

Abstract
Macrolides represent a large family of protein synthesis inhibitors of great clinical interest due to their applicability to human medicine. Macrolides are composed of a macrocyclic lactone of different ring sizes, to which one or more deoxy‐sugar or amino sugar residues are attached. Macrolides act as antibiotics by binding to bacterial 50S ribosomal subunit and interfering with protein synthesis. The high affinity of macrolides for bacterial ribosomes, together with the highly conserved structure of ribosomes across virtually all of the bacterial species, is consistent with their broad‐spectrum activity. Since the discovery of the progenitor macrolide, erythromycin, in 1950, many derivatives have been synthesised, leading to compounds with better bioavailability and acid stability and improved pharmacokinetics. These efforts led to the second generation of macrolides, including well‐known members such as azithromycin and clarithromycin. Subsequently, in order to address increasing antibiotic resistance, a third generation of macrolides displaying improved activity against many macrolide resistant strains was developed. However, these improvements were accompanied with serious side effects, leading to disappointment and causing many researchers to stop working on macrolide derivatives, assuming that this procedure had reached the end. In contrast, a recent published breakthrough introduced a new chemical platform for synthesis and discovery of a wide range of diverse macrolide antibiotics. This chemical synthesis revolution, in combination with reduction in the side effects, namely, ‘Ketek effects’, has led to a macrolide renaissance, increasing the hope for novel and safe therapeutic agents to combat serious human infectious diseases.
Abbreviations
- CAP
community acquired pneumonia
- MIC
minimum inhibitory concentration
- MLSB
macrolide–lincosamide–streptogramin B
- NPET
nascent peptide exit tunnel
- PTC
peptidyl transferase centre
Isolation of natural macrolides and their chemical structure
The first macrolide antibiotic was isolated from a Streptomyces strain in 1950 and was named pikromycin due to its bitter taste (from the ancient Greek word pikro meaning bitter) (Brockmann and Hekel, 1951). The main chemical characteristic of pikromycin which is common to all later isolated macrolides is the presence of a macrocyclic lactone ring from which the macrolide name derives, as proposed by Woodward in 1950 (see Omura, 2002). Macrolide antibiotics are classified according to the size of the macrocyclic lactone ring as being either 12‐, 14‐, 15‐ or 16‐membered ring macrolides (Figure 1). The majority of macrolides contain amino sugar and/or neutral sugar moieties connected to the lactone ring via a glycosylic bond.
Figure 1.
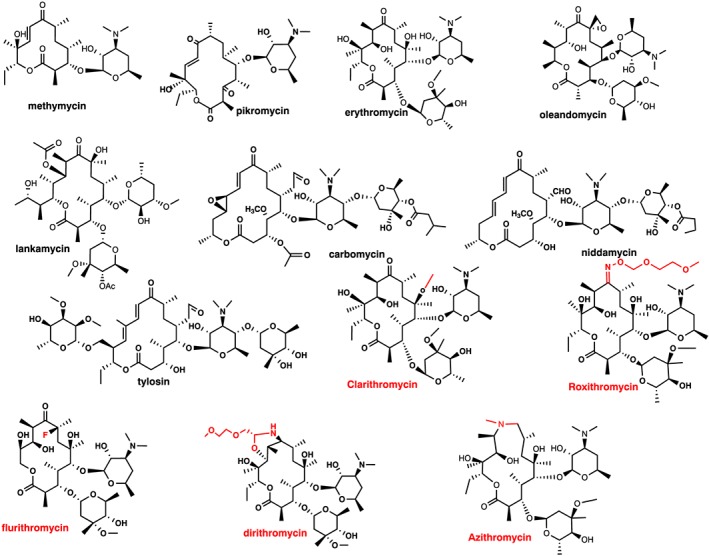
Macrolide structures. First generation: 12‐membered (methymycin), 14‐membered (pikromycin, erythromycin, oleandomycin and lankamycin) and 16‐membered (carbomycin, niddamycin and tylosin), all natural products. Second generation: 14‐membered (clarithromycin, roxithromycin, flurithromycin dirithromycin) and 15‐membered (azithromycin). Red colour indicates modifications inserted in the erythromycin molecule to generate the second generation of 14‐ and 15‐membered macrolides.
Methymycin produced by Streptomyces sp. is the main representative of the 12‐membered macrolides, with only a few other compounds in this class (Figure 1) (Donin et al., 1953). Erythromycin (Figure 1) is the best known member of the 14‐membered group and was isolated from the Streptomyces erythraeus or Arthrobacter sp. (McGuire et al., 1952). Oleandomycin (Sobin et al., 1955), lankamycin (Gäumann et al., 1960) and pikromycin (Brockmann and Hekel, 1951) (Figure 1) are also important members of this group. The last group comprises the 16‐membered macrolides, with the most important members being tylosin (Hamill et al., 1961), carbomycin (Wagner et al., 1953) and niddamycin (Huber et al., 1962) (Figure 1). In addition to the size of the lactone ring, macrolides can also differ from one another by containing either a disaccharide or a monosaccharide attached to the lactone ring.
Almost all macrolides are produced by strains of Streptomyces. However, several species of the genus Micromonospora were found to produce either 14‐ or 16‐membered macrolides (Weinstein et al., 1969; Wagman et al., 1972). Because the antibiotic productivity of Actinomyces isolated from a soil sample is very low, higher yields were obtained by examination of various cultural conditions and by improvement of the producing strain using mutational approaches. Industrial yields of macrolide antibiotics are presumed to reach 10 mg·mL−1, although the exact details are not known due to company secrecy. Today, although the total synthesis of erythromycin has been reported (Woodward et al., 1981), the fermentation production is preferred due to higher yields.
In this short review, we describe the historical development of macrolides and their mode of action, which has been completely revised during the past few years. Moreover, all resistance mechanisms that render pathogens resistant to macrolides and are responsible for their decreased usage are presented. Finally, the latest developments that have returned this antibiotic family to the forefront of science are discussed, leading to the conclusion that the next‐generation macrolide family members will be highly active with reduced toxicity and will therefore re‐enter the market in the near future.
Antimicrobial activity and chemical derivatization
In general, macrolide antibiotics are active mainly against Gram‐positive bacteria and have only limited activity against Gram‐negative bacteria (Nakayama, 1984). Macrolides are very active against Staphylococcus, Streptococcus and Diplococcus Gram‐positive bacteria, and among Gram‐negative cocci, Neisseria gonorrhoea, Haemophilus influenzae, Bordetella pertussis and Neisseria meningitis. Additionally, they are also extremely active against various Mycoplasmas, although there are some susceptibility differences between 14‐ and 16‐membered macrolides (Bébéar et al., 1997; Doucet‐Populaire et al., 1998; Morozumi et al., 2008). They have very low activity against eukaryotes due to their low affinity for binding to eukaryotic ribosomes (Corcoran, 1984; Böttger et al., 2001). Additionally, eukaryote rRNAs carry a guanosine in the equivalent position A2058 of prokaryotes (Böttger et al., 2001), although this difference is not the only determinant responsible for the difference in macrolide susceptibility between yeast and prokaryotes (Bommakanti et al., 2008).
Although macrolides display excellent antibacterial activity, their generally poor bioavailability, unpredictable pharmacokinetics and low stability in the acidic pH of the stomach prompted early searches for new derivatives with improved properties. This resulted in the second generation of macrolides, which were semisynthetic derivatives of the first, natural product, generation. Five derivatives of erythromycin were developed and marketed, namely, clarithromycin (Omura et al., 1992), dirithromycin (Counter et al., 1991), roxithromycin (Chantot et al., 1986), flurithromycin (Toscano et al., 1983; Gialdroni‐Grassi et al., 1986) and azithromycin (Girard et al., 1987; Retsema et al., 1987) (Figure 1). Miokamycin (Omoto et al., 1976; Borzani et al., 1989) and rokitamycin (Sakakibara et al., 1981) were the only 16‐membered second‐generation compounds developed for human use (Figure 2). Tilmicosin (Debono et al., 1989), a semisynthetic derivative of tylosin, was developed solely for veterinary use (Figure 2). Clarithromycin and azithromycin are highly marketed worldwide, whereas dirithromycin (Brogden and Peters, 1994; Kirst, 1995), flurithromycin (Benazzo et al., 1998) and roxithromycin (Jain and Danziger, 2004) have had a much more limited distribution.
Figure 2.
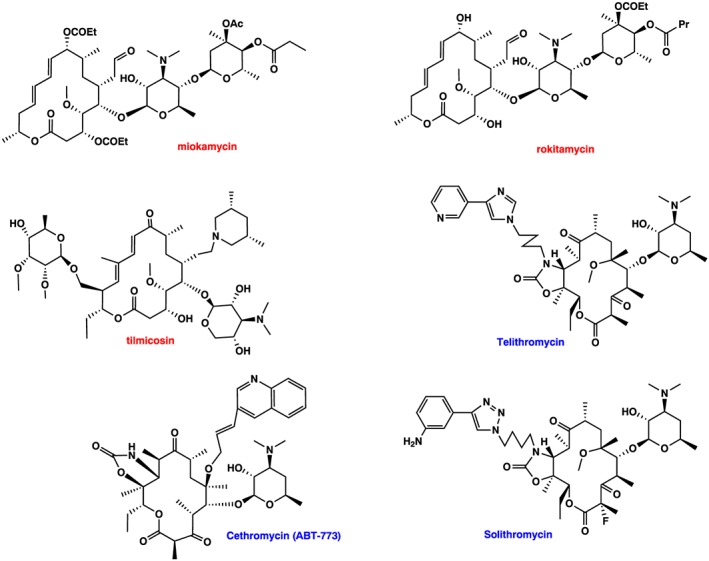
Macrolides structure. Second generation: 16‐membered (miokamycin, rokitamycin and tilmicosin). Third generation: ketolides: Telithromycin, cethromycin and solithromycin. Red and blue colour indicates second‐ and third‐generation macrolides respectively.
Clarithromycin and azithromycin were prepared from erythromycin A in a short, four‐ to six–step, sequence of chemical transformations (Morimoto et al., 1984; Bright et al., 1988). The second‐generation erythromycin derivatives contain all modifications at the C6 or C9 positions of the lactone ring, thereby preventing the formation of the 9,12‐ and/or the 6,9‐hemiketal forms, which degrade to spiroketal inactive derivatives and therefore exhibited immunity to acid‐catalysed inactivation. Clarithromycin is still degraded under acidic conditions to form such derivatives, albeit at reduced rates relative to erythromycin A (Nakagawa et al., 1992; Mordi et al., 2000). The above mentioned second‐generation derivatives (Figures 1 and 2) have each improved oral bioavailability and increased half‐life in plasma, enabling the oral dosage to be reduced to once or twice a day (Foulds et al., 1990; Bahal and Nahata, 1992; Piscitelli et al., 1992; Rodvold, 1999). These compounds also exhibited enhanced tissue penetration because of their higher lipophilicities, relative to that of the parent compound erythromycin A and hence were more effective for treatment of intracellular pathogens such as H. influenzae (Alvarez‐Elcoro and Yao, 2002). Although the search for the second generation of macrolides was undertaken with the desire to discover compounds with expanded spectra and improved activity, the compounds selected did not exhibit improved activity against Gram‐positive bacteria, and some, in fact, such as azithromycin had reduced potency compared with the mother compound erythromycin (Barry et al., 1988, 2001; Fernandes and Hardy, 1988). Nevertheless, they were selected for development mainly because of their enhanced pharmacokinetic profiles, in particular, the ability to accumulate to high levels within lung tissue (Wise, 1989; Foulds et al., 1990; Retsema et al., 1990; Hardy et al., 1992). Clarithromycin is also used generally in combination with other antibiotics, for the treatment of gastric ulcers caused by Helicobacter pylori and for AIDS‐related respiratory infections caused by the Mycobacterium avium complex (Haefner et al., 1999).
While the second generation of macrolides provided solutions with respect to improved pharmacokinetics and acidic inactivation, they provided few answers with respect to antibiotic resistance. As macrolide resistance was becoming increasing dangerous, as was happening with all other antibiotic classes, this prompted research into the development of the next generation of macrolides to combat macrolide resistant strains. This effort yielded the third generation of macrolides, termed ketolides, where the 3‐keto group in the lactone ring replaces the l‐cladinose present in erythromycin (reviewed by Katz and Ashley, 2005). In addition, nearly all ketolides contained the addition of a fused 11,12‐cyclic carbamate as well as an alkyl–aryl side chain tethered to different positions of the lactone ring (Figure 2). Initially, all chemical efforts were focused on the structure and length of the alkyl–aryl side chains and the tethering position on the lactone ring. The first position tethered was the C11 carbon of the lactone ring (Denis et al., 1999; Putnam et al., 2011), although in parallel, C6‐tethered ketolides were also prepared (Or et al., 2000; Ma et al., 2001; Wu and Su, 2001; Plata et al., 2004). Later, a series of C9‐oximes ether ketolides bearing N‐aryl–alkyl acetamides were also synthesized, where the length of the alkyl group differed by up to five atoms (Iwaki et al., 2005; Nomura et al., 2005; Nomura et al., 2006). Other structure modifications included the following: modified 5‐O‐desosamine ketolides (Chen et al., 2012), fluorination at the C2‐ and/or C12 positions (Denis and Bonnefoy, 2001; Krokidis et al., 2014), variation of the cyclic carbonate and hydrazono‐carbamate at 11,12 positions (Hunziker et al., 2004; Andreotti et al., 2007; Zhu et al., 2007) or variation of the aglycon ring (Shaw et al., 2005; Ashley et al., 2006; Sugimoto and Tanikawa, 2010). Lastly, modifications also included replacement of desosamine with different sugars (Liang et al., 2005; Romero et al., 2005; Chen et al., 2012). A detailed review covering all the chemical efforts to improve the ketolide activity is presented in the review of Liang and Han (2013). The only third‐generation macrolide in the market today is telithromycin (Figure 2), commercialized as Ketek by Aventis, a 14‐membered ‘ketolide’ that was derived from clarithromycin using eight chemical steps (Denis et al., 1999). Another ketolide cethromycin (Figure 2) (Ma et al., 2001) with similar activity to telithromycin was denied FDA approval in 2009. Lastly, solithromycin (Pereira and Fernandes, 2011) (Figure 2) is currently undergoing phase III clinical trials (Farrell et al., 2016) and seems to be the most promising ketolide.
These ketolides (telithromycin, cethromycin and solithromycin) (Figure 2) have outstanding activity against Gram‐positive aerobic pathogens, including macrolide‐resistant strains of Streptococcus pneumoniae (Shortridge et al., 2002; Farrell et al., 2015). In addition, they display good activity against some Gram‐negative aerobes, such as Moraxella catarrhalis and H. influenzae, and gratifying activity against atypical/intracellular CAP pathogens such as C. pneumoniae, Mycoplasma pneumoniae, and Legionella pneumophila (Bébéar et al., 1997; Hammerschlag et al., 2001; Fernandes et al., 2016). Unlike azithromycin and clarithromycin, telithromycin is not a substrate for the efflux pumps found in S. pneumoniae and Streptococcus pyogenes and does not induce ribosomal methylation associated with inducible macrolide–lincosamine–streptogramin B (MLSB) resistance in Streptococci and Staphylococci (Bryskier, 2000). However, Staphylococcal and S. pyogenes strains that carry constitutively methylated ribosomes are not susceptible to telithromycin. Although rare, ketolide (telithromycin)‐resistant strains have been isolated worldwide (Doern, 2006; Felmingham et al., 2007). Unlike macrolides, which are considered as time‐dependent bacteriocides, ketolides show concentration‐dependent killing (Zhanel and Hoban, 2002; Woosley et al., 2010). In addition to the three ketolides mentioned above, other ketolide compounds have been developed, although none of them have so far attracted much attention (Fu et al., 2006; Karahalios et al., 2006; Kouvela et al., 2009; Liang and Han, 2013; Ruan et al., 2013; Krokidis et al., 2014).
Side effects
Although first and second generation of macrolides were safe and well tolerated, the third generation of macrolides and more specifically, the ketolide telithromycin exhibited rare but serious irreversible hepatotoxicity named ‘Ketek effects’ (Young, 2007; Georgopapadakou, 2014; Telithromycin 2014; Fernandes et al., 2016). This is why telithromycin use was restricted and led to pharmaceutical companies focusing on improving the safety of macrolides. According to Fernandes and colleagues, it is the pyridine ring included in telithomycin's alkyl–aryl‐side chain (Figure 2) that blocks nicotinic acetylcholine receptors, resulting under specific conditions in serious hepatotoxicity (Fernandes et al., 2016). Therefore, the first requirement to avoid hepatotoxicity is the absence of a pyridine ring from the alkyl–aryl side chain of a new macrolide.
Procedures for development of the next generation macrolides
A breakthrough in macrolide development and synthesis occurred last year when Seiple and colleagues presented a new approach to total synthesis, capable of generating virtually any kind of macrolide (Seiple et al., 2016). Specifically, they developed a platform of unprecedented versatility for the development and synthesis of novel macrolide antibiotics, employing a design strategy that involved a multi‐convergent assembly of macrolides from simple chemical building blocks. The assembly utilizes eight initial building blocks into which they can introduce huge diversities, and follows a sequence of convergent coupling reactions to form two fundamental intermediates, which participate in the next key reaction, the macrocyclization reaction step (Boeckman and Pruitt, 1989). The success of macrocyclization is part of the main contribution of this new procedure because many previous pioneer attempts had all failed (Seiple et al., 2016). In addition to the modifications inserted in the initial building blocks, further modifications are possible either at a later step or after lactone ring complemention. As a result, a library of macrolide antibiotics was constructed containing more than three hundred different compounds. Some of the novel macrolides exhibited high antimicrobial activity against many macrolide‐resistant pathogenic bacteria and are undergoing further evaluation. Following this new procedure, many compounds of clinical importance were also obtained, such as azithromycin, telithromycin and solithromycin, bypassing the established semisynthetic method of chemical modification, which starts from the erythromycin molecule as a fermentation product. Furthermore, this procedure enables success in designed macrolide compounds that could not have been synthesized earlier using the derivatization method.
Other tools in the macrolide renaissance include the structure‐based drug design procedure (Lounnas et al., 2013) as well as molecular simulations (Pavlova and Gumbart, 2015; Pavlova et al., 2017) for the optimization of a chemical structure, both with the same goal of identifying a compound suitable for clinical testing as a drug candidate. These procedures are based on the knowledge of the three‐dimensional structure of the macrolides and how its shape and charge cause it to interact with the 50S ribosomal subunit. Lastly, genetic engineering procedures need to be mentioned, especially with respect to polyketide synthases (Khosla and Zawada, 1996; Khosla, 2009; Walsh, 2017), the multienzyme systems responsible for macrolide biosynthesis, which have substantially increased the potential to develop new macrolides.
Mode of action
Regardless of whether first, second or third generation, all macrolide antibiotics bind to the large ribosomal subunit of the prokaryotic ribosome, occupying a site within the nascent peptide exit tunnel (NPET) adjacent to the peptidyl transferase centre (PTC). The binding site of a diverse range of macrolide antibiotics on different bacterial (and archeal) ribosomes has been revealed using X‐ray crystallography (Schlünzen et al., 2001; Berisio et al., 2003; Tu et al., 2005; Bulkley et al., 2010; Dunkle et al., 2010) (Figure 3). There was initially a controversy concerning the conformation of the macrolide‐bound lactone ring and the contacts they make with the ribosome (Schlünzen et al., 2001; Hansen et al., 2002; Berisio et al., 2003; Tu et al., 2005). However, subsequent studies using Escherichia coli (Dunkle et al., 2010) and Thermus thermofilus ribosomes (Bulkley et al., 2010) resolved the situation and confirmed that the macrolactone ring of all macrolides is similarly oriented in the ribosomal tunnel, regardless of the type of macrolide or the size of the lactone ring. Macrolides interact with the nucleobase of A2058 of the 23S rRNA, which involves a hydrogen bond between the desosamine hydroxyl and the N1 atom of A2058. In addition, the binding of macrolides is stabilized by tight packing of the hydrophobic face of the lactone ring against rRNA nucleotides 2611 and 2057 (Figure 3C). However, species‐specific differences do appear to exist with respect to macrolides or ketolides bearing the alkyl–aryl side chain. In the case of telithromycin, this heterocyclic side chain was observed in different positions when comparing the drug bound to the archaeal Haloarcula marismortui 50S subunit (Tu et al., 2005), Deinococcus radiodurans 50S subunit (Berisio et al., 2003; Schlünzen et al., 2003), T. thermophilus (Bulkley et al., 2010) or E. coli 70S ribosomes (Dunkle et al., 2010), as previously described (Wilson et al., 2005) (Figure 3D).
Figure 3.
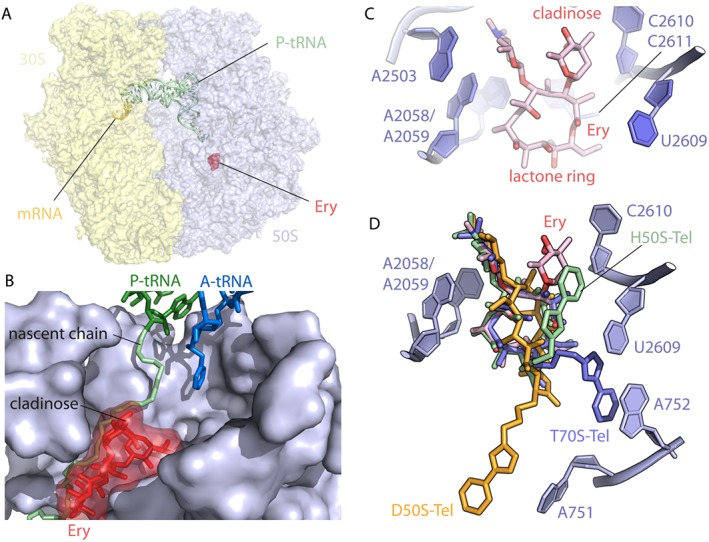
Interaction of macrolides with the ribosome: (A) Overview of the antibiotic erythromycin (Ery, red) bound to the 70S E. coli ribosome (PDB entry 4V7U; Dunkle et al., 2010). (B) Close‐up view of the erythromycin binding site in the ribosomal exit tunnel in the presence of the P‐site peptidyl‐tRNA (green) and the A‐site tRNA (blue) (prepared with modifications from Wilson 2014). (C) Erythromycin binding pocket within the E. coli 70S ribosome is located adjacent to bases A2058, A2059, A2503 and U2609. The desosamine amino sugar of Ery at position 5 of the lactone ring contains a dimethyl amine that makes pivotal contact with the base A2058 (PDB entry 4V7U; Dunkle et al., 2010). (D) Superposition of telithromycin (Tel) bound to the ribosomes from different species. All structures of ribosome‐bound Tel were aligned based on domain V of the 23S rRNA. Note that although the lactone rings almost perfectly match in all cases, the position of the alkyl–aryl groups varies significantly depending on the species. Shown are Haloarcula marismortui (green, PDB entry 1YIJ; Tu et al., 2005), D. radiodurans (orange, PDB entry 1P9X; Berisio et al., 2003) and T. thermophilus (blue, PDB entry 4V7Z; Bulkley et al., 2010). All figures were prepared using PyMol software.
For a long time, macrolides were considered as general inhibitors of translation by simply obstructing the ribosomal exit tunnel and thereby preventing the progress of the synthesis of the nascent polypeptide chain (Menninger and Otto, 1982; Tenson et al., 2003; Mankin, 2008). In contrast to this view, Mankin and coworkers have demonstrated that the mode of action of these drugs is more complicated (Kannan et al., 2012; Kannan et al., 2014; Sothiselvam et al., 2016). For the majority of proteins, the binding of the drug within the tunnel does cause synthesis to be aborted when the nascent peptide chain reaches a nominal length of 5–11 amino acids where prolongation is prevented, leading to dissociation of the peptidyl‐tRNA (drop‐off) from the ribosome (Menninger and Otto, 1982; Menninger, 1995; Tenson et al., 2003). A small number of short specific nascent peptides, such as those encoded in the regulatory cistrons of genes rendering macrolides ineffective, can induce ribosome stalling by interacting with the NPET. In this case, the peptidyl‐tRNA remains bound, but peptide bond formation with the arriving A‐site bound aminoacyl‐tRNA is prevented (Horinouchi and Weisblum, 1980; Vazquez‐Laslop et al., 2008; Ramu et al., 2011). Moreover, Mankin and coworkers recently demonstrated that some peptide sequences have the ability to bypass the antibiotic within the NPET which leads either to the synthesis of long polypeptides on drug‐bound ribosomes or to the interruption at a later state when the length of the nascent chain has already passed the antibiotic binding site (Kannan et al., 2012). Macrolides, such as erythromycin, appear to allow fewer proteins to bypass compared with ketolides, such as telithromycin (Kannan et al., 2012; Davis et al., 2014; Kannan et al., 2014), presumably because erythromycin contains a C3‐bound cladinose sugar (Figure 1) that projects into the lumen of the tunnel (Schlünzen et al., 2001; Bulkley et al., 2010; Dunkle et al., 2010).
Ribosome profiling studies have also been used to dissect macrolide action (Davis et al., 2014; Kannan et al., 2014). Ribosome profiling or Ribo‐seq is a recently developed high‐throughput sequencing technique that allows the identification of RNA fragments resistant to RNAse digestion by translating ribosomes. Therefore, it provides a snapshot of ribosomal movement on the template mRNA (Ingolia et al., 2009; Li et al., 2012). A high number of ribosome‐protected fragments mapping to the transcriptome is indicative of prolonged ribosome occupancy at a given position, also known as ribosome stalling/pausing. Ribo‐seq carried out recently in Gram‐positive and Gram‐negative bacteria treated with different macrolides identified the major sites of late translation arrest and allowed classification of problematic sequences for the first time (Davis et al., 2014; Kannan et al., 2014). Several amino acid motifs favourable to macrolide‐induced arrest were revealed by these studies. The most dominant motif contained the tripeptide sequence motif R/K‐X‐R/K, where R and K denote arginine and lysine amino acids, respectively, and X represents any amino acid. In vitro biochemical experiments supported the conclusion drawn from the mRNA profiling analysis that the ribosome stalls when the codon representing the middle amino acid (X) of the motif enters the P site. Accordingly, the first residue of the consensus (R or K) represents the position before the last amino acid of the nascent peptide chain, whereas the last consensus sequence residue (also R or K) corresponds to the amino acid attached to the A site‐bound aminoacyl‐tRNA (Davis et al., 2014; Kannan et al., 2014; Sothiselvam et al., 2014, 2016). These findings suggest that rather than inducing a universal arrest of protein synthesis, macrolides and ketolides actually allow translation of only a small subset of proteins. Taking into account that telithromycin, which is bactericidal, allows more nascent peptides bypass than erythromycin (which is bacteriostatic), it has been suggested that an inequity in protein inhibition is more harmful for the cell compared with uniform inhibition of protein synthesis (Kannan et al., 2012).
Kinetic analysis of many different macrolides revealed that most of them act as slow‐binding inhibitors (Morrison and Walsh, 1988). Moreover, while their association rate constants (k on) are nearly identical, there are large differences in their dissociation rate constants (k off values). The k off values for some macrolides are extremely low, in the range of 10−5 s−1, indicating that these macrolides are almost irreversibly bound to the ribosome (Di Giambattista et al., 1987; Dinos and Kalpaxis, 2000; Dinos et al., 2003; Krokidis et al., 2016). Mankin and colleagues have suggested that such very low macrolide dissociation constants from the ribosome may also contribute to their bactericidal effect (Svetlov and Mankin, personal communication).
Champney and co‐workers have shown that macrolides inhibit ribosome assembly (Chittum and Champney, 1995; Champney and Miller, 2002; Champney and Pelt, 2002), but this results from a secondary effect due to the inhibition of synthesis of ribosomal proteins (Siibak et al., 2009). Lastly, a novel action of macrolides previously unknown is the promotion of ribosome frameshifting (Gupta et al., 2013).
Macrolide resistance
The two most common resistance mechanisms in the bacterial pathogens are the reduced binding affinity of the drug, firstly, due to modification of either the bacterial ribosome or the antibiotic molecule and, secondly, due to efflux of macrolides from the bacterial cell, arising via altering either the membrane permeability or efflux pump expression (Wilson, 2014; Fyfe et al., 2016). Efflux proteins belong mainly to Mef and Msr families, and ribosome modification mechanisms include either ribosomal 23S rRNA or large ribosomal subunit proteins, while drug‐inactivating mechanisms include phosphorylation of the 2′‐hydroxyl of the sugar by phosphotransferases and hydrolysis of the macrocyclic lactone by esterases.
Mef family
Mef pumps are proteins that are members of the major facilitator superfamily, consisting of 12 transmembrane domains linked by hydrophilic loops (Pao et al., 1998). Mef pumps work as antiporters, exchanging the bound macrolide with a proton (Law et al., 2008). mef genes are found in Gram‐positive bacteria but have also been reported in some Gram‐negative species (Ojo et al., 2004). There are two major subclasses, mef(A) and mef(E). Although there is higher than 80% homology, they are carried on distinct genetic elements. Both genes confer resistance to 14‐ and 15‐membered, but not to 16‐membered, macrolides, lincosamides and streptogramins B, affording the so‐called ‘M phenotype’ but not the ‘MLSB phenotype’. mef(E), as well as the Staphylococcus aureus msr(A) family of genes, carries an adjacent ATP‐binding cassette‐type transporter gene known as the msr(D) gene. Msr(D) and Mef(E) co‐expression is required for high‐level macrolide resistance in S. pneumoniae, and both proteins interact synergistically to increase macrolide resistance in E. coli (Nunez‐Samudio and Chesneau, 2013). Recently, additional mef genes were described, namely, mef(B) and mef(E), with medium homology to mef(A) and mef(I), as well as mef(O) with high homology to mef(A) (see Fyfe et al., 2016). mef genes are regulated by transcription attenuation, with the induction of the mef(E)/msr(D) operon occurring by anti‐attenuation of transcription in the presence of inducing macrolides. There is also evidence, however, that an additional regulation mechanism is existing with a leader peptide encoded upstream of mef(E) (Subramaniam et al., 2011; Chancey et al., 2015).
Msr family
These proteins displace macrolide antibiotics from the ribosome, offering ribosome protection by binding and chasing the bound drug from the ribosome (Sharkey et al., 2016; Wilson, 2016). There are four classes of Msr proteins, namely, types A, C, D and E, with each class having an ATP‐binding motif and sequence homology with the ATP‐binding superfamily (Ross et al., 1990). The Msr family confer resistance to 14‐ and 15‐membered macrolides and low level to ketolides (Ross et al., 1995; Canton et al., 2005; Reynolds and Cove, 2005; Vimberg et al., 2015). The msr genes related to macrolide resistance have been isolated from Staphylococcus epidermidis (Ross et al., 1990), Staphylococcus xylosus (Milton et al., 1992), St. aureus (Matsuoka et al., 1999, 2003), Enterococcus (Portillo et al., 2000), Streptococcus (Varaldo et al., 2009), Pseudomonas and Corynebacterium (Ojo et al., 2006). For a long time, the Msr family was thought to confer macrolide resistance by acting as efflux pumps (Fyfe et al., 2016). Now, it seems that these proteins act in a similar way to TetM/TetO proteins, chasing the bound macrolide from the ribosome (Sharkey et al., 2016; Wilson, 2016). Tet(M) and Tet(O) are paralogs of the translational GTPase EF‐G and remove tetracycline from the ribosome in a GTP hydrolysis‐dependent manner causing tetracycline (not macrolides) resistance (Burdett, 1996; Connell et al., 2003).
23S rRNA modification
Gram‐positive bacteria, as well as E. coli, can acquire genes that modify the 23S rRNA and confer high‐level MLSB resistance to macrolides, lincosamides and some members of the streptogramin B family (Weisblum, 1995a, 1995b; Roberts et al., 1999). The genes encode methyltransferase enzymes that either mono‐ or di‐methylate the N6 position of nucleotide A2058 (E. coli numbering) of the 23S rRNA (Katz et al., 1987). The enzyme class was named Erm for erythromycin resistance methylase, and individual genes as ermA, ermB, etc. (Roberts et al., 1999). The erm genes have been found on high‐ and low‐copy plasmids and within transposons, usually in association with other genes responsible for resistance expression to other antibiotics (Alekshun and Levy, 2007). The ermE gene from the erythromycin‐producer Saccharopolyspora erythraea has been found in commercial preparations of the drug, causing one to wonder whether resistance in clinical isolates originated from the producing strain and whether it was spread directly because of drug usage (Webb and Davies, 1993, 1994). Some of the enzymes, such as ErmN, catalyse only monomethylation (Liu and Douthwaite, 2002), whereas others, such as ErmE (Katz et al., 1987) and ErmC (Denoya and Dubnau, 1989), catalyse dimethylation, but it is not known whether dimethylation takes place through a concerted two‐step process since these latter enzymes can use monomethylated RNA as a substrate. Moreover, induction is dependent upon the presence of the antibiotic with the correct structures of a 14‐ or 15‐membered macrolides that contain a neutral sugar at C3. In contrast, 16‐membered macrolides and 14‐membered‐ketolides are not inducers. Erm‐mediated resistance exists in two forms: inducible and constitutive. In the inducible form of resistance, the ribosomal methylation is established only after the macrolide is transported into the cells (Ramu et al., 2009). In hosts that are constitutively resistant to macrolides, Erm‐catalysed methylation of the ribosomes does not require the presence of the macrolides (Poehlsgaard and Douthwaite, 2003). Both inducible and constitutive MLSB resistance require the complete sequence encoding the erm gene (Weisblum, 1995a, 1995b).
Recently, it was shown that the tunnel acts as a regulatory compartment where the sequence of the nascent peptide acts together with the drug to slow the rate of translation elongation or even stop translation completely (Otaka and Kaji, 1975; Tenson et al., 2003; Davis et al., 2014; Kannan et al., 2014). A number of genes regulated by recognition of the nascent peptide have been identified in bacteria and eukaryotes (see Wilson et al., 2016). The inducible macrolide resistance genes remain silent in the absence of the antibiotic but are activated in its presence. Activation of the inducible gene is regulated by ribosome stalling at a precise position on programmed, evolutionarily defined site of the regulatory upstream open reading frame (uORF), which precedes the resistance gene (Weisblum, 1995a, 1995b; Ramu et al., 2009; Subramaniam et al., 2011). The ribosomes, which are arrested at the uORF of the regulatory gene, alter the folding of the mRNA, activating the expression of the downstream resistance cistron (Figure 4). The chemical structure of the antibiotic and the sequence and/or the structure of the leader peptide are the two main factors that regulate the position of translation arrest on the mRNA of the leader peptide (Gryczan et al., 1980; Horinouchi and Weisblum, 1980; Vazquez‐Laslop et al., 2008, 2010; Ramu et al., 2011; Arenz et al., 2014a, 2014b). As mentioned above, the amino acid sequence motif R/K‐X‐R/K was recognized as one of the major motifs that induce translation arrest in the presence of macrolides (Davis et al., 2014; Kannan et al., 2014). The R/K‐X‐R/K motif has been detected in many regulatory uORFs of macrolide resistance genes, including also the ermDL ORF that controls expression of the ermD gene that has been studied extensively (Kwak et al., 1991; Kwon et al., 2006; Sothiselvam et al., 2014). Many regulatory peptides like ermCL and ErmBL do not feature this motif and direct translation arrest via distinct stalling sequences (Wilson, 2016). Ketolides which do not induce ribosome stalling at the uORF of the ermC resistance gene trigger its translation through frameshifting, allowing so the translating ribosome to invade the intergenic spacer (Gupta et al., 2013). In all tested regulatory uORFs with the R/K‐X‐R/K motif, the ribosome stalls when the second codon of the consensus enters the ribosomal P site (Sothiselvam et al., 2014; Almutairi et al., 2015). RNA chemical probing has shown that binding of macrolides to the NPET of the vacant ribosome was sufficient to allosterically induce structural changes in the PTC (Sothiselvam et al., 2014, 2016). This is confirmed by Mankin's experiments where translation of the 5′terminally truncated ermDL ORF, which encodes the peptide starting with the MRLR (methionine–arginine–leucine–arginine) sequence, was efficiently arrested by macrolides at the third codon responsible for leucine, when the length of the nascent peptide chain was only three amino acids and is predicted to establish only limited contacts with the drug (Sothiselvam et al., 2014). These results suggested that the inhibition of translation elongation by macrolides does not block the progress of the nascent chain in the drug‐engaged exit tunnel but rather that binding of the antibiotic allosterically modifies the PTC which is unable to catalyse peptide bond formation when certain combinations of donor and acceptor substrates have occupied the A‐ and P‐sites (Kannan et al., 2014; Sothiselvam et al., 2014, 2016). Additionally, the efficiency of stalling by a minimal MRLR sequence allowed Mankin and colleagues to conclude that the amino acids preceding the stalling motif in the ErmDL peptide do not make a significant contribution to the translation arrest. Rather, the length of the side chains and the positive charge of the amino acids occupying the A‐ and P‐sites appear to be more important for the efficiency of stalling (Sothiselvam, 2016). Such an allosteric modification of PTC after macrolide binding on the ribosome was presented many years before, when addition of erythromycin in a mixture of ribosomes with bound radioactive chloramphenicol caused complete release of the latter despite the apparent lack of overlap in the binding sites of the two antibiotics (Pestka and LeMahieu, 1974; Dunkle et al., 2010).
Figure 4.
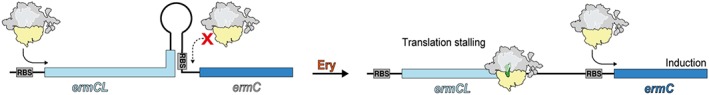
Regulation of gene expression by ribosomal stalling. In the absence of erythromycin, there is no stalling and no translation of ermC. In the presence of erythromycin, there is stalling during translation of the leader peptide ermCL, which causes the ribosome to block stem‐loop formation and exposes the ribosome binding site (RBS) of the downstream cistrons, allowing its expression. (Figure was prepared with modifications from Wilson et al., 2016).
Mutations in ribosomal RNA
All published data indicate that A2058 is the key nucleotide of the 23S rRNA for macrolide resistance (Vester and Douthwaite, 2001; Canu et al., 2002; Berisio et al., 2006). The mutation A2058G confers high‐level resistance to all macrolides including many ketolides (Vester and Douthwaite, 2001; Canu et al., 2002; Misyurina et al., 2004; Berisio et al., 2006). The exception is S. pneumonia where the A2058G mutation confers resistance to macrolides but low level to ketolides (Canu et al., 2002; Farrell et al., 2003). This unusual behaviour is explained by the 2057–2611 base pair, which is part of the cradle housing the lactone ring of the macrolide (Pfister et al., 2005; Kannan and Mankin, 2011). The 2057–2611 base pair is conserved across bacteria (Akshay et al., 2011), and the polymorphism of this base pair (i.e. the presence of G‐C vs. A‐U) determines the ketolide susceptibility of A2058G mutants (Pfister et al., 2005). Another resistance mutation occurs at the A2059 position and has been found in vivo in Mycobacteria, Propionibacteria, H. pylori and S. pneumoniae (Vester and Douthwaite, 2001). Mutations at position 2057 have also been observed in clinical isolates although with a limited frequency (Fyfe et al., 2016). The U2609C mutation in E. coli was selected for resistance to telithromycin and cethromycin (Garza‐Ramos et al., 2002). In E. coli, nucleotides A752 and U2609 form a base pair that connects domains II and V in the 23S rRNA (Schuwirth et al., 2005). In the E. coli 70S‐telithromycin structure (Dunkle et al., 2010), this base pair offers a surface for the alky–aryl arm to engage in stacking interactions that favour drug binding. The protection of A752 from chemical probing by telithromycin therefore most likely arises through stabilization of the A752–U2609 base pair. The C6 alkyl–aryl side chain of cethromycin bound to the D. radiodurans 50S subunit adopts a distinct conformation compared with that observed in E. coli, most likely because D. radiodurans has C752 instead of A752 and therefore cannot form a canonical base pair with U2609 (Figure 3D) (Vester and Douthwaite, 2001; Schlünzen et al., 2003; Franceschi et al., 2004). Additional mutations in S. pneumoniae have been reported, namely, C2610U and C2611U, that also confer macrolide resistance, whereas deletion of A752 results in high resistance to both macrolides and ketolides (Canu et al., 2002).
Mutations in ribosomal proteins
Mutations in genes encoding ribosomal proteins L4 and L22 can confer erythromycin resistance and reduce telithromycin susceptibility (Tait‐Kamradt et al., 2000a; Pihlajamaki et al., 2002). In addition to E. coli laboratory isolates, a variety of clinical isolates have also been identified with ribosomal protein mutations that confer resistance to macrolides, including S. pneumoniae (Tait‐Kamradt et al., 2000a, 2000b; Farrell et al., 2004), S. pyogenes (Bingen et al., 2002), St. aureus (Prunier et al., 2005), H. influenzae (Peric et al., 2003) and Mycoplasma genitalium (Shimada et al., 2011). In addition to the changes detailed below, a list of L4 and L22 mutations can be found in Franceschi et al. (2004). Changes within a highly conserved sequence of S. pneumoniae L4 (63KPWRQKGTGRAR74), resulted in decreased susceptibility to macrolides or ketolides (a 500‐fold increase to a telithromycin MIC of 3.12 mg·mL−1 for one variation), as well as a reduction in fitness (Tait‐Kamradt et al., 2000a, 2000b; Farrell et al., 2004). Mutations encoding amino acid changes in the C‐terminal region of ribosomal protein L22 (e.g. G95D, P99Q, A93E, P91S, G83E, A101P and 109RTAHIT114 tandem duplication) also led to decreased susceptibility to macrolides and ketolides, although the MICs were not greater than 1 mg·L−1 in S. pneumoniae (Farrell et al., 2003). In M. pneumoniae, all 14‐membered macrolide‐resistant isolates harboured a T508C mutation in L22, and for most, either an A2058G or A2059G mutation in 23S rRNA was also present (Cao et al., 2010; Jensen et al., 2014). Resistance to telithromycin in S. pneumoniae significantly increases when 23S rRNA methylation/mutations are combined with ribosomal protein mutations. For example, a combination of a truncated leader peptide leading to constitutive synthesis of erm(B) conferred a telithromycin MIC of 16 mg·L−1 (Wolter et al., 2008), whereas clinical isolates with both a constitutive erm(B) and a 69GTG71 to TPS substitution in L4 (Wolter et al., 2007), or a combined A2058G mutation and a three‐amino acid deletion in L22 (Faccone et al., 2005), provided high‐level telithromycin resistance (256 mg·L−1). It is also important to mention that, since all previous protein mutation positions are rather distal from the macrolide binding pocket (9–10 Å), it was concluded that resistance may be triggered by induction of structural changes in the rRNA nucleotides that propagate to the binding pocket of the antibiotics (Tu et al., 2005).
Modification by macrolide esterases
Macrolide aglycons are converted to cyclic lactones via an ester bond that is formed during the final ring synthesis step and is catalysed by the thioesterase activity module of the polyketide synthase, responsible for the ring closure step that generates 6‐deoxyerythronolide B (Donadio et al., 1991). It is therefore expected that this key bond would have been targeted by macrolide resistance enzymes operating by reversing the ring closure reaction. The first erythromycin esterase was reported in 1984 and was isolated from a macrolide‐resistant E. coli strain (Barthelemy et al., 1984; Wright, 2005). Cloning of this ereA gene revealed a protein of 406 amino acids with an expected mass of 44.8 kDa (Ounissi and Courvalin, 1985). Subsequently, another orthologue, ereB, was cloned from another E. coli isolate (Arthur et al., 1986). Ere(A) and Ere(B) both hydrolyze the lactone ring in 14‐membered macrolides; however, the two enzymes are only weakly related with 25% protein sequence identity. Through the use of a genomic enzymology approach, the catalytic mechanisms of the ‘erythromycin esterase superfamily’ enzymes were compared (Morar et al., 2012). Ere(A), Ere(B) and two related enzymes from Bacillus cereus, Bcr135 and Bcr136, whose three‐dimensional structures had previously been determined, were studied. Only Ere(A) and Ere(B) were predicted to cleave the macrocyclic ester, and their resolved enzymic hydrolytic mechanism was shown to pass through an hemiketal intermediate, while Bcr136 was confirmed as an esterase that is, however, unable to inactivate macrolides (Morar et al., 2012). The presence of these genes on mobile genetic elements implies the ability to become widespread in the microbial community, and the presence of esterases has been confirmed in at least one clinical isolate of St. aureus (Wondrack et al., 1996) and in environmental isolates of Pseudomonas sp. (Kim et al., 2002). Hydrolytic inactivation of macrolides by esterases specifically involves 14‐ and 15‐membered macrolides, whereas ketolides and 16‐membered macrolides, such as josamycin, midecamycin, rosaramycin and spiramycin, are not substrates (Arthur and Courvalin, 1986; Arthur et al., 1987; Morar et al., 2012).
Modification by kinases (or phosphotransferases)
Macrolide phosphotransferases are macrolide‐inactivating enzymes widespread in Gram‐negative and Gram‐positive bacteria (Sutcliffe and Leclercq, 2002; Roberts, 2008; Fyfe et al., 2016) that, by in silico analysis, are classified in the same family as aminoglycoside and macrolide protein kinases (Shakya and Wright, 2010). The first reported purifications of macrolide phosphotransferases were from macrolide‐resistant E. coli, and this mechanism was soon shown to be prevalent in clinical isolates of E. coli Tf481A, in Japan (O'Hara et al., 1989; Kono et al., 1992; Taniguchi et al., 2004). Macrolide 2′‐phosphotransferases, commonly found on mobile genetic elements, are inducible (e.g. mph(A)) or constitutively expressed (e.g. mph(B)) intracellular enzymes capable of transferring the γ‐phosphate of nucleotide triphosphate to the desosamine 2′‐OH group of 14‐, 15‐, and 16‐membered ring macrolide antibiotics, thereby disrupting the key interaction of macrolides with A2058 (Shakya and Wright, 2010). Mphs can be divided into two classes distinguished by differences in primary sequence and substrate specificity, while their structures in complexes with many macrolides have been resolved in atomic resolution (Fong et al., 2017). Although early studies showed that Mph enzymes could use ATP, more recent work with Mph(A) has demonstrated a preference for GTP under physiologically relevant in vitro assay conditions (Shakya and Wright, 2010). Expression of mph(A) is induced by erythromycin, and recently, the structure of the MphR(A) repressor protein, a negative regulator of mph(A) expression, was solved, uncomplexed and complexed with erythromycin to 2.00 and 1.76 Å resolutions respectively (Zheng et al., 2009). Erythromycin binds with a stoichiometry of 1:1 to each monomer of the functional MphR(A) dimer in a large hydrophobic cavern composed of residues from an α‐helix of one monomer and the dimeric interface of the other monomer that appears too close around the ligand as it binds (Zheng et al., 2009). Macrolide phosphotransferases are widespread in bacteria of clinical, veterinary, agricultural and environmental origins. Genes encoding Mph enzymes are usually found on mobile genetic elements containing other macrolide resistance genes and genes conferring resistance to other antibiotic classes. The most recently identified macrolide phosphotransferase, mph(G), has been found in Vibrio spp. and photobacteria in the seawater of fish farms (Nonaka et al., 2015).
Glycosyltransferases modification
Glycosyl transfer is not a widespread mechanism of antibiotic resistance, although it certainly plays an important role in self‐protection of antibiotic‐producing organisms. Therefore, the antibiotic does not strongly interfere with the synthesizing machineries or the producer organism, which explains why not all antibiotic producers are resistant against their produced drug. Macrolide resistance due to 2΄‐glucosylation has not been reported yet in a bacterial pathogen but has been found in Streptomyces antibioticus, which produces the known macrolide oleandomycin (Vilches et al., 1992; Fyfe et al., 2016). In this macrolide self‐resistance mechanism, the intracellular glycosylation inactivates the antibiotic, and after secretion is reactivated by an extracellular‐glycosidase (Vilches et al., 1992). The glycosylation of macrolides is mediated by glycosyltransferases, which transfer activated donor sugars to acceptor species. These enzymes are grouped into families based on their sequence, and to date they display only two major folds defined as GT‐A and GT‐B (Lombard et al., 2014). The oleandomycin inactivation takes place by transfer of a glucose molecule from a donor/UDP‐glucose to oleandomycin, a process catalysed by an intracellular glucosyl transfer (Quirós and Salas, 1995). The extracellular glucosidase that activates oleandomycin, OleR, converts the glycosylated form of oleandomycin into the active antibiotic (Quirós et al., 1998). Two glycosyltransferase proteins OleI and OleD and one glucosidase OleR were isolated and studied from S. antibioticus, and a model was proposed for oleandomycin intracellular inactivation, secretion and extracellular reactivation (Quirós et al., 1998). Unlike OleI, which only glycosylates oleandomycin, OleD displays broad acceptor specificity and hence will inactivate a wider spectrum of macrolide antibiotics, including also tylosin and erythromycin (Coutinho et al., 2003).
Conclusions
The resistance of pathogens to antibiotics has become a serious and persistent therapeutic problem today, with a rapid development of new effective and safe antibiotics being the only answer to this problem. Macrolides are a family of valuable first choice antibiotics with great contribution to therapy, which is gradually becoming ineffective due to increasing resistance. Thus, the development of new generations of macrolides is required, as soon as possible. Additionally, this development has to be associated with safety issues to overcome problems related to the use of the last generation of macrolides (Kim et al., 2012; Telithromycin, 2014). In the beginning of the previous decade, there was a revolution concerning the development of new macrolides and a great enthusiasm prevailed after approval of telithromycin by the FDA. However, telithromycin usage was accompanied by serious side effects. This led to disappointment and caused many researchers to stop working on macrolide derivatization, assuming that this procedure had reached the end. In parallel, the discovery of modular macrolide polyketide synthases initiated efforts to alter the specificities and activities of the enzyme domains, for the purpose of changing the structure of the corresponding aglycone and/or the linked sugars (Khosla and Zawada, 1996; Khosla, 2009). This procedure permitted recombinant genes to be introduced into the macrolide producers in order to create desired changes to the structure of macrolides produced (Katz and McDaniel, 1999; McDaniel et al., 2005). The few fully elaborated novel macrolides produced by genetic engineering have not yet fulfilled the original promise (Park et al., 2010). Therefore, it is still too early to assess whether this avenue of discovery will be effective. The findings that many of engineered PKSs either do notproduce the expected compounds, or do so at levels too low to be useful, indicate that greater understanding of the biochemical details of polyketide biosynthesis is required before full exploitation of their chemical potential can be realized. Fortunately, this inability of genetic manipulation to rapidly produce new compounds designed on the huge amount of available crystal structure data has been recently overcome by chemical macrolide synthesis procedures, which have opened new horizons into the synthesis of novel macrolide compounds (Seiple et al., 2016). On the other hand, the recent resolution of the ‘ketek effects’ also provided answers to the second critical issue, namely, the safety of macrolides in clinical use (Andrade and Tulkens, 2011; Fernandes et al., 2016), and therefore, macrolide antibiotics are back in the forefront of science, and new, important, effective and safe macrolides are expected very soon to re‐enter the market.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Conflict of interest
The authors declare no conflicts of interest.
Dinos, G. P. (2017) The macrolide antibiotic renaissance. British Journal of Pharmacology, 174: 2967–2983. doi: 10.1111/bph.13936.
References
- Akshay S, Bertea M, Hobbie SN, Oettinghaus B, Shcherbakov D, Böttger EC et al. (2011). Phylogenetic sequence variations in bacterial rRNA affect species‐specific susceptibility to drugs targeting protein synthesis. Antimicrob Agents Chemother 55: 4096–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekshun MN, Levy SB (2007). Molecular mechanisms of antibacterial multidrug resistance. Cell 128: 1037–1050. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almutairi MM, Park SR, Rose S, Hansen DA, Vazquez‐Laslop N, Douthwaite S et al. (2015). Resistance to ketolide antibiotics by coordinated expression of rRNA methyltransferases in a bacterial producer of natural ketolides. Proc Natl Acad Sci U S A 112: 12956–12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Elcoro S, Yao JDC (2002). Antimicrobial macrolides in clinical practice In: Omura S. (ed). Macrolide Antibiotics, Chemistry, Biology and Practice, 2nd edn. Academic press: Orlando, FL, pp. 363–402. [Google Scholar]
- Andrade RJ, Tulkens PM (2011). Hepatic safety of antibiotics used in primary care. J Antimicrob Chemother 66: 1431–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti D, Bientinesi I, Biondi S, Donati D, Erbetti I, Lociuro S et al. (2007). A novel ketolide class: synthesis and antibacterial activity of a lead compound. Bioorg Med Chem Lett 17: 5265–5269. [DOI] [PubMed] [Google Scholar]
- Arenz S, Meydan S, Starosta AL, Berninghausen O, Beckmann R, Vazquez‐Laslop N et al. (2014a). Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol Cell 56: 446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenz S, Ramu H, Gupta P, Berninghausen O, Beckmann R, Vazquez‐Laslop N et al. (2014b). Molecular basis for erythromycin‐dependent ribosome stalling during translation of the ErmBL leader peptide. Nat Commun 5: 3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur M, Courvalin P (1986). Contribution of two different mechanisms to erythromycin resistance in Escherichia coli. Antimicrob Agents Chemother 30: 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur M, Autissier D, Courvalin P (1986). Analysis of the nucleotide sequence of the ereB gene encoding the erythromycin esterase type II. Nucleic Acids Res 14: 4987–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur M, Brisson‐Noel A, Courvalin P (1987). Origin and evolution of genes specifying resistance to macrolide, lincosamide and streptogramin antibiotics: data and hypotheses. J Antimicrob Chemother 20: 783–802. [DOI] [PubMed] [Google Scholar]
- Ashley GW, Burlingame M, Desai R, Fu H, Leaf T, Licari PJ et al. (2006). Preparation of erythromycin analogs having functional groups at C‐15. J Antibiot 59: 392–401. [DOI] [PubMed] [Google Scholar]
- Bahal N, Nahata MC (1992). The new macrolide antibiotics: azithromycin, clarithromycin, dirithromycin, and roxithromycin. Ann Pharmacother 26: 46–55. [DOI] [PubMed] [Google Scholar]
- Barry AL, Jones RN, Thornsberry C (1988). In vitro activities of azithromycin (CP 62,993), clarithromycin (A‐56268; TE‐031), erythromycin, roxithromycin, and clindamycin. Antimicrob Agents Chemother 32: 752–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AL, Fuchs PC, Brown SD (2001). Relative potency of telithromycin, azithromycin and erythromycin against recent clinical isolates of gram‐positive cocci. J Clin Microbiol Infect Dis 20: 494–497. [DOI] [PubMed] [Google Scholar]
- Barthelemy P, Autissier D, Gerbaud G, Courvalin P (1984). Enzymic hydrolysis of erythromycin by a strain of Escherichia coli. A new mechanism of resistance. J Antibiot (Tokyo) 37: 1692–1696. [DOI] [PubMed] [Google Scholar]
- Bébéar CM, Renaudin H, Aydin MD, Chantot JF, Bébéar C (1997). In‐vitro activity of ketolides against mycoplasmas. J Antimicrob Chemother 39: 669–670. [DOI] [PubMed] [Google Scholar]
- Benazzo M, Giacopini G, Oldini C, Scheiber E, Tombolini A, Mira E (1998). Flurithromycin versus clarithromycin in upper respiratory tract infections. Curr Ther Res 59: 28–38. [Google Scholar]
- Berisio R, Harms J, Schluenzen F, Zarivach R, Hansen HA, Fucini P et al. (2003). Structural insight into the antibiotic action of telithromycin against resistant mutants. J Bacteriol 185: 4276–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berisio R, Corti N, Pfister P, Yonath A, Böttger EC (2006). 23S rRNA A2058G alteration mediates ketolide resistance in combination with deletion in L22. Antimicrob Agents Chemother 50: 3816–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingen E, Leclercq R, Fitoussi F, Brahimi N, Malbruny B, Deforche D et al. (2002). Emergence of group A streptococcus strains with different mechanisms of macrolide resistance. Antimicrob Agents Chemother 46: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckman RK Jr, Pruitt JR (1989). A new, highly efficient, selective methodology for formation of medium‐ring and macrocyclic lactones via intramolecular ketene trapping: an application to a convergent synthesis of (−)‐kromycin. J Am Chem Soc 111: 8286–8288. [Google Scholar]
- Bommakanti AS, Lindahl L, Zengel JM (2008). Mutation from guanine to adenine in 25S rRNA at the position equivalent to E. coli A2058 does not confer erythromycin sensitivity in Sacchromyces cerevisae. RNA 14: 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borzani M, Varotto F, Garlaschi L, Conio F, Dell'Olio M, Careddu P (1989). Clinical and microbiological evaluation of miocamycin activity against group A beta‐hemolytic streptococci in pediatric patients. Three years' incidence of erythromycin‐resistant group A streptococci. J Chemother 1: 35–38. [DOI] [PubMed] [Google Scholar]
- Böttger EC, Springer B, Prammananan T, Kidan Y, Sander P (2001). Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep 2: 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright GM, Nagel AA, Bordner J, Desai KA, Dibrino JN, Nowakowska J et al. (1988). Synthesis, in vitro and in vivo activity of novel 9‐deoxo‐9a‐AZA‐9a‐homoerythromycin A derivatives; a new class of macrolide antibiotics, the azalides. J Antibiot (Tokyo) 41: 1029–1047. [DOI] [PubMed] [Google Scholar]
- Brockmann H, Hekel W (1951). Pikromycin, ein bitter schmeckendes antibioticum aus actinomyeceten. Chem Ber 84: 284–288. [Google Scholar]
- Brogden RN, Peters DH (1994). Dirithromycin. A review of its antimicrobial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 48: 599–616. [DOI] [PubMed] [Google Scholar]
- Bryskier A (2000). Ketolides‐telithromycin, an example of a new class of antibacterial agents. Clin Microbiol Infect 6: 661–669. [DOI] [PubMed] [Google Scholar]
- Bulkley D, Innis CA, Blaha G, Steitz TA (2010). Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci U S A 107: 17158–17163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdett V (1996). Tet(M)‐promoted release of tetracycline from ribosomes is GTP dependent. J Bacteriol 178: 3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton R, Mazzariol A, Morosini MI, Baquero F, Cornaglia G (2005). Telithromycin activity is reduced by efflux in Streptococcus pyogenes . J Antimicrob Chemother 55: 489–495. [DOI] [PubMed] [Google Scholar]
- Canu A, Malbruny B, Coquemont M, Davies TA, Appelbaum PC, Leclercq R (2002). Diversity of ribosomal mutations conferring resistance to macrolides, clindamycin, streptogramin, and telithromycin in Streptococcus pneumoniae . Antimicrob Agents Chemother 46: 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Zhao CJ, Yin YD, Zhao F, Song SF, Bai L et al. (2010). High prevalence of macrolide resistance in Mycoplasma pneumoniae isolates from adult and adolescent patients with respiratory tract infection in China. Clin Infect Dis 51: 189–194. [DOI] [PubMed] [Google Scholar]
- Champney WS, Miller M (2002). Inhibition of 50S ribosomal subunit assembly in Haemophilus influenzae cells by azithromycin and erythromycin. Curr Microbiol 44: 418–424. [DOI] [PubMed] [Google Scholar]
- Champney WS, Pelt J (2002). The ketolide antibiotic ABT‐773 is a specific inhibitor of translation and 50S ribosomal subunit formation in Streptococcus pneumoniae cells. Curr Microbiol 45: 155–160. [DOI] [PubMed] [Google Scholar]
- Chancey ST, Bai X, Kumar N, Drabek EF, Daugherty SC, Colon T et al. (2015). Transcriptional attenuation controls macrolide inducible efflux and resistance in Streptococcus pneumoniae and in other Gram‐positive bacteria containing mef/mel(msr(D)) elements. PLoS One 10 e0116254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantot JF, Bryskier A, Gasc JC (1986). Antibacterial activity of roxithromycin: a laboratory evaluation. J Antibiot (Tokyo) 39: 660–668. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu P, Xu Y, Liu L, Liu Y, Zhu D et al. (2012). Synthesis and antibacterial activity of novel modified 5‐O‐desosamine ketolides. Bioorg Med Chem Lett 22: 7402–7405. [DOI] [PubMed] [Google Scholar]
- Chittum HS, Champney WS (1995). Erythromycin inhibits the assembly of the large ribosomal subunit in growing Escherichia coli cells. Curr Microbiol 30: 273–279. [DOI] [PubMed] [Google Scholar]
- Connell SR, Trieber CA, Dinos G, Einfeldt E, Taylor DE, Nierhaus KH (2003). Mechanism of Tet(O)‐mediated tetracycline resistance. EMBO J 22: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran JW (1984). Mode of action and resistance mechanisms of macrolides In: Omura S. (ed). Macrolide Antibiotics: Chemistry, Biology and Practice, 1st edn. Academic press: Orlando, FL USA. [Google Scholar]
- Counter FT, Ensminger PW, Preston DA, Wu CY, Greene JM, Felty‐Duckworth AM et al. (1991). Synthesis and antimicrobial evaluation of dirithromycin (AS‐E 136; LY237216), a new macrolide antibiotic derived from erythromycin. Antimicrob Agents Chemother 35: 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B (2003). An evolving hierarchical family classification for glycosyltransferases. J Mol Biol 328: 307–317. [DOI] [PubMed] [Google Scholar]
- Davis AR, Gohara DW, Yap MN (2014). Sequence selectivity of macrolide‐induced translational attenuation. Proc Natl Acad Sci U S A 111: 15379–15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debono M, Willard KE, Kirst HA, Wind JA, Crouse GD, Tao EV et al. (1989). Synthesis and antimicrobial evaluation of 20‐deoxo‐20‐(3,5‐dimethylpiperidin‐1‐yl)desmycosin (tilmicosin, EL‐870) and related cyclic amino derivatives. J Antibiot (Tokyo) 42: 1253–1267. [DOI] [PubMed] [Google Scholar]
- Denis A, Agouridas C, Auger JM, Benedetti Y, Bonnefoy A, Bretin F et al. (1999). Synthesis and antibacterial activity of HMR 3647 a new ketolide highly potent against erythromycin‐resistant and susceptible pathogens. Bioorg Med Chem Lett 9: 3075–3080. [DOI] [PubMed] [Google Scholar]
- Denis A, Bonnefoy A (2001). Novel fluoroketolides: synthesis and antibacterial activity. Drug Future 26: 975–984. [Google Scholar]
- Denoya C, Dubnau D (1989). Mono‐ and dimethylating activities and kinetic studies of the ermC 23 S rRNA methyltransferase. J Biol Chem 264: 2615–2624. [PubMed] [Google Scholar]
- Di Giambattista M, Engelborghs Y, Nyssen E, Cocito C (1987). Kinetics of binding of macrolides, lincosamides, and synergimycins to ribosomes. J Biol Chem 262: 8591–8597. [PubMed] [Google Scholar]
- Dinos G, Kalpaxis D (2000). Studies on the interaction between a ribosomal complex active in peptide bond formation and the macrolide antibiotics tylosin and erythromycin. Biochemistry 39: 11621–11628. [DOI] [PubMed] [Google Scholar]
- Dinos G, Connell S, Nierhaus K, Kalpaxis D (2003). Erythromycin, roxithromycin and clarithromycin: use of slow‐binding kinetics to compare their in vitro interaction with a ribosomal complex active in peptide bond formation. Mol Pharmacol 63: 617–623. [DOI] [PubMed] [Google Scholar]
- Doern GV (2006). Macrolide and ketolide resistance with Streptococcus pneumoniae . Med Clin North Am 90: 1109–1124. [DOI] [PubMed] [Google Scholar]
- Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L (1991). Modular organization of genes required for complex polyketide biosynthesis. Science 252: 675–679. [DOI] [PubMed] [Google Scholar]
- Donin MN, Pagano J, Dutcher JD, McKee CM (1953. ‐1954). Methymycin, a new crystalline antibiotic. Antibiot Annu 1: 179–185. [Google Scholar]
- Doucet‐Populaire F, Capobianco JO, Zakula D, Jarlier V, Goldman RC (1998). Molecular basis of clarithromycin activity against Mycobacterium avium and Mycobacterium smegmatis . J Antimicrob Chemother 41: 179–187. [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Xiong L, Mankin AS, Cate JH (2010). Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci U S A 107: 17152–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccone D, Andres P, Galas M, Tokumoto M, Rosato A, Corso A (2005). Emergence of a Streptococcus pneumoniae clinical isolate highly resistant to telithromycin and fluoroquinolones. J Clin Microbiol 43: 5800–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell DJ, Douthwaite S, Morrissey I, Bakke S, Poehlsgaard J, Jakobsen L et al. (2003). Macrolide resistance by ribosomal mutation in clinical isolates of Streptococcus pneumoniae from the PROTEKT 1999–2000 study. Antimicrob Agents Chemother 47: 1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell DJ, Morrissey I, Bakker S, Buckridge S, Felmingham D (2004). In vitro activities of telithromycin, linezolid, and quinupristin–dalfopristin against Streptococcus pneumoniae with macrolide resistance due to ribosomal mutations. Antimicrob Agents Chemother 48: 3169–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell DJ, Mendes RE, Jones RN (2015). Antimicrobial activity of solithromycin against serotyped macrolide‐resistant Streptococcus pneumoniae isolates collected from U.S. medical centers in 2012. Antimicrob Agents Chemother 59: 2432–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell DJ, Flamm RK, Sade HS, Jones RN (2016). Results from the Solithromycin International Surveillance Program. Antimicrob Agents Chemother 60: 3662–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmingham D, Cantón R, Jenkins SG (2007). Regional trends in beta‐lactam, macrolide, fluoroquinolone and telithromycin resistance among Streptococcus pneumoniae isolates 2001‐2004. J Infect 55: 111–118. [DOI] [PubMed] [Google Scholar]
- Fernandes PB, Hardy DJ (1988). Comparative in vitro potencies of nine new macrolides. Drugs Exp Clin Res 14: 445–451. [PubMed] [Google Scholar]
- Fernandes P, Martens E, Bertrand D, Pereira D (2016). The solithromycin journey—it is all in the chemistry. Bioorg Med Chem 24: 6420–6428. [DOI] [PubMed] [Google Scholar]
- Fong DH, Burk DL, Blanchet J, Yan AY, Berghuis AM (2017). Structural basis for kinase‐mediated macrolide antibiotic resistance. Structure 25: 750–761.e5. [DOI] [PubMed] [Google Scholar]
- Foulds G, Shepard RM, Johnson RB (1990). The pharmacokinetics of azithromycin in human serum and tissues. J Antimicrob Chemother 25 (Suppl A): 73–82. [DOI] [PubMed] [Google Scholar]
- Franceschi F, Kanyo Z, Sherer EC, Sutcliffe J (2004). Macrolide resistance from the ribosome perspective. Curr Drug Targets Infect Disord 4: 177–191. [DOI] [PubMed] [Google Scholar]
- Fu H, Marquez S, Gu X, Katz L, Myles DC (2006). Synthesis and in vitro antibiotic activity of 16‐membered 9‐O‐arylalkyloxime macrolides. Bioorg Med Chem Lett 16: 1259–1266. [DOI] [PubMed] [Google Scholar]
- Fyfe C, Grossman TH, Kerstein K, Sutcliffe J (2016). Resistance to macrolide antibiotics in public health pathogens. Cold Spring Harb Perspect Med 6 pii: a025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza‐Ramos G, Xiong L, Zhong P, Mankin A (2002). Binding site of macrolide antibiotics on the ribosome: new resistance mutation identifies a specific interaction of ketolides with rRNA. J Bacteriol 183: 6898–6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gäumann E, Hütter R, Keller‐Schierlein W, Neipp L, Prelog V, Zähner H (1960). Lankamycin und lankacidin. Helv Chim Acta 80: 601–606. [Google Scholar]
- Georgopapadakou N (2014). The wobbly status of ketolides: where do we stand? Expert Opin Investig Drugs 23: 1313–1319. [DOI] [PubMed] [Google Scholar]
- Gialdroni‐Grassi G, Alesina R, Bersani C, Ferrara A, Fietta A, Peona V (1986). In vitro activity of flurithromycin, a novel macrolide antibiotic. Chemioterapia 5: 177–184. [PubMed] [Google Scholar]
- Girard AE, Girard D, English AR, Gootz TD, Cimochowski CR, Faiella JA et al. (1987). Pharmacokinetic and in vivo studies with azithromycin (CP‐62,993), a new macrolide with an extended half‐life and excellent tissue distribution. Antimicrob Agents Chemother 31: 1948–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryczan TJ, Grandi G, Hahn J, Grandi R, Dubnau D (1980). Conformational alteration of mRNA structure and the posttranscriptional regulation of erythromycin‐induced drug resistance. Nucleic Acids Res 8: 6081–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Kannan K, Mankin A, Vazquez‐Laslop N (2013). Regulation of gene expression by macrolide‐induced ribosomal frameshifting. Mol Cell 52: 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haefner M, Funke‐Kissling P, Pfyffer GE, Luthy R, Opravil M (1999). Clarithromycin, rifabutin and clofazimine for treatment of disseminated Mycobacterium avium complex disease in AIDS patients. Clin Drug Investig 17: 171–178. [Google Scholar]
- Hamill RL, Haney ME, Stamper M, Wiley PF (1961). Tylosin a new antibiotic. II. Isolation, properties and preparation of desmycosin, a microbiologically active degradation product. Antibiot Chemother (Northfield) 11: 328–334. [PubMed] [Google Scholar]
- Hammerschlag MR, Roblin PM, Bebear CM (2001). Activity of telithromycin, a new ketolide antibacterial, against atypical and intracellular respiratory pathogens. J Antimicrob Chemother 48 (Suppl T1): 25–31. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB, Steitz TA (2002). The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol Cell 10: 117–128. [DOI] [PubMed] [Google Scholar]
- Hardy DJ, Guay DR, Jones RN (1992). Clarithromycin, a unique macrolide. A pharmacokinetic, microbiological, and clinical overview. Diagn Microbiol Infect Dis 15: 39–53. [DOI] [PubMed] [Google Scholar]
- Horinouchi S, Weisblum B (1980). Posttranscriptional modification of mRNA conformation: mechanism that regulates erythromycin‐induced resistance. Proc Natl Acad Sci U S A 77: 7079–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber G, Wallhaeusser KH, Fries L, Steigler A, Weidenmuller H (1962). Niddamycin, a new macrolide antibiotic. Arzneimittel‐Forsch 12: 1191–1195. [PubMed] [Google Scholar]
- Hunziker D, Wyss PC, Angehrn P, Mueller A, Marty HP, Halm R et al. (2004). Novel ketolide antibiotics with a fused five‐membered lactone ring‐synthesis, physicochemical and antimicrobial properties. Bioorg Med Chem 12: 3503–3519. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009). Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki T, Nomura T, Narukawa Y, Uotani K (2005). A simple method for deprotection of the N‐ and O‐carbobenzoxy groups and N‐methylation of the desosamine sugar moiety of ketolides. Application to the synthesis of ketolide analogues with various 9‐iminoether moieties and their antibacterial activities. J Antibiot (Tokyo) 58: 679–685. [DOI] [PubMed] [Google Scholar]
- Jain R, Danziger LH (2004). The macrolide antibiotics: a pharmacokinetic and pharmacodynamic overview. Curr Pharm Des 10: 3045–3053. [DOI] [PubMed] [Google Scholar]
- Jensen JS, Fernandes P, Unemo M (2014). In vitro activity of the new fluoroketolide solithromycin (CEM‐101) against macrolide‐resistant and ‐susceptible Mycoplasma genitalium strains. Antimicrob Agents Chemother 58: 3151–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Mankin AS (2011). Macrolide antibiotics in the ribosome exit tunnel: species‐specific binding and action. Ann N Y Acad Sci 1241: 33–47. [DOI] [PubMed] [Google Scholar]
- Kannan K, Vazquez‐Laslop N, Mankin AS (2012). Selective protein synthesis by ribosomes with a drug‐obstructed exit tunnel. Cell 151: 508–520. [DOI] [PubMed] [Google Scholar]
- Kannan K, Kanabar P, Schryer D, Florin T, Oh E, Bahroos N et al. (2014). The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci U S A 111: 15958–15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karahalios P, Kalpaxis D, Fu H, Katz L, Wilson D, Dinos G (2006). On the mechanism of action of 9‐O‐arylalkyloxime derivatives of 6‐O‐mycaminosyltylonolide, a new class of 16‐membered macrolide antibiotics. Mol Pharmacol 70: 1271–1280. [DOI] [PubMed] [Google Scholar]
- Katz L, Brown D, Boris K, Tuan J (1987). Expression of the macrolide–lincosamide–streptogramin‐B‐resistance methylase gene, ermE, from Streptomyces erythraeus in Escherichia coli results in N6‐monomethylation and N6, N6‐dimethylation of ribosomal RNA. Gene 55: 319–325. [DOI] [PubMed] [Google Scholar]
- Katz L, McDaniel R (1999). Novel macrolides through genetic engineering. Med Res Rev 19: 543–558. [DOI] [PubMed] [Google Scholar]
- Katz L, Ashley GW (2005). Translation and protein synthesis: macrolides. Chem Rev 105: 499–528. [DOI] [PubMed] [Google Scholar]
- Khosla C (2009). Structures and mechanisms of polyketide synthases. J Org Chem 74: 6416–6420. [DOI] [PubMed] [Google Scholar]
- Khosla C, Zawada RJ (1996). Generation of polyketide libraries via combinatorial biosynthesis. Trends Biotechnol 14: 335–341. [DOI] [PubMed] [Google Scholar]
- Kim SH, Song JH, Chung DR, Thamlikitkul V, Yang Y, Wang H et al. (2012). Changing trends in antimicrobial resistance and serotypes of Streptococcus pneumoniae isolates in Asian countries: an Asian Network for Surveillance of Resistant Pathogens (ANSORP) study. Antimicrob Agents Chemother 56: 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Cha CJ, Cerniglia CE (2002). Purification and characterization of an erythromycin esterase from an erythromycin resistant Pseudomonas sp. FEMS Microbiol Lett 210: 239–244. [DOI] [PubMed] [Google Scholar]
- Kirst HA (1995). Dirithromycin: introduction and historical development. Drugs Today 31: 89–97. [Google Scholar]
- Kono M, O'Hara K, Ebisu T (1992). Purification and characterization of macrolide 2′‐phosphotransferase type II from a strain of Escherichia coli highly resistant to macrolide antibiotics. FEMS Microbiol Lett 76: 89–94. [DOI] [PubMed] [Google Scholar]
- Kouvela EC, Kalpaxis DL, Wilson DN, Dinos G (2009). A distinct mode of interaction of a novel ketolide antibiotic that displays enhanced antimicrobial activity. Antimicrob Agents Chemother 53: 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokidis M, Bougas A, Stavropoulou M, Kalpaxis D, Dinos G (2016). The slow dissociation rate of K‐1602 contributes to the enhanced inhibitory activity of this novel alkyl–aryl‐bearing fluoroketolide. J Enzyme Inhib Med Chem 31: 276–282. [DOI] [PubMed] [Google Scholar]
- Krokidis M, Márquez V, Wilson DN, Kalpaxis D, Dinos G (2014). Insights into the mode of action of novel fluoroketolides, potent inhibitors of bacterial protein synthesis. Antimicrob Agents Chemother 58: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JH, Choi EC, Weisblum B (1991). Transcriptional attenuation control of ermK, a macrolide–lincosamide–streptogramin B resistance determinant from Bacillus licheniformis . J Bacteriol 173: 4725–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon AR, Min YH, Yoon EJ, Kim JA, Shim MJ, Choi EC (2006). ErmK leader peptide: amino acid sequence critical for induction by erythromycin. Arch Pharm Res 29: 1154–1157. [DOI] [PubMed] [Google Scholar]
- Law CJ, Maloney PC, Wang DN (2008). Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62: 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GW, Oh E, Weissman JS (2012). The anti‐Shine‐Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484: 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CH, Yao SL, Chiu YH, Leung PY, Robert N, Seddon J et al. (2005). Synthesis and biological activity of new 5‐O‐sugar modified ketolide and 2‐fluoro‐ketolide antibiotics. Bioorg Med Chem Lett 15: 1307–1310. [DOI] [PubMed] [Google Scholar]
- Liang JH, Han X (2013). Structure–activity relationships and mechanism of action of macrolides derived from erythromycin as antibacterial agents. Curr Top Med Chem 13: 3131–3164. [DOI] [PubMed] [Google Scholar]
- Liu M, Douthwaite S (2002). Activity of the ketolide telithromycin is refractory to Erm monomethylation of bacterial rRNA. Antimicrob Agents Chemother 46: 1629–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014). The carbohydrate‐active enzymes database (CAZy) in 2013. Nucleic Acids Res 42 (Database issue): D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounnas V, Ritschel T, Kelder J, McGuire R, Bywater RP, Foloppe N (2013). Current progress in structure‐based rational drug design marks a new mindset in drug discovery. Comput Struct Biotechnol J 5 e201302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Clark RF, Brazzale A, Wang S, Rupp MJ, Li L et al. (2001). Novel erythromycin derivatives with aryl groups tethered to the C‐6 position are potent protein synthesis inhibitors and active against multidrug‐resistant respiratory pathogens. J Med Chem 44: 4137–4156. [DOI] [PubMed] [Google Scholar]
- Mankin AS (2008). Macrolide myths. Curr Opin Microbiol 11: 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Janosi L, Endou K, Nakajima Y (1999). Cloning and sequences of inducible and constitutive macrolide resistance genes in Staphylococcus aureus that correspond to an ABC transporter. FEMS Microbiol Lett 181: 91–100. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Inoue M, Endo Y, Nakajima Y (2003). Characteristic expression of three genes, msr(A), mph(C) and erm(Y), that confer resistance to macrolide antibiotics on Staphylococcus aureus . FEMS Microbiol Lett 220: 287–293. [DOI] [PubMed] [Google Scholar]
- McDaniel R, Welch M, Hutchinson CR (2005). Genetic approaches to polyketide antibiotics. Chem Rev 105: 543–558. [DOI] [PubMed] [Google Scholar]
- McGuire JM, Bunch RL, Anderson RC, Boaz HE, Flynn EH, Powell HM et al. (1952). Ilotycin, a new antibiotic. Antibiot Chemother (Northfield) 2: 281–283. [PubMed] [Google Scholar]
- Menninger JR (1995). Mechanism of inhibition of protein synthesis by macrolide and lincosamide antibiotics. J Basic Clin Physiol Pharmacol 6: 229–250. [DOI] [PubMed] [Google Scholar]
- Menninger JR, Otto DP (1982). Erythromycin, carbomycin, and spiramycin inhibit protein synthesis by stimulating the dissociation of peptidyl‐tRNA from ribosomes. Antimicrob Agents Chemother 21: 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton ID, Hewitt CL, Harwood CR (1992). Cloning and sequencing of a plasmid‐mediated erythromycin resistance determinant from Staphylococcus xylosus . FEMS Microbiol Lett 76: 141–147. [DOI] [PubMed] [Google Scholar]
- Misyurina OY, Chipitsyna EV, Finashutina YP, Lazarev VN, Akopian TA, Savicheva AM et al. (2004). Mutations in a 23S rRNA gene of Chlamydia trachomatis associated with resistance to macrolides. Antimicrob Agents Chemother 48: 1347–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morar M, Pengelly K, Koteva K, Wright GD (2012). Mechanism and diversity of the erythromycin esterase family of enzymes. Biochemistry 51: 1740–1751. [DOI] [PubMed] [Google Scholar]
- Mordi MN, Pelta MD, Boote V, Morris GA, Barber J (2000). Acid‐catalyzed degradation of clarithromycin and erythromycin B: a comparative study using NMR spectroscopy. J Med Chem 43: 467–474. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Takahashi Y, Watanabe Y, Omura S (1984). Chemical modification of erythromycins. I. Synthesis and antibacterial activity of 6‐O‐methylerythromycins A. J Antibiot (Tokyo) 37: 187–189. [DOI] [PubMed] [Google Scholar]
- Morrison JF, Walsh C (1988). The behavior and significance of slow binding inhibitors. Adv Enzymol Relat Areas Mol Biol 61: 201–301. [DOI] [PubMed] [Google Scholar]
- Morozumi M, Iwata S, Hasegawa K, Chiba N, Takayanagi R, Matsubara K et al. (2008). Increased macrolide resistance of Mycoplasma pneumoniae in pediatric patients with community‐acquired pneumonia. Antimicrob Agents Chemother 52: 348–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, Itai S, Yoshida T, Nagai T (1992). Physicochemical properties and stability in the acidic solution of a new macrolide antibiotic, clarithromycin, in comparison with erythromycin. Chem Pharm Bull (Tokyo) 40: 725–728. [DOI] [PubMed] [Google Scholar]
- Nakayama I (1984). Macrolides in clinical practice In: Omura S. (ed). Macrolide Antibiotics: Chemistry, Biology and Practice, 1st edn. Academic press: Orlando, FL USA. [Google Scholar]
- Nomura T, Iwaki T, Yasukata T, Nishi K, Narukawa Y, Uotani K et al. (2005). A new type of ketolides bearing an N‐aryl–alkyl acetamide moiety at the C‐9 iminoether synthesis and structure–activity relationships. Bioorg Med Chem 13: 6615–6628. [DOI] [PubMed] [Google Scholar]
- Nomura T, Iwaki T, Narukawa Y, Uotani K, Hori T, Miwa H (2006). A new type of ketolide bearing an N‐aryl–alkyl acetamide moiety at the C‐9 iminoether: synthesis and structure–activity relationships (2). Bioorg Med Chem 14: 3697–3711. [DOI] [PubMed] [Google Scholar]
- Nonaka L, Maruyama F, Suzuki S, Masuda M (2015). Novel macrolide‐resistance genes, mef(C) and mph(G), carried by plasmids from Vibrio and Photobacterium isolated from sediment and seawater of a coastal aquaculture site. Lett Appl Microbiol 61: 1–6. [DOI] [PubMed] [Google Scholar]
- Nunez‐Samudio V, Chesneau O (2013). Functional interplay between theATP binding cassette Msr(D) protein and the membrane facilitator superfamily Mef(E) transporter for macrolide resistance in Escherichia coli . Res Microbiol 164: 226–235. [DOI] [PubMed] [Google Scholar]
- O'Hara K, Kanda T, Ohmiya K, Ebisu T, Kono M (1989). Purification and characterization of macrolide 2′‐phosphotransferase from a strain of Escherichia coli that is highly resistant to erythromycin. Antimicrob Agents Chemother 33: 1354–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo KK, Ulep C, Van Kirk N, Luis H, Bernardo M, Leitao J et al. (2004). The mef(A) gene predominates among seven macrolide resistance genes identified in Gram‐negative strains representing 13 genera, isolated from healthy Portuguese children. Antimicrob Agents Chemother 48: 3451–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo KK, Striplin MJ, Ulep C, Close NS, Zittle J, Luis H et al. (2006). Staphylococcus efflux msr(A) gene characterized in Streptococcus, Enterococcus, Corynebacterium, and Pseudomonas isolates. Antimicrob Agents Chemother 50: 1089–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto S, Iwamatsu K, Inouye S, Niida T (1976). Modifications of a macrolide antibiotic midecamycin (SF‐837). I. Synthesis and structure of 9,3″‐diacetylmidecamycin. J Antibiot (Tokyo) 29: 536–548. [DOI] [PubMed] [Google Scholar]
- Omura S (2002). Macrolide Antibiotics: Chemistry, Biology and Practice, 2nd edn. Academic press: Orlando, FL USA. [Google Scholar]
- Omura S, Morimoto S, Nagate T, Adachi T, Kohno Y (1992). Research and development of clarithromycin. Yakugaku Zasshi 112: 593–614. [DOI] [PubMed] [Google Scholar]
- Or YS, Clark RF, Wang S, Chu DT, Nilius AM, Flamm RK et al. (2000). Design, synthesis, and antimicrobial activity of 6‐O‐substituted ketolides active against resistant respiratory tract pathogens. J Med Chem 43: 1045–1049. [DOI] [PubMed] [Google Scholar]
- Otaka T, Kaji A (1975). Release of (oligo) peptidyl‐tRNA from ribosomes by erythromycin A. Proc Natl Acad Sci U S A 72: 2649–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounissi H, Courvalin P (1985). Nucleotide sequence of the gene ereA encoding the erythromycin esterase in Escherichia coli . Gene 35: 271–278. [DOI] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH Jr (1998). Major facilitator superfamily. Microbiol Mol Biol Rev 62: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Han AR, Ban YH, Yoo YJ, Kim EJ, Yoon YJ (2010). Genetic engineering of macrolide biosynthesis: past advances, current state, and future prospects. Appl Microbiol Biotechnol 85: 1227–1239. [DOI] [PubMed] [Google Scholar]
- Pavlova A, Gumbart JC (2015). Parametrization of macrolide antibiotics using the force field toolkit. J Comput Chem 36: 2052–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova A, Parks JM, Oyelere AK, Gumbart JC (2017). Toward the rational design of macrolide antibiotics to combat resistance. Chem Biol Drug Des . https://doi.org/10.1111/cbdd.13004. [DOI] [PubMed] [Google Scholar]
- Peric M, Bozdogan B, Jacobs MR, Appelbaum PC (2003). Effects of an efflux mechanism and ribosomal mutations on macrolide susceptibility of Haemophilus influenza clinical isolates. Antimicrob Agents Chemother 47: 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira D, Fernandes P (2011). Synthesis and antibacterial activity of novel 4‐aryl‐[1,2,3]‐triazole containing macrolides. Bioorg Med Chem Lett 21: 510–513. [DOI] [PubMed] [Google Scholar]
- Pestka S, LeMahieu RA (1974). Inhibition of [14C]chloramphenicol binding to Escherichia coli ribosomes by erythromycin derivatives. Antimicrob Agents Chemother 6: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister P, Corti N, Hobbie S, Bruell C, Zarivach R, Yonath A et al. (2005). 23S rRNA base pair 2057‐2611 determines ketolide susceptibility and fitness cost of the macrolide resistance mutation 2058A‐‐>G. Proc Natl Acad Sci U S A 102: 5180–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlajamaki M, Kataja J, Seppala H, Elliot J, Leinonen M, Huovinen P et al. (2002). Ribosomal mutations in Streptococcus pneumoniae clinical isolates. Antimicrob Agents Chemother 46: 654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscitelli SC, Danziger LH, Rodvold KA (1992). Clarithromycin and azithromycin: new macrolide antibiotics. Clin Pharm 11: 137–152. [PubMed] [Google Scholar]
- Plata DJ, Leanna MR, Rasmussen M, McLaughlin MA, Condon SL, Kerdesky FAJ et al. (2004). The synthesis of ketolide antibiotic ABT‐773 (cethromycin). Tetrahedron 60: 10171–10180. [Google Scholar]
- Poehlsgaard J, Douthwaite S (2003). Macrolide antibiotic interaction and resistance on the bacterial ribosome. Curr Opin Investig Drugs 4: 140–148. [PubMed] [Google Scholar]
- Portillo A, Ruiz‐Larrea F, Zarazaga M, Alonso A, Martinez JL, Torres C (2000). Macrolide resistance genes in Enterococcus spp. Antimicrob. Agents Chemother 44: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier AL, Trong HN, Tande D, Segond C, Leclercq R (2005). Mutation of L4 ribosomal protein conferring unusual macrolide resistance in two independent clinical isolates of Staphylococcus aureus . Microb Drug Resist 11: 18–20. [DOI] [PubMed] [Google Scholar]
- Putnam SD, Sader HS, Farrell DJ, Biedenbach DJ, Castanheira M (2011). Antimicrobial characterisation of solithromycin (CEM‐101), a novel fluoroketolide: activity against staphylococci and enterococci. Int J Antimicrob Agents 37: 39–45. [DOI] [PubMed] [Google Scholar]
- Quirós LM, Aguirrezabalaga I, Olano C, Méndez C, Salas JA (1998). Two glycosyltransferases and a glycosidase are involved in oleandomycin modification during its biosynthesis by Streptomyces antibioticus. Mol Microbiol 28: 1177–1185. [DOI] [PubMed] [Google Scholar]
- Quirós LM, Salas JA (1995). Biosynthesis of the macrolide oleandomycin by Streptomyces antibioticus. Purification and kinetic characterization of an oleandomycin glucosyltransferase. J Biol Chem 270: 18234–18239. [DOI] [PubMed] [Google Scholar]
- Ramu H, Mankin A, Vazquez‐Laslop N (2009). Programmed drug‐ dependent ribosome stalling. Mol Microbiol 71: 811–824. [DOI] [PubMed] [Google Scholar]
- Ramu H, Vazquez‐Laslop N, Klepacki D, Dai Q, Piccirilli J, Micura R et al. (2011). Nascent peptide in the ribosome exit tunnel affects functional properties of the A‐site of the peptidyl transferase center. Mol Cell 41: 321–330. [DOI] [PubMed] [Google Scholar]
- Retsema JA, Girard AE, Girard D, Milisen WB (1990). Relationship of high tissue concentrations of azithromycin to bactericidal activity and efficacy in vivo. J Antimicrob Chemother 25 (Suppl A): 83–89. [DOI] [PubMed] [Google Scholar]
- Retsema J, Girard A, Schelkly W, Manousos M, Anderson M, Bright G et al. (1987). Spectrum and mode of action of azithromycin (CP‐62,993), a new 15‐membered‐ring macrolide with improved potency against gram‐negative organisms. Antimicrob Agents Chemother 31: 1939–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ED, Cove JH (2005). Resistance to telithromycin is conferred by msr(A), msrC and msr(D) in Staphylococcus aureus . J Antimicrob Chemother 56: 1179–1180. [DOI] [PubMed] [Google Scholar]
- Roberts MC, Sutcliffe J, Courvalin P, Jensen LB, Rood J, Seppala H (1999). Nomenclature for macrolide and macrolide–lincosamide–streptogramin B resistance determinants. Antimicrob Agents Chemother 43: 2823–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MC (2008). Update on macrolide–lincosamide–streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett 282: 147–159. [DOI] [PubMed] [Google Scholar]
- Rodvold KA (1999). Clinical pharmacokinetics of clarithromycin. Clin Pharmacokinet 37: 385–398. [DOI] [PubMed] [Google Scholar]
- Romero A, Liang CH, Chiu YH, Yao SL, Duffield J, Sucheck SJ et al. (2005). An efficient entry to new sugar modified ketolide antibiotics. Tetrahedron Lett 46: 1483–1487. [Google Scholar]
- Ross JI, Eady EA, Cove JH, Cunliffe WJ, Baumberg S, Wootton JC (1990). Inducible erythromycin resistance in staphylococci, is encoded by a member of the ATP‐binding transport super‐gene family. Mol Microbiol 4: 1207–1214. [DOI] [PubMed] [Google Scholar]
- Ross JI, Eady EA, Cove JH, Baumberg S (1995). Identification of a chromosomally encoded ABC‐transport system with which the Staphylococcal erythromycin exporter MsrA may interact. Gene 153: 93–98. [DOI] [PubMed] [Google Scholar]
- Ruan ZX, Huangfu DS, Xu XJ, Sun PH, Chen WM (2013). 3D‐QSAR and molecular docking for the discovery of ketolide derivatives. Expert Opin Drug Discov 8: 427–444. [DOI] [PubMed] [Google Scholar]
- Sakakibara H, Okekawa O, Fujiwara T, Otani M, Omura S (1981). Acyl derivatives of 16‐membered macrolides. I. Synthesis and biological properties of 3″‐O‐propionylleucomycin A5 (TMS‐19‐Q). J Antibiot (Tokyo) 34: 1001–1010. [DOI] [PubMed] [Google Scholar]
- Schlünzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R et al. (2001). Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 413: 814–821. [DOI] [PubMed] [Google Scholar]
- Schlünzen F, Harms JM, Franceschi F, Hansen HA, Bartels H, Zarivach R et al. (2003). Structural basis for the antibiotic activity of ketolides and azalides. Structure 11: 329–338. [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila‐Sanjurjo A, Holton JM et al. (2005). Structures of the bacterial ribosome at 3.5 A resolution. Science 310: 827–834. [DOI] [PubMed] [Google Scholar]
- Seiple IB, Zhang Z, Jakubec P, Langlois‐Mercier A, Wright PM, Hog DT et al. (2016). A platform for the discovery of new macrolide antibiotics. Nature 533: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya T, Wright GD (2010). Nucleotide selectivity of antibiotic kinases. Antimicrob Agents Chemother 54: 1909–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey LK, Edwards TA, O'Neill AJ (2016). ABC‐F proteins mediate antibiotic resistance through ribosomal protection. MBio 7 e01975‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SJ, Abbanat D, Ashley GW, Bush K, Foleno B, Macielag M et al. (2005). 15‐Amido erythromycins: synthesis and in vitro activity of a new class of macrolide antibiotics. J Antibiot (Tokyo) 58: 167–177. [DOI] [PubMed] [Google Scholar]
- Shimada Y, Deguchi T, Nakane K, Yasuda M, Yokoi S, Ito S et al. (2011). Macrolide resistance‐associated 23S rRNA mutation in Mycoplasma genitalium, Japan. Emerg Infect Dis 17: 1148–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortridge VD, Zhong P, Cao Z, Beyer JM, Almer LS, Ramer NC et al. (2002). Comparison of in vitro activities of ABT‐773 and telithromycin against macrolide‐susceptible and ‐resistant Streptococci and Staphylococci . Antimicrob Agents Chemother 46: 783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siibak T, Peil L, Xiong L, Mankin A, Remme J, Tenson T (2009). Erythromycin‐ and chloramphenicol‐induced ribosomal assembly defects are secondary effects of protein synthesis inhibition. Antimicrob Agents Chemother 53: 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobin B, English AR, Celmer WD (1955). PA‐105 a new antibiotic. Antibiot Annu 1954‐1955. New York, 1955, Medical Encyclopedia Inc, pp. 827–830. [Google Scholar]
- Sothiselvam S, Liu B, Han W, Ramu H, Klepacki D, Atkinson GC et al. (2014). Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proc Natl Acad Sci U S A 111: 9804–9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sothiselvam S, Neuner S, Rigger L, Klepacki D, Micura R, Vázquez‐Laslop N et al. (2016). Binding of macrolide antibiotics leads to ribosomal selection against specific substrates based on their charge and size. Cell Rep 16: 1789–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam SL, Ramu H, Mankin AS (2011). Inducible resistance to macrolide antibiotics In: Dougherty TJ, Pucci MJ. (eds). Antibiotic Drug Discovery and Development. Springer: New York, pp. 455–484. [Google Scholar]
- Sugimoto T, Tanikawa T (2010). Synthesis and antibacterial activity of a novel class of 15‐membered macrolide antibiotics, “11a‐Azalides”. ACS Med Chem Lett 2: 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe J, Leclercq R (2002). Mechanisms of resistance to macrolides, lincosamides, and ketolides In: Schonfeld W, Kirst HA. (eds). Macrolide Antibiotics. Birkhauser Verlag: Basel, Switzerland, pp. 281–317. [Google Scholar]
- Tait‐Kamradt A, Davies T, Appelbaum PC, Depardieu F, Courvalin P, Petitpas J et al. (2000a). Two new mechanisms of macrolide resistance in clinical strains of Streptococcus pneumoniae from Eastern Europe and North America. Antimicrob Agents Chemother 44: 3395–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait‐Kamradt A, Davies T, Cronan M, Jacobs MR, Appelbaum PC, Sutcliffe J (2000b). Mutations in 23S rRNA and ribosomal protein L4 account for resistance in pneumococcal strains selected in vitro by macrolide passage. Antimicrob Agents Chemother 44: 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Nakamura A, Tsurubuchi K, O'Hara K, Sawai T (2004). The role of histidine residues conserved in the putative ATP‐binding region of macrolide 2′‐phosphotransferase II. FEMS Microbiol Lett 232: 123–126. [DOI] [PubMed] [Google Scholar]
- Telithromycin: review of adverse effects (2014). Prescrire Int 23: 264–266. [PubMed] [Google Scholar]
- Tenson T, Lovmar M, Ehrenberg M (2003). The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J Mol Biol 330: 1005–1014. [DOI] [PubMed] [Google Scholar]
- Toscano L, Fioriello G, Spagnoli R, Cappelletti L (1983). New semisynthetic fluorinated “hybrid” macrolides. J Antibiot (Tokyo) 36: 1585–1588. [DOI] [PubMed] [Google Scholar]
- Tu D, Blaha G, Moore PB, Steitz TA (2005). Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 121: 257–270. [DOI] [PubMed] [Google Scholar]
- Varaldo PE, Montanari MP, Giovanetti E (2009). Genetic elements responsible for erythromycin resistance in streptococci. Antimicrob Agents Chemother 53: 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez‐Laslop N, Thum C, Mankin AS (2008). Molecular mechanism of drug‐dependent ribosome stalling. Mol Cell 30: 190–202. [DOI] [PubMed] [Google Scholar]
- Vazquez‐Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS (2010). The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J 29: 3108–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester B, Douthwaite S (2001). Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob Agents Chemother 45: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilches C, Hernandez C, Mendez C, Salas JA (1992). Role of glycosylation and deglycosylation in biosynthesis of and resistance to oleandomycin in the producer organism, Streptomyces antibioticus . J Bacteriol 174: 161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimberg V, Lenart J, Janata J, Balikova Novotna G (2015). ClpP‐independent function of ClpX interferes with telithromycin resistance conferred by Msr(A) in Staphylococcus aureus . Antimicrob Agents Chemother 59: 3611–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagman GH, Waitz JA, Marquez J, Murawaski A, Oden EM, Testa RT et al. (1972). A new Micromonospora‐produced macrolide antibiotic, rosamicin. J Antibiot (Tokyo) 25: 641–646. [DOI] [PubMed] [Google Scholar]
- Wagner RL, Hochstein FA, Murai K, Messina N, Regna PP (1953). Magnamycin. A new antibiotic. J Amer Chem Soc 75: 4684–4687. [Google Scholar]
- Walsh CT (2017). At the Intersection of chemistry, biology, and medicine. Annu Rev Biochem . https://doi.org/10.1146/annurev‐biochem‐110716‐121241. [DOI] [PubMed] [Google Scholar]
- Webb V, Davies J (1993). Antibiotic preparations contain DNA: a source of drug resistance genes? Antimicrob Agents Chemother 37: 2379–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb V, Davies J (1994). Accidental release of antibiotic‐resistance genes. Trends Biotechnol 12: 74–75. [DOI] [PubMed] [Google Scholar]
- Weinstein MJ, Wagman GH, Marquez JA, Testa RT, Oden E, Waitz JA (1969). Megalomicin, a new macrolide antibiotic complex produced by Micromonospora . J Antibiot (Tokyo) 22: 253–258. [DOI] [PubMed] [Google Scholar]
- Weisblum B (1995a). Erythromycin resistance by ribosome modification. Antimicrob Agents Chemother 39: 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum B (1995b). Insights into erythromycin action from studies of its activity as inducer of resistance. Antimicrob Agents Chemother 39: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DN (2014). Ribosome‐targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol 12: 35–48. [DOI] [PubMed] [Google Scholar]
- Wilson DN (2016). The ABC of ribosome‐related antibiotic resistance. MBio 7 e00598‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DN, Harms JM, Nierhaus KH, Schluenzen F, Fucini P (2005). Species‐specific antibiotic‐ribosome interactions: implications for drug development. Biol Chem 386: 1239–1252. [DOI] [PubMed] [Google Scholar]
- Wilson DN, Arenz S, Beckmann R (2016). Translation regulation via nascent polypeptide‐mediated ribosome stalling. Curr Opin Struct Biol 37: 123–133. [DOI] [PubMed] [Google Scholar]
- Wise R (1989). The development of macrolides and related compounds. J Antimicrob Chemother 23: 299–300. [DOI] [PubMed] [Google Scholar]
- Wolter N, Smith AM, Low DE, Klugman KP (2007). High level telithromycin resistance in a clinical isolate of Streptococcus pneumoniae . Antimicrob Agents Chemother 51: 1092–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter N, Smith AM, Farrell DJ, Northwood JB, Douthwaite S, Klugman KP (2008). Telithromycin resistance in Streptococcus pneumoniae is conferred by a deletion in the leader sequence of erm(B) that increases rRNA methylation. Antimicrob Agents Chemother 52: 435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au‐Yeung BW et al. (1981). Asymmetric total synthesis of erythromycin. 3. Total synthesis of erythromycin. J Am Chem Soc 103: 3215–3217. [Google Scholar]
- Woosley LN, Castanheira M, Jones RN (2010). CEM‐101 activity against Gram‐positive organisms. Antimicrob Agents Chemother 54: 2182–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondrack L, Massa M, Yang BV, Sutcliffe J (1996). Clinical strain of Staphylococcus aureus inactivates and causes efflux of macrolides. Antimicrob Agents Chemother 40: 992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GD (2005). Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv Drug Deliv Rev 57: 1451–1470. [DOI] [PubMed] [Google Scholar]
- Wu YJ, Su WG (2001). Recent developments on ketolides and macrolides. Curr Med Chem 8: 1727–1758. [DOI] [PubMed] [Google Scholar]
- Young D (2007). Limit Ketek to pneumonia, experts advise: advisers urge black‐box warning. Am J Health Syst Pharm 64: 124–125. [DOI] [PubMed] [Google Scholar]
- Zhanel GG, Hoban DJ (2002). Ketolides in the treatment of respiratory infections. Expert Opin Pharmacother 3: 277–297. [DOI] [PubMed] [Google Scholar]
- Zheng J, Sagar V, Smolinsky A, Bourke C, LaRonde‐LeBlanc N, Cropp TA (2009). Structure and function of the macrolide biosensor protein, MphR(A), with and without erythromycin. J Mol Biol 387: 1250–1260. [DOI] [PubMed] [Google Scholar]
- Zhu B, Marinelli BA, Abbanat D, Foleno BD, Bush K, Macielag MJ (2007). Synthesis and antibacterial activity of 3‐keto‐6‐O‐carbamoyl‐11,12‐cyclic thiocarbamate erythromycin A derivatives. Bioorg Med Chem Lett 17: 3900–3904. [DOI] [PubMed] [Google Scholar]