

Abstract
Background and Purpose
Human ether‐a‐go‐go‐related gene (hERG; Kv11.1) channel inhibition is a widely accepted predictor of cardiac arrhythmia. hERG channel inhibition alone is often insufficient to predict pro‐arrhythmic drug effects. This study used a library of dofetilide derivatives to investigate the relationship between standard measures of hERG current block in an expression system and changes in action potential duration (APD) in human‐induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CMs). The interference from accompanying block of Cav1.2 and Nav1.5 channels was investigated along with an in silico AP model.
Experimental Approach
Drug‐induced changes in APD were assessed in hiPSC‐CMs using voltage‐sensitive dyes. The IC50 values for dofetilide and 13 derivatives on hERG current were estimated in an HEK293 expression system. The relative potency of each drug on APD was estimated by calculating the dose (D150) required to prolong the APD at 90% (APD90) repolarization by 50%.
Key Results
The D150 in hiPSC‐CMs was linearly correlated with IC50 of hERG current. In silico simulations supported this finding. Three derivatives inhibited hERG without prolonging APD, and these compounds also inhibited Cav1.2 and/or Nav1.5 in a channel state‐dependent manner. Adding Cav1.2 and Nav1.2 block to the in silico model recapitulated the direction but not the extent of the APD change.
Conclusions and Implications
Potency of hERG current inhibition correlates linearly with an index of APD in hiPSC‐CMs. The compounds that do not correlate have additional effects including concomitant block of Cav1.2 and/or Nav1.5 channels. In silico simulations of hiPSC‐CMs APs confirm the principle of the multiple ion channel effects.
Abbreviations
- APD
action potential duration
- CiPA
comprehensive in vitro proarrhythmia assay
- hERG
human ether‐a‐go‐go‐related gene
- hiPSC‐CMs
human‐induced pluripotent stem cell‐derived cardiomyocytes
- IKr
delayed rectifier potassium current
- TdP
torsade de pointes
Introduction
The current paradigm of assessing drug‐induced pro‐arrhythmic risk is based on a link between drug‐induced human ether‐a‐go‐go‐related gene (hERG also known as Kv11.1) channel blockade and QT‐interval prolongation; for review, see Sanguinetti and Tristani‐Firouzi (2006). The abnormal activity of cardiac myocytes such as early after‐depolarizations (EADs) is more likely to occur when the cardiac action potential (AP) is prolonged. EADs manifest as a single spike or oscillations of the membrane potential at the repolarising phase of the AP (Keating and Sanguinetti, 2001; Morita et al., 2008; Liu et al., 2012) and are commonly seen in patients with an acquired long‐QT syndrome (Veldkamp et al., 2001; Pogwizd and Bers, 2004). EADs are pro‐arrhythmic because of their potential to induce dispersed refractory periods in cardiac tissue, which is a vital condition for the precipitation of arrhythmias. A link between EADs and torsade de pointes (TdP) has been previously studied (Volders et al., 2000), and it is widely accepted that the prolongation of the QT interval is the precursor of EADs and TdP caused by many drugs (Hancox et al., 2008; Sager et al., 2014).
Many experimental and theoretical studies have been performed to investigate the ionic mechanisms of EADs in isolated cardiomyocytes (Zeng and Rudy, 1995; Sato et al., 2010; Liu et al., 2012). The repolarization phase of cardiac AP results from a complex interplay between several ionic currents such as inward sodium current (INa), inward calcium current (ICaL) and several potassium currents mainly rapid delayed rectifier potassium current (Ikr). EADs can be produced either by increasing the inward currents, mainly L‐type calcium current (ICaL), or reducing the outward currents (IKr), or both. So, for example, a cell can be made susceptible to EADs by inhibiting IKr through hERG with dofetilide, activating the late sodium current (INaLate) with veratridine or by increasing the conductance of ICaL through Cav1.2 channels with BAY K8644 (Horváth et al., 2015). Drugs with unidirectional inhibition of inward and outward currents are generally unable to prolong AP duration (APD) and thus unlikely to induce EADs. Verapamil is one example that simultaneously inhibit ICal and hERG current without prolonging the QT interval (Zhang et al., 1999).
Kramer J et al. (2013) have found that prediction of pro‐arrhythmogenity may be improved by considering the effect of drugs on currents from three key ion channels: hERG potassium channels (Kv11.1), sodium channels (Nav1.5) and calcium channels (Cav1.2). The development of multiple ion channel effect models leads to a significant reduction in false‐positive and false‐negative predictions when compared with hERG assays alone. Recently, the Cardiac Safety Research Consortium and the Food and Drug Administration proposed a new cardiac safety paradigm labelled as ‘comprehensive in vitro pro‐arrhythmia assay’ (CiPA). The new CiPA guidelines advocate studying the pharmacological effects of drugs on multiple ion channels that play an important role in shaping the ventricular AP (hERG, Nav1.5, Cav1.2) instead of only hERG screening, and confirmation of electrophysiological effects using myocyte assays such as human‐induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CMs).
Previous studies of pro‐arrhythmic effects of hERG inhibitors used a variety of chemical classes with different potencies and different selectivity. In this study, minor changes in the chemical structure of the highly potent and selective hERG inhibitor dofetilide generate compounds with a wide range of IC50 values. A remarkable linear relationship was observed between the IC50 value and the degree of AP duration change observed in hiPSC‐CM a relationship confirmed using an in silico model. The few derivatives not adhering to this linear relationship showed significant effects on Nav1.5 and Cav1.2 ion channels.
Methods
Group sizes
Numbers (n) for all experiments are provided and refer to independent single measurements. Data subjected to statistical analysis have n of at least five per group. In the case of the APD, experiments on hiPSC‐CMs have a minimum of n = 4 in some cases. The n = 4 can discriminate a 15% change in APD90 (APD at 90% repolarization) with α = 0.95 and β = 0.2, from power calculations. The variability in APD values on a well‐to‐well basis (in 96‐well plate) was measured and can be expressed in terms of a coefficient of variation for CDI cells [commercially available from Cellular Dynamics International (CDI), Madison, WI, USA] after rate correction (1 Hz) is 0.08.
Randomization
Randomization was not applicable, hence not performed.
Blinding
Blinding of experiments is not applicable.
Human‐induced pluripotent stem cell‐derived cardiomyocytes cell culture
Cryopreserved iCell hiPSC‐CMs (CDI, Lot no 1093711) were plated using iCell‐Plating Media (CDI, CMM‐100‐110‐001) by following the manufacturer's instructions. The cells were seeded at 25 000 cells per well in 96‐well glass‐bottomed plates (MatTek, p96G‐1.5‐5‐F) pre‐coated with 1:100 fibronectin (Sigma, F1141) in DPBS (Gibco, ThermoFisher Scientific, UK, 14 040–133) for 3 h at 37°C before cell plating. The plates were then incubated at 37°C, 5% CO2. Forty‐eight hours post‐thaw, 100% of the plating medium was replaced with CDI Maintenance Medium (CDI, CMC‐100‐120‐001), and further, 100% media changes were performed every 2 days after that. Optical recordings were performed 10–14 days post‐thaw at 37°C (Hortigon‐Vinagre et al., 2016).
Optical measurement of transmembrane potential signals using voltage‐sensitive dyes
Two hours before the experiments, the cells were transiently loaded with the voltage‐sensitive dye (VSD) di‐4‐ANEPPS (6 μM, 1 min at room temperature) in serum‐free media (DMEM, Gibco 11 966, supplemented with galactose 10 mM and sodium pyruvate 1 mM). Afterwards, the medium containing VSD was replaced by fresh serum‐free medium, and the cells were returned to the incubator. The multi‐well plate was placed in an environmentally controlled stage incubator (37°C, 5% CO2, water‐saturated air atmosphere) (Okolab Inc, Burlingame, CA, USA) of the CelIOPTIQ platform (Clyde Biosciences Ltd, Glasgow, Scotland). The di‐4‐ANEPPS fluorescence signal was recorded from a 0.2 × 0.2 mm area using a 40× (NA 0.6) objective lens. Excitation wavelength was 470 ± 10 nm using a light‐emitting diode (LED), and emitted light was collected by two photomultipliers (PMTs) at 510–560 and 590–650 nm respectively. LED, PMT, associated power supplies and amplifiers were supplied by Cairn Research Ltd (Kent, UK). The two channels of fluorescence signals were digitized at 10 kHz, and the ratio of florescence (short wavelength/long wavelength) was used to assess the time course of the transmembrane potential independent of cell movement (Knisley et al., 2001). Baseline spontaneous electrical activity was recorded by capturing a 20 s segment of fluorescent signal prior to compound (drug) addition. Acute effects of dofetilide and derivatives were assessed by exposure to increasing drug concentration with matched vehicle controls for each concentration. A 20 s recording was then taken 30 min after exposure to the drug or vehicle with only one concentration applied per well. The records were subsequently analysed offline using proprietary software (CellOPTIQ). The procedure was repeated from four to five times, and parallel matched control (vehicle) measurements were taken on cardiomyocytes with equivalent concentrations of vehicle (DMSO). AP parameters were measured, including APD at 50, 75 and 90% repolarization (APD50, APD75 and APD90 respectively). Data are given as % change from control for the treated groups (vehicle, control and drug). This allowed a single comparison to be made at each concentration, and every experiment was performed with its own set of controls (vehicle). No data were used more than once.
Cell culture and transient transfection tsA‐201 cells
HEK tsA‐201 cells were grown at 5% CO2 and 37°C to 80% confluence in Dulbecco's modified Eagle's/F‐12 medium supplemented with 10% (v·v−1) FCS and 100 U·mL−1 penicillin/streptomycin. Cells were split with trypsin/EDTA and plated on 35 mm Petri dishes (Falcon) at 30–50% confluence ~16 h before transfection.
Patch‐clamp studies on hERG, Nav1.5 and Cav1.2 channels
Currents through hERG channels (Anaxon GmbH) and Nav1.5 channels stably expressed in HEK293 cells were studied within 8 h of harvest in the whole‐cell configuration of the planar patch clamp technique (NPC‐16 Patchliner, Nanion Technologies GmbH, Munich, Germany), using an EPC 10 patch‐clamp amplifier (HEKA Elektronik Dr. Schulze GmbH, Lambrecht/Pfalz, Germany) (Milligan et al., 2009). Currents were low‐pass filtered at 10 kHz using the internal Bessel filter and sampled at 25 kHz. The extracellular solution for hERG current recording contained 140 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2,5 mM D‐glucose and 10 mM HEPES (pH 7.4) (Sigma‐Aldrich). The intracellular solution for hERG current recording contained 50 mM KCl, 10 mM NaCl, 60 mM KF, 20 mM EGTA and 10 mM HEPES (pH 7.2). The extracellular solution for measuring sodium currents in HEK cells stably expressing the human clone of Nav1.5 (GenBank M77235) contained 4 mM KCl, 20 mM NaCl, 1.8 mM CaCl2, 0.75 mM MgCl2, 5 mM HEPES, 120 mM choline chloride and pH 7.4 using NaOH. The intracellular solution for sodium current recording contained 120 mM CsF, 20 mM CsCl, 5 mM EGTA, 5 mM HEPES and pH 7.4 using CsOH. All chemicals were obtained from Sigma‐Aldrich Chemie GmbH (Taufkirchen, Germany). The compound solutions were applied by means of the automated NPC‐16 Patchliner planar patch‐clamp platform. Data acquisition was done using the PatchMaster software version 2.65 (HEKA Elektronik Dr. Schulze GmbH).
For barium current (IBa) measurements through voltage‐gated Ca2+ channels, HEK tsA‐201 cells were co‐transfected with cDNAs encoding the rabbit CaV1.2 α1‐subunit (GenBank X15539) with auxiliary β2a (Perez‐Reyes et al., 1992) as well as α2‐δ1 (Ellis et al., 1988) subunits and GFP to identify transfected cells (see Beyl et al., 2012, for details). The transfection of tsA‐201 cells was performed using the FUGENE6 Transfection Reagent (Roche Diagonstics GmbH, Mannheim, Germany) following standard protocols. The tsA‐201 cells were used until passage number 15. No variation in channel gating related to different cell passage numbers was observed. IBa were studied by manual patch‐clamping (Hamill et al., 1981) using an Axopatch 200A patch clamp amplifier (Axon Instruments, Foster City) 36–48 h after transfection. The extracellular bath solution (in mM: BaCl2 20, MgCl2 1, HEPES 10, choline‐Cl 90) was titrated to pH 7.4 with methanesulfonic acid. Patch pipettes with resistances of 1 to 4 MΩ were made from borosilicate glass (Clark Electromedical Instruments, UK) and filled with pipette solution (in mM: CsCl 145, MgCl2 3, HEPES 10, EGTA 10), titrated to pH 7.25 with CsOH. The drugs were applied to cells under voltage clamp using a microminifold perfusion system. Ca2+ channel block was estimated as peak IBa inhibition during a train of short (50 ms) test pulses from −80 mV at a frequency of 0.2 Hz. Patch clamp experiments to study hERG, Nav1.5 and Cav1.2 currents were performed at room temperature (22–25°C). All data were digitized and saved to disc. Current traces were filtered at 5 kHz and sampled at 10 kHz. The pClamp software package (Version 7.0 Axon Instruments, Inc.) was used for data acquisition and preliminary analysis. Microcal Origin 7.0 was used for analysis, and sigmoidal curves were fitted using the Hill equation.
In silico studies of hiPSC‐CMs’ action potentials
The cellular AP model of Paci et al. (2012) for ventricular hiPSC‐CMs was used for comparative computational studies of APD90 prolongation caused by dofetilide and its derivatives. These effects were described by the common pore block model in which the currents through the channels potentially sensitive to drugs were calculated with a coefficient equal to a fraction of channels free of drug:
All computations were performed in MATLAB R2015b. AP simulations were performed for a temperature of 310 K (37°C).
Data processing and normalization
Origin 7.0 (Origin Lab Corp., Northampton, MA, USA) was employed for data analysis and curve fitting. The cumulative concentration–inhibition curves were fitted using the Hill equation:
where IC50 is the concentration at which hERG inhibition is half‐maximal; C is the applied drug concentration; A is the fraction of hERG current that is not blocked; and nH is the Hill coefficient (Windisch et al., 2011). Data are presented as mean ± SEM for at least five cells from two different batches or for three independent measurements with HEK293 cells.
Statistical comparison
Statistically significant differences were calculated using Student's t‐tests and one‐way ANOVA and data from independent recordings. Only P‐values <0.05 were accepted as statistically significant. Linear correlation was used to confirm a linear relationship between hERG IC50 and APD data. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Drugs
Dofetilide was obtained from Sigma, and its derivatives were prepared as previously described (Shagufta et al., 2009). All derivatives were dissolved in DMSO to prepare a 10 mM stock and stored at −20°C. Drug stocks were diluted to the required concentration in extracellular solution on the day of each experiment. The maximal DMSO concentration in the bath (1%) did not affect Cav1.2 or Nav1.5 currents in any of the preparations. (Supporting Information Fig. S1).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Dofetilide derivatives library
The small library of derivatives used in this study was previously described by Shagufta et al. (2009). The chemical structures of dofetilide and its 13 derivatives are shown in Figure 1. The structural modifications conserved the phenyl rings on both sides of the molecules and comprised the following: (i) attaching different substituents to the rings (all excluding Dofe30); (ii) changing the substituents on the protonated nitrogen (Dofe54, Dofe60); and (iii) varying chain length (Dofe78, Dofe81).
Figure 1.
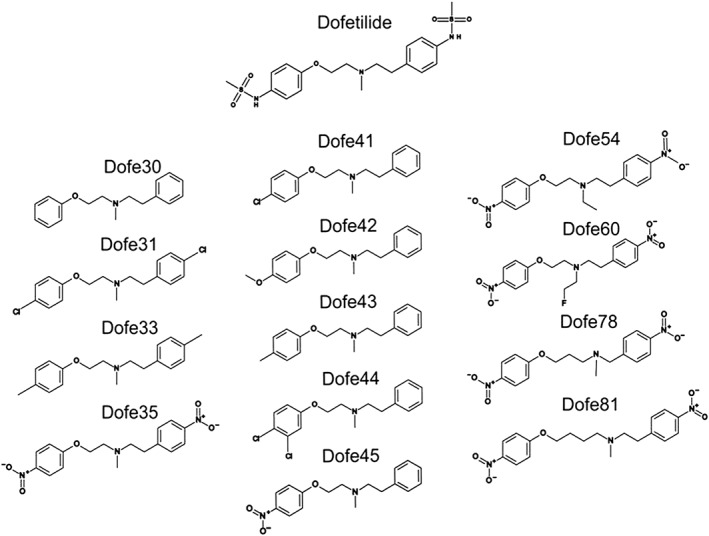
Chemical structures of dofetilide and its derivatives.
Drug‐induced prolongation of APs in hiPSC‐CMs
Effects of different concentrations of dofetilide and 13 derivatives on AP parameters were studied in hiPSC‐CMs. The changes in APD (as % of control) are given in Table 1. Figure 2 shows representative effects of dofetilide and two of its derivatives on spontaneous APs in cardiomyocytes. The derivative Dofe54 represents a potent pro‐arrhythmic compound and Dofe33 is an example with weak (if any) pro‐arrhythmic activity. Dofetilide‐induced concentration‐dependent lengthening of the AP was accompanied by incidence of EADs at concentrations of 10, 30 and 100 nM. The highest concentration used (100 nM) dramatically increased the spontaneous rate of myocyte contraction (Figure 2A).
Table 1.
Changes in APD90 in hiPSC‐CMs after application of dofetilide and derivatives
| Compound | 0.1 nM | 1 nM | 10 nM | 30 nM | 100 nM | 300 nM | 1000 nM |
|---|---|---|---|---|---|---|---|
| Dofetilide | 191(n = 6) | 246(n = 4) | 641(n = 4) | 827(n = 4) | 1032(n = 5) | – | – |
| Dofe54 | – | 107(n = 4) | 651(n = 4) | 702(n = 4) | 710(n = 4) | – | – |
| Dofe81 | – | 117(n = 4) | 389(n = 4) | 858(n = 4) | 1048(n = 5) | – | – |
| Dofe60 | – | 95(n = 4) | 112(n = 4) | 185(n = 6) | 771(n = 4) | 413(n = 6) | – |
| Dofe35 | – | 102(n = 4) | 159(n = 4) | 770(n = 4) | 746(n = 4) | 650(n = 5) | – |
| Dofe78 | – | – | 116(n = 4) | – | 420(n = 4) | 300(n = 4) | 417(n = 4) |
| Dofe45 | – | – | 71(n = 4) | – | 89(n = 4) | 95(n = 4) | 215(n = 4) |
| Dofe33 | – | 122(n = 4) | 96(n = 4) | 101(n = 4) | 112(n = 4) | 177(n = 4) | 263(n = 4) |
| Dofe31 | – | 127(n = 4) | 100(n = 4) | 103(n = 4) | 108(n = 4) | 214(n = 4) | 296(n = 4) |
| Dofe30 | – | – | 99(n = 4) | 92(n = 4) | 105(n = 4) | 190(n = 4) | 140(n = 4) |
| Dofe41 | – | – | 140(n = 6) | – | 144(n = 5) | 162(n = 5) | 262(n = 4) |
| Dofe42 | – | – | 113(n = 4) | – | 92(n = 4) | 95(n = 4) | 98(n = 4) |
| Dofe43 | – | – | 167(n = 5) | – | 156(n = 5) | 175(n = 5) | 390(n = 4) |
| Dofe44 | – | – | 90(n = 4) | – | 83(n = 4) | 87(n = 4) | 92(n = 4) |
The values are presented as a % of control.
Figure 2.
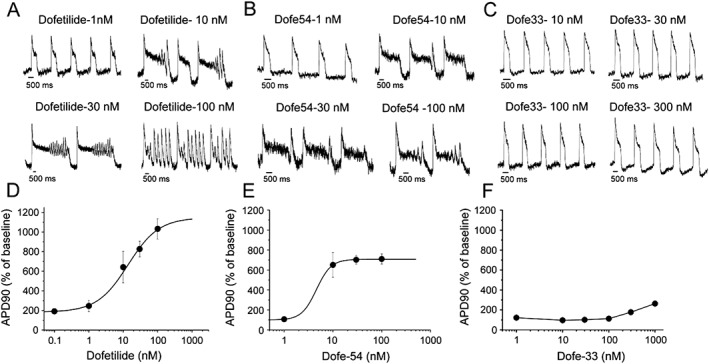
Effects of dofetilide and its derivatives Dofe54 and Dofe33 on AP characteristics in hiPSC‐CMs. Representative AP recordings of hiPSC cardiomyocytes after incubating with dofetilide, n = 4–5 (A), the high affinity derivative Dofe54, n = 4 (B) and the low affinity derivative Dofe33, n = 4 (C) and plots of APD90 as % of control versus concentrations of dofetilide, n = 4–5 (D), Dofe54, n = 4 (E) and Dofe33, n = 4 (F). The data points represent the mean ± SEM (see Table 1) and were fitted by a Hill equation for dofetilide and Dofe54. The data points for Dofe33 were connected by lines.
The potent derivative Dofe54 produced a slightly different pattern of AP distortion: smaller amplitude of oscillation during EADs and prolongation of APs at relatively low concentrations was observed. The 10 nM concentration induced approximately 700% prolongation of the AP (Figure 2B). The Dofe33 exhibited a negligible effect on APD90 prolongation until 100 nM. At a concentration of 300 nM, the APD90 was increased by approximately 170% of control. The maximal AP prolongation of 250% was observed at micromolar concentrations (Figure 2C). The concentration dependence of APD90 (in % to control) for dofetilide, Dofe54 and Dofe33 are shown in Figure 2D–F. The sigmoidal curves (Figure 2D, E) were fitted to the Hill equation.
Derivatives Dofe54, Dofe81, Dofe35, Dofe60 and Dofe78 had the most potent effects on APD (maximal level up to approximately 1000%), with incidence of EADs at the higher concentrations. In contrast, derivatives Dofe30, Dofe31, Dofe33, Dofe43, Dofe41 and Dofe45 exhibited relatively less effect on the APD90 without (if any) incidence of EADs. Derivatives Dofe42 (Figure 3A, D) and Dofe44 (Figure 3B, E) did not affect APD90 even at 1 μM while Dofe45 (Figure 3C, F) only slightly prolonged the AP.
Figure 3.
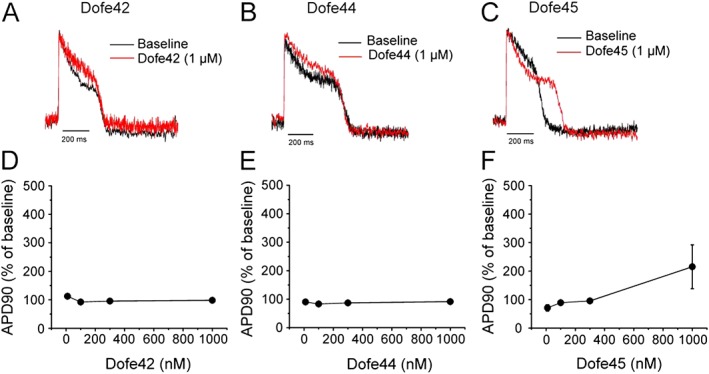
Effect of Dofe42, Dofe44 and Dofe45 on AP. (A–C) Representative AP traces of controls and in the presence of the indicated drugs. (D–F) Show dependence of APD90 on the concentration of indicated derivatives (n = 4–6, see Table 1).
hERG channel inhibition by dofetilide and its derivatives
hERG channel inhibition by dofetilide and derivatives was studied in HEK293 cell lines stably expressing hERG channels using an automated planar patch system (see Methods). After application of a given drug concentration, 0.3 Hz pulse trains were applied until a steady‐state of hERG current inhibition occurred. hERG current inhibition by Dofe54 is illustrated in Figure 4A. The concentration–inhibition relationships were analysed by plotting the normalized values of peak tail current versus peak tail steady current in the presence of the respective cumulatively applied compound concentrations (Figure 4B, C). Data points were fitted using Hill equation.
Figure 4.
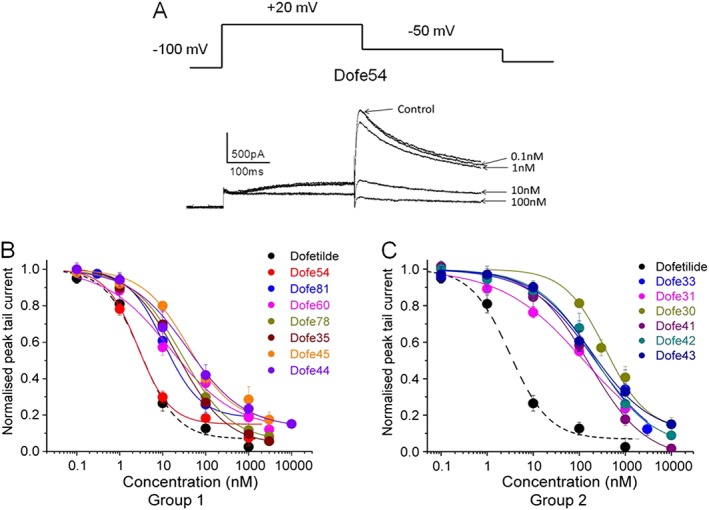
Effect of dofetilide and derivatives on potassium currents mediated through hERG channels expressed in HEK293 cells. (A) Representative current traces of control current (in the absence of drug) and in the presence of Dofe54 after steady state was reached at each concentration applied. The voltage protocol illustrated was applied every 3 s (A, upper panel). (B) Concentration‐inhibition curves for dofetilide (n = 5) and high affinity derivatives: Dofe54 (n = 5), Dofe81 (n = 6), Dofe60 (n = 5), Dofe78 (n = 7), Dofe35 (n = 8), Dofe45 (n = 5) and Dofe44 (n = 6). (C) Concentration‐inhibition curves for dofetilide and low affinity derivatives: Dofe33 (n = 6), Dofe31 (n = 7), Dofe30 (n = 7), Dofe41 (n = 8), Dofe42 (n = 8) and Dofe43 (n = 5). Peak tail current values (mean ± SEM, see Table 2) were fitted by the Hill equation.
Figure 4 illustrates that dofetilide derivatives can be subdivided into the following: (i) high affinity derivatives hERG current inhibition with IC50 values ranging between 3 and 40 nM (Figure 4B); and (ii) low affinity derivatives with an IC50s of >100 nM (Figure 4C). The concentration–inhibition curves of group 1 derivatives were close to the dofetilide curve while curves of group 2 derivatives indicated reduced potency (approximately 10‐fold) of channel inhibition.
Prolongation of AP correlates with potency to block hERG
The potency of dofetilide derivatives to prolong AP was related to their apparent affinity for hERG potassium channels. The drugs inhibiting hERG at lower concentrations prolonged the AP and induced EADs at lower concentrations (Table 2). In a first attempt, we failed, however, to observe a quantitative correlation between IC50 of hERG inhibition and the concentration that increased APD (D150) by 50%. Dofe42 and Dofe44 did not induce prolongation of AP and Dofe45 slightly prolonged the AP at high concentrations.
Table 2.
Dofetilide and its derivatives: affinity for hERG potassium channels and concentration (D150) that prolongs the AP by 50%
| Compound | MW | hERG IC50 (nM) | D150 (nM) |
|---|---|---|---|
| Dofetilide | 441.567 | 3.1 ± 0.6 (n = 5) | 0.04 |
| Dofe54 | 395.84 | 2.6 ± 0.4 (n = 5) | 4.3 |
| Dofe81 | 409.87 | 10.7 ± 1.4 (n = 6) | 2.5 |
| Dofe60 | 413.83 | 15.3 ± 8.4 (n = 5) | 21.3 |
| Dofe78 | 345.35 | 28.2 ± 4.9 (n = 7) | 20 |
| Dofe35 | 381.82 | 22.1 ± 5.5 (n = 8) | 8.3 |
| Dofe45 | 336.82 | 38.6 ± 9.2 (n = 5) | 538 |
| Dofe33 | 319.88 | 221.6 ± 40.8 (n = 6) | 215 |
| Dofe31 | 360.71 | 125.2 ± 19.4 (n = 7) | 151 |
| Dofe30 | 291.82 | 296.9 ± 77.5 (n = 7) | 213 |
| Dofe41 | 326.26 | 164.6 ± 31.8 (n = 8) | 157.2 |
| Dofe42 | 321.84 | 213 ± 83.5 (n = 8) | 650 |
| Dofe43 | 305.84 | 184.3 ± 66.9 (n = 5) | 99.3 |
| Dofe44 | 360.71 | 38.1 ± 12.6(n = 6) | 650 |
A plot of D150 versus drug affinities (IC50) is shown in Figure 5 (see also Table 2). Data points corresponding to derivatives that were not efficient at prolonging the AP (Dofe42, Dore44 and Dofe45; Figure 3) are illustrated as red circles in Figure 5. Excluding these data points from analysis led to a strong correlation (r = 0.94, P < 0.05) (Figure 5, black line) while taking them into account made the correlation non‐significant. The predicted relationship between IC50 and D150 by mathematical AP model is indicated by the red line. This model will be discussed in more detail later. In order to examine the possibility that additional block of inward currents may have counterbalanced hERG inhibition, we investigated effects of Dofe42, Dofe44 and Dofe45 on calcium (Cav1.2) and sodium (Nav1.5) channels.
Figure 5.
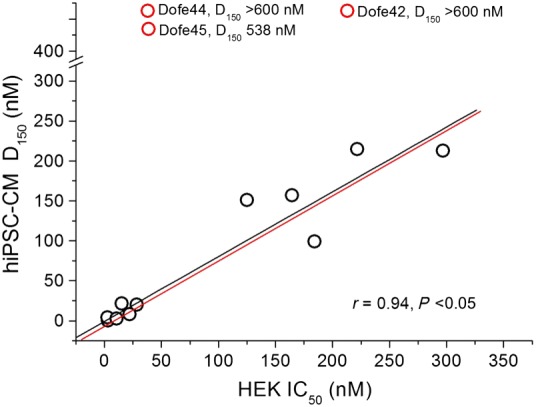
Correlation between D150 (concentration that prolongs AP in hiPSC‐CM by 50%) and IC50 (half‐maximal concentration inhibiting hERG channels in HEK293 cells). A significant linear correlation (r = 0.94, P < 0.05) was observed for 12 data points (black circles) including dofetilide and 11 derivatives. Derivatives Dofe45, Dofe44 and Dofe42 (red circles) were not included in the correlation analysis. Dofe45 prolonged the AP in hiPSC‐CM by 50% only at 538 nM and Dofe42 and 44 at >600 nM. The red line represents a prediction of the mathematical simulation of the hiPSC‐CM's AP (see Figure 8).
Inhibition of Cav1.2 by dofetilide and derivatives
Dofetilide itself does not inhibit Cav1.2 even at a high concentration of 100 μM (Supporting Information Fig. S1a). Figure 6A illustrates the effects of Dofe42, Dofe44 and Dofe45 on Cav1.2 at the indicated concentrations, and Figure 6B shows the corresponding concentration–inhibition curves obtained during continuous pulsing at 0.2 Hz. Dofe45 was identified as a potent Cav1.2 blocker (IC50 = 190 ± 3 nM, Figure 6B, right panel) while Dofe42 and Dofe44 inhibited Cav1.2 with comparably low potencies [IC50 of 38 ± 9.3 μM (Dofe42) and >100 μM (Dofe44)]. Use‐dependent channel inhibition was studied during trains of 1 Hz and 50 ms test pulses (from −80 to +10 mV). After the application of 20 test pulses in control (absence of drug), the cells were incubated for 3 min with drug at rest. Peak current inhibition during the first pulse (1st, Figure 6C) in the presence of the drug reflects ‘resting state’ block. Additional current inhibition during a subsequently applied pulse train illustrates use‐dependent block. Peak current inhibition in control and drug are compared in Figure 6D. Dofe42 and Dofe45 induced pronounced resting state block and additional use‐dependent block (compare last currents of the train in control and in drug). No use‐dependent block was observed for Dofe44 (Figure 6C, D middle panel).
Figure 6.
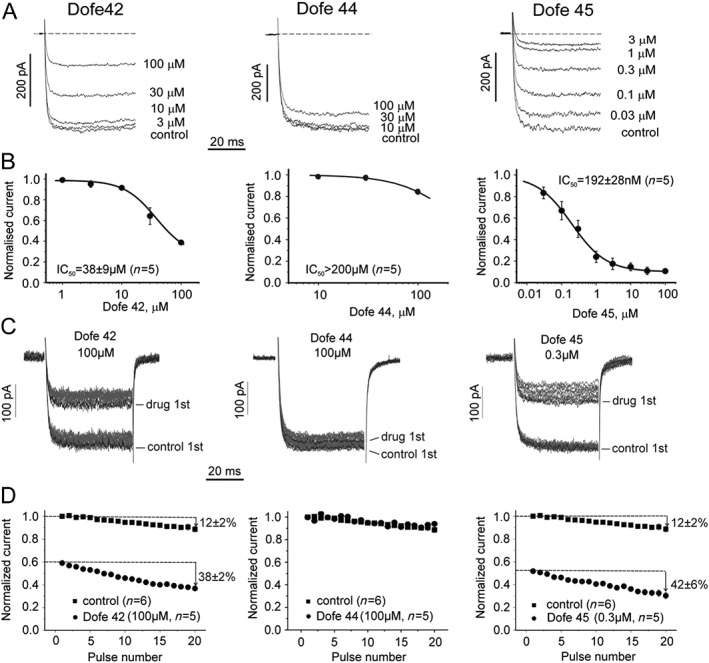
Inhibition of Cav1.2 channel by dofetilide derivatives. (A) Superimposed barium currents through rabbit Cav1.2 in control (black) and in the presence of indicated concentrations of Dofe42 (left), Dofe44 (middle) and Dofe45 (right). Barium currents were recorded in response to 50 ms pulses (0.2 Hz) from the holding potential of −80 to +10 mV. (B) Concentration‐dependence of peak IBa inhibition by Dofe42 (IC50 = 38 ± 9.3 μM, n = 5, left), Dofe44 (IC50 > 200 μM, n = 5, middle) and Dofe45 (IC50 = 192 ± 28 nM, n = 5, right). The IC50 values were obtained by fitting the data by the Hill equation. (C) Barium currents through Cav1.2 during 1 Hz trains of 50 ms pulses from −80 to +10 mV under control conditions (absence of drug) and after 3 min incubation in the presence of the indicated concentrations of dofetilide derivatives. The first current in drug reflects the resting state inhibition. (D) Mean peak current amplitudes during 50 ms pulse trains in control and the presence of the indicated concentration of Dofe42, Dofe44 and Dofe45. The peak current decay after 20 pulses at 1 Hz in control indicates the development of inactivation. Peak current decay in the presence of Dofe42 (100 μM, 38 ± 2%, n = 5) and Dofe45 (100 nM, 42 ± 6%, n = 5) versus in control (12 ± 2%, n = 6) illustrates additional significant (P < 0.05) use‐dependent block.
Inhibition of Nav1.5 by dofetilide derivatives
Dofetilide (100 μM) did not inhibit Nav1.5 (Supporting Information Fig. S1b) while block was observed for Dofe42, Dofe44 and Dofe45. Figure 7A shows representative current traces illustrating the inhibition of Nav1.5 by derivatives at indicated concentrations. The concentration–inhibition curves for all three derivatives were first estimated at a holding potential of −140 mV where all Nav1.5 are available (Figure 7B, Wang et al., 2015). Dofe44 inhibited cardiac sodium channels with an IC50 of 23.3 ± 1.9 μM (n = 6) compared with statistically less potent Dofe45 (IC50s of 69.7 ± 1.0 μM, n = 6, P < 0.05) and Dofe42 (77.9 ± 9.7 μM, n = 6, P < 0.05). However, the reported resting potentials of iPSC‐CM range between −75 and −63 mV (Hoekstra et al., 2012) would induce substantial inactivation. In order to evaluate block of inactivated Nav1.5, we investigated INa inhibition at a holding potential of −80 mV where more than 60% of Nav1.5 were in an inactivated state (Wang et al., 2015). Interestingly, the concentration–response curves where significantly shifted towards lower drug concentrations (Figure 7B, Dofe42: 5.6‐fold, Dofe44: fivefold and Dofe45: 7.7‐fold), suggesting that inactivated Nav1.5 are blocked with higher affinity. The application of test pulses at a higher frequency (1 Hz) did not induce additional channel inhibition.
Figure 7.
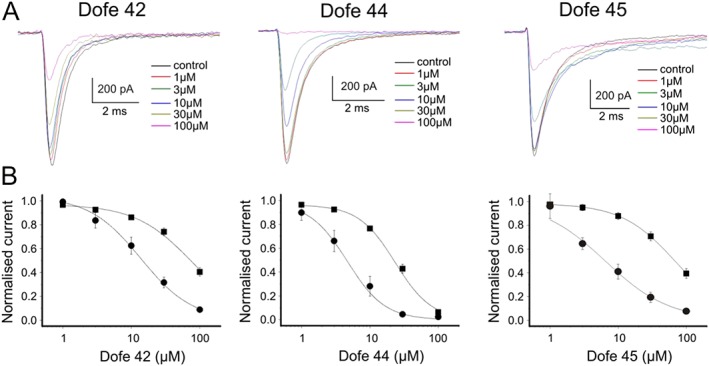
Inhibition of Nav1.5 by dofetilide derivatives. (A) Superimposed INa through human Nav1.5 in control (black) and in the presence of indicated concentrations of Dofe42 (left), Dofe44 (middle) or Dofe45 (right). Sodium currents were recorded in response to 20 ms pulses (0.2 Hz) from a holding potential of −140 to −10 mV. (B) Concentration‐dependence of peak INa inhibition at a holding potential of −140 mV (squares) and −80 mV (circles) yielding IC50 values for Dofe 42 of IC50 = 77.9 ± 9.7 (at −140 mV, n = 6) and IC50 = 13.8 ± 1.9 (at −80 mV, n = 5), Dofe 44 of IC50 = 23.3 ± 1.9 (at −140 mV, n = 6) and IC50 = 4.7 ± 2.0 (at −80 mV, n = 6) and Dofe 45 of IC50 = 69.7 ± 1.0 (at −140 mV, n = 6) and IC50 = 6.4 ± 1.0 μM (at −80 mV, n = 5).
Computational studies support experimental findings
The in silico AP model (Paci et al. 2012) for ventricular hiPSC‐CM was run with a pacing of 1 Hz until limit cycling was achieved in order to determine control APD90. In the first series of calculations, we have described a prolongation of AP under inhibition of hERG potassium channels. The drug dose D was set as a multiple of IC50, that is, D = x × IC50, enabling the use of the nonlinear forward mapping F: x → APD90(x). The factor x corresponding to a prolongation of the control APD90 by 50% was determined by solving (Engl et al., 2009) the nonlinear inverse problem F(x) = 1.5 × (control APD90). The predicted relationship between IC50 and D150 is shown in Figure 5 (red line). Figure 8A displays AP simulations at different levels of inhibition of the hERG channel. The APD exhibited a linear correlation with logarithm of concentration of hERG channel blocker (Figure 8B).
Figure 8.
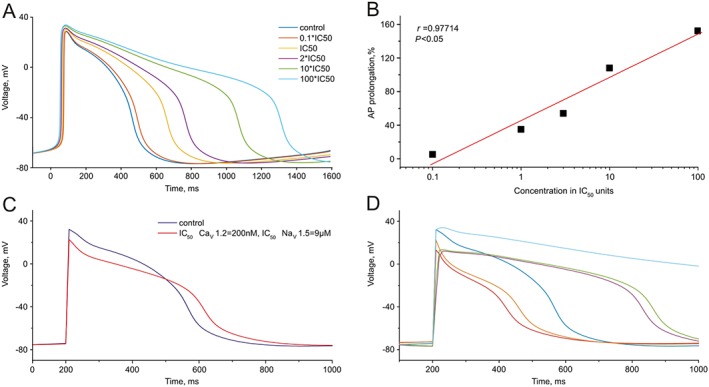
Simulation of hiPSC‐CM AP at indicated levels of hERG, Cav1.2 and Nav1.5 channel inhibition. (A) Simulation of hiPSC‐CM APs for different levels of selective hERG channel inhibition. (B) Dependence of the calculated APD90 (as % of control) on the concentration of a selective hERG channel inhibitor. (C) Simulated APs at a Dofe45 concentration of 300 nM accounting for hERG inhibition (IC50 = 40 nM) and simultaneous inhibition of Cav1.2 (IC50 = 200 nM) and Nav1.5 (IC50 = 8.9 μM). (D) Comparison of simulated APs at different IC50s of Cav1.2 and Nav1.5 inhibition. Control AP is shown in dark blue and AP for selective hERG channel inhibition (IC50 = 40 nM) in light blue. Red 100 nM (Cav1.2) and 1 μM (Nav1.5), orange 100 nM (Cav1.2) and 10 μM (Nav1.5), magenta 500 nM (Cav1.2) and 1 μM (Nav1.5), green 500 nM (Cav1.2) and 10 μM (Nav1.5). See also Supporting Information Table S1 comparing the values used in silico AP models of adult ventricular myocytes and hiPSC‐CM.
In order to test whether inhibition of inward currents compensates for the APD changes seen with hERG channel block, we simulated APs for different concentrations of half‐maximal Cav1.2 and Nav1.5 inhibition (for Cav1.2, IC50 = 200 nM and Nav1.5, IC50 = 10 μM). Both IC50s are characteristic for Dofe 45 (Table 2, Figures 6 and 7). The simulation (Figure 8C) surprisingly coincides with experimental records (Figure 3C). The ‘selective inhibition’ of the hERG channels by Dofe45 would induce a substantial prolongation of the AP. Figure 8D illustrates the sensitivity of the APD to Cav1.2 and Nav1.5 inhibition at different IC50s. Inhibition of either Cav1.2 or Nav1.5 caused shortening of APD, the largest effects seen on Cav1.2 inhibition (Figure 8D red and orange AP).
Discussion and conclusion
Potential pro‐arrhythmic effects in early stages of drug development have often been assessed solely by examining hERG channel block. The principle role of hERG channel block for AP repolarization and its consequences have been extensively discussed (Sanguinetti and Tristani‐Firouzi, 2006). The new CiPA paradigm proposes that drugs should be tested by screening multiple ion channels including IKr, IKs and IK1 as well as INaLate and ICaL and predicting their effect on the human APD using in silico models to integrate the effects of a number of ion channels (Sager et al., 2014). The CiPA scheme also suggests analysing pro‐arrhythmic effects using human iPSC‐derived cardiac muscle as a surrogate for human myocardium. However, not all ion channels expressed in human myocardium are equally well represented in hiPSC‐CM. In particular, studies suggest that IK1, IKs and INaLate currents have minimal contributions to the electrophysiology of the iPS cells (Paci et al., 2012). The Nav1.5 channel that generates the upstroke phase and the Cav 1.2 responsible for maintaining the plateau phase of AP are known to be active. At the end of the plateau phase and beginning of repolarization, inward currents are small (largely inactivated) and countered by the activation of outward K+ currents, predominately hERG, which is responsible for initiating the repolarization phase, is well represented in iPS cell. We have previously reported the absence of INaLate effect in the presence of ranolazine in hiPSC‐CM (Hortigon‐Vinagre et al., 2016), which shows that presence of INaLate in hiPSC is unlikely. Yang et al. (2014) reported an enhancement of INa‐L by dofetilide after chronic (5 h) drug exposure. Drug effects in our experiments were, however, studied a after short‐time (several minutes) of application and no increase in INaLate was observed. It is under discussion whether commercial hiPSC cell lines contain a range of cell types or simply broad‐spectrum features. The majority of hiPSC cells appear to have ventricular phenotype, and they are likely to operate as a functional syncytium via gap junction links (Bett et al., 2013; Kane et al., 2016).
Combining in silico studies with hERG (and other ion channels) inhibition and effects on APD should enable a more profound understanding of pro‐arrhythmic potential. To test this concept, we compared the prolongation of APs of hiPSC‐CM by the selective hERG inhibitor dofetilide and 13 derivatives with respect to their potencies to inhibit hERG (Figure 5). Derivatives retained the common scaffold of dofetilide while changing the functional group on both the ends or modifying the central nitrogen or altering the length of the molecule. The 13 derivatives inhibited hERG potassium channels with IC50s ranging from 3 to 300 nM (Figure 4B, C). Examining the effects of these derivatives on hiPSC‐derived cardiac myocyte APD revealed a correlation between the concentration (D150) inducing a 50% increase of APD90 of the cardiac AP with half‐maximal concentrations (IC50s) of hERG channel inhibition (Figure 5 and Supporting Information Fig. S2).
There was no correlation between the K i values (affinity of derivatives to hERG estimated in radioligand studies; Shagufta et al., 2009), and IC50s measured in electrophysiological experiments (see Supporting Information Table S2) was observed. All derivatives (except Dofe30) were similarly active in the binding study while IC50s measured in patch clamp experiments varied over two orders of magnitude (from 2.6 to 296 nM, Table 2). The lack of correlation between K i and IC50 indicates that the interaction of these derivatives with their binding pocket is not the only determinant of hERG channel inhibition (Saxena et al., 2016). The K i value reflects the affinity of a derivative for the binding pocket putatively located in the channel pore while the IC50s estimated in patch‐clamp studies are affected by the following: (i) channel state‐dependent drug effects (Fernandez et al., 2004; Sanguinetti and Tristani‐Firouzi, 2006; Stork et al., 2007; Perry et al., 2010; Windisch et al., 2011); (ii) their ability to pass the entrance barrier or leave the channel cavity; and (iii) their affinity to the binding pocket within the channel. In this regard, it is interesting to note that the IC50s estimated from hERG inhibition in functional studies are in a reciprocal relation to the molecular weight of the tested derivatives (Supporting Information Fig. S3). It is tempting to speculate that the dependence of IC50 on the molecular size is caused by an energetic barrier at the channel pore entrance. In such a scenario, bulkier molecules with higher molecular weight leave the channel with lower probability, resulting in lower off rates and correspondingly in lower IC50 values.
Three derivatives (Dofe42, 44 and 45) failed, however, to fit a linear correlation (Figure 5). We hypothesize that the ineffectiveness of derivatives Dofe45, Dofe44 and Dofe42 to prolong the AP might be due to their interference with Cav1.2, Nav1.5 and potentially other ion channels. Dofe45 was subsequently shown to be a potent inhibitor of Cav1.2. In a first series of experiments, performed at a low stimulus frequency (0.2 Hz), this derivative inhibited Cav1.2 with an IC50 of 190 ± 3 nM (Figure 6A, B). It is well established that open and inactivated channels may have a higher affinity for inhibitors than channels in the resting state (Hondeghem and Katzung, 1977). Therefore, additional measurements were made at a higher frequency of (1 Hz), which is comparable with the beating frequency of iPSC‐CM. The shorter (50 ms) pulses (1 Hz) revealed some additional use‐dependent channel inhibition by Dofe42 and 45 (Figure 6C, D). Thus, 1 Hz pulsing can enhance Cav1.2 inhibition due to additional block of open and/or inactivated channels. However, both derivatives inhibited Cav1.2 predominantly in the resting state (Figure 6). A comparison of INa inhibition at −140 and −80 mV close to the resting potential of iPSC‐CM, where more than 60% of Nav1.5 channels are in an inactivated state (Hoekstra et al., 2012; Wang et al., 2015), revealed that Dofe42, Dofe44 and Dofe45 preferentially inhibit inactivated Nav1.5. This study is mainly focused on primary targets of these derivatives like INa and ICaL, and it is very unlikely that these derivatives would have an effect on secondary targets such as IKs, Na/K pump, NCX and/or SR Ca2+ release.
As shown in Figures 5 and 8, our in silico studies on the AP model (Paci et al. 2012) at a resting potential of −80 mV reproduced the link between hERG inhibition (IC50) and prolongation of the AP (D150). Furthermore, accounting for inhibition of hERG, Cav1.2 and Nav1.5 by Dofe45 reproduced the principal features of AP changes observed on hiPSC‐CM (compare Figures 3C and 8C). The acceleration of early repolarization (phase 1) and inhibition of the AP overshoot are obviously caused by simultaneous inhibition of sodium channels while the prolongation of the AP was predominantly balanced by simultaneous block of Cav1.2. Hence, as illustrated in Figure 8D, selective hERG inhibition by 300 nM Dofe45 would induce a more pronounced AP prolongation. The inability of Dofe42 and Dofe44 to prolong the AP is hard to explain exclusively by inhibition of Cav1.2 and Nav1.5 as these channels appear to be blocked only at high concentrations. But the conditions of the ion channel assay are not the same as those of the iPSC‐CMs. The oscillatory voltage changes and the temperature will almost certainly alter the level of activation/inactivation of the currents. Both Nav1.5 and Cav1.2 show voltage‐ and time‐dependant effects of drugs, and inhibition of inactivated Nav1.5 by Dofe42 and Dofe44 was stronger at −80 mV relative to −140 mV (Figure 7). Furthermore, Cav1.2 showed use‐dependent block of by Dofe42 (Figure 6C, D). Therefore, the precise effect of drugs on both of these channels in the context of an AP in IPSC‐CMs is difficult to assess. Also, while Cav1.2 and Nav1.5 are the most likely candidates for alternative drug actions, these derivatives may also modulate other ion channels that contribute to the AP shape.
The implications of this work are that potency of hERG current inhibition correlates linearly with an index of APD in hiPSC‐CMs. This simple relationship, confirmed in silico, allows data gained in one standard assay to predict the effect on another, that is, the IC50 of a drug in an ion channel hERG screen predicts the dose required to increased APD90 in iPSC‐CMs or vice versa. Furthermore, compounds that do not correlate will have additional effects including concomitant block of Cav1.2 and/or Nav1.5 channels. Finally, the study shows that while in silico simulations can confirm the principle of the effects of Cav1.2 and Nav1.5 inhibition on APD90, more comprehensive voltage clamp data are required to accurately predict the consequences of Cav1.2 and Nav1.5 block on AP shape and duration in silico.
Limitations
In the myocardium and hiPSC‐CM hERG, Cav1.2 and Nav1.5 channels function under different conditions than in patch clamp experiments on mammalian cells. In order to relate patch clamp data to the hiPSC‐CM assay, it would be desirable to study these ionic currents at the beating frequency of hiPSC‐CM (~1 Hz) at 37°C. But most patch clamp ion channel assays place constraints on the design of the pulse protocol. As illustrated in Figure 6C, D, continuous 1 Hz pulsing with even short (50 ms) test pulses results in peak current decay of calcium currents caused by channel inactivation. Application of longer test pulses (e.g. 300 ms, corresponding to the length of the ventricular cardiac AP) at 1 Hz leads to inactivation by 30 and 40%, during a train of 20 pulses. Further optimization of experimental (temperature, test pulse length and shape, holding potential, pacing frequency, etc.) and theoretical conditions (analysis of participation of additional ionic currents in AP shaping) is required to achieve a higher level of congruence between the different assay data.
Author contributions
P.S., M.P.V.H., A.C., S.B., I.B. and S.M.I. performed the experiments; P.S., M.P.V.H., A.C., S.B., A.P.I., P.K., E.T., G.L.S. and S.H. designed the study; P.S., M.P.V.H., A.C., S.B., A.P.I., P.K., E.T., G.L.S. and S.H. analysed data; P.S., M.P.V.H., E.T., P.K., G.L.S. and S.H. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Dofetilide (100 μM) does not inhibit Cav1.2 or Nav1.5.
Figure S2 Estimation of dose required to prolong the action potential by 150% (D150).
Figure S3 Relationship between potencies of dofetilide derivatives to inhibit hERG (IC50) and their MW (MW).
Table S1 Major maximal conductance of ion channels used for AP simulations of human embryonic stem cell‐derived myocytes described in Paci et al. (2012) and corresponding values used for adult ventricular cardiomyocyte models.
Table S2 Potencies of dofetilide derivatives to inhibit hERG potassium channels estimated in patch clamp experiments and Ki values from binding studies (from Shagufta et al. 2009) in relation to MW.
Acknowledgements
We thank Clyde Biosciences, UK, for financial support. This work is supported by Austrian Science Fund (FWF; http://www.fwf.ac.at). P.S. was supported by a doctoral programme ‘molecular drug targets’ funded by FWF W1232. M.P.H.V. is recipient of Fundacion Alfonso Martin Escudero (SPAIN) postdoctoral fellowship. S.B. is supported by FWF grant P27729.
Saxena, P. , Hortigon‐Vinagre, M. P. , Beyl, S. , Baburin, I. , Andranovits, S. , Iqbal, S. M. , Costa, A. , IJzerman, A. P. , Kügler, P. , Timin, E. , Smith, G. L. , and Hering, S. (2017) Correlation between human ether‐a‐go‐go‐related gene channel inhibition and action potential prolongation. British Journal of Pharmacology, 174: 3081–3093. doi: 10.1111/bph.13942.
Contributor Information
G L Smith, Email: godfrey.smith@glasgow.ac.uk.
S Hering, Email: steffen.hering@univie.ac.at.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett GCL, Kaplan AD, Lis A, Cimato TR, Tzanakakis ES, Zhou Q et al. (2013). Electronic ‘expression’ of the inward rectifier in cardiocytes derived from human‐induced pluripotent stem cells. Hear Rhythm 10: 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyl S, Depil K, Hohaus A, Stary‐Weinzinger A, Linder T, Timin E et al. (2012). Neutralisation of a single voltage sensor affects gating determinants in all four pore‐forming S6 segments of CaV1.2: a cooperative gating model. Pflügers Arch ‐ Eur J Physiol 464: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S, Williams M, Ways N, Brenner R, Sharp AH, Leung AT et al. (1988). Sequence and expression of mRNAs encoding the alpha 1 and alpha 2 subunits of a DHP‐sensitive calcium channel. Science 241: 1661–1664. [DOI] [PubMed] [Google Scholar]
- Engl H, Flamm C, Kuegler P, Lu J, Mueller S, Schuster P (2009). Inverse problems in systems biology. IOP Sci 25: 1–51. [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC (2004). Physicochemical features of the HERG channel drug binding site. J Biol Chem 279: 10120–10127. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflügers Arch Eur J Physiol 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Hancox JC, McPate MJ, Harchi AE, Zhang YH (2008). The hERG potassium channel and hERG screening for drug‐induced torsades de pointes. Pharmacol Ther 119: 118–132. [DOI] [PubMed] [Google Scholar]
- Hoekstra M, Mummery CL, Wilde AAM, Bezzina CR, Verkerk AO (2012). Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol 3: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG (1977). Time‐ and voltage‐dependent interactions of antiarrhythmic drugs with cardiac sodium channels. BBA ‐ Rev Biomembr 472: 373–398. [DOI] [PubMed] [Google Scholar]
- Hortigon‐Vinagre MP, Zamora V, Burton FL, Green J, Gintant GA, Smith GL (2016). The use of ratiometric fluorescence measurements of the voltage sensitive dye Di‐4‐ANEPPS to examine action potential characteristics and drug effects on human induced pluripotent stem cell‐derived cardiomyocytes. Toxicol Sci 154: 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth B, Hegyi B, Kistamás AK, Váczi K, Bányász T, Magyar J et al. (2015). Cytosolic calcium changes affect the incidence of early afterdepolarizations in canine ventricular myocytes. Can J Physiol Pharmacol 93: 527–534. [DOI] [PubMed] [Google Scholar]
- Kane C, Du DTM, Hellen N, Terracciano CM (2016). The fallacy of assigning chamber specificity to iPSC cardiac myocytes from action potential morphology. Biophys J 110: 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC (2001). Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104: 569–580. [DOI] [PubMed] [Google Scholar]
- Knisley SB, Justice RK, Kong W, Johnson PL (2001). Ratiometry of transmembrane voltage‐sensitive fluorescent dye emission in hearts. Am J Physiol Hear Circ Physiol 73104 : 1616–1622. [DOI] [PubMed] [Google Scholar]
- Kramer J, Obejero‐Paz CA, Myatt G, Kuryshev YA, Bruening‐Wright A, Verducci JS et al. (2013). MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci Rep 3: 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Choi B, Ziv O, Li W, Lange ED, Qu Z et al. (2012). Differential conditions for early after‐depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J Physiol 5: 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, Li J, Sukumar P, Majeed Y, Dallas ML, English A et al. (2009). Robotic multiwell planar patch‐clamp for native and primary mammalian cells. Nat Protoc 4: 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, Wu J, Zipes DP (2008). The QT syndromes: long and short. Lancet 372: 750–763. [DOI] [PubMed] [Google Scholar]
- Paci M, Sartiani L, Lungo MD, Jaconi M, Mugelli A, Cerbai E et al. (2012). Mathematical modelling of the action potential of human embryonic stem cell derived cardiomyocytes. Biomed Eng Online 11: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E, Castellano A, Kim H, Bertrand P, Baggstrom E, Lacerda A et al. (1992). Cloning and expression of a cardiac/brain beta subunit of the L‐type calcium channel. J Biol Chem 267: 1792–1797. [PubMed] [Google Scholar]
- Perry M, Sanguinetti M, Mitcheson J (2010). Revealing the structural basis of action of hERG potassium channel activators and blockers. J Physiol 588: 3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM (2004). Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 14: 61–66. [DOI] [PubMed] [Google Scholar]
- Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 167: 292–300. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani‐Firouzi M (2006). hERG potassium channels and cardiac arrhythmia. Nature 440: 463–469. [DOI] [PubMed] [Google Scholar]
- Sato D, Xie LH, Nguyen TP, Weiss JN, Qu Z (2010). Irregularly appearing early afterdepolarizations in cardiac myocytes: random fluctuations or dynamical chaos? Biophys J 99: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena P, Zangerl‐Plessl E, Linder T, Hohaus A, Windisch A, Knape et al. (2016). New poterntial binding determinant of hERG channel inhibitors. Sci Rep 6: 24182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagufta, Guo D, Klaasse E, De Vries H, Brussee J, Nalos L et al. (2009). Exploring chemical substructures essential for hERG K+ channel blockade by synthesis and biological evaluation of dofetilide analogues. ChemMedChem 4: 1722–1732. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M et al. (2007). State dependent dissociation of HERG channel inhibitors. Br J Pharmacol 151: 1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp MW, Verkerk AO, Ginneken van ACG, Baartscheer A, Schumacher C, de Jonge N et al. (2001). Norepinephrine induces action potential prolongation and early afterdepolarizations in ventricular myocytes isolated from human end‐stage failing hearts. Eur Heart J 22: 955–963. [DOI] [PubMed] [Google Scholar]
- Volders PGA, Vos MA, Szabo B, Sipido KR, De Groot SHM, Gorgels APM et al. (2000). Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. 46: 376–392. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mi J, Lu K, Lu Y, Wang K (2015). Comparison of gating properties and use‐dependent block of Nav1.5 and Nav1.7 channels by anti‐arrhythmics mexiletine and lidocaine. PLoS One 10: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windisch A, Timin E, Schwarz T, Stork‐Riedler D, Erker T, Ecker G et al. (2011). Trapping and dissociation of propafenone derivatives in HERG channels. Br J Pharmacol 162: 1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C et al. (2014). Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation 130: 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Rudy Y (1995). Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J 68: 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT (1999). Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res 84: 989–998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Dofetilide (100 μM) does not inhibit Cav1.2 or Nav1.5.
Figure S2 Estimation of dose required to prolong the action potential by 150% (D150).
Figure S3 Relationship between potencies of dofetilide derivatives to inhibit hERG (IC50) and their MW (MW).
Table S1 Major maximal conductance of ion channels used for AP simulations of human embryonic stem cell‐derived myocytes described in Paci et al. (2012) and corresponding values used for adult ventricular cardiomyocyte models.
Table S2 Potencies of dofetilide derivatives to inhibit hERG potassium channels estimated in patch clamp experiments and Ki values from binding studies (from Shagufta et al. 2009) in relation to MW.