
Quantitative real‐time PCR and low‐density array
Gene expression of multiple fibrosis‐related markers was assessed by quantitative real‐time PCR and low‐density array (LDA). Total RNA was isolated from pooled snap‐frozen PCLS (rPCLS and hPCLS: 3; chPCLS: 6) using the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands). Reverse transcription was performed with 1 μg total RNA using the Reverse Transcription System (Promega, Leiden, The Netherlands) at 25°C for 10 min, 45°C for 60 min and 95°C for 5 min.
Gene expression was determined using specific primers (Supporting Information Table S1) and the SensiMix™ SYBR Hi‐ROX kit (Bioline, Luckenwalde, Germany) on a 7900HT qPCR system (Applied Biosystems, California, USA) with a cycle at 95°C for 10 min followed by 45 cycles of 95°C for 15 s and 60°C for 25 s. Expression levels were corrected using GAPDH as reference gene (ΔCt) and compared with the control group (ΔΔCt). Results are displayed as fold induction (2−ΔΔCt).
LDA or custom‐made Taqman Array Microfluidic Cards (Applied Biosystems) with preloaded primers in 384‐wells plates were used to elucidate the effects of galunisertib on 40 genes‐related to fibrosis (Remst et al., 2014) (Supporting Information Table S2). cDNA, 10 ng·μL−1, was mixed with 2X Taqman PCR mastermix (Applied Biosystems). Thermal cycling and fluorescence detection were performed on a ViiA™ 7 Real‐Time PCR system (Applied Biosystems) with a cycle of 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 90°C for 15 s and 60°C for 1 min. Expression levels were corrected using GAPDH as reference gene (ΔCt) and compared with the control group (ΔΔCt). Results are displayed as fold induction (2−ΔΔCt).
Western blotting
TGF‐β1 signalling proteins, that is, phosphorylated SMAD1 and SMAD2, and protein expression of key fibrosis markers were quantified by Western blotting. Three PCLS were pooled, snap frozen and lysed. Total protein (100 μg) of PCLS lysates was separated on a 10% sodium dodecyl sulfate polyacrylamide gel and transferred to a polyvinylidene fluoride membrane. Immunodetection was performed by incubating the membranes with specific antibodies (Supporting Information Table S3). Targeted proteins were visualized with VisiGlo™ Prime HRP Chemiluminescent Substrate Kit (Amresco, Ohio, USA). Equal protein loading was confirmed by using GAPDH as internal control protein. Signal intensity was measured using ImageJ software (US National Institutes of Health). The data are expressed as relative value compared to the intensity of the control group.
Western blotting of procollagen I C‐peptide (PICP) was performed under non‐reduced conditions by precipitation of culture medium using 90% methanol. The total precipitated protein was redissolved and separated on a 7.5% sodium dodecyl sulfate polyacrylamide gel.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Three to five livers were individually used for each experiment, using triplicate slices from each liver. The results are expressed as means ± SEM and compared to the control group, using either Student's t‐test or ANOVA followed by either Dunnett's or Tukey's post hoc analysis. Statistical differences were determined on relative value of ATP, ΔCt for mRNA expression, and relative signal intensity of the proteins. P‐values less than 0.05 were considered significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Spontaneous fibrosis in cultured PCLS
ATP content of the PCLS increased at the start of culture and remained constant for 48 h (hPCLS and chPCLS) and 72 h (rPCLS) (Figure 1A–C) indicating that the viability of the PCLS was maintained during culture.
Figure 1.
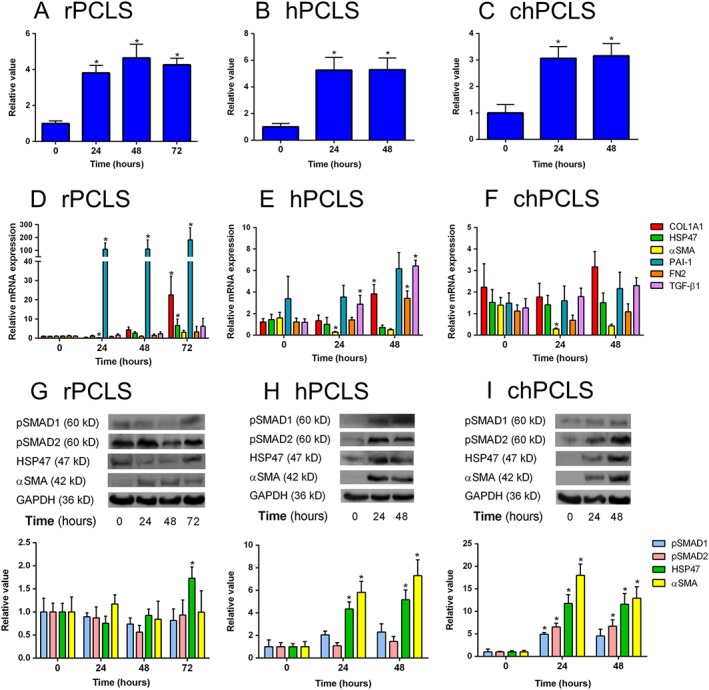
General features of PCLS during incubation. (A–C) ATP level/protein, (D–F) gene and (G–I) protein expression of r(rat)PCLS (A, D, G; n = 5–6 per group), h(human)PCLS (B, E, H; n = 4–8 per group) and ch(cirrhotic human)PCLS (C, F, I; n = 3–6 per group) respectively. *P < 0.05, significantly different from 0 h. Representative sets of Western blots and average protein expression (means ± SEM) of all experimental groups shown as bar graphs after normalization to GAPDH protein.
Quantitative PCR revealed that, at the start of culture (0 h), the basal expression of collagen type I, α 1 (COL1A1), α‐smooth muscle actin (αSMA) and plasminogen activator inhibitor 1 (PAI‐1) in hPCLS and chPCLS was significantly higher than those of rPCLS. Furthermore, the expression of COL1A1 and TGF‐β1 was significantly higher in chPCLS as compared to hPCLS (Supporting Information Figure S1).
Nonetheless, we observed a spontaneous onset of fibrogenesis during culture, as revealed by an increase in several markers of fibrosis. In rPCLS, Pai‐1 levels were markedly elevated at 24 h up till 72 h. At the latter time point, gene expression of Col1a1, heat shock protein 47 (Hsp47) and αSma was also significantly increased (Figure 1D). In hPCLS, TGF‐β1 levels increased at 24 h up till 48 h, at which time COL1A1 and fibronectin (FN2) expression was also up‐regulated (Figure 1E). In contrast, none of the fibrosis markers were modulated in chPCLS (Figure 1F).
On a protein level, expression of HSP47 and αSMA was markedly increased up to 48 h in both hPCLS and chPCLS (Figure 1H, I). Moreover, TGF‐β1 signalling was active in these PCLS, as illustrated by the presence of phosphorylated SMAD1 and SMAD2. Conversely, no changes were observed in rPCLS (Figure 1G). Taken together, it is clear that fibrosis can be induced in different types of PCLS by culture activation.
Effect of galunisertib on spontaneous fibrosis
The effects of galunisertib on the fibrotic response in cultured PCLS was subsequently investigated. None of the tested concentrations of galunisertib affected PCLS viability (Figure 2).
Figure 2.
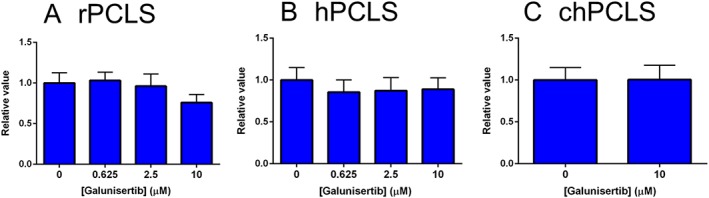
ATP level/protein of PCLS after treatment with galunisertib. (A; n = 5) 72 h in rPCLS, 48 h in (B; n = 8–9) hPCLS and (C; n = 6) chPCLS.
In rPCLS, galunisertib appeared to concentration‐dependently inhibit gene expressions of Col1a1, Hsp47 and αSma, up to 92, 50 and 83%, respectively, whereas Pai‐1, Fn2 and Tgf‐β1 levels remained unchanged. However, statistically significant differences were found at the highest tested concentration (10 μM; Figure 3A). In hPCLS, COL1A1 and TGF‐β1 gene expression were concentration‐dependently inhibited, up to 78 and 40% respectively (Figure 3B). Moreover, galunisertib significantly inhibited COL1A1 (81%), HSP47 (34%), PAI‐1 (54%) and TGF‐β1 (47%), gene expression in chPCLS (Figure 3C).
Figure 3.
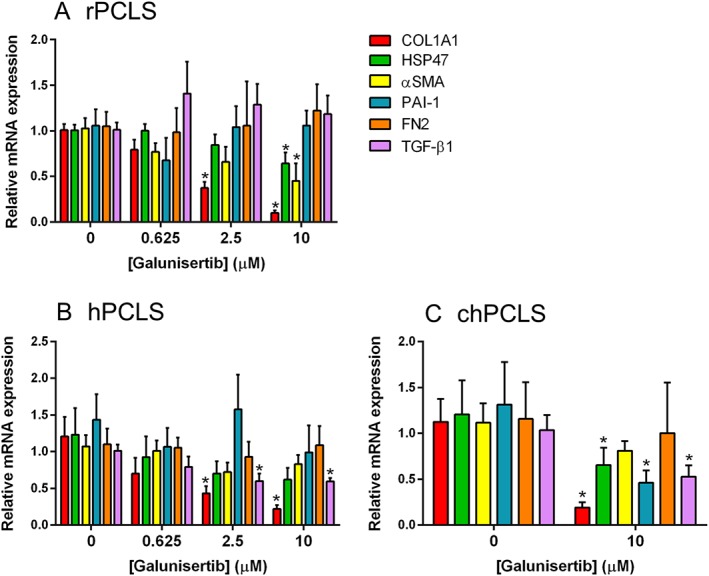
Fibrosis‐related gene expression after treatment with galunisertib. (A; n = 5) for 72 h in rPCLS, 48 h in (B; n = 5–8) hPCLS and (C; n = 4–6) chPCLS. *P < 0.05, significantly different from control.
Effect of galunisertib on TGF‐β1‐induced fibrosis
Next, we studied whether the antifibrotic effect of galunisertib could also be observed in the presence of exogenous TGF‐β1. Exposure to TGF‐β1 significantly up‐regulated the expression of Col1α1, Hsp47 and αSma, at least two‐fold in rPCLS, and these up‐regulations were significantly mitigated by galunisertib treatment (Figure 4A). Conversely, TGF‐β1 did not increase the expression of the fibrosis markers in hPCLS (Figure 4B) and chPCLS (Figure 4C). Nonetheless, in the presence TGF‐β1, galunisertib still reduced the gene expression of COL1A1, HSP47, αSMA and TGF‐β1 in both hPCLS and chPCLS (Figure 4B, C).
Figure 4.
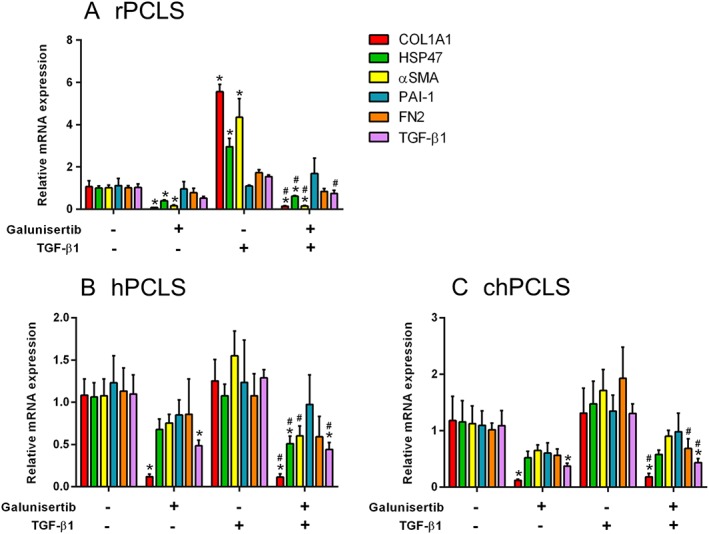
Fibrosis‐related gene expression after treatment with 10 μM galunisertib in the presence or absence of 1 ng·mL−1 TGF‐β1. (A; n = 5) 72 h in rPCLS, 48 h in (B; n = 5–6) hPCLS and (C; n = 4) chPCLS. * P < 0.05, significantly different from control; # P < 0.05, significantly different from the TGF‐β1‐treated group.
Furthermore, Western blotting revealed that treatment with TGF‐β1 up‐regulated Hsp47 and αSma protein expression in rPCLS, which was significantly antagonized by galunisertib (Figure 5A). This antifibrotic effect was not generally observed in hPCLS (Figure 5B) and chPCLS (Figure 5C); however, the effect of TGF‐β1 and galunisertib in human PCLS was extremely variable (Supporting Information Figure S2).
Figure 5.
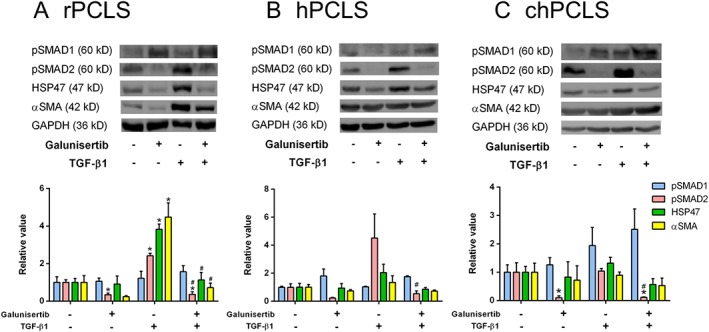
Phosphorylated SMADs and fibrogenic protein expression after treatment with 10 μM galunisertib in the presence or absence of 1 ng·mL−1 TGF‐β1. (A; n = 6) 72 h in rPCLS, 48 h in (B; n = 4) hPCLS and (C; n = 4) chPCLS. * P < 0.05, significantly different from control; # P < 0.05, significantly different from the TGF‐β1‐treated group. Representative sets of Western blots and average protein expression (means ± SEM) of all experimental groups shown as bar graphs after normalization to GAPDH protein.
Activation of TGF‐β1 signalling in rPCLS by TGF‐β1 was illustrated by a marked increase of SMAD2 phosphorylation (Figure 5A). Again, this downstream activation was not clearly seen in hPCLS (Figure 5B) and chPCLS (Figure 5C). Still, galunisertib clearly inhibited SMAD2 phosphorylation in the presence and absence of TGF‐β1 in rat and human PCLS (Figure 5).
TGF‐β1 did not affect SMAD1 phosphorylation in rPCLS, hPCLS and chPCLS (Figure 5). However, galunisertib concentration‐dependently activated SMAD1 phosphorylation in rPCLS (Supporting Information Figure S3), in contrast to hPCLS and chPCLS, in which galunisertib had no effect on SMAD1 activation (Figure 5B, C).
Effect of galunisertib on collagens and ECM
To further determine the effect of galunisertib on collagens and ECM in human PCLS, an LDA was performed. Galunisertib significantly inhibited the expression of almost all collagen subtypes (collagen types I, III, IV, V and VI), as well as numerous genes associated with collagen maturation and homeostasis. The expression levels of non‐collagenous ECM components commonly found in fibrosis were also decreased (Table 1). These antifibrotic effects of galunisertib were observed in both hPCLS and chPCLS, although the impact was more profound in chPCLS. Noteworthy, in chPCLS, galunisertib increased the expression of MMP13, which is involved in ECM degradation (Table 1).
Table 1.
Expression of transcriptions related to different collagens, collagen maturation, non‐collagenous ECM components, ECM remodelling and select ECM receptors, by hPCLS and chPCLS after treatment with 10 μM galunisertib for 48 h. LDA results are expressed as average fold induction ± SEM relative to GAPDH (n = 4)
| Gene | hPCLS | chPCLS |
|---|---|---|
| Type of collagen | ||
| COL1A1 | ↓ 0.13 ± 0.03* | ↓ 0.13 ± 0.02* |
| COL1A2 | ↓ 0.40 ± 0.12* | ↓ 0.26 ± 0.09* |
| COL3A1 | ↓ 0.46 ± 0.01* | ↓ 0.33 ± 0.06* |
| COL4A1 | ↓ 0.31 ± 0.04* | ↓ 0.28 ± 0.03* |
| COL5A1 | ↓ 0.37 ± 0.10* | ↓ 0.20 ± 0.03* |
| COL6A1 | ↓ 0.67 ± 0.07* | ↓ 0.63 ± 0.14 |
| Collagen maturation | ||
| PLOD1 | ↓ 0.54 ± 0.07* | ↓ 0.75 ± 0.15 |
| PLOD2 | ↓ 0.54 ± 0.22 | ↓ 0.49 ± 0.15 |
| PLOD3 | ↓ 0.87 ± 0.17 | ↑ 1.12 ± 0.31 |
| P4HA1 | ↑ 1.16 ± 0.26 | ↔ 0.95 ± 0.43 |
| P4HA2 | ↔ 1.02 ± 0.37 | ↑ 1.45 ± 0.18 |
| P4HA3 | ↓ 0.47 ± 0.13 | ↓ 0.22 ± 0.04* |
| P4HB | ↓ 0.78 ± 0.08 | ↑ 1.17 ± 0.34 |
| LEPRE1 | ↓ 0.75 ± 0.09 | ↓ 0.55 ± 0.12 |
| LEPREL1 | ↓ 0.71 ± 0.13 | ↓ 0.51 ± 0.10* |
| LEPREL2 | ↓ 0.60 ± 0.15 | ↓ 0.82 ± 0.32 |
| LOX | ↔ 0.91 ± 0.58 | ↓ 0.51 ± 0.15 |
| LOXL1 | ↓ 0.42 ± 0.13* | ↓ 0.62 ± 0.19 |
| LOXL2 | ↓ 0.49 ± 0.06* | ↓ 0.34 ± 0.05* |
| LOXL3 | ↓ 0.81 ± 0.32 | ↓ 0.55 ± 0.16 |
| LOXL4 | ↓ 0.40 ± 0.16 | ↓ 0.31 ± 0.09* |
| SERPINH | ↓ 0.59 ± 0.12 | ↓ 0.43 ± 0.04* |
| ADAMTS2 | ↓ 0.70 ± 0.14 | ↓ 0.47 ± 0.16 |
| ADAMTS3 | ↑ 1.97 ± 1.44 | ↓ 0.42 ± 0.09* |
| ADAMTS14 | No expression | ↓ 0.43 ± 0.17 |
| BMP1 | ↓ 0.71 ± 0.07* | ↓ 0.40 ± 0.07* |
| PCOLCE | ↓ 0.75 ± 0.06* | ↓ 0.30 ± 0.04* |
| PCOLCE2 | ↓ 0.83 ± 0.15 | ↓ 0.84 ± 0.44 |
| FKBP10 | ↓ 0.47 ± 0.04* | ↓ 0.38 ± 0.05* |
| SLC39A13 | ↔ 0.91 ± 0.08 | ↓ 0.79 ± 0.10 |
| COLGALT1 | ↓ 0.78 ± 0.12 | ↓ 0.67 ± 0.14 |
| Extracellular matrix component | ||
| Fibronectin 1 | ↓ 0.13 ± 0.03* | ↓ 0.13 ± 0.02* |
| Elastin | ↓ 0.40 ± 0.12* | ↓ 0.26 ± 0.09* |
| Decorin | ↓ 0.46 ± 0.01* | ↓ 0.33 ± 0.06* |
| Biglycan | ↓ 0.31 ± 0.04* | ↓ 0.28 ± 0.03* |
| Fibromodulin | ↓ 0.37 ± 0.10* | ↓ 0.20 ± 0.03* |
| Extracellular matrix remodelling | ||
| MMP1 | ↑ 1.31 ± 0.44 | ↑ 1.95 ± 0.72 |
| MMP13 | No expression | ↑ 6.37 ± 1.47* |
| MMP14 | ↓ 0.42 ± 0.02* | ↓ 0.38 ± 0.05* |
| TIMP1 | ↓ 0.47 ± 0.03* | ↓ 0.40 ± 0.10* |
| CTSK | ↓ 0.70 ± 0.10 | ↓ 0.62 ± 0.15 |
| Extracellular matrix protein receptor | ||
| DDR1 | ↓ 0.65 ± 0.05* | ↓ 0.55 ± 0.07* |
| DDR2 | ↔ 1.00 ± 0.21 | ↓ 0.42 ± 0.07* |
| MRC2 | ↓ 0.24 ± 0.07* | ↓ 0.15 ± 0.02* |
↓ indicates down‐regulation (fold induction < 0.9).
↑ indicates up‐regulation (fold induction > 0.9).
↔ indicates no difference (0.9 ≤ fold induction ≤ 1.1).
P < 0.05, significantly different from control.
In addition, galunisertib significantly decreased the release of PICP by both hPCLS and chPCLS, indicating a reduction in collagen type I production (Figure 6).
Figure 6.
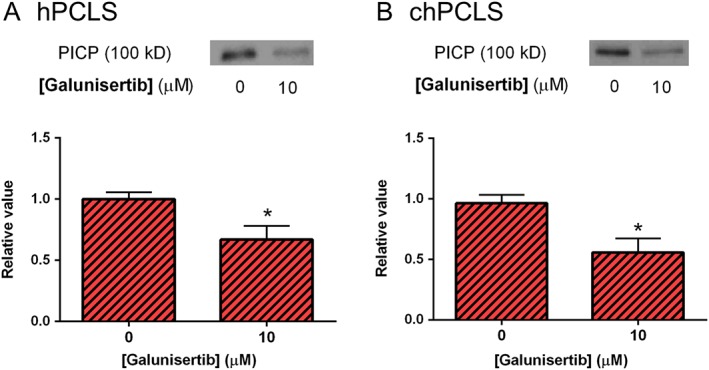
PICP released after treatment with galunisertib for 24 h. (A; n = 5) hPCLS and (B; n = 3) chPCLS. *P < 0.05, significantly different from control. Representative sets of Western blots and average protein expression (means ± SEM) of all experimental groups shown as bar graphs.
Discussion
PCLS are an effective ex vivo model to study the potency and mechanism of action of putative antifibrotic compounds (Olinga and Schuppan, 2013; Westra et al., 2016). As shown here, PCLS prepared from human and rat tissues are viable up to 48 and 72 h respectively. Furthermore, key fibrotic genes and proteins are spontaneously up‐regulated during culture. Therefore, the potency of antifibrotic compounds can be explored on both transcriptional and translational level. In addition, PCLS represent a model for the early‐onset and end‐stage of fibrosis, since they can be prepared from both healthy and diseased human livers. Results obtained with human PCLS could reasonably be extrapolated without concerns regarding species differences, a major caveat of the usual animal studies (Stribos et al., 2016; Westra et al., 2016).
Toxicity of galunisertib
Systemic inhibition of TGF‐β signalling in vivo might interfere with various beneficial biological processes, which is an important concern for drug development. A severe adverse effect in animals treated with small molecule inhibitors against ALK5 (TGF‐β receptor type I) was haemorrhagic, degenerative and inflammatory lesions in heart valves (de Gramont et al., 2017). Nevertheless, based on impressive toxicity profiles in animal (Herbertz et al., 2015) and pharmacokinetic/pharmacodynamic relationship study (Gueorguieva et al., 2014), galunisertib was selected for clinical investigation.
A human pharmacokinetic study showed that at steady state on day 14, the average maximum plasma concentration of galunisertib following 300 mg·day−1 was 990 ng·mL−1 or 2.68 μM (Rodon et al., 2015). This therapeutic dosage was deemed safe and is currently used in several clinical studies for the treatment of cancers. To date, these studies are ongoing without premature termination due to galunisertib‐related toxicities (Eli‐Lilly, 2010; 2015). And also in our study, galunisertib appeared to be non‐toxic to the liver as shown by stable ATP levels during experiments. Although the ex vivo concentrations cannot be compared to physiological portal drug concentrations, the effective concentrations of galunisertib used in our study seem to be in line with the actual therapeutic concentrations attainable in patients.
Galunisertib mode of action in fibrosis
Our results demonstrated that galunisertib significantly inhibited TGF‐β1 gene expression in the absence and especially in the presence of exogenous TGF‐β1 in both hPCLS and chPCLS. In rat PCLS, the inhibitory effect was solely observed in the presence of TGF‐β1. Due to the important role of TGF‐β1 in fibrosis development and progression (Hellerbrand et al., 1999; Dooley et al., 2000; Akhurst and Hata, 2012; Dooley and ten Dijke, 2012), our findings suggested that galunisertib which pharmacologically inhibits the actions of TGF‐β1 should exert activity appropriate for the treatment of liver fibrosis.
Galunisertib acts via inhibition of TGF‐β receptor type I kinase activation. Therefore, the phosphorylation status of downstream signalling proteins, mainly SMAD1 and SMAD2, was used in our study to indicate the activation state of the TGF‐β receptor. Galunisertib inhibited SMAD2 phosphorylation in rat and human PCLS, and these results are in line with previous studies targeting cancer cells (Maier et al., 2015; Serova et al., 2015). The important role of TGF‐β1‐meditated ALK5/SMAD2 phosphorylation in liver fibrosis and HSCs activation has been acknowledged by others (Hellerbrand et al., 1999; Dooley et al., 2000). Thus, as observed in our study, the decrease in SMAD2 phosphorylation further underlines the antifibrotic potency of galunisertib. In contrast, after the treatment with galunisertib, SMAD1 phosphorylation was not inhibited in human PCLS; while significant up‐regulation of phosphorylated SMAD1 was observed in rat PCLS. Even though the role of TGF‐β1‐meditated ALK1/SMAD1 activation in liver fibrosis is poorly understood (Munoz‐Felix et al., 2013), the up‐regulation of SMAD1 phosphorylation in rats, but not in human, may be a compensatory pathway for the inhibition of ALK5/SMAD2. Nevertheless, the activation of ALK1/SMAD1 seems to be less significant for fibrogenesis when compared to the net decrease of the fibrosis markers.
As galunisertib clearly showed antifibrotic efficacy in the presence of exogenous TGF‐β1 in rPCLS, interestingly, the antifibrotic potency of galunisertib was still displayed in human PCLS although exogenous TGF‐β1 could not further stimulate the expression levels of fibrosis‐related markers. In fact, it was anticipated because a previous study showed that a further increase of the fibrosis‐related markers would not be observed in human PCLS in the presence of TGF‐β1 (Westra et al., 2016). Nevertheless, in our study, we aimed to show that galunisertib might exhibit antifibrotic potency via the inhibition of profibrotic spontaneous SMAD2 expression regardless of augmentative stimuli. On the other hand, galunisertib may possibly possess other mechanisms, apart from TGF‐β1‐related actions.
Effect of galunisertib on fibrosis markers
αSMA is a well‐known marker of activated HSCs. A previous study showed that the transcription level of αSMA did not change during incubation in human PCLS (Westra et al., 2016). Here, we show for the first time that, in contrast, αSMA protein levels are increased during culture of PCLS. This controversy needs to be further investigated. Nevertheless, post‐transcriptional regulation of αSMA in activated HSCs of human PCLS as observed in Dupuytren's nodular cells might be possible (Verjee et al., 2010). Interestingly, TGF‐β1 did not further increase gene and protein expression of αSMA in human PCLS, nor was the expression affected by galunisertib. On the other hand, in rat PCLS, αSma gene and protein expression were increased by TGF‐β1 treatment, and galunisertib reduced the expression both in the absence and presence of TGF‐β1. This discrepancy might indicate that the activation status of HSCs differs in human and rat PCLS at the start of experiments. This difference might arise due to partial activation of HSCs before or during surgery by stress and injury (Lau et al., 2005). Importantly, this finding highlights the notion that observations in animal models may not be readily translatable to the human liver.
Our results further demonstrated that the expression of HSP47, a chaperone protein involved in intracellular collagen maturation (Mala and Rose, 2010; Ishida and Nagata, 2011), was also affected by galunisertib. In rat PCLS, galunisertib clearly reduced both the gene and protein levels of HSP47, while only its gene expression was lowered in human PCLS. This discrepancy might again be caused by species differences and also by a variant exposure to perioperative stress, injury or constitutive expression of the human samples (Brown et al., 2005). It is also possible that longer incubation times are needed to see effects of galunisertib on HSP47 protein levels in human PCLS.
Most importantly, the antifibrotic efficacy of galunisertib was clearly illustrated by the reduced collagen type I production in human hPCLS and chPCLS both on a transcriptional and translational level. Our results indicated that secretion of PICP, a by‐product of collagen type I production, could be used as a marker of collagen production and fibrosis (Brown et al., 2005). Although there are many types of collagen present in fibrotic lesions, collagen type I is the main ECM protein produced in liver fibrosis (Voss et al., 1980; Yamamoto et al., 1984).
We demonstrated that galunisertib down‐regulated not only collagen type I but also collagen types III, IV, V and VI. In addition, the expression of genes that encode enzymes associated with collagen maturation and fibril formation such as LOXL2, FKBP10, BMP1 and PCOLCE was inhibited by galunisertib. These results suggest a unique effect of galunisertib on production, maturation and formation of collagen fibrils. Moreover, galunisertib decreased the expression of FN2, which is recognized as an important ECM protein involved in liver fibrosis (Altrock et al., 2015) as well as the levels of non‐collagenous ECM proteins, that is, elastin, decorin, biglycan and fibromodulin. In addition, collagen and FN2 receptors, DDR1 and MRC2, were also decreased by galunisertib. These results highlight the uniquely broad effect of galunisertib on ECM formation.
The imbalance between ECM production and degradation is an important determinant in liver fibrosis progression (Schuppan and Afdhal, 2008). Following treatment with galunisertib, a marked increase in the expression of MMP13, an enzyme that promotes collagen cleavage (Endo et al., 2011), was observed in chPCLS. Thus, galunisertib might also accelerate the degradation of ECM components in an ECM‐rich environment as found in cirrhotic livers, which can be beneficial as well as detrimental. On the other hand, proteins that are normally increased during liver fibrosis, that is, tissue inhibitor of metalloproteinase‐1 (TIMP1) and MMP14 (Takahara et al., 1997; Busk et al., 2014), were decreased by the action of galunisertib, supporting the beneficial property of this compound in the resolution of excessive ECM.
Conclusions
As illustrated in our human ex vivo study, galunisertib is a promising drug to be further investigated for the treatment of human liver fibrosis. Although, species‐specific biological activity of compounds is widespread and a major hurdle in the development of effective therapeutics (Iswandana et al., 2016; Westra et al., 2016), encouragingly, galunisertib exhibited its antifibrotic effects not only in rodent but also in humans PCLS. The inhibition of SMAD2 phosphorylation is probably the main mechanism underlying the antifibrotic effect of galunisertib. Moreover, the unique antifibrotic efficacy of galunisertib seems to be related to the production, maturation, formation and degradation of ECM.
Author contributions
T.L. was responsible for study design; experiments and procedures; acquisition, analysis and interpretation of data; drafting of the manuscript; and statistical analysis. S.S., E.B. and M.B. did the work on the experiments and procedures. D.O. also worked on the experiments and procedures as well as material support. K.P.d.J. and D.S. revised the manuscript for important intellectual content. H.A.M.M. did the analysis and interpretation of data and drafting and revision of the manuscript for important intellectual content. P.O. also worked on the analysis and interpretation of data; drafting and revision of the manuscript for important intellectual content; obtained funding; and study supervision.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Primers used in quantitative real‐time PCR.
Table S2 Specifications of genes‐encoding collagens, collagen maturation, non‐collagenous ECM components, ECM remodelling and select ECM receptors, in Low Density Array (LDA).
Table S3 Buffer and antibodies used in Western blotting.
Figure S1 Comparison of fibrosis‐related gene expression at 0 h. (A; n = 4–8) among rPCLS, hPCLS and chPCLS, and (B; n = 4–8) between hPCLS and chPCLS. *P < 0.05, significantly different from control (A) rPCLS or (B) hPCLS.
Figure S2 Scatter plots of HSP47 and αSMA protein expression after treatment with 10 μM galunisertib in the presence or absence of 1 ng·mL−1 TGF‐β1 for 48 h. (A; n = 4) hPCLS and (B; n = 4) chPCLS. Examples of Western blots, averages of all experimental groups and scatter plots indicating individual band intensities after normalization to GAPDH protein.
Figure S3 Phosphorylated SMADs after treatment with 0.625, 2.5 and 10 μM galunisertib. (A; n = 3) 72 h in rPCLS and (B; n = 3–4) 48 h in hPCLS. *P < 0.05, significantly different from control. Representative sets of Western blots, and average protein expression (means ± SEM) of all experimental groups shown as bar graphs after normalization to GAPDH protein.
Acknowledgements
This work was kindly supported by ZonMw, grant number 114021010. We greatly thank all liver donors and recipients for dedication of liver specimen. Our research was supported by the Department of Hepato‐Pancreato‐Biliary Surgery and Liver Transplantation, University of Medical Center Groningen, and Department of Pharmacokinetics Toxicology and Targeting, University of Groningen. We are also largely grateful to valuable comments from everyone during experiments and data discussions.
Luangmonkong, T. , Suriguga, S. , Bigaeva, E. , Boersema, M. , Oosterhuis, D. , de Jong, K. P. , Schuppan, D. , Mutsaers, H. A. M. , and Olinga, P. (2017) Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. British Journal of Pharmacology, 174: 3107–3117. doi: 10.1111/bph.13945.
References
- Akhurst RJ, Hata A (2012). Targeting the tgfbeta signalling pathway in disease. Nat Rev Drug Discov 11: 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altrock E, Sens C, Wuerfel C, Vasel M, Kawelke N, Dooley S et al. (2015). Inhibition of fibronectin deposition improves experimental liver fibrosis. J Hepatol 62: 625–633. [DOI] [PubMed] [Google Scholar]
- Brown KE, Broadhurst KA, Mathahs MM, Brunt EM, Schmidt WN (2005). Expression of hsp47, a collagen‐specific chaperone, in normal and diseased human liver. Lab Invest 85: 789–797. [DOI] [PubMed] [Google Scholar]
- Busk TM, Bendtsen F, Nielsen HJ, Jensen V, Brunner N, Moller S (2014). Timp‐1 in patients with cirrhosis: relation to liver dysfunction, portal hypertension, and hemodynamic changes. Scand J Gastroenterol 49: 1103–1110. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf IA, Olinga P, de Jager MH, Merema MT, de Kanter R, van de Kerkhof EG et al. (2010). Preparation and incubation of precision‐cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc 5: 1540–1551. [DOI] [PubMed] [Google Scholar]
- de Gramont A, Faivre S, Raymond E (2017). Novel tgf‐beta inhibitors ready for prime time in onco‐immunology. Oncoimmunology 6: e1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley S, Delvoux B, Lahme B, Mangasser‐Stephan K, Gressner AM (2000). Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 31: 1094–1106. [DOI] [PubMed] [Google Scholar]
- Dooley S, ten Dijke P (2012). Tgf‐beta in progression of liver disease. Cell Tissue Res 347: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eli‐Lilly (2010). A study of ly2157299 in participants with hepatocellular carcinoma . [ Online ] Available from https://clinicaltrials.gov/ct2/show/NCT01246986. [Accessed: January 13 2017].
- Eli‐Lilly (2015). A study of galunisertib (ly2157299) in combination with nivolumab in advanced refractory solid tumors and in recurrent or refractory nsclc, hepatocellular carcinoma, or glioblastoma . [ Online ] Available from https://clinicaltrials.gov/ct2/show/study/NCT02423343. [Accessed: January 13 2017].
- Endo H, Niioka M, Sugioka Y, Itoh J, Kameyama K, Okazaki I et al. (2011). Matrix metalloproteinase‐13 promotes recovery from experimental liver cirrhosis in rats. Pathobiology 78: 239–252. [DOI] [PubMed] [Google Scholar]
- Giannelli G, Villa E, Lahn M (2014). Transforming growth factor‐beta as a therapeutic target in hepatocellular carcinoma. Cancer Res 74: 1890–1894. [DOI] [PubMed] [Google Scholar]
- Gueorguieva I, Cleverly AL, Stauber A, Pillay NS, Rodon JA, Miles CP et al. (2014). Defining a therapeutic window for the novel tgf‐β inhibitor ly2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br J Clin Pharmacol 77: 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA (1999). The role of tgfbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 30: 77–87. [DOI] [PubMed] [Google Scholar]
- Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST et al. (2015). Clinical development of galunisertib (ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor‐beta signaling pathway. Drug Des Devel Ther 9: 4479–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Nagata K (2011). Hsp47 as a collagen‐specific molecular chaperone. Methods Enzymol 499: 167–182. [DOI] [PubMed] [Google Scholar]
- Iswandana R, Pham BT, van Haaften WT, Luangmonkong T, Oosterhuis D, Mutsaers HA et al. (2016). Organ‐ and species‐specific biological activity of rosmarinic acid. Toxicol In Vitro 32: 261–268. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau DT, Luxon BA, Xiao SY, Beard MR, Lemon SM (2005). Intrahepatic gene expression profiles and α‐smooth muscle actin patterns in hepatitis c virus induced fibrosis. Hepatology 42: 273–281. [DOI] [PubMed] [Google Scholar]
- Maier A, Peille AL, Vuaroqueaux V, Lahn M (2015). Anti‐tumor activity of the tgf‐beta receptor kinase inhibitor galunisertib (ly2157299 monohydrate) in patient‐derived tumor xenografts. Cell Oncol (Dordr) 38: 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mala JG, Rose C (2010). Interactions of heat shock protein 47 with collagen and the stress response: an unconventional chaperone model? Life Sci 87: 579–586. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Felix JM, Gonzalez‐Nunez M, Lopez‐Novoa JM (2013). Alk1‐smad1/5 signaling pathway in fibrosis development: friend or foe? Cytokine Growth Factor Rev 24: 523–537. [DOI] [PubMed] [Google Scholar]
- Olinga P, Schuppan D (2013). Precision‐cut liver slices: a tool to model the liver ex vivo. J Hepatol 58: 1252–1253. [DOI] [PubMed] [Google Scholar]
- Remst DF, Blom AB, Vitters EL, Bank RA, van den Berg WB, Blaney Davidson EN et al. (2014). Gene expression analysis of murine and human osteoarthritis synovium reveals elevation of transforming growth factor beta‐responsive genes in osteoarthritis‐related fibrosis. Arthritis Rheumatol 66: 647–656. [DOI] [PubMed] [Google Scholar]
- Rodon J, Carducci M, Sepulveda‐Sanchez JM, Azaro A, Calvo E, Seoane J et al. (2015). Pharmacokinetic, pharmacodynamic and biomarker evaluation of transforming growth factor‐beta receptor i kinase inhibitor, galunisertib, in phase 1 study in patients with advanced cancer. Invest New Drugs 33: 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmierer B, Hill CS (2007). Tgfbeta‐smad signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8: 970–982. [DOI] [PubMed] [Google Scholar]
- Schuppan D, Afdhal NH (2008). Liver cirrhosis. The Lancet 371: 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serova M, Tijeras‐Raballand A, Dos Santos C, Albuquerque M, Paradis V, Neuzillet C et al. (2015). Effects of tgf‐beta signalling inhibition with galunisertib (ly2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 6: 21614–21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The iuphar/bps guide to pharmacology in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stribos EG, Luangmonkong T, Leliveld AM, de Jong IJ, van Son WJ, Hillebrands JL et al. (2016). Precision‐cut human kidney slices as a model to elucidate the process of renal fibrosis. Transl Res 170: 8–16 e11. [DOI] [PubMed] [Google Scholar]
- Takahara T, Furui K, Yata Y, Jin B, Zhang LP, Nambu S et al. (1997). Dual expression of matrix metalloproteinase‐2 and membrane‐type 1‐matrix metalloproteinase in fibrotic human livers. Hepatology 26: 1521–1529. [DOI] [PubMed] [Google Scholar]
- Torok NJ, Dranoff JA, Schuppan D, Friedman SL (2015). Strategies and endpoints of antifibrotic drug trials: summary and recommendations from the aasld emerging trends conference, chicago, june 2014. Hepatology 62: 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein C, Friedman SL, Schuppan D, Pinzani M (2015). Hepatic fibrosis: concept to treatment. J Hepatol 62: S15–S24. [DOI] [PubMed] [Google Scholar]
- Verjee LS, Midwood K, Davidson D, Eastwood M, Nanchahal J (2010). Post‐transcriptional regulation of alpha‐smooth muscle actin determines the contractile phenotype of dupuytren's nodular cells. J Cell Physiol 224: 681–690. [DOI] [PubMed] [Google Scholar]
- Voss B, Rauterberg J, Allam S, Pott G (1980). Distribution of collagen type i and type iii and of two collagenous components of basement membranes in the human liver. Pathol Res Pract 170: 50–60. [DOI] [PubMed] [Google Scholar]
- Westra IM, Mutsaers HA, Luangmonkong T, Hadi M, Oosterhuis D, de Jong KP et al. (2016). Human precision‐cut liver slices as a model to test antifibrotic drugs in the early onset of liver fibrosis. Toxicol In Vitro 35: 77–85. [DOI] [PubMed] [Google Scholar]
- Westra IM, Oosterhuis D, Groothuis GM, Olinga P (2014). The effect of antifibrotic drugs in rat precision‐cut fibrotic liver slices. PLoS One 9: e95462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiercinska E, Wickert L, Denecke B, Said HM, Hamzavi J, Gressner AM et al. (2006). Id1 is a critical mediator in tgf‐beta‐induced transdifferentiation of rat hepatic stellate cells. Hepatology 43: 1032–1041. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sumiyoshi H, Nakagami K, Tahara E (1984). Distribution of collagen types i, iii, and v in fibrotic and neoplastic human liver. Acta Pathol Jpn 34: 77–86. [DOI] [PubMed] [Google Scholar]
- Yang L, Inokuchi S, Roh YS, Song J, Loomba R, Park EJ et al. (2013). Transforming growth factor‐beta signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte‐specific deletion of tak1. Gastroenterology 144: 1042–1054 e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Busuttil RW (2013). Liver transplantation: past, present and future. Nat Rev Gastroenterol Hepatol 10: 434–440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used in quantitative real‐time PCR.
Table S2 Specifications of genes‐encoding collagens, collagen maturation, non‐collagenous ECM components, ECM remodelling and select ECM receptors, in Low Density Array (LDA).
Table S3 Buffer and antibodies used in Western blotting.
Figure S1 Comparison of fibrosis‐related gene expression at 0 h. (A; n = 4–8) among rPCLS, hPCLS and chPCLS, and (B; n = 4–8) between hPCLS and chPCLS. *P < 0.05, significantly different from control (A) rPCLS or (B) hPCLS.
Figure S2 Scatter plots of HSP47 and αSMA protein expression after treatment with 10 μM galunisertib in the presence or absence of 1 ng·mL−1 TGF‐β1 for 48 h. (A; n = 4) hPCLS and (B; n = 4) chPCLS. Examples of Western blots, averages of all experimental groups and scatter plots indicating individual band intensities after normalization to GAPDH protein.
Figure S3 Phosphorylated SMADs after treatment with 0.625, 2.5 and 10 μM galunisertib. (A; n = 3) 72 h in rPCLS and (B; n = 3–4) 48 h in hPCLS. *P < 0.05, significantly different from control. Representative sets of Western blots, and average protein expression (means ± SEM) of all experimental groups shown as bar graphs after normalization to GAPDH protein.