

Abstract
Background and Purpose
Patients with active rheumatoid arthritis (RA) have increased cardiovascular mortality, paradoxically associated with reduced circulating lipid levels. The JAK inhibitor tofacitinib ameliorates systemic and joint inflammation in RA with a concomitant increase in serum lipids. We analysed the effect of tofacitinib on the lipid profile of hyperlipidaemic rabbits with chronic arthritis (CA) and on the changes in reverse cholesterol transport (RCT) during chronic inflammation.
Experimental Approach
CA was induced in previously immunized rabbits, fed a high‐fat diet, by administering four intra‐articular injections of ovalbumin. A group of rabbits received tofacitinib (10 mg·kg−1·day−1) for 2 weeks. Systemic and synovial inflammation and lipid content were evaluated. For in vitro studies, THP‐1‐derived macrophages were exposed to high lipid concentrations and then stimulated with IFNγ in the presence or absence of tofacitinib in order to study mediators of RCT.
Key Results
Tofacitinib decreased systemic and synovial inflammation and increased circulating lipid levels. Although it did not modify synovial macrophage density, it reduced the lipid content within synovial macrophages. In foam macrophages in culture, IFNγ further stimulated intracellular lipid accumulation, while the JAK/STAT inhibition provoked by tofacitinib induced lipid release by increasing the levels of cellular liver X receptor α and ATP‐binding cassette transporter (ABCA1) synthesis.
Conclusions and Implications
Active inflammation could be associated with lipid accumulation within macrophages of CA rabbits. JAK inhibition induced lipid release through RCT activation, providing a plausible explanation for the effect of tofacitinib on the lipid profile of RA patients.
Abbreviations
- ABCA1
ATP‐binding cassette A1
- ABCG1
ATP‐binding cassette G1
- CA
chronic arthritis
- CE
cholesterol efflux
- CRP
C‐reactive protein
- CVD
cardiovascular disease
- DMARDs
disease modifying antirheumatic drugs
- HDL‐C
HDL cholesterol
- HFD
high‐fat diet
- h‐oxLDL
oxidized human LDL
- IL‐6
Interleukin‐6
- LDL‐C
LDL cholesterol
- LXRα
liver X receptor α
- ORO
Oil Red‐O
- OVA
ovalbumin
- RA
rheumatoid arthritis
- RCT
reverse cholesterol transport
- SR‐BI
scavenger receptor class B type I
- TC
total cholesterol
- TOFA
tofacitinib
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic, inflammatory autoimmune disorder causing symmetrical polyarthritis and affects at least 1% of the world population (Majithia and Geraci, 2007). The most frequent comorbidity of RA is cardiovascular disease (CVD), which is also the leading cause of mortality (Meune et al., 2009). Paradoxically, active RA is associated with reduced levels of LDL cholesterol (LDL‐C), HDL cholesterol (HDL‐C) and total cholesterol (TC; Choy and Sattar, 2009; Johnsson et al., 2013), changes that hinder the assessment of CVD risk (Choy et al., 2014). In fact, an inverse relationship between C‐reactive protein (CRP) and circulating lipid levels has been observed (Johnsson et al., 2013). Furthermore, some disease‐modifying antirheumatic drugs (DMARDs) have been described as increasing serum LDL‐C and HDL‐C levels (Robertson et al., 2013; Charles‐Schoeman et al., 2016b). Although the reduction of chronic systemic inflammation seems to be responsible for these phenomena (Khovidhunkit et al., 2004; Robertson et al., 2013; Tall and Yvan‐Charvet, 2015), the mechanisms associated are still poorly understood. In fact, similar reductions in disease activity and systemic inflammatory parameters can be achieved by different treatments, although their effects on the lipid profile are variable (Souto et al., 2015). Therefore, aside from a dampening of inflammation, additional and specific mechanisms may be responsible for the lipid balance induced by different treatments (Robertson et al., 2013; Souto et al., 2015).
Macrophage activation is central to atherosclerosis and the development of CVD and is responsible for lipid uptake and foam‐cell formation within the atherosclerotic plaque (Tall and Yvan‐Charvet, 2015). Cholesterol efflux (CE) from macrophages is critical not only in preventing atherosclerotic lesions but also in avoiding the toxic effects of elevated cholesterol concentration at a cellular level. The initial step in reverse cholesterol transport (RCT) is the CE from the cell to acceptor particles through specific transporters. ATP‐binding cassette A1 (ABCA1) on macrophages promotes phospholipid and CE onto pre‐β‐HDL particles, whereas ATP‐binding cassette G1 (ABCG1) transfers cholesterol onto mature HDL. Then, free cholesterol is further esterified and can be either taken up by the liver via the scavenger receptor class B type I (SR‐BI) or transferred to different lipoproteins (Luo et al., 2010). Patients with RA show specific alterations in different circulating lipoproteins related to their composition and in the ability of these particles to elicit cholesterol efflux from macrophages (Charles‐Schoeman et al., 2012; Ronda et al., 2014). Recent data indicate that the plasma cholesterol ester fractional catabolic rate is increased in RA patients (Charles‐Schoeman et al., 2015). However, little is known of specific alterations in the macrophage CE during RA.
Tofacitinib (TOFA, CP‐690550) is an oral inhibitor of the Janus kinase (JAK) family, which has been approved by the US Food and Drug Administration and the European Medicines Agency for the treatment and prevention of RA and other immune‐mediated diseases. A proportion of patients receiving tofacitinib exhibited increases in mean HDL‐C, LDL‐C, TC, apolipoprotein A‐I and apolipoprotein B levels within the first 4–8 weeks of treatment (Fleischmann et al., 2012; Kremer et al., 2012; van Vollenhoven et al., 2012) with a low incidence of cardiovascular events (Charles‐Schoeman et al., 2016a). The JAK/STAT pathway, and particularly IFNγ, has been proposed to be a key pathway in the regulation of CE from macrophages (Hao et al., 2009). This cytokine has been broadly recognized as an activator of synovial macrophages and a key contributor to the pathogenesis of rheumatoid arthritis and other autoimmune diseases (Dolhain et al., 1996; Schulze‐Koops and Kalden, 2001; Milman et al., 2010; Kennedy et al., 2011). In addition, it has previously been shown to be of relevance in the early response to tofacitinib in RA patients (Maeshima et al., 2012; Boyle et al., 2015). However, the specific mechanisms underlying the effect of JAK inhibition on RCT and its significance are currently unclear.
We hypothesized that inflammation would favour the accumulation of lipids within tissue macrophages such as those from the inflamed synovium. Such an accumulation could account, at least partially, for the reduction in circulating cholesterol observed in RA patients. Tofacitinib may prevent this process, conceivably by acting on RCT pathways, thus further explaining how serum lipid levels increase with the treatment.
We developed a rabbit model of chronic arthritis (CA) in which the animals are fed a high‐fat diet (HFD) that mimics the lipid paradox seen in RA patients (Romero et al., 2010). We aimed to study the effects of tofacitinib on the serum lipid profiles of these rabbits and the changes occurring in the synovium. Furthermore, we went deeper into the role of tofacitinib‐induced JAK/STAT blockade on the CE from macrophages.
Methods
Animal model
New Zealand male white rabbits of 3 months of age, weighing 2.7 ± 0.15 kg (Granja San Bernardo, Navarra, Spain), were housed individually in cages with transparent walls (0.50 m of cage height and 0.6 m2 of floor space) exposed to a 12 h light/dark cycle. Rabbits were randomly assigned to three groups: healthy rabbits (control group, n = 6), CA rabbits (CA group, n = 9) and CA rabbits receiving tofacitinib (CA + TOFA group, n = 9). All animals were fed ad libitum with a HFD (0.5% cholesterol and 4% peanut oil). Antigen‐induced CA was induced in 18 animals as described previously (Largo et al., 2008). Briefly, four weekly intra‐articular injections of ovalbumin (OVA; Sigma‐Aldrich, St. Louis, MO, USA) were given to previously immunized rabbits in both knees in order to resemble the disease flares. After the second intra‐articular injection, nine CA rabbits received tofacitinib (10 mg·kg−1·day−1, early in the morning) and nine CA rabbits received placebo, p.o. via gavage as previously described for mice and rats (Dowty et al., 2013; Fujii and Sengoku, 2013; Thacker et al., 2017), until the end of the study (Supporting Information Figure S1). Six saline‐injected animals were used as controls. In order to eliminate bias, blinding was performed by using a non‐consecutive numerical code for each animal. The scientist in charge of preparing the intra‐articular injection (OVA or saline) was different from the one performing the injections, and the same applies for the administering of tofacitinib or placebo, which were given without knowing whether the animal was arthritic or not. In addition, the outcomes were assessed blinded to group assignment.
Body weight and food intake were monitored at baseline and once per week throughout the study. All the rabbits were killed by an overdose of intra‐cardiac pentobarbital (50 mg·kg−1; tiobarbital; Braun Medical SA, Barcelona, Spain) 4 weeks after the first intra‐articular injection and 1 day after the last intra‐articular injection. Blood was collected, just before the animals were killed, from the marginal ear vein of the rabbits after overnight fasting. Blood was allowed to clot, centrifuged, and serum was frozen in aliquots. Synovial tissue was collected and cut in two equal pieces for paraffin and OCT embedding.
Rabbits have been widely used as a model of CA since it resembles RA in its chronicity, pathogenesis and histological manifestations (Jasin, 1988). In addition, they have been claimed as the model of choice for the study of lipoprotein metabolism (Yanni, 2004; Fan et al., 2015). As opposed to rats and mice, lipoprotein metabolism in rabbits is similar to humans. Specifically, the main cholesterol pool is from hepatic origin, LDL is the major plasma lipoprotein and they also have an abundant plasma cholesteryl ester transfer protein activity, which is essential for cholesterol metabolism (Tall, 1993).
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All animal care and experimental protocols for this study complied with the Spanish regulations and the Guidelines for the Care and Use of Laboratory Animals drawn up by the National Institutes of Health (USA) and were approved by the Institutional Ethics Committee of the Fundación Jiménez Díaz Hospital.
Determination of CRP and lipid profile in rabbit serum
Levels of TC and HDL‐C were measured in serum by Advia® 2400 Chemistry System (Siemens Healthcare Diagnostics, Tarrytown, NY, USA). LDL‐C was estimated by the Friedewald equation. CRP was measured with a specific commercial enzyme‐linked immunosorbent assay (ab157726; Abcam, Cambridge, UK).
Histology
Synovial histopathology was evaluated in haematoxylin–eosin (H&E) stained sections of 4 μm by two blinded observers as previously described (Krenn et al., 2002; Alvarez‐Soria et al., 2006). Briefly, lining hyperplasia, fibrovascular alterations at the interstitium, and the tissue infiltration were independently evaluated using 0‐ to 3‐point subscales, where 0 indicates absence, 1 mild, 2 intermediate and 3 strong. The total score was obtained from the sum of partial grades with a maximum total score of 9. In order to assess the lipid content within the synovium, two consecutive sections of 10 μm were obtained with a Leica CM1850 cryostat (Leica Co., Germany) and then air dried and fixed in 10% formalin. Subsequently, one slice was stained with H&E, and the consecutive slice was rinsed in deionized water and immersed in a 60% Oil Red‐O (ORO) working solution (Sigma‐Aldrich) for 30 min. After three rinses, the tissue was counterstained with haematoxylin and mounted in Glycergel Aqueous Mounting Medium (Dako, Glostrup, Denmark). Sections were photographed using an automated iScan Coreo slide scanner (Ventana Medical Systems, USA). Five random areas per slide, excluding adipocytes, were selected blinded to group assignment and quantified with ImageJ software, and the percentage of positive stained area was calculated. In order to show the percentage of change seen with the treatment, the Y axis was set so the CA group value is 100% and results are expressed as the percentage of the mean value of the CA group.
Immunohistochemistry of rabbit macrophages in synovium
Synovial macrophages were identified by immunohistochemistry as previously described (Prieto‐Potín et al., 2013). Briefly, tissue sections (4 μm) were deparaffinized and rehydrated in an ethanol series. Sections were blocked for non‐specific binding with 4% BSA and 3% sheep serum and then incubated overnight with a monoclonal anti‐rabbit macrophage antibody (clone RAM11; Dako, Glostrup, Denmark). The antibody was detected with a biotinylated goat anti‐mouse IgG (GE Healthcare, Little Chalfont, Buckinghamshire, UK) visualized with a horseradish peroxidase/ABC complex using 3,3 diaminobenzidine tetra‐hydrochloride as the chromogen (Dako, Golstrup, Denmark). The tissues were counterstained with haematoxylin and mounted in DPX medium (VWR International, Leuven, Belgium). Sections were photographed using an automated iScan Coreo slide scanner (Ventana Medical Systems, USA). Five random areas per slide (including or not adipocytes) were selected blinded to group assignment and quantified with Image J software. Results are expressed as a percentage of positive stained area. An IgG isotype was used as a negative control.
Isolation and oxidation of LDL
Human LDL was isolated and oxidized from a fresh plasma pool by sequential ultracentrifugation as previously described (Moreno et al., 2009).
Cell culture
THP‐1 monocytes were obtained from the American Type Culture Collection (Manassas, Virginia, USA). They were grown at 37°C and 5% CO2 in RPMI 1640 (Gibco BRL, Grand Island, NY, USA) supplemented with 10% decomplemented FBS, 50 U·mL−1 penicillin, 50 U·mL−1 streptomycin and 2 mM L‐glutamine (Gibco BRL). THP‐1 monocytes were differentiated into macrophages in the presence of 100 nM PMA (Sigma‐Aldrich) for 24 h before being used in experiments.
Silencing of ABCA1 in THP‐1 cells
THP‐1‐differentiated macrophages were nucleofected with 0.75 nM of Silencer Select siRNA targeting human ABCA1 (ID: s848, Thermo Scientific, Madrid, Spain) or vehicle‐only following an existing protocol (Maeß et al., 2014).
Experimental conditions
After 24 h of starvation, cells were incubated with oxidized human LDL (h‐oxLDL; 50 μg·mL−1) or with a pool of serum from the control group of our rabbit model (4% HFD serum) for 24 h to become foam cells. The final amount of TC and LDL in the cell culture was in the same range as those expected in the synovial fluid of RA patients (65 mg·mL−1 of TC and 60 mg·mL−1 of LDL) (Oliviero et al., 2009; Oliviero et al., 2012; De Seny et al., 2015). Cells were then stimulated for 24 h with recombinant human IFNγ (50 ng·mL−1) in presence or absence of tofacitinib dissolved in DMSO at different doses (CP‐690550; Pfizer International, Peapack, NJ, USA). The final concentration of DMSO in the culture medium was less than 0.1%. Lipid release and protein expression were assessed.
Assessment of lipid release in THP‐1 cells
THP‐1‐treated cells were incubated in RPMI containing 0.2% (w·v−1) BSA plus 20 μg·mL−1 Apo‐AI for 6 h to facilitate the lipid release. Thereafter, cells were rinsed in PBS and fixed in 10% formalin solution (Sigma‐Aldrich) for 5 min. After washing the cells twice in distilled H2O, 60% isopropanol was added for 5 min and then cells were stained with 60% Oil Red O working solution (Sigma‐Aldrich). Cells were observed in a Leica DFC420C microscope with 400× magnification and then photographed using a Leica DFC420 C Digital Camera blinded to group assignment. ImageJ was used to quantify Oil Red O intensity as previously described (Mehlem et al., 2013). The given results are the mean percentage of the positive stained area from five random fields per condition. Normalization per number of cells has been performed in order to ensure that the changes shown are due to differences in the lipid content regardless of cellularity.
Western blotting
Total cell lysates, 35 μg, were loaded onto an 8% SDS‐polyacrylamide gel, electrophoresed for 2 h at 100 V and then transferred to a nitrocellulose membrane in a semi‐dry Trans‐Blot device (Bio‐Rad, Madrid, Spain) for 30 min at 25 V. Membranes were blocked in 3% skimmed milk for 1 h at room temperature and then incubated with antibodies against ABCA1 [AB.H10] at 2 μg·mL−1, ABCG1 [EP1366Y] at 100 ng·mL−1 and liver X receptor α (LXRα) [EPR6508(N)] at 0.063 μg·mL−1 (Abcam, Cambridge, UK), phospho‐STAT1 [KIKSI0803] at 5 μg·mL−1 (Affymetrix, Santa Clara, CA, USA) and α‐tubulin at 0.65 μg·mL−1 (Sigma‐Aldrich) as the loading control. Antibody binding was detected by chemiluminescence using peroxidase‐linked species‐specific secondary antibodies purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Results are corrected by α‐tubulin in order to normalize the loading, and by the control group (HDF serum‐ or oxLDL‐only) in order to avoid bias due to differences in the intensity of the chemiluminescent signal between blots.
RNA isolation and RT‐ PCR
RNA was isolated using TRIzol reagent (Roche Diagnostics, Barcelona, Spain), dissolved in nuclease‐free water and quantified using a NanoDrop ND1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was obtained from 1 μg of total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer's instructions. RNA expression was quantified by single‐reporter real‐time PCR using the Step One Plus Detection system (Applied Biosystems). The specific TaqMan probe for ABCA1 (Assay ID: Hs01059118_m1) was purchased from Applied Biosystems. Quantitative measurements were determined using the ΔΔCt method, and expression of 18S was used as the internal control.
Statistical analysis
Data were analysed by non‐parametric Kruskal–Wallis test followed by Dunn's multiple comparison test for comparisons between multiple groups and Mann–Whitney U‐test by pairwise comparisons between two groups. Correlation coefficients were calculated using a non‐parametric Spearman correlation. Non‐parametric tests were used because of the small sample size and the lack of normality and homogeneity, as tested by Shapiro–Wilk test and Levene's test respectively. A P‐value of less than 0.05 was considered significant. Statistical analysis was performed using Windows SPSS 21.0 (SPSS, Inc., Chicago, IL, USA). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Systemic alterations in a rabbit model of CA that reproduces the lipid paradox
CA rabbits gained significantly less weight than the corresponding HFD‐fed controls (Table 1). tofacitinib partially restored weight gain in CA rabbits. Furthermore, tofacitinib attenuated the increase in circulating CRP observed in CA animals. Regarding serum lipids, CA rabbits showed lower levels of TC and LDL‐C than control rabbits. TC and LDL‐C concentrations tended to increase with tofacitinib, while TC/HDL‐C was significantly increased in CA + TOFA rabbits when compared with untreated CA animals (Table 1). Our data also show that serum TC and CRP concentration were inversely correlated in our rabbits, (P < 0.05, r = −0.454; Supporting Information Figure S2), as has been observed in RA patients (Charles‐Schoeman et al., 2016b).
Table 1.
Systemic alterations in a rabbit model of CA
| Control (n = 6) | CA (n = 9) | CA + TOFA (n = 9) | |
|---|---|---|---|
| Weight gain (kg) | 0.7 ± 0.07 | 0.1 ± 0.05a | 0.4 ± 0.06a , b |
| CRP (μg·mL−1) | 21 ± 14 | 173 ± 18a | 94 ± 17a, b |
| TC (mg·mL−1) | 25.7 ± 3.2 | 13.4 ± 1a | 16 ± 1.2a |
| HDL‐C (mg·mL−1) | 0.12 ± 0.03 | 0.1 ± 0.01 | 0.07 ± 0.01a |
| LDL‐C (mg·mL−1) | 24.8 ± 3.4 | 12.8 ± 0.9a | 15.5 ± 1.1a |
| TC/HDL‐C ratio | 277 ± 71 | 150 ± 15 | 231 ± 17b |
Net body weight gain of rabbits at the end of the study. Concentration of CRP, TC, HDL‐C, LDL‐C, and TC/HDL‐C ratio. Data are shown as mean and SEM. Mann–Whitney test.
P < 0.05 versus Control;
P < 0.05 versus CA.
Synovial histopathology
CA induced an increase in synovial inflammation, with an enlargement of the synovial lining layer and a marked infiltration of inflammatory cells with lymphoid aggregates. CA synovium also showed stromal hypercellularity with densely located fibroblast‐like cells. Tofacitinib partially reversed the increased lining hyperplasia and reduced the inflammatory infiltrate in CA animals. As a result, the global score was diminished in the treated group (Figure 1A–C, G).
Figure 1.
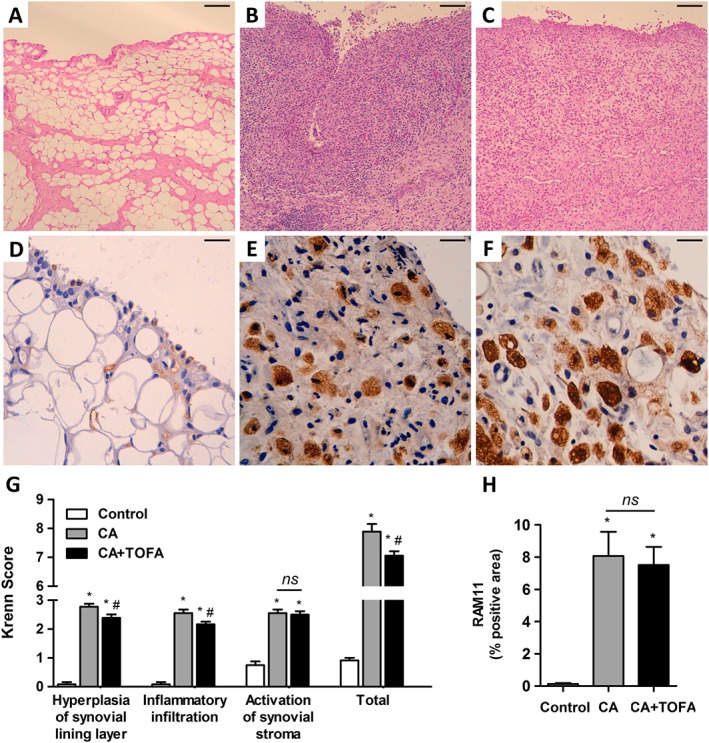
Assessment of synovitis and macrophage infiltration. (A–C) Representative sections of synovium stained with haematoxylin and eosin. Control (A), CA (B) and CA + TOFA (C). Scale bar = 100 μm. (D–F) Immunohistochemistry of representative sections of synovium using the monoclonal anti‐rabbit macrophage antibody RAM11. Control (D), CA (E) and CA + TOFA (F). Scale bar = 25 μm. (G) Global synovitis score, quantified as described in Methods. Bars show the mean and SEM (n Control = 12 paws, n CA = 18 paws and n CA+TOFA = 18 paws). Mann–Whitney test, *P < 0.05 versus Control, #P < 0.05 versus CA, ns = non‐significant. (H) Densitometric analysis of RAM11 staining as a percentage in the synovium of each group of animals. Bars show the mean and SEM (n Control = 12 paws, n CA = 18 paws and n CA+TOFA = 18 paws). Mann–Whitney test; *P < 0.05 versus Control, ns = non‐significant.
Macrophage infiltration and presence of lipids within the synovium
The synovium of both treated and non‐treated CA rabbits exhibited extensive infiltration of lipid‐loaded macrophages, both in the synovial lining and sublining (Figure 1E, F). Quantification of macrophage density revealed that tofacitinib did not have a significant effect on macrophage infiltration (Figure 1H).
We also quantified the amount of lipids within synovial cells (verified by H&E staining in consecutive sections) with the exception of the adipose tissue of the synovium. Quantification of Oil Red O staining revealed that CA + TOFA rabbits showed a 58% reduction in the amount of lipids within synovial cells in comparison with untreated CA rabbits (Figure 2).
Figure 2.
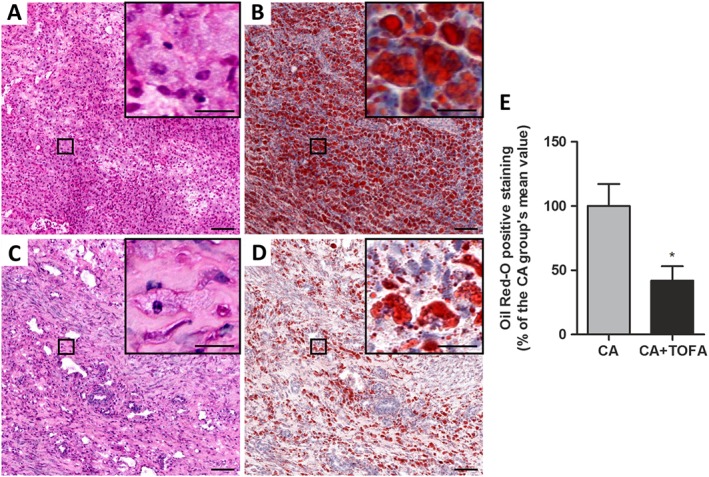
Assessment of the amount of lipids within the inflamed synovium. (A–D) Haematoxylin and eosin (A, C) and Oil Red‐O staining (B, D) of representative sections of the synovium. CA (A, B) and CA + TOFA (C, D). Scale bar = 100 and 25 μm in the inset. (E) Densitometric analysis of Oil Red‐O staining in the synovium presented as a % of the CA group of animals, excluding adipocytes. Bars show the mean and SEM (n = 6 rabbits per group). Mann–Whitney test; *P < 0.05 versus CA group.
Cholesterol efflux impairment in IFNγ‐treated foam THP‐1 cells and reversal by tofacitinib
We next explored whether tofacitinib was able to reduce intracellular lipid content in in vitro lipid‐loaded macrophages. THP‐1‐derived macrophages were incubated with increasing concentrations of a pool of hyperlipidaemic serum from HFD‐fed rabbits (0.5–8%), without subsequent exposure to the efflux medium. Intracellular Oil Red O staining of lipid‐loaded THP‐1 cells increased in a dose‐dependent manner (Figure 3A, F). The serum concentration for further experiments was set to 4%.
Figure 3.
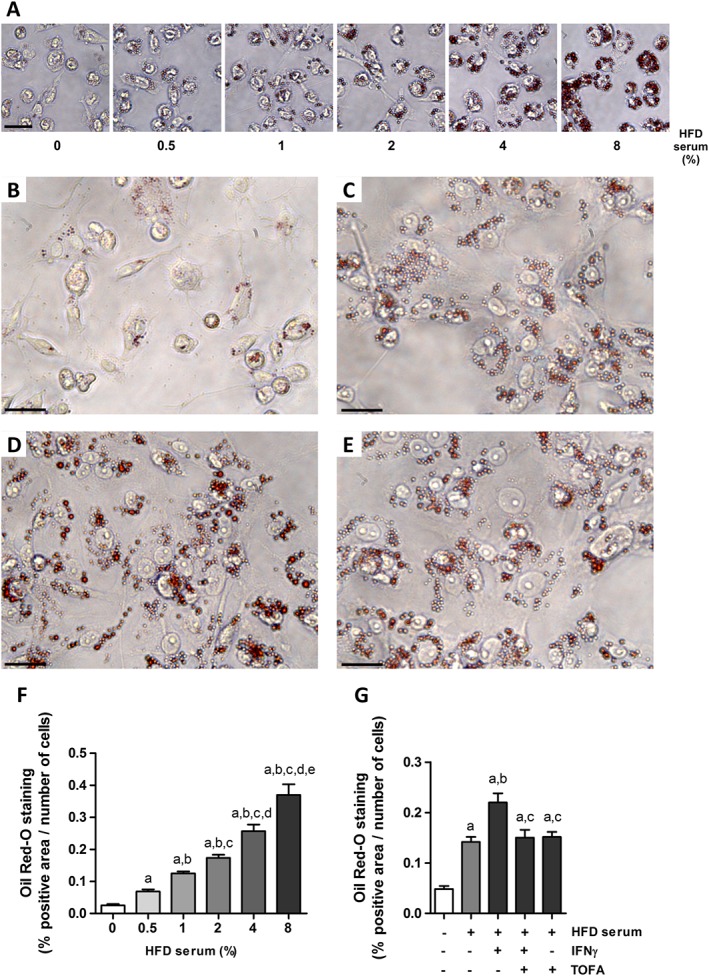
Cholesterol efflux impairment in IFNγ‐treated foam THP‐1 cells is prevented by tofacitinib. (A) Representative images of THP‐1 cells treated with increasing doses of HFD rabbit serum ranging from 0.5 to 8% and stained with Oil Red‐O. Scale bar = 50 μm. (F) Quantification of Oil Red‐O positive staining in THP‐1 cells treated with increasing doses of HFD rabbit serum. Results normalized per number of cells as described in Methods. Mann–Whitney test, P < 0.05 a: versus 0%, b: versus 0.5%, c: versus 1%, d: versus 2% and e: versus 4%. Bars show the mean and SEM (n = 6). (B–E) Representative images of treated THP‐1 cells stained with Oil Red‐O after a 6 h incubation period with the efflux medium. (B) Basal media, (C) 4% HFD serum, (D) 4% HFD serum + 50 ng·mL−1 IFNγ, (E) 4% HFD serum + 50 ng·mL−1 IFNγ + 2 μM tofacitinib. Scale bar = 25 μm. (G) Quantification of Oil Red‐O positive staining and normalized per number of cells as described in Methods. Mann–Whitney test; a: P < 0.05 versus basal media, b: P < 0.05 versus HFD serum, c: P < 0.05 versus HFD serum + IFNγ. Bars show the mean and SEM (n = 6).
Thereafter, we studied the effects of IFNγ on the lipid content of THP‐1 macrophages and the role of the JAK/STAT pathway. After a 6 h period of incubation in the efflux medium containing Apo‐AI, we observed that the amount of lipids remaining inside the HFD rabbit serum‐stimulated cells in the presence of IFNγ (Figure 3D) was higher than that seen in the corresponding control (Figure 3C, G). This impaired lipid release was prevented when cells were also treated with tofacitinib (Figure 3E, G.). Results for cells stimulated with tofacitinib only were similar to those for cells treated with the hyperlipidaemic serum only (Figure 3G).
Tofacitinib restores IFNγ‐induced down‐regulation of ABCA1 protein expression in THP‐1 cells pretreated with HFD rabbit serum or with h‐oxLDL
After 24 h of treatment, IFNγ reduced the protein levels of ABCA1 in HFD serum‐treated THP‐1 cells, whereas the levels of ABCG1 remained unchanged. This phenomenon was accompanied by an increase in STAT1 phosphorylation. Tofacitinib was able to increase ABCA1 protein levels after IFNγ exposure, thus indicating that the JAK/STAT pathway plays a key role in the impaired CE induced by this cytokine. The protein expression of LXRα did not change in these conditions (Figure 4A).
Figure 4.
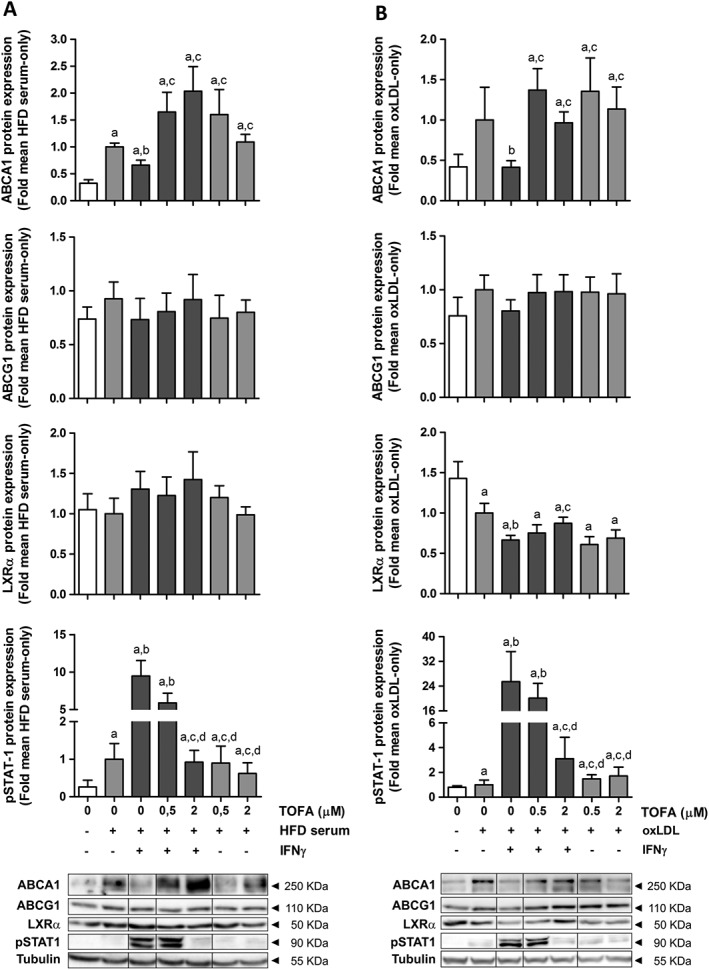
Tofacitinib restores down‐regulation of ABCA1 protein expression induced by IFNγ in THP‐1 cells pretreated with HFD rabbit serum or h‐oxLDL. Western blot of ABCA1, ABCG1, LXRα and phospho‐STAT1 in THP‐1 foam cells obtained after incubation with 4% HFD rabbit serum (A, left panel, n = 6) or 50 μg·mL−1 h‐oxLDL (B, right panel, n = 7) for 24 h. Cells were exposed to IFNγ (50 ng·mL−1) and/or tofacitinib (0.5 or 2 μM) for an additional 24 h. Results are normalized to those of tubulin and expressed as fold change of the HFD serum‐only or oxLDL‐only group. Bars show the mean and SEM. Mann–Whitney test; a: P < 0.05 versus basal media, b: P < 0.05 versus HFD serum (A) or oxLDL (B), c: P < 0.05 versus HFD serum + IFNγ (A) or oxLDL + IFNγ (B); d: P < 0.05 versus HFD serum + IFNγ + 0.5 μM TOFA (A) or oxLDL + IFNγ + 0.5 μM TOFA (B).
We also studied the effect of tofacitinib in lipid‐loaded macrophages transformed with h‐oxLDL in order to clarify whether these effects were due to an increase in the lipid concentration, independently of other mediators present in the rabbit serum. IFNγ reduced the amount of ABCA1 in h‐oxLDL‐treated cells, while ABCG1 protein levels tended to decrease. In parallel, the expression of LXRα was diminished in this inflammatory milieu. In response to tofacitinib treatment, ABCA1 protein levels increased and ABCG1 levels tended to increase, although statistical significance was not reached in the latter. Remarkably, this pan‐JAK inhibitor only restored LXRα protein levels when STAT1 phosphorylation was completely abolished (Figure 4B).
ABCA1 mRNA levels increased in the presence of IFNγ and decreased after JAK blockade (Supporting Information Figure S3) both in HFD serum‐ and h‐oxLDL‐treated cells.
Cholesterol efflux impairment in IFNγ‐treated foam THP‐1 cells is prevented by tofacitinib via ABCA1
In order to assess whether the effect of tofacitinib on CE was ABCA1‐dependent, we silenced ABCA1. We observed that the amount of lipids remaining inside the vehicle + IFNγ‐treated cells in the presence of the hyperlipidaemic serum was higher than that in the corresponding controls. Tofacitinib diminished the intracellular lipid content by 36% in vehicle + IFNγ‐stimulated cells. Results for cells stimulated with vehicle + tofacitinib only were similar to those for cells treated with vehicle + hyperlipidaemic serum only (Figure 5A; Figure 5B, upper panels). The lipid content within ABCA1‐silenced cells was significantly increased in comparison with their vehicle‐treated counterparts, except for the cells that had been incubated with the basal medium only. Tofacitinib showed no effect in preventing lipid accumulation in IFNγ‐stimulated ABCA1 silenced cells (Figure 5A; Figure 5B, lower panels).
Figure 5.
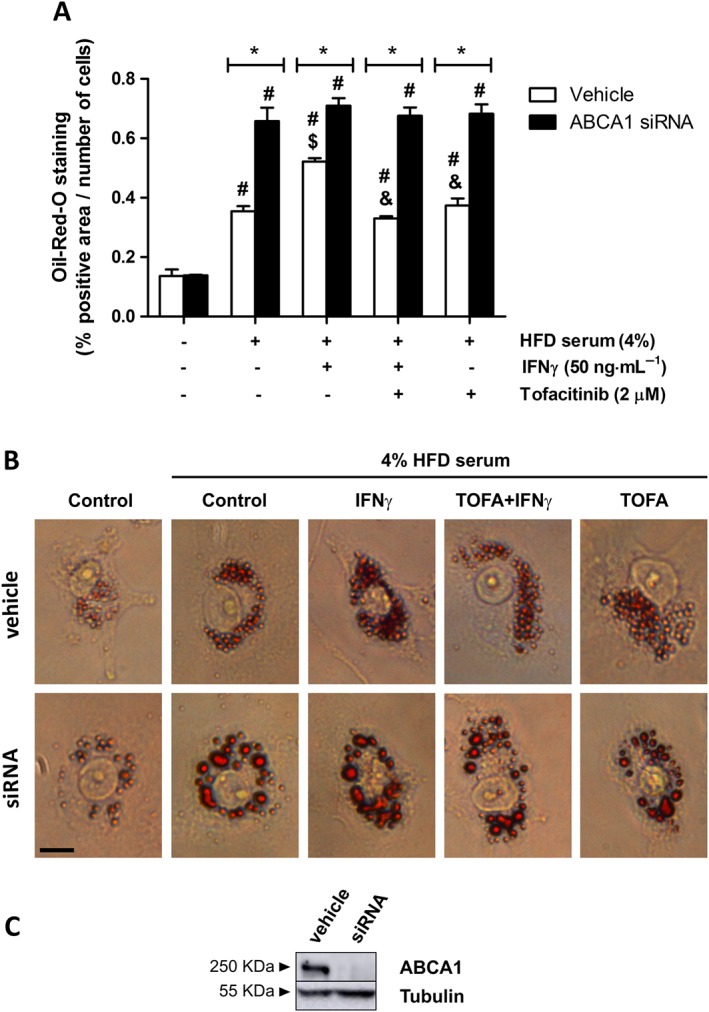
Cholesterol efflux impairment in IFNγ‐treated foam THP‐1 cells is prevented by tofacitinib via its effect on ABCA1. (A) Quantification of Oil Red‐O positive staining in THP‐1 cells (vehicle‐ or ABCA1 siRNA‐treated) in the presence or absence of HFD rabbit serum, 50 ng·mL−1 IFNγ and 2 μM tofacitinib for 24 h and after a 6 h incubation period with the efflux medium.* P < 0.05 versus vehicle, # P < 0.05 versus basal media, $ P < 0.05 versus HFD rabbit serum and & P < 0.05 versus HFD rabbit serum + IFNγ. Bars show the mean and SEM (n = 5). (B) Representative images of THP‐1 cells stained with Oil Red‐O. Scale bar = 10 μm. (C) Representative image of ABCA1 protein expression assessed by Western blot in THP‐1 cells at the end of the experiment.
Discussion
Here, we have demonstrated that HFD‐fed CA rabbits had lower serum LDL‐C and TC than control HFD‐fed animals and showed an increase in serum CRP levels and synovitis. Tofacitinib treatment diminished the serum CRP level and ameliorated synovial inflammation along with an increase in TC/HDL‐C ratio. Therefore, our model adequately reproduced the lipid paradox described in RA patients, characterized by an inverse correlation between circulating lipids and disease severity.
Different studies have focused on serum soluble factors to explain the driving forces behind dyslipidaemia in RA. Circulating HDL particles from RA patients seem to have a decreased ability to elicit CE from macrophages (Charles‐Schoeman et al., 2012; Ronda et al., 2014). Furthermore, an increase in cholesterol ester catabolism in RA patients has been associated with a decrease in cholesterol concentration, and this effect was abolished by tofacitinib (Charles‐Schoeman et al., 2015).
We hypothesized that additional cellular mechanisms could account for the altered lipid metabolism in RA. In particular, activated macrophages could accumulate lipids during chronic inflammation, thus being partially responsible for the reduction in circulating cholesterol. Previous work by our group showed that foam macrophage infiltration boosted synovitis in CA rabbits (Prieto‐Potín et al., 2013). Both in that study and in the current work, we observed that rabbits with increased systemic inflammation showed an accumulation of lipids within synovial cells, especially within macrophages. In addition, we found that a decrease in CRP was associated with a decrease in the lipid content within synovial macrophages. It has been suggested that other cells, such as adipocytes, exhibit a dysfunction in lipid metabolism in response to chronic inflammation (Bag‐Ozbek and Giles, 2015). Adipose and synovial tissue share common features in terms of cell composition and macrophage infiltration during tissue inflammation. In this regard, pro‐inflammatory cytokines could induce lipid accumulation within tissue compartments due to alterations in RCT pathways as described in inflamed adipose tissue (Lu et al., 2006).
Using this experimental approach, we have been able to reproduce the early stages of a very active form of human RA in the initial phases of DMARD therapy. Our results showed a subtle improvement in joint inflammation, a decreased lipid content within the synovium and no changes in the number of synovial macrophages in tofacitinib‐treated animals, indicating that structural changes in the synovium are not among the initial effects induced by this inhibitor. These outcomes are not surprising considering that the last OVA injection was performed only one day before the rabbit was killed. In addition, similar results have been described for RA patients after 28 days of treatment (Boyle et al., 2015). Nonetheless, 2 weeks of tofacitinib treatment were sufficient to induce a decrease in the amount of lipids within the inflamed synovium, concomitant with a tendency towards an increase in circulating lipids. This prompt effect indicates that lipid changes could be a direct effect of the treatment, rather than a global consequence of dampening inflammation. The whole scenario suggests that JAK inhibition could reduce lipid accumulation in the synovium, thus favouring an increase in circulating cholesterol.
Both acute and chronic inflammation modulate RCT pathways. The acute phase response induced following an injection of LPS into mice interferes with the expression of ABCA1 and other RCT mediators (Tall and Yvan‐Charvet, 2015). Voloshyna et al. observed that plasma from RA patients promoted foam cell transformation of THP‐1 macrophages, increased SR‐BI expression and down‐regulated the mediators of RCT (Voloshyna et al., 2014).
Our in vitro studies showed that a pro‐inflammatory milieu, such as that induced by IFNγ, further increased the lipid conctration inside activated macrophages in the presence of hyperlipidaemic rabbit serum. IFNγ also induced a decrease in ABCA1 protein expression in foam macrophages, alongside an increase in STAT1 phosphorylation. Therefore, a down‐regulation of ABCA1 could be responsible for the augmented lipid accumulation within IFNγ‐treated THP‐1 cells. Tofacitinib re‐established the protein levels of ABCA1 in foam macrophages and brought about a decrease in the lipid content within macrophages that was not observed when ABCA1 was silenced. These data indicate a key role for ABCA1 in RCT from IFNγ‐stimulated foam macrophages mediated through the activation of the JAK/STAT pathway. Surprisingly, the lowest dose of tofacitinib did not fully abolish STAT1 phosphorylation despite the increase in ABCA1 levels, thus suggesting the existence of further alternative mechanisms in ABCA1 regulation. In this regard, Frisdal et al. demonstrated that Interleukin‐6 (IL‐6) secretion was augmented after lipid loading in human macrophages, especially when ABCA1 was down‐regulated. In turn, IL‐6 induces ABCA1 up‐regulation via the JAK‐2/STAT3 pathway (Frisdal et al., 2011). Considering that the IC50 for blocking IL‐6‐dependent STAT3 phosphorylation is higher than that needed for abolishing IFN‐induced STAT1 phosphorylation, it seems plausible that there could be a balance towards STAT3 signalling at this treatment point, thus allowing the IL‐6‐induced up‐regulation of ABCA1 protein. This finding is of particular interest considering the preliminary results from our laboratory (in preparation), in which we observed a decrease in synovial phospho‐STAT1 after tofacitinib treatment, whereas STAT3 remained phosphorylated. Therefore, the possibility that IL‐6‐induced STAT3 phosphorylation, which then favours cholesterol efflux in the synovium, occurs in vivo cannot be dismissed. In this regard, further studies are needed to clarify the role of the different JAKs and STATs in our particular system.
ABCA1 protein expression has been demonstrated to be both LXRα‐dependent and independent, and transcriptional and post‐transcriptional mechanisms have also been shown to be involved in its modulation (Alfaro Leon et al., 2005; Chen et al., 2007; Pascual‐García et al., 2013). In this regard, LXRα protein expression remained unchanged in HFD rabbit serum‐treated THP‐1 cells. This could be due to the existence of multiple factors in the serum, which could modulate this system. When THP‐1 cells were incubated with h‐oxLDL, ABCA1 and LXRα synthesis were changed in parallel both in the absence and presence of tofacitinib. Additionally, LXRα protein levels were only fully restored at a dose at which STAT1 phosphorylation was completely abolished. This suggests that a complete blockade of STAT1 phosphorylation is pivotal in the up‐regulation of LXRα protein expression after IFNγ treatment. Noteworthy, gene expression studies revealed a possible negative feedback at the time of study. Gene expression did not parallel protein presence, probably reflecting the transcriptional and post‐transcriptional regulation of ABCA1. Furthermore, different feedback regulatory mechanisms could account for this discrepancy.
The crosstalk between STAT1 and the LXRα has been demonstrated previously (Pascual‐García et al., 2013), although its involvement in the synovial macrophages during RA has not yet been fully explored. Asquith et al. reported that the LXR pathway is up‐regulated in synovial macrophages from RA patients and showed a pro‐inflammatory role for this nuclear receptor in a murine collagen‐induced arthritis model (Asquith et al., 2009; 2013). However, LXR agonists have been shown to ameliorate inflammation in several autoimmune models in vivo (Joseph et al., 2003; Birrell et al., 2007; Chintalacharuvu et al., 2007; A‐Gonzalez et al., 2009; Park et al., 2010; Cui et al., 2011), suggesting that LXR agonists could have an anti‐inflammatory effect in some contexts. Whether the induction of LXRs by tofacitinib has a pro‐ or an anti‐inflammatory effect in the synovium remains to be determined.
The potential weaknesses of our work include, on the one hand, a plausible confounding effect of the high cholesterol levels found in our animals. Hypercholesterolaemic rabbits have been claimed as the animal model of choice for the study of lipoprotein metabolism (Yanni, 2004; Fan et al., 2015). However, such high concentrations could appear to be ‘artificial’. Nonetheless, the effects of CA‐related inflammation on reducing these high circulating lipid levels and the prompt effects of the treatment on lipid management justify the reliability of our model in mimicking the lipid paradox seen in RA patients.
On the other hand, our results in vitro only confirm that tofacitinib restores the IFNγ‐induced RCT‐inhibition. Our focus on this cytokine relies on its well‐described role in RA pathogenesis (Dolhain et al., 1996; Schulze‐Koops and Kalden, 2001; Milman et al., 2010; Kennedy et al., 2011) and its effects on RCT in macrophages (Reiss et al., 2004; Alfaro Leon et al., 2005; Hao et al., 2009; Pascual‐García et al., 2013; Yu et al., 2015). In addition, Boyle and co‐workers found that IFN‐regulated chemokines had a role in the initial synovial response to JAK blockade (Boyle et al., 2015). Nonetheless, it would be of interest to study different JAK‐dependent cytokines, such as IL‐6, to better characterize the link between the JAK/STAT pathway and dyslipidaemia.
In conclusion, our results suggest that chronic inflammation favours the accumulation of lipids into macrophages – in the synovium and probably in other tissues – thus decreasing serum lipid levels. Tofacitinib may prevent this phenomenon, at least partially, by acting on RCT pathways in macrophages. In fact, it has been recently proposed that the anti‐atherosclerotic activity associated with different DMARDs that are known to increase serum lipid levels in RA may be mediated by effects on cholesterol handling in macrophages (Ronda et al., 2015).
Our study is of clinical relevance as it provides a plausible explanation for the reduction in serum cholesterol during RA and partially explains the effect of tofacitinib on the lipid profile of RA patients. A better understanding of lipid changes that occur during inflammation and with anti‐inflammatory therapies will aid the management of patients at risk of cardiovascular complications.
Author contributions
S.P.‐B., G.H.‐B. and R.L. were in charge of the conception, design, analysis and interpretation of data. S.P.‐B. and J.I.B. performed the animal experiments. S.P.‐B., J.I.B., P.G., A.L.‐V. and I.P.‐P. contributed to the acquisition of data. S.P.‐B., J.I.B., P.G., A.L.‐V., I.P.‐P., G.H.‐B. and R.L. were involved in drafting the article or revising it critically for important intellectual content and approved the final version. G.H.‐B. and R.L. had full access to overall data and take responsibility for the integrity and accuracy of the data analysis.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Schematic representation of the experimental model. Antigen‐induced chronic arthritis (CA) was induced in 18 rabbits. Four weekly intra‐articular injections of ovalbumin (OVA) were given to previously immunized rabbits. Tofacitinib was administered orally in a dose of 10 mg·kg−1·day−1 for 2 weeks until the end of the study. Six saline‐injected animals were used as controls.
Figure S2 Correlation between serum TC and CRP. Spearman correlation, r = −0.454, P < 0.05.
Figure S3 ABCA1 gene expression in THP‐1 cells stimulated with HFD rabbit serum (or h‐oxLDL) and IFNγ in absence/presence of tofacitinib. Semi quantitative PCR of ABCA1 in THP‐1 foam cells obtained after incubation with 4% HFD rabbit serum (A, left panel, n = 6) or 50 μg·mL−1 h‐oxLDL (B, right panel, n = 6) for 24 h. Cells were exposed to IFNγ (50 ng·mL−1) and/or tofacitinib (0.5 or 2 μM) for additional 24 h. Results are expressed as fold change of the HFD serum‐only or oxLDL‐only group. Bars show the mean and SEM. Mann‐Withney test, a: P < 0.05 versus basal media, b: P < 0.05 versus HFD serum (A) or oxLDL (B), c: P < 0.05 versus HFD serum + IFNγ (A) or oxLDL + IFNγ (B).
Acknowledgements
We are especially grateful to Oliver Shaw for his proofreading of English and to Dr Juan A. Moreno for his kind help with LDL isolation and THP‐1 culture protocols. This work was partially supported by a Pfizer Investigator Initiated Research (IIR) Competitive Grant (ASPIRE, #XZJ‐IIR‐01‐1) and grants from the Instituto de Salud Carlos III (PI13/00570; PI15/00340, PI16/00065 and RETICEF RD12/0043/0008), co‐funded by Fondo Europeo de Desarrollo Regional (FEDER).
Pérez‐Baos, S. , Barrasa, J. I. , Gratal, P. , Larrañaga‐Vera, A. , Prieto‐Potin, I. , Herrero‐Beaumont, G. , and Largo, R. (2017) Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: understanding the lipid paradox associated with rheumatoid arthritis. British Journal of Pharmacology, 174: 3018–3031. doi: 10.1111/bph.13932.
References
- A‐Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradly MN, Zelcer N et al. (2009). Apoptotic cells promote their own clearance and immune tolerance through activation of LXR. Immunity 31: 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro Leon ML, Evans GF, Farmen MW, Zuckerman SH (2005). Post‐transcriptional regulation of macrophage ABCA1, an early response gene to IFN‐gamma. Biochem Biophys Res Commun 333: 596–602. [DOI] [PubMed] [Google Scholar]
- Alvarez‐Soria MA, Largo R, Santillana J, Sánchez‐Pernaute O, Calvo E, Hernández M et al. (2006). Long term NSAID treatment inhibits COX‐2 synthesis in the knee synovial membrane of patients with osteoarthritis: differential proinflammatory cytokine profile between celecoxib and aceclofenac. Ann Rheum Dis 65: 998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asquith DL, Ballantine LE, Nijjar JS, Makdasy MK, Patel S, Wright PB et al. (2013). The liver X receptor pathway is highly upregulated in rheumatoid arthritis synovial macrophages and potentiates TLR‐driven cytokine release. Ann Rheum Dis 72: 2024–2031. [DOI] [PubMed] [Google Scholar]
- Asquith DL, Miller AM, Hueber AJ, McKinnon HJ, Sattar N, Graham GJ et al. (2009). Liver X receptor agonism promotes articular inflammation in murine collagen‐induced arthritis. Arthritis Rheum 60: 2655–2665. [DOI] [PubMed] [Google Scholar]
- Bag‐Ozbek A, Giles JT (2015). Inflammation, adiposity, and atherogenic dyslipidemia in rheumatoid arthritis: is there a paradoxical relationship? Curr Allergy Asthma Rep 15: 497. [DOI] [PubMed] [Google Scholar]
- Birrell MA, Catley MC, Hardaker E, Wong S, Willson TM, McCluskie K et al. (2007). Novel role for the liver X nuclear receptor in the suppression of lung inflammatory responses. J Biol Chem 282: 31882–31890. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P et al. (2015). The JAK inhibitor tofacitinib suppresses synovial JAK1‐STAT signalling in rheumatoid arthritis. Ann Rheum Dis 74: 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles‐Schoeman C, Fleischmann R, Davignon J, Schwartz H, Turner SM, Beysen C et al. (2015). Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheum 67: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles‐Schoeman C, Gonzalez‐Gay MA, Kaplan I, Boy M, Geier J, Luo Z et al. (2016b). Effects of tofacitinib and other DMARDs on lipid profiles in rheumatoid arthritis: implications for the rheumatologist. Semin Arthritis Rheum 46: 71–80. [DOI] [PubMed] [Google Scholar]
- Charles‐Schoeman C, Lee YY, Grijalva V, Amjadi S, Fitzgerald J, Ranganath VK et al. (2012). Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis 71: 1157–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles‐Schoeman C, Wicker P, Gonzalez‐Gay MA, Boy M, Zuckerman A, Soma K et al. (2016a). Cardiovascular safety findings in rheumatoid arthritis patients treated with tofacitinib, a novel, oral janus kinase inhibitor. Arthritis Rheum 46: 261–271. [DOI] [PubMed] [Google Scholar]
- Chen M, Li W, Wang N, Zhu Y, Wang X, Chen M et al. (2007). ROS and NF‐kB but not LXR mediate IL‐1 signaling for the downregulation of ATP‐binding cassette transporter A1. Am J Physiol Cell Physiol 292: 1493–1501. [DOI] [PubMed] [Google Scholar]
- Chintalacharuvu SR, Sandusky GE, Burris TP, Burmer GC, Nagpal S (2007). Liver X receptor is a therapeutic target in collagen‐induced arthritis. Arthritis Rheum 56: 1365–1367. [DOI] [PubMed] [Google Scholar]
- Choy E, Ganeshalingam K, Semb AG, Szekanecz Z, Nurmohamed M (2014). Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology (Oxford) 53: 2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy E, Sattar N (2009). Interpreting lipid levels in the context of high‐grade inflammatory states with a focus on rheumatoid arthritis: a challenge to conventional cardiovascular risk actions. Ann Rheum Dis 68: 460–469. [DOI] [PubMed] [Google Scholar]
- Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q et al. (2011). Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Investig 121: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolhain RJ, ter Haar NT, Hoefakker S, Tak PP, de Ley M, Claassen E et al. (1996). Increased expression of interferon (IFN)‐gamma together with IFN‐gamma receptor in the rheumatoid synovial membrane compared with synovium of patients with osteoarthritis. Br J Rheumatol 35: 24–32. [DOI] [PubMed] [Google Scholar]
- Dowty ME, Jesson MI, Ghosh S, Lee J, Meyer DM, Krishnaswami S et al. (2013). Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J Pharmacol Exp Ther 348: 165–173. [DOI] [PubMed] [Google Scholar]
- Fan J, Kitajima S, Watanabe T, Xu J, Zhang J, Liu E et al. (2015). Rabbit models for the study of human atherosclerosis: from pathophysiological mechanisms to translational medicine. Pharmacol Ther 146: 104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann R, Kremer J, Cush J, Schulze‐Koops H, Connell C a, Bradley JD et al. (2012). Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 367: 495–507. [DOI] [PubMed] [Google Scholar]
- Frisdal E, Lesnik P, Olivier M, Robillard P, Chapman MJ, Huby T et al. (2011). Interleukin‐6 protects human macrophages from cellular cholesterol accumulation and attenuates the proinflammatory response. J Biol Chem 286: 30926–30936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Sengoku T (2013). Effects of the Janus kinase inhibitor CP‐690550 (tofacitinib) in a rat model of oxazolone‐induced chronic dermatitis. Pharmacology 91: 207–213. [DOI] [PubMed] [Google Scholar]
- Hao XR, Cao DL, Hu YW, Li XX, Liu XH, Xiao J et al. (2009). IFN‐gamma down‐regulates ABCA1 expression by inhibiting LXR‐alpha in a JAK/STAT signaling pathway‐dependent manner. Atherosclerosis 203: 417–428. [DOI] [PubMed] [Google Scholar]
- Jasin H (1988). Chronic arthritis in rabbits. Methods Enzymol 162: 379–385. [DOI] [PubMed] [Google Scholar]
- Johnsson H, Panarelli M, Cameron A, Sattar N (2013). Analysis and modelling of cholesterol and high‐density lipoprotein cholesterol changes across the range of C‐reactive protein levels in clinical practice as an aid to better understanding of inflammation‐lipid interactions. Ann Rheum Dis 73: 1495–1499. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P (2003). Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med 9: 213–219. [DOI] [PubMed] [Google Scholar]
- Kennedy A, Fearon U, Veale DJ, Godson C (2011). Macrophages in synovial inflammation. Front Immunol 2: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR et al. (2004). Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 45: 1169–1196. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JM, Cohen S, Wilkinson BE, Connell C a, French JL, Gomez‐Reino J et al. (2012). A phase IIb dose‐ranging study of the oral JAK inhibitor tofacitinib (CP‐690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum 64: 970–981. [DOI] [PubMed] [Google Scholar]
- Krenn V, Morawietz L, Häupl T, Neidel J, Petersen I, König A (2002). Grading of chronic synovitis – a histopathological grading system for molecular and diagnostic pathology. Pathol Res Pract 198: 317–325. [DOI] [PubMed] [Google Scholar]
- Largo R, Sánchez‐Pernaute O, Marcos ME, Moreno‐Rubio J, Aparicio C, Granado R et al. (2008). Chronic arthritis aggravates vascular lesions in rabbits with atherosclerosis: a novel model of atherosclerosis associated with chronic inflammation. Arthritis Rheum 58: 2723–2734. [DOI] [PubMed] [Google Scholar]
- Lu B, Moser AH, Shigenaga JK, Feingold KR, Grunfeld C (2006). Type II nuclear hormone receptors, coactivator, and target gene repression in adipose tissue in the acute‐phase response. J Lipid Res 47: 2179–2190. [DOI] [PubMed] [Google Scholar]
- Luo D, Cao D, Xiong Y, Peng X, Liao D (2010). A novel model of cholesterol efflux from lipid‐loaded cells. Acta Pharmacol Sin 31: 1243–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, Yamaoka K, Kubo S, Nakano K, Iwata S, Saito K et al. (2012). The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon‐γ and interleukin‐17 production by human CD4+ T cells. Arthritis Rheum 64: 1790–1798. [DOI] [PubMed] [Google Scholar]
- Maeß MB, Wittig B, Lorkowski S (2014). Highly efficient transfection of human THP‐1 macrophages by nucleofection. J Vis Exp 91 e51960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majithia V, Geraci SA (2007). Rheumatoid arthritis: diagnosis and management. Am J Med 120: 936–939. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A (2013). Imaging of neutral lipids by Oil Red O for analyzing the metabolic status in health and disease. Nat Protoc 8: 1149–1154. [DOI] [PubMed] [Google Scholar]
- Meune C, Touzé E, Trinquart L, Allanore Y (2009). Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta‐analysis of cohort studies. Rheumatology (Oxford) 48: 1309–1313. [DOI] [PubMed] [Google Scholar]
- Milman N, Karsh J, Booth RA (2010). Correlation of a multi‐cytokine panel with clinical disease activity in patients with rheumatoid arthritis. Clin Biochem 43: 1309–1314. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Muñoz‐García B, Martín‐Ventura JL, Madrigal‐Matute J, Orbe J, Páramo JA et al. (2009). The CD163‐expressing macrophages recognize and internalize TWEAK: potential consequences in atherosclerosis. Atherosclerosis 207: 103–110. [DOI] [PubMed] [Google Scholar]
- Oliviero F, Lo Nigro A, Bernardi D, Giunco S, Baldo G, Scanu A et al. (2012). A comparative study of serum and synovial fluid lipoprotein levels in patients with various arthritides. Clin Chim Acta 413: 303–307. [DOI] [PubMed] [Google Scholar]
- Oliviero F, Sfriso P, Baldo G, Dayer JM, Giunco S, Scanu a et al. (2009). Apolipoprotein A‐I and cholesterol in synovial fluid of patients with rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Clin Exp Rheumatol 27: 79–83. [PubMed] [Google Scholar]
- Park M‐C, Kwon Y‐J, Chung S‐J, Park Y‐B, Lee S‐K (2010). Liver X receptor agonist prevents the evolution of collagen‐induced arthritis in mice. Rheumatology (Oxford) 49: 882–890. [DOI] [PubMed] [Google Scholar]
- Pascual‐García M, Rué L, León T, Julve J, Carbó JM, Matalonga J et al. (2013). Reciprocal negative cross‐talk between liver X receptors (LXRs) and STAT1: effects on IFN‐γ‐induced inflammatory responses and LXR‐dependent gene expression. J Immunol (Baltimore, Md : 1950) 190: 6520–6532. [DOI] [PubMed] [Google Scholar]
- Prieto‐Potín I, Roman‐Blas JA, Martínez‐Calatrava MJ, Gómez R, Largo R, Herrero‐Beaumont G (2013). Hypercholesterolemia boosts joint destruction in chronic arthritis. An experimental model aggravated by foam macrophage infiltration. Arthritis Res Ther 15: R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AB, Patel C a, Rahman MM, Chan ESL, Hasneen K, Montesinos MC et al. (2004). Interferon‐gamma impedes reverse cholesterol transport and promotes foam cell transformation in THP‐1 human monocytes/macrophages. Med Sci Monit 10: BR420–BR425. [PubMed] [Google Scholar]
- Robertson J, Peters MJ, McInnes IB, Sattar N (2013). Changes in lipid levels with inflammation and therapy in RA: a maturing paradigm. Nature reviews . Rheumatology 9: 513–523. [DOI] [PubMed] [Google Scholar]
- Romero FI, Martínez‐Calatrava MJ, Sánchez‐Pernaute O, Gualillo O, Largo R, Herrero‐Beaumont G (2010). Pharmacological modulation by celecoxib of cachexia associated with experimental arthritis and atherosclerosis in rabbits. Br J Pharmacol 161: 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronda N, Favari E, Borghi MO, Ingegnoli F, Gerosa M, Chighizola C et al. (2014). Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 73: 609–615. [DOI] [PubMed] [Google Scholar]
- Ronda N, Greco D, Adorni MP, Zimetti F, Favari E, Hjeltnes G et al. (2015). Newly identified antiatherosclerotic activity of methotrexate and adalimumab: Complementary effects on lipoprotein function and macrophage cholesterol metabolism. Arthritis Rheumatol 67: 1155–1164. [DOI] [PubMed] [Google Scholar]
- Schulze‐Koops H, Kalden JR (2001). The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol 15: 677–691. [DOI] [PubMed] [Google Scholar]
- De Seny D, Cobraiville G, Charlier E, Neuville S, Lutteri L, Le Goff C et al. (2015). Apolipoprotein‐A1 as a damage‐associated molecular patterns protein in osteoarthritis: ex vivo and in vitro pro‐inflammatory properties. PLoS One 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souto A, Salgado E, Maneiro JR, Mera A, Carmona L, Gómez‐Reino JJ (2015). Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: a systematic review and meta‐analysis. Arthritis Rheumatol 67: 117–127. [DOI] [PubMed] [Google Scholar]
- Tall AR (1993). Plasma cholesteryl ester transfer protein. J Lipid Res 34: 1255–1274. [PubMed] [Google Scholar]
- Tall AR, Yvan‐Charvet L (2015). Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15: 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker SG, Abdalrahman Z, Sciumè G, Tsai WL, Anna M (2017). Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol 69: 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S et al. (2012). Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 367: 508–519. [DOI] [PubMed] [Google Scholar]
- Voloshyna I, Modayil S, Littlefield MJ, Belilos E, Belostocki K, Rosenblum G et al. (2014). Plasma from rheumatoid arthritis patients promotes pro‐atherogenic cholesterol transport gene expression in THP‐1 human macrophages. Exp Biol Med (Maywood, NJ) 238: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanni AE (2004). The laboratory rabbit: an animal model of atherosclerosis research. Lab Anim 38: 246–256. [DOI] [PubMed] [Google Scholar]
- Yu X‐H, Zhang J, Zheng X‐L, Yang Y‐H, Tang C‐K (2015). Interferon‐γ in foam cell formation and progression of atherosclerosis. Clin Chim Acta 441: 33–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Schematic representation of the experimental model. Antigen‐induced chronic arthritis (CA) was induced in 18 rabbits. Four weekly intra‐articular injections of ovalbumin (OVA) were given to previously immunized rabbits. Tofacitinib was administered orally in a dose of 10 mg·kg−1·day−1 for 2 weeks until the end of the study. Six saline‐injected animals were used as controls.
Figure S2 Correlation between serum TC and CRP. Spearman correlation, r = −0.454, P < 0.05.
Figure S3 ABCA1 gene expression in THP‐1 cells stimulated with HFD rabbit serum (or h‐oxLDL) and IFNγ in absence/presence of tofacitinib. Semi quantitative PCR of ABCA1 in THP‐1 foam cells obtained after incubation with 4% HFD rabbit serum (A, left panel, n = 6) or 50 μg·mL−1 h‐oxLDL (B, right panel, n = 6) for 24 h. Cells were exposed to IFNγ (50 ng·mL−1) and/or tofacitinib (0.5 or 2 μM) for additional 24 h. Results are expressed as fold change of the HFD serum‐only or oxLDL‐only group. Bars show the mean and SEM. Mann‐Withney test, a: P < 0.05 versus basal media, b: P < 0.05 versus HFD serum (A) or oxLDL (B), c: P < 0.05 versus HFD serum + IFNγ (A) or oxLDL + IFNγ (B).