

Abstract
Background and Purpose
One of the protective actions of angiotensin converting enzyme‐2 (ACE2) is the inactivation of angiotensin II. Expression and activity of ACE2 was reduced in glomeruli of diabetic patients and in animal models of diabetes. Recently the potential role of recombinant ACE2 administration in preventing diabetic nephropathy (DN) has been shown. Here we have tested the effects of the ACE2 activator, diminazene aceturate (DIZE), in a model of DN.
Experimental Approach
Male Wistar rats were rendered diabetic using a single dose of streptozotocin (55 mg·kg−1, i.p.). After 4 weeks, diabetic animals were divided into experimental groups and treated with DIZE, at a low dose (5 mg·kg−1·day−1), a high dose (15 mg·kg−1·day−1) and the high dose with of the AT2 receptor antagonist PD123319 (10 mg·kg−1·day−1). At the end of the treatment , kidneys from all the groups were collected and processed separately for glomerular isolation, protein isolation, mRNA extraction and for immunohistochemical studies.
Key Results
Treatment with DIZE restored ACE2 expression in glomeruli and increased expression of AT2 receptors in whole kidney and isolated glomeruli of diabetic animals. DIZE administration reduced angiotensin II levels and increased angiotensin‐(1–7) levels in diabetic kidney. However, PD123319 treatment reversed all these actions of DIZE.
Conclusions and Implications
DIZE treatment reduced diabetes‐induced renal damage as shown by reduction of fibrosis and apoptosis. These protective actions of DIZE were blocked by the AT2 receptor antagonist. Taken together, these results suggest that DIZE protected against DN through the ACE2/angiotensin‐(1–7)/ AT2 receptor axis.
Abbreviations
- Ang 1–7
angiotensin‐(1–7)
- Ang II
angiotensin II
- BUN
blood urea nitrogen
- DN
diabetic nephropathy
- NC
normal control
- PAL
plasma albumin
- PCr
plasma creatinine
- PGL
plasma glucose
- STZ
streptozotocin
Introduction
Diabetic nephropathy (DN) is one of the most common causes of the development of end‐stage renal disease globally (Giacco et al., 2014). The pathogenesis of DN is mainly due to uncontrolled or chronic hyperglycaemia (TA, 2014). DN is characterized pathologically by glomerular hypertrophy, thickening of the basement membrane, apoptosis and fibrosis due to accumulation of extracellular matrix proteins such as collagen and fibronectin (Deshpande et al., 2013). Proteinuria, albuminuria and glomerular hyperfiltration rate are the clinical features of DN (Gross et al., 2005).
Activation of the renal renin–angiotensin system (RAS) plays an important role in the progression of DN through generation of the peptide angiotensin II (Ang II) and, consequently, blockade of the RAS protected against the development of diabetic kidney injury (Burns, 2000). Ang II acts through two angiotensin receptors, AT1 and AT2 receptors. In the kidney, Ang II mediates its pathological actions through AT1 receptors and these actions include cellular differentiation, proliferation, hypertrophy, fibrosis, renal vasoconstriction and increased tubular sodium reabsorption (Ruggenenti et al., 2010), whereas the functional role of the AT2 receptors in the development of DN is not completely understood. Recent reports suggest that some actions, such as the anti‐inflammatory, antifibrotic, vasodilatory, anti‐hypertrophic and anti‐apoptotic effects are due to activation of AT2 receptors(Cha et al., 2013; Ocaranza et al., 2014; Pandey and Gaikwad, 2017). Although chronic treatments with angiotensin receptor antagonists and ACE inhibitors are effective in retarding the progression of DN, such treatments are not a cure (Bilous et al., 2009; Mauer et al., 2009), indicating requirement for identifying additional pathways within the RAS system as potential drug targets.
A newly recognized component of the RAS is the enzyme ACE2, which shares 42% homology with ACE but with different biochemical activities. ACE and ACE2 are co‐expressed in many tissues and the effects of ACE2 activity are usually opposed to those of ACE. In the kidney, ACE2 activity is reno‐protective by cleaving and thus inactivating Ang II and, at the same time, generating the protective peptide angiotensin‐(1–7) (Ang 1–7) (Tikellis and Thomas, 2012). In several animal models of diabetes, ACE2 expression and activity was decreased in renal tissue (Soler et al., 2006; Leehey et al., 2008). In male Akita mice, a model of type 1 diabetes, treatment with human recombinant ACE2 reduced albuminuria, mild hypertension, plasma Ang II levels, activation of NADPH oxidase, glomerular hypertrophy and mesangial matrix expansion, thereby preventing the progression of DN (Oudit et al., 2010). In another study, injection of adenoviral‐ACE2 in streptozotocin (STZ)‐induced diabetic rats for 4 weeks improved the signs of DN (Liu et al., 2011). In addition, in patients with type 2 diabetes, expression of glomerular and tubular ACE2 were also reduced (Mizuiri et al., 2008). Taken together, these studies suggest that ACE2 plays a protective role against the development of DN.
In a range of disease models, the ACE2 activator, diminazene aceturate (DIZE), plays a protective role. Thus, short‐term treatment with DIZE prevented the reduction in ACE2 activity in rats with subtotal nephrectomy (Velkoska et al., 2015). In rats, treatment with DIZE improved the myocardial infarction through inhibiting cardiac inflammation and apoptosis (Qi et al., 2013). In another study, DIZE prevented hypoxia‐induced death of cardiomyocytes through inhibiting high‐mobility group box 1 protein (Qi et al., 2016). There is also evidence that DIZE is beneficial in several diabetes‐induced pathologies. Chronic treatment with DIZE prevented the oxidative stress and endothelial damage in db/db mice by increasing ACE2 activity and Ang 1–7 levels (Zhang et al., 2015b). DIZE also prevented diabetes‐induced cardiac electrical changes in STZ‐induced diabetic rats (Coutinho et al., 2014). However, so far little is known about the effects of DIZE on the molecular mechanisms involved in the progression of DN.
Methods
Animal studies and drug treatment
All animal care and experimental procedures were approved by the Institutional Animal Ethics Committee, Birla Institute of Technology and Science, Pilani (BITS Pilani) under Protocol No: IAEC/RES/18/05. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male adult Wistar rats (180–220 g) were supplied by the central animal facility of BITS Pilani. Animals were maintained under standard environmental conditions and provided with food and water ad libitum.
The study was performed according to the protocol for Type 1 diabetes, as described previously (Goru et al., 2016). Briefly, diabetes was induced by injecting a single dose of STZ (55 mg·kg−1, i.p.), dissolved in ice‐cold sodium citrate buffer (0.01 M, pH 4.4) and the normal animals were injected only with ice‐cold sodium citrate buffer. Animals with plasma glucose (PGL) levels >16 mmol·L−1 after induction of diabetes were included in the study as diabetic animals. All the diabetic animals received an i.p. injection of insulin (2–3 U) every 3 days to maintain blood glucose levels between 16 and 25 mmol·L−1 in order to prevent mortality induced by excessively high blood glucose levels. At 4 weeks after the injection so STZ or saline, renal functional parameters were estimated in all the animals before separation in to experimental groups (8 rats per group). Normal animals (injected with saline) were divided into normal control (NC), NC treated with high dose of DIZE (NC + HD) and diabetic animals ( STZ‐treated) grouped into diabetic control (DC), DC treated with low dose of DIZE (DC + LD), DC treated with high dose of DIZE (DC + HD) and DC treated with high dose DIZE and PD123319 (DC + HD + PD). The low dose (5 mg·kg−1·day−1) and the high dose (15 mg·kg−1·day−1) of DIZE were administered as single daily i.p. injections (Dhawale et al., 2016) for 4 weeks; PD123319 (10 mg·kg−1·day−1) was administered as single daily s.c. injections for the last 2 weeks (Pandey et al., 2015) as an addition to the treatment with DIZE. At the end of 8 weeks, kidneys from all the individual groups were processed separately for protein isolation, RNA extraction and for immunohistochemical studies.
Assessment of biochemical parameters in plasma
Biochemical estimations were performed as described by Goru et al. (2016). Venous blood (0.5mL) was taken at the time of group allocation (4weeks after STZ)and then again at the end of the whole experiment (8 weeks after STZ). All animals were fasted overnight before blood collection. Heparin (200 IU mL‐1 of blood) was used as an anticoagulant. The plasma was separated from the blood by centrifuging the blood samples at 2000 x g for 15 min, at 4oC. Plasma samples were analysed for glucose (PGL), BUN, albumin (PAL) and creatinine (PCr) by using commercially available kits (Accurex).
Immunohistochemistry
Immunohistochemistry was performed as described previously (Pandey et al., 2015). Briefly, kidney sections (5 μm) were taken from paraffin blocks and deparaffinized with xylene, followed by antigen retrieval by heating in citrate buffer (10 mmol·L−1). The following primary antibodies were used: anti‐collagen IV, anti‐fibronectin, anti‐AT1 receptor , anti‐AT2 receptor (rabbit, 1:200 dilution; Santa Cruz Biotechnology, Dallas, Texas, USA), anti‐TGF‐β (rabbit, 1:200 dilution; Cell Signaling Technology, Danvers, Massachusetts, USA) and anti‐MAS1 receptor (goat, 1:200 dilution; Santa Cruz Biotechnology), and anti‐rabbit and anti‐goat HRP‐conjugated secondary antibody was used, followed by detection with diaminobenzidine (DAB) as a chromogen. The slides were counterstained with haematoxylin, dehydrated with alcohol and xylene and mounted in DPX (Sigma‐Aldrich). At least 25 kidney sections from each group were observed, and images were captured at 100× magnification by using Olympus microscope (model no. BX51, Olympus, Tokyo, Japan). All the images were analysed using ImageJ software (NIH, Bethesda, MD, USA) for calculating DAB‐positive area.
Glomeruli isolation from whole kidney
Kidneys from both normal and diabetic animals were collected and placed in ice‐cold PBS, pH 7.4, and glomeruli were isolated as described previously (Goru et al., 2016). Briefly, kidneys were cut into thin slices, the medullary portion was removed carefully and the cortical area was minced with a scalpel blade to a paste‐like consistency. This was passed through a stainless steel sieve with a pore size of 250 μm. The material that passed through this sieve was suspended in ice‐cold PBS and passed through three consecutive cell strainers (200, 100 and 70 μm), pluriStrainer set 3 (pluriSelect, Leipzig, Germany). The glomeruli retained on the 100 and 70 μm cell strainers were washed with ice‐cold PBS and resuspended in ice‐cold PBS. These glomeruli were assessed under a light microscope and used for Western blotting analysis.
Protein isolation and Western blotting
Western blotting was carried out as described previously (Goru et al., 2016). Immunoblotting was performed by using rabbit monoclonal antibodies against: TGF‐β, cleaved caspase‐3, cleaved PARP, Smurf2 (Cell Signaling Technology), ACE2 and AT2 receptors (Santa Cruz Biotechnology). All antibodies were used in 1:1000 (v/v) dilutions. As secondary, anti‐rabbit IgG, HRP‐linked antibody, was used in 1:20 000 (v/v) dilution. Proteins were detected by using the ECL system and Hyperfilm. Immunoblots were quantified by densitometric analysis using ImageJ software, and the exposures were in the linear dynamic range; TGF‐β, cleaved caspase‐3, cleaved PARP, ACE2 and AT2 receptor proteins were normalized using β‐actin.
Estimation of systemic and tissue‐specific levels of ACE, ACE2, Ang II and Ang 1–7
Plasma, urine, whole kidney and isolated glomeruli samples were obtained from rats (n = 6 per group) of all the groups. Levels of ACE, ACE2, Ang II and Ang 1–7 were assayed with ELISA kits following the manufacturer's recommendations (Wuhan Fine Biological Technology Co., Ltd., Wuhan, China) (Zhang et al., 2015a).
RNA isolation and real‐time polymerase chain reaction (RT‐PCR)
RNA isolation and RT‐PCR were performed as described previously (Goru et al., 2016). Briefly, RNA was reverse transcribed, and RT‐PCR was performed using the FastStart Essential DNA Green MAS1ter (Roche, Penzberg, Germany). Relative expression of each gene of interest was evaluated on LightCycler® 96 Real‐Time PCR System (Roche) using LightCycler Software (Roche). Abundance of targeted mRNA was normalized against 18 s mRNA. All the primers used in the study are listed in Table S1.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean ± SEM, and n refers to number of animals in a particular group. Statistical analysis was performed using GraphPad Prism, version 5.01 (GraphPad Software Inc., La Jolla, CA, USA). Statistical significance was determined with the one‐way ANOVA followed by the Tukey's test for multiple comparisons when F achieved P < 0.05. Results were considered significant if P < 0.05.
Materials
STZ and DIZE were obtained from Sigma. Glucose (glucose oxidase–peroxidase), blood urea nitrogen (BUN), creatinine and albumin kits were purchased from Accurex (Mumbai, India). All the other chemicals were purchased from Sigma, unless otherwise mentioned.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Changes in body weight, kidney weight and kidney weight/body weight ratio by treatment with diminazene aceturate (DIZE) and DIZE in presence of the AT2 receptor antagonist PD123319
STZ‐induced diabetic animals showed significantly decreased body weight and increased kidney weight, leading to an increase in the kidney /body weight ratio (Table 1). This ratio is a marker for the development of DN. Treatment of diabetic rats with the higher dose of DIZE (15 mg·kg−1) reversed the increases in kidney weight and decreased the kidney /body weight ratio (Table 1). However, adding the AT2 receptor antagonist PD123319 for the last two weeks reversed these effects of DIZE (Table 1). Further, treatment of normal rats with the high dose of DIZE did not alter theses parameters (Table 1).
Table 1.
Effect of DIZE alone or with the AT2 receptor antagonist PD123319, on morphometric parameters
| Group | Body weight (g) | Kidney weight (g) | Kidney weight/body weight × 100 |
|---|---|---|---|
| NC | 208 ± 4.17 | 0.64 ± 0.02 | 0.31 ± 0.02 |
| NC + HD | 214 ± 6.8 | 0.59 ± 0.03 | 0.28 ± 0.01 |
| DC | 149 ± 5.6a | 0.8 ± 0.03a | 0.54 ± 0.03a |
| DC + LD | 162 ± 12 | 0.7 ± 0.04 | 0.44 ± 0.05 |
| DC + HD | 170 ± 7.2 | 0.56 ± 0.02b | 0.34 ± 0.03b |
| DC + HD + PD | 155 ± 7.5 | 0.82 ± 0.027a , c | 0.62 ± 0.04a , c |
Values shown are means ± SEM; n = 8 rats per group.
P < 0.05, significantly different from NC and NC + HD.
P < 0.05 , significantly different from DC.
P < 0.05, significantly different from DC + HD.
Effect of DIZE on plasma biochemical parameters in diabetic rats
After 8 weeks, levels of plasma glucose in diabetic rats were significantly higher than in the NC. Treatment with DIZE did not show any significant effects on plasma glucose levels in NC and in diabetes‐induced rats (Table 2). Increased PCr and BUN levels are the indicators of the development of DN in rats. DIZE at both doses (5 and 15 mg·kg−1) decreased the increased PCr and BUN levels in diabetic rats. These effects were not dose‐dependent (Table 2). When compared with control animals, PAL levels were significantly decreased in diabetic control rats and this decrease was significantly inhibited by both doses of DIZE, again without dose‐dependence (Table 2). This normalisation of these biochemical parameters by DIZE suggests that DIZE protects against renal damage in diabetic animals. However, DIZE treatment in the presence of PD123319 failed to normalize the diabetes‐induced changes in plasma (Table 2). In normal rats, DIZE did not alter any of the plasma biochemical parameters measured (Table 2).
Table 2.
Effect of DIZE alone or with PD123319 on plasma biochemical parameters
| Group | PGL (mmol·L−1) | PCr (μmol·L−1) | BUN (mmol·L−1) | PAL (g·L−1) |
|---|---|---|---|---|
| NC | 6 ± 0.5 | 132 ± 7.6 | 7.7 ± 0.9 | 42 ± 1 |
| NC + HD | 7 ± 0.5 | 134 ± 5.8 | 5 ± 1.6 | 36.2 ± 1.2 |
| DC | 26 ± 1a | 214 ± 12a | 16.35 ± 0.7a | 21.76 ± 1.4a |
| DC + LD | 24.4 ± 1a | 171 ± 6.2b | 11 ± 0.8b | 31.7 ± 2.3b |
| DC + HD | 24 ± 1.2a | 145 ± 7.4b | 10.2 ± 0.5b | 34 ± 2.5b |
| DC + HD + PD | 27 ± 1.7a | 235 ± 5.2c | 18 ± 0.9c | 20.48 ± 2c |
Values shown are means ± SEM; n = 8 rats per group.
P < 0.05, significantly different from NC and NC + HD.
P < 0.05, significantly different from DC.
P < 0.05, significantly different from DC + HD.
ACE2 activation prevented renal fibrosis and apoptosis
Renal fibrosis and apoptosis are considered to be the underlying causes for the development of diabetic kidney disease. In this study, we found increased expression of the profibrotic marker, TGF‐β, and increased markers of apoptosis such as cleaved PARP and cleaved caspase‐3, in diabetic kidneys. These changes were normalized significantly by the higher dose of DIZE (Figure 1A–D).
Figure 1.
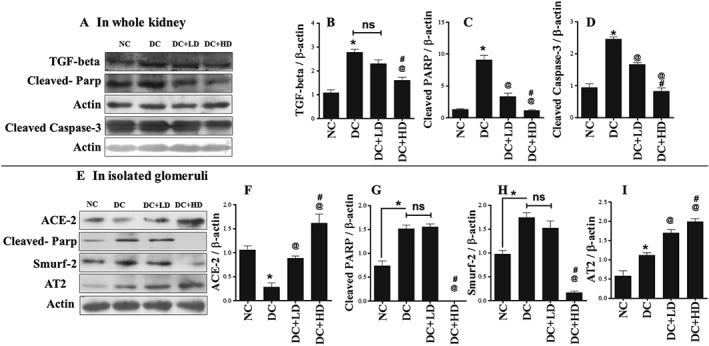
DIZE inhibited diabetes‐induced renal fibrosis and apoptosis through increasing glomerular ACE2 and expression of AT2 receptor protein. (A) Western blot analysis of TGF‐β, cleaved PARP and cleaved caspase‐3 in whole kidney and (B–D) their respective quantitative analysis using ImageJ software. (E) Western blot analysis of ACE2, cleaved PARP, Smurf2 and AT2 receptors in isolated glomeruli and (F–I) their respective quantitative analysis using ImageJ software. Values shown are means ± SEM. n = 6 rats per group; *P < 0.05, significantly different from NC, @ P < 0.05, significantly different from DC. # P < 0.05, significantly different from DC + LD.
DIZE treatment increased glomerular ACE2, and AT2 receptor expression decreased the expression of cleaved PARP and Smurf2
Further, to check the molecular changes in glomeruli, we performed Western blotting experiments on isolated glomeruli. In glomeruli from diabetic animals, expression of ACE2 protein was reduced and this was restored, dose‐dependently, by DIZE treatment (Figure 1E, F). The protein expression of cleaved PARP and Smurf2 was also increased in diabetic glomeruli, and this was reversed by the higher dose of DIZE (Figure 1E, G, H). Smurf2 is an ubiquitin E3 ligase that degrades SMAD7, a negative regulator of TGF‐β, and is the key molecule involved in the development of fibrosis. We also observed increased expression of AT2 receptor protein in diabetic glomeruli, which was further increased dose‐dependently by DIZE administration (Figure 1E, I).
Effect of treatment with DIZE alone and in presence of PD123319 on systemic and tissue‐specific changes in RAS components (ACE, ACE2, Ang II and Ang 1–7)
Changes in the RAS are critical features of the pathogenesis of diabetes. In this study, we measured the systemic (plasma and urinary) levels of major RAS components including ACE and ACE2 and also in whole kidney and in isolated glomeruli (Figure 2). In diabetic animals, expression of ACE was increased in plasma, whole kidneys and isolated glomeruli (Figure 2A, C, D), and DIZE treatment reduced ACE in plasma (Figure 2A) but not in whole kidney and isolated glomeruli. The AT2 receptor antagonist, PD123319, combined with DIZE further increased these levels in whole kidney and isolated glomeruli (Figure 2C, D). Levels of ACE protein in urine were normal in diabetic animals, and in diabetic animals treated with DIZE. However in diabetic rats treated with DIZE and PD123319, urinary ACE protein was increased (Figure 2B). For ACE2, levels in plasma were the same in all experimental groups (Figure 2E), whereas in urine, ACE2 levels were increased in diabetic animals, and these levels were normalized by treatment with DIZE alone and again increased when DIZE was combined with PD123319 (Figure 2F). In whole kidney, levels of ACE2 protein were elevated in diabetic animals, remained high after DIZE alone and were further increased by treatment with DIZE and PD123319 (Figure 2G). In isolated glomeruli, levels of ACE2 protein were lower in diabetic kidneys and this low level was restored after DIZE alone. Adding the AT2 receptor antagonist to DIZE reversed the effect of DIZE and reduced the protein levels of ACE2 (Figure 2H).
Figure 2.
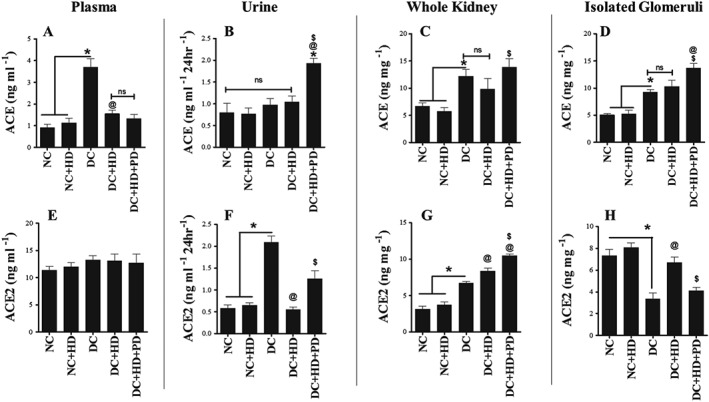
Systemic and tissue‐specific protein expression of RAS components in plasma, urine, whole kidney and isolated glomeruli. (A–D) Levels of ACE protein in plasma, urine, whole kidney and glomeruli; (E–H) Levels of ACE2 protein in plasma, urine, whole kidney and glomeruli. Values shown are means ± SEM. n = 6; *P < 0.05, significantly different from NC and NC + HD, @ P < 0.05, significantly different from DC. $ P < 0.05, significantly different from DC + HD; ns, not significant.
We also assayed levels of Ang II and Ang 1–7 levels in plasma, whole kidney and isolated glomeruli. Ang II is formed from Ang I through the enzymic actions of ACE and ACE2 acts on Ang II to generate the reno‐protective peptide Ang 1–7. In our study, levels of Ang II in plasma, whole kidney and isolated glomeruli were increased by diabetes and this increase was reversed after treatment with DIZE alone. Adding PD123319 to DIZE had no effect on this reversal in plasma and whole kidney (Figure 3A, B), whereas in isolated glomeruli, the AT2 receptor antagonist again increased the levels of Ang II (Figure 3C). For Ang 1‐7, levels in plasma, whole kidney and in isolated glomeruli were decreased by diabetes and DIZE treatment improved these levels. Adding PD123319 to DIZE did not alter plasma levels of Ang 1–7 but did reverse the effects of DIZE on levels of Ang 1–7 in whole kidney and isolated glomeruli (Figure 3D–F). Importantly, in normal rats, DIZE did not affect any of the components of the RAS measured (Figures 2 and 3).
Figure 3.
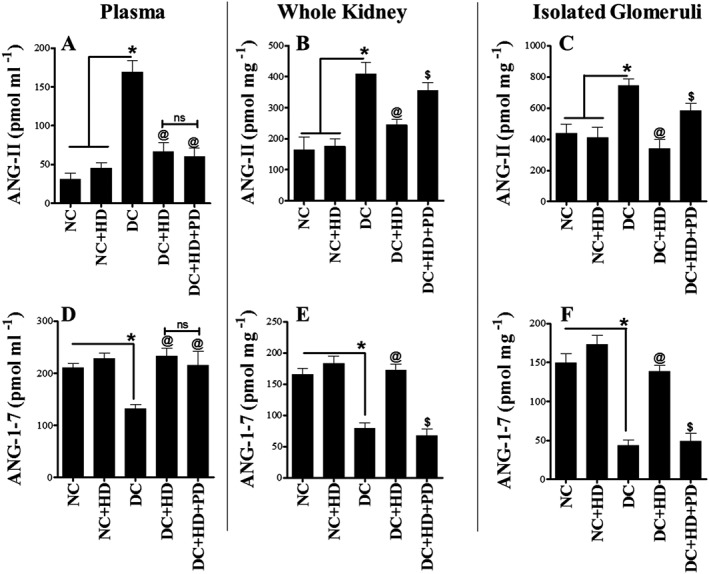
Levels of Ang II and Ang 1–7 peptides in plasma, whole kidney and isolated glomeruli. (A–C) Levels of Ang II peptide in plasma, whole kidney and glomeruli; (D–F) Levels of Ang 1–7 peptide in plasma, whole kidney and glomeruli. Values shown are means ± SEM. n = 6; *P < 0.05, significantly different from NC and NC + HD. @ P < 0.05, significantly different from DC. $ P < 0.05, significantly different from DC + HD. ns, not significant.
Changes in the expression of mRNA for ACE, ACE2, AT1, AT2 and MAS1 receptors, in whole kidney and glomeruli, after treatment with DIZE alone and with PD123319
To determine the effect of our experimental conditions on the transcription of genes for ACE, ACE2, AT1, AT2 and MAS1 receptors, we quantified expression of their mRNA, in whole kidney and isolated glomeruli. In whole kidney, mRNA for ACE was increased in diabetic animals, DIZE treatment normalized this increase and the AT2 receptor antagonist PD123319 reversed the effect of DIZE (Figure 4A). However, for ACE2, gene expression was reduced in whole kidneys from diabetic rats and DIZE treatment increased these levels. This effect of DIZE was not affected by adding the AT2 receptor antagonist (Figure 4G). The mRNA for AT1 receptors was increased in whole kidneys from diabetic animals, DIZE alone had no effect on this increase but adding PD123319 further elevated these levels (Figure 4C). For AT2 receptors, mRNA expression was increased in whole kidneys by diabetes, DIZE alone further increased these levels which wer not changed by adding PD123319 (Figure 4D). Surprisingly, the mRNA for MAS1 receptors was significantly reduced in whole kidneys of diabetic rats and treatment with DIZE alone or combined with PD123319 did not affect this reduction (Figure 4E). In isolated glomeruli, expression of these mRNAs showed a pattern similar to that in whole kidney (Figure 4F–J). Note that DIZE did not affect expression of these genes in normal rats (Figure 4).
Figure 4.
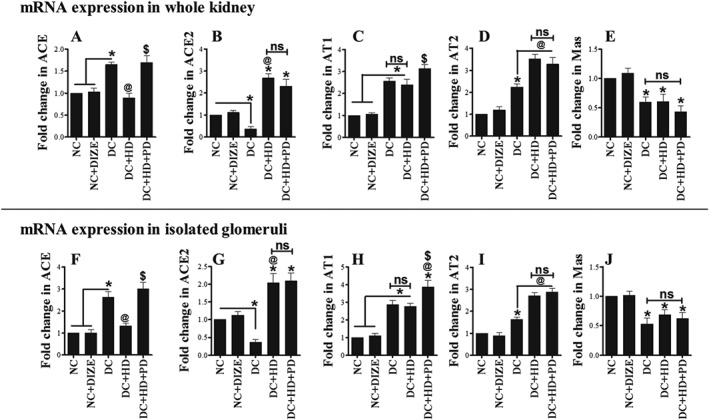
Expression of mRNA for ACE, ACE2, AT1, AT2 and MAS1 receptors in whole kidney and glomeruli. (A, F) mRNA expression of ACE, (B, G) mRNA expression of ACE2, (C, H) mRNA expression of AT1 receptors, (D, I) mRNA expression of AT2 receptors and (E, J) mRNA expression of MAS1 receptors. Values shown are means ± SEM. n = 6; *P < 0.05, significantly different from NC and NC + HD. @ P < 0.05, significantly different from DC. $ P < 0.05, significantly different from DC + HD. ns, not significant.
Expression of AT1, AT2 and MAS1 receptor protein in whole kidney after treatment with DIZE alone and combined with PD123319
Next, we used immunohistochemistry to monitor the expression of AT1, AT2 and MAS1 receptor protein, in our experimental groups. Expression of AT1 receptor protein was increased in diabetic kidney, DIZE alone significantly normalized this change and combining DIZE and PD123319 reversed the effects of DIZE (Figure 5A, B). For AT2 receptors, expression in diabetic kidney was up‐regulated and DIZE treatment further increased this expression. This effect of DIZE was blocked by adding PD123319, returning expression of AT2 receptor protein to the same level as that of diabetic control rats (Figure 5C, D). Interestingly, we found down‐regulation of MAS1 receptors in diabetic kidney. However, administration of DIZE alone or in presence of PD123319 had no effect on MAS1 receptor expression in diabetic kidneys (Figure 5E, F).
Figure 5.
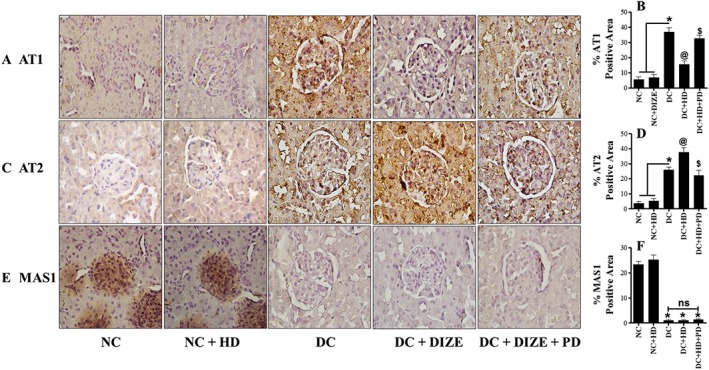
Renal expression of AT1, AT2 and MAS1 receptor proteins. (A, C, E) Immunohistochemical staining of AT1, AT2 and MAS1 receptors in kidney sections and (B, D, F) their respective semi‐quantitative analysis using ImageJ software. Values shown are means ± SEM. n = 25. * P < 0.05, significantly different from NC and NC + HD, @ P < 0.05 significantly different from DC. $ P < 0.05, significantly different from DC + HD. ns, not significant.
AT2 receptor blockade re‐induced renal fibrosis and apoptosis and prevented the protective actions of DIZE
Further, to assess the effects of AT2 receptor blockade with PD123319 on reno‐protection mediated by DIZE, we quantified expression of three markers of fibrosis ‐ TGF‐β, fibronectin and collagen IV‐ along with two markers of apoptosis ‐ cleaved PARP and cleaved caspase‐3 ‐ in our experimental groups. We observed increased expression of TGF‐β, fibronectin and collagen IV in diabetic kidneys, compared with kidneys from normal rats. Treatment of diabetic rats with DIZE significantly attenuated this increased expression of fibrotic markers and adding PD123319 to DIZE, reversed the DIZE‐mediated attenuation (Figure 6A–F). We measured the markers of apoptosis in isolated glomeruli and found cleaved PARP and cleaved caspase‐3 were increased by diabetes and this increase was normalized by administering DIZE alone. However, DIZE combined with PD123319 was no longer able to reduce these apoptotic markers, and expression of cleaved PARP and cleaved caspase‐3 was again markedly increased (Figure 7A–C).
Figure 6.
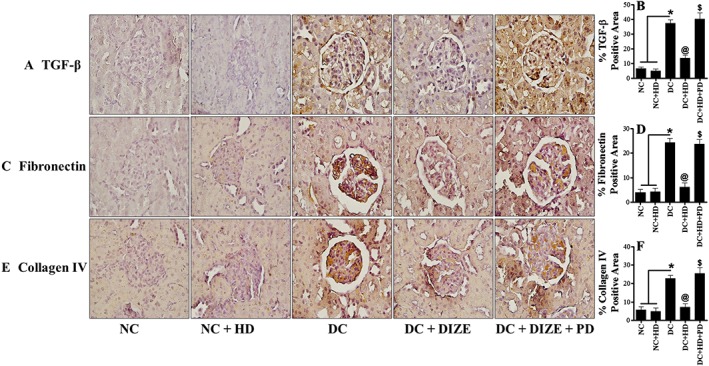
Blockade of protective actions of DIZE in presence of AT2 receptor antagonist and re‐expression of fibrotic markers. (A, C, E) Immunohistochemical staining of TGF‐β, fibronectin and collagen IV in kidney sections and (B, D, F) their respective semi‐quantitative analysis using ImageJ software. Values shown are means ± SEM. n = 25. * P < 0.05, significantly different from NC and NC + HD. @ P < 0.05, significantly different from DC. $ P < 0.05, significantly different from DC + HD.
Figure 7.
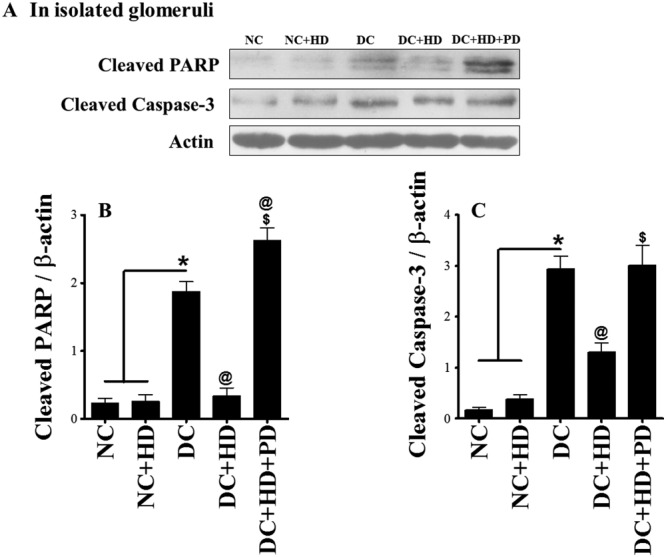
Prevention of DIZE mediated protection and increased apoptosis in isolated glomeruli by AT2 receptor antagonist. (A) Western blot analysis of cleaved PARP, cleaved caspase‐3 in isolated glomeruli and (B, C) their respective quantitative analysis using ImageJ software. Values shown are means ± SEM. n = 26. * P < 0.05, significantly different from NC and NC + HD. @ P < 0.05, significantly different from DC. $ P < 0.05, significantly different from DC + HD.
Discussion
To study the effect of ACE2 activation on the progression of DN, we have used a well‐known off‐target (Gjymishka et al., 2010) ACE2 activator, DIZE (Kulemina and Ostrov, 2011). In our study, DIZE treatment significantly improved the renal morphometric parameters ‐ kidney weight and kidney weight/body weight ratio ‐ at higher doses but it did not affect the reduced body weights in diabetic animals (Table 1). DIZE treatment also normalized plasma biochemistry ‐ BUN, PCr and PAL ‐ in diabetic animals (Table 2). Further, at molecular level, DIZE treatment significantly reduced the expression of fibrotic markers such as TGF‐β in whole kidney and Smurf2 in isolated glomeruli, at the higher dose (15 mg·kg−1·day−1) (Figure 1). Similarly, DIZE also prevented diabetes‐induced renal apoptosis through reducing the expression of cleaved PARP and cleaved caspase‐3 in whole kidney, as well as in glomeruli (Figure 1). Moreover, in isolated glomeruli from diabetic animals, we observed decreased expression of ACE2 and increased expression of AT2 receptors, and DIZE treatment restored the low ACE2 levels and further increased expression of AT2 receptors, dose dependently (Figure 1). These results led us to a conclusion that reno‐protection of DIZE involves increased expression of ACE2 and AT2 receptors.
Our results contrast with recent studies of direct ACE2 activation and expression by DIZE. Recently, Haber et al. (2014) demonstrated that DIZE did not activate ACE2 either in in vitro or in ex vivo experiments. Similarly, Raffai et al. (2014) also failed to show ACE2 activation by DIZE in an ex vivo experiment using porcine coronary artery rings. However, these studies were designed to study the acute effects of DIZE and did not assess the effects of chronic administration of DIZE on ACE2 activation in models in vivo. For instance, Zhang et al. (2015b) reported that chronic treatment with DIZE prevented oxidative stress and endothelial damage in db/db mice by increasing ACE2 activity and Ang 1–7 levels. Moreover, Ali et al. (2013) demonstrated that chronic AT2 receptor activation induced increased ACE2 activity and reduced BP in Zucker obese rats. In the same study, AT2 receptor agonists significantly increased the ACE2 activity in HK‐2 cells in vitro, and this effect was blocked by a AT2 receptor antagonist. These reports raised the possibility that DIZE‐mediated reno‐protection in our study might not be through increased ACE2 expression but through an AT2 receptor‐mediated pathway. To test this, we treated diabetic animals with a high dose of DIZE, with and without the AT2 receptor antagonist PD123319.
Further, to check the effects of chronic treatment with DIZE on several components of the RAS, we have quantified the tissue‐specific levels of ACE and ACE2. Although there are several reports on tissue‐specific expression of ACE and ACE2 in diabetic kidney, these findings are still a matter of debate. Ye et al. (2006) demonstrated reduced tubular ACE and increased glomerular ACE in 8‐week‐old female db/db mice. In the same study, they observed increased levels of ACE2 in renal tubules and decreased glomerular ACE2 in diabetic mice. In another study, the same group showed reduced mRNA levels, protein levels and activity of ACE and increased ACE2 protein levels and activity, but not its mRNA, in renal tubules of female db/db mice (Ye et al., 2004). Similarly, Wysocki et al. (2006) also showed reduced mRNA levels, protein levels and activity of ACE and increased ACE2 protein levels and activity but not its mRNA levels in renal tubules of female db/db mice and STZ‐induced female diabetic mice. In contrast to this, Tikellis et al. (2008) demonstrated the induction of diabetes in male C57Bl6 mice decreased renal cortical expression of ACE2 mRNA and ACE2 protein, and this was associated with a significant reduction in cortical levels of Ang 1–7. In addition, Moon et al. (2008) showed increased mRNA for ACE in both tubules and glomeruli of STZ‐induced male diabetic rats. ACE2 expression was increased only in tubules and decreased in glomeruli, whereas mRNA expression was unaltered in these diabetic rats. Previously, we have shown increased expression of ACE2 in kidney sections from type 2 diabetic rats (Pandey et al., 2015). These reports clearly indicate the site‐specific expression of ACE and ACE2 protein and of their mRNA, but their activities are not well correlated in diabetic kidney, and even species, strain, age, sex and the extent of glucose levels may also affect the expression of these two enzymes. However, drugs or agents that inhibit ACE activity and increase ACE2 activity or expression have proved to be effective in DN, irrespective of gender (Amann et al., 2003; Gross et al., 2003; Alderson et al., 2004; Oudit et al., 2010; Riera et al., 2016).
In our study, DIZE treatment only reduced the plasma levels of ACE but not that in the whole kidney or glomeruli, whereas further increase was observed in both whole kidney and glomeruli when DIZE was combined with the AT2 receptor antagonist (Figure 2A, C, D). Urinary ACE levels were increased only in the group treated with DIZE and PD123319 and remained unaltered in other groups (Figure 2B). Further, ACE2 protein was increased in whole kidneys from groups treated with DIZE or DIZE with PD123319 (Figure 2G). On the other hand, DIZE treatment restored the decreased levels of ACE2 and, combined with PD123319, DIZE failed to restore these levels in isolated glomeruli (Figure 2H). In addition, DIZE treatment reduced the mRNA expression of ACE in whole kidney and increased the mRNA expression of ACE2 in glomeruli of diabetic animals, whereas adding PD123319 had no effect on these actions of DIZE (Figure 4A, B, F, G). Recent studies have demonstrated that the increased urinary levels of ACE2 and activity in animal models of DN were due to increased activity of ADAM17 and that inhibition of ADAM17 reduced urinary ACE2 levels and activity (Salem et al., 2014; Riera et al., 2016). In our study, DIZE significantly reduced levels of urinary ACE2, whereas DIZE combined with PD123319 lost this effect to some extent and resulted in increased urinary ACE2 levels (Figure 2A). These results suggest that the protective actions of DIZE were exerted mainly through increasing renal ACE2 levels and the termination of the protective actions of DIZE by the AT2 receptor antagonist may correlate with the increased expression of ACE in whole kidney, glomeruli and the reduced expression of ACE2 in glomeruli.
Next, we checked the activity of ACE and ACE2 indirectly through quantifying the levels of Ang II and Ang 1–7 in plasma, whole kidney and isolated glomeruli. RAS activation is critical for the development of DN (Burns, 2000). Increased levels of Ang II is a major outcome of the activation of the RAS, through the actions of ACE (Kobori et al., 2013). Ang II was up‐regulated in plasma and renal tissue of DN (Mezzano et al., 2003), whereas Ang 1–7 was down‐regulated in plasma and renal tissue of diabetic animals (Zhang et al., 2015a). In line with these reports, we observed increased levels of Ang II and reduced levels of Ang 1–7 in plasma, whole kidney and isolated glomeruli from diabetic animals (Figure 3). This can be attributed to the increased and decreased ACE and ACE2 activity in plasma, whole kidney and isolated glomeruli of diabetic animals. However, the high dose of DIZE decreased Ang II and increased Ang 1–7 in plasma, whole kidney and isolated glomeruli, and further, AT2 receptor blockade reversed the effect of DIZE in whole kidney and isolated glomeruli, but not in plasma (Figure 3A–F). These results indicate that ACE2 activity in renal tissue was increased after DIZE administration, and that this increase was lost in presence of AT2 receptor blockade, implying a role for AT2 receptors in the activation of ACE2.
There are many reports of the protective actions of Ang 1–7, which acts through MAS1 receptors and counteracts the actions of Ang II at AT1 receptors. Recently, Zhang et al. (2015a) demonstrated that a large dose of Ang 1–7 (800 ng·kg−1·min−1) administered s.c. by an embedded mini‐osmotic pump in STZ‐induced diabetic rats, for 4 weeks, significantly improved renal function, attenuated glomeruli sclerosis, reduced oxidative stress markers (NOX4 and p47phox) and decreased the expression of ECM markers (collagen IV and TGF‐β1) through increasing the expression of renal MAS1 receptors, and all these effects were blocked by the MAS1 receptor antagonist (A779). In another recent study, Shi et al. (2015) demonstrated similar effects of Ang 1–7 in DN, when administered at a dose of 500 μg·kg−1·day−1 for 6 weeks in Akita mice, and these effects were blocked by the MAS1 receptor antagonist. However, both the studies showed down‐regulation of MAS1 receptors in diabetic animals, and a large amount of Ang 1–7 was needed to restore the MAS1 receptor expression. (Shi et al., 2015; Zhang et al., 2015a). Down‐regulation of Ang 1–7 and MAS1 receptors have also been reported in human diabetic kidneys (Mizuiri et al., 2012). In our study, we found a loss of MAS1 receptors in diabetic kidneys,but DIZE administration did not restore these levels even after increasing the Ang 1–7 levels, at both mRNA and protein levels in diabetic kidney (Figure 5E, F). This suggests that the large doses of Ang 1–7 used by Zhang et al. (2015a) and Shi et al. (2015) may be required for the transcriptional and translational activation of MAS1 receptors. Thus, in our study, the DIZE‐mediated reno‐protective actions in DN were not mediated through the Ang 1–7/MAS1 pathway. However, we did observe increased mRNA and protein levels of AT2 receptors in diabetic kidney after treatment with DIZE (Figures 1E, I and 5C, D). Previously, Sourris et al. (2010) showed that the protective effect of RAGE blockade in a mouse model of DN is through increased expression of AT2 receptors. Ang 1–7 is known to show its protective actions through activating AT2 receptors, actions which were blocked by AT2 receptor antagonists (Ohshima et al., 2014). Therefore, our results strongly suggest that the DIZE‐mediated reno‐protection we have observed here is more likely to be mediated through the Ang 1–7/ AT2 receptor‐mediated pathway (Figure 8). Ang 1–7 and AT2 receptors also regulate and counteract the actions of AT1 receptor agonists (Miura et al., 2010; Ohshima et al., 2014; Galandrin et al., 2016). Further, in our study, DIZE administration significantly decreased the levels of diabetes‐induced AT1 receptor protein, but not the mRNA . Blockade of AT2 receptors again increased AT1 receptor protein and mRNA levels in diabetic kidneys (Figure 5A, B, C, H). Moreover, chronic activation of AT2 receptors prevented caspase‐3‐mediated apoptosis and renal fibrosis in type 2 DN (Pandey and Gaikwad, 2017). Interestingly, in support of these results, DIZE‐mediated reno‐protection was lost in presence of AT2 receptor blockade which re‐induced renal fibrosis, with an increase in fibrotic markers (TGF‐β, fibronectin and collagen IV) (Figure 6) and increased glomerular apoptosis, with elevated levels of cleaved PARP and cleaved caspase‐3 (Figure 7) in glomeruli. In conclusion, the DIZE‐mediated reno‐protection in type 1 diabetic animals was mediated through the ACE2/Ang 1–7/ AT2 receptor pathway (Figure 8), and hence, treatment with DIZE should be beneficial in human DN as well as in other abnormalities where the ACE2/Ang 1–7/ AT2 receptor pathway is dysregulated.
Figure 8.
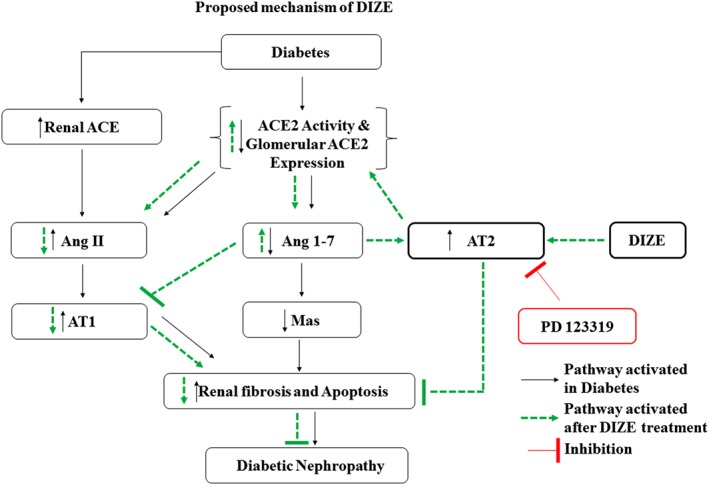
Proposed mechanism of action of DIZE. DIZE prevented diabetes‐induced renal abnormalities like decreased ACE2 activity, ACE2 expression in glomeruli and Ang 1–7 levels and increased AT1 receptors, fibrotic and apoptotic markers by acting through ACE2/Ang 1–7/AT2 receptor axis.
Author contributions
S.K.G. conceived the idea, and A.B.G. designed the experiments. S.K.G., A.K., A.P., V.M. and N.S. performed all the experiments and completed the manuscript writing.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Study plan.
Figure S2 Negative staining for A. TGF, Collagen IV, AT1, AT2, Fibronectin and B. MAS1.
Table S1 Primers list for qRT‐PCR.
Acknowledgements
The authors thank Professor Kulbhushan Tikoo, National Institute of Pharmaceutical Education and Research, SAS Nagar, Punjab, India, for providing their microscope facility.
This work was financially supported by the Council of Scientific & Industrial Research, Government of India [via sanction no. 37(1644)/15/EMR‐II].
Goru, S. K. , Kadakol, A. , Malek, V. , Pandey, A. , Sharma, N. , and Gaikwad, A. B. (2017) Diminazene aceturate prevents nephropathy by increasing glomerular ACE2 and AT2 receptor expression in a rat model of type1 diabetes. British Journal of Pharmacology, 174: 3118–3130. doi: 10.1111/bph.13946.
References
- Alderson N, Chachich M, Frizzell N, Canning P, Metz T, Januszewski A et al (2004). Effect of antioxidants and ACE inhibition on chemical modification of proteins and progression of nephropathy in the streptozotocin diabetic rat. Diabetologia 47: 1385–1395. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Q, Wu Y, Hussain T (2013). Chronic AT2 receptor activation increases renal ACE2 activity, attenuates AT1 receptor function and blood pressure in obese Zucker rats. Kidney Int 84: 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann B, Tinzmann R, Angelkort B (2003). ACE inhibitors improve diabetic nephropathy through suppression of renal MCP‐1. Diabetes Care 26: 2421–2425. [DOI] [PubMed] [Google Scholar]
- Bilous R, Chaturvedi N, Sjølie AK, Fuller J, Klein R, Orchard T et al (2009). Effect of candesartan on microalbuminuria and albumin excretion rate in diabetes: three randomized trials. Ann Intern Med 151: 11–20. [DOI] [PubMed] [Google Scholar]
- Burns KD (2000). Angiotensin II and its receptors in the diabetic kidney. Am J Kidney Dis 36: 449–467. [DOI] [PubMed] [Google Scholar]
- Cha SA, Park BM, Gao S, Kim SH (2013). Stimulation of ANP by angiotensin‐(1‐9) via the angiotensin type 2 receptor. Life Sci 93: 934–940. [DOI] [PubMed] [Google Scholar]
- Coutinho DC, Monnerat‐Cahli G, Ferreira AJ, Medei E (2014). Activation of angiotensin‐converting enzyme 2 improves cardiac electrical changes in ventricular repolarization in streptozotocin‐induced hyperglycaemic rats. Europace 16: 1689–1696. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande SD, Putta S, Wang M, Lai JY, Bitzer M, Nelson RG et al (2013). Transforming growth factor‐β‐induced cross talk between p53 and a microRNA in the pathogenesis of diabetic nephropathy. Diabetes 62: 3151–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawale VS, Amara VR, Karpe PA, Malek V, Patel D, Tikoo K (2016). Activation of angiotensin‐converting enzyme 2 (ACE2) attenuates allergic airway inflammation in rat asthma model. Toxicol Appl Pharmacol 306: 17–26. [DOI] [PubMed] [Google Scholar]
- Galandrin S, Denis C, Boularan C, Marie J, M'Kadmi C, Pilette C et al (2016). Cardioprotective angiotensin‐(1–7) peptide acts as a natural‐biased ligand at the angiotensin II type 1 receptorNovelty and significance. Hypertension 68: 1365–1374. [DOI] [PubMed] [Google Scholar]
- Giacco F, Du X, D'Agati VD, Milne R, Sui G, Geoffrion M et al (2014). Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 63: 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjymishka A, Kulemina LV, Shenoy V, Katovich MJ, Ostrov DA, Raizada MK (2010). Diminazene aceturate is an ACE2 activator and a novel antihypertensive drug. The FASEB Journal 24 (1 Supplement): 1032–1033. [Google Scholar]
- Goru SK, Kadakol A, Pandey A, Malek V, Sharma N, Gaikwad AB (2016). Histone H2AK119 and H2BK120 mono‐ubiquitination modulate SET7/9 and SUV39H1 in type 1 diabetes‐induced renal fibrosis. Biochem J 473: 3937–3949. [DOI] [PubMed] [Google Scholar]
- Gross JL, De Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T (2005). Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care 28: 164–176. [DOI] [PubMed] [Google Scholar]
- Gross M‐L, El‐Shakmak A, Szabo A, Koch A, Kuhlmann A, Münter K et al (2003). ACE‐inhibitors but not endothelin receptor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia 46: 856–868. [DOI] [PubMed] [Google Scholar]
- Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D (2014). Angiotensin‐converting enzyme 2–independent action of presumed angiotensin‐converting enzyme 2 activatorsNovelty and significance. Hypertension 63: 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori H, Kamiyama M, Harrison‐Bernard LM, Navar LG (2013). Cardinal role of the intrarenal renin‐angiotensin system in the pathogenesis of diabetic nephropathy. J Invest Med 61: 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulemina LV, Ostrov DA (2011). Prediction of off‐target effects on angiotensin‐converting enzyme 2. J Biomol Screen. 16: 878–885. [DOI] [PubMed] [Google Scholar]
- Leehey DJ, Singh AK, Bast JP, Sethupathi P, Singh R (2008). Glomerular renin angiotensin system in streptozotocin diabetic and Zucker diabetic fatty rats. Transl Res 151: 208–216. [DOI] [PubMed] [Google Scholar]
- Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB et al (2011). Angiotensin‐converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med 17: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T et al (2009). Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med 361: 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzano S, Droguett A, Burgos ME, Ardiles LG, Flores CA, Aros CA et al (2003). Renin‐angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int 64: S64–S70. [DOI] [PubMed] [Google Scholar]
- Miura S, Matsuo Y, Kiya Y, Karnik SS, Saku K (2010). Molecular mechanisms of the antagonistic action between AT1 and AT2 receptors. Biochem Biophys Res Commun 391: 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuiri S, Hemmi H, Arita M, Ohashi Y, Tanaka Y, Miyagi M et al (2008). Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am J Kidney Dis 51: 613–623. [DOI] [PubMed] [Google Scholar]
- Mizuiri S, Nishizawa Y, Hamanoue M, Hemmi H, Arita M, Shibuya K et al (2012). ACE2‐Ang 1‐7‐MAS1 axis in human diabetic nephropathy. J Nephrol Therapeutic S 2: 005. [Google Scholar]
- Moon J‐Y, Jeong K‐H, Lee S‐H, Lee T‐W, Ihm C‐G, Lim SJ (2008). Renal ACE and ACE2 expression in early diabetic rats. Nephron Exp Nephrol 110: e8–e16. [DOI] [PubMed] [Google Scholar]
- Ocaranza MP, Moya J, Barrientos V, Alzamora R, Hevia D, Morales C et al (2014). Angiotensin‐(1–9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J Hypertens 32: 771–783. [DOI] [PubMed] [Google Scholar]
- Ohshima K, Mogi M, Nakaoka H, Iwanami J, Min L‐J, Kanno H et al (2014). Possible role of angiotensin‐converting enzyme 2 and activation of angiotensin II type 2 receptor by angiotensin‐(1–7) in improvement of vascular remodeling by angiotensin II type 1 receptor blockadeNovelty and significance. Hypertension 63: e53–e59. [DOI] [PubMed] [Google Scholar]
- Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J et al (2010). Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 59: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A, Gaikwad AB (2017). Compound 21 and Telmisartan combination mitigates type 2 diabetic nephropathy through amelioration of caspase mediated apoptosis. Biochem Biophys Res Commun 487: 827–833. [DOI] [PubMed] [Google Scholar]
- Pandey A, Goru SK, Kadakol A, Malek V, Gaikwad AB (2015). Differential regulation of angiotensin converting enzyme 2 and nuclear factor‐κB by angiotensin II receptor subtypes in type 2 diabetic kidney. Biochimie 118: 71–81. [DOI] [PubMed] [Google Scholar]
- Qi Y, Zhang J, Cole‐Jeffrey CT, Shenoy V, Espejo A, Hanna M et al (2013). Diminazene aceturate enhances angiotensin‐converting enzyme 2 activity and attenuates ischemia‐induced cardiac pathophysiology. Hypertension 62: 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi YF, Zhang J, Wang L, Shenoy V, Krause E, Oh SP et al (2016). Angiotensin‐converting enzyme 2 inhibits high‐mobility group box 1 and attenuates cardiac dysfunction post‐myocardial ischemia. J Mol Med 94: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffai G, Khang G, Vanhoutte PM (2014). Angiotensin‐(1‐7) augments endothelium‐dependent relaxations of porcine coronary arteries to bradykinin by inhibiting angiotensin‐converting enzyme 1. J Cardiovasc Pharmacol 63: 453–460. [DOI] [PubMed] [Google Scholar]
- Riera M, Anguiano L, Clotet S, Roca‐Ho H, Rebull M, Pascual J et al (2016). Paricalcitol modulates ACE2 shedding and renal ADAM17 in NOD mice beyond proteinuria. Am J Physiol Renal Physiol 310: F534–F546. [DOI] [PubMed] [Google Scholar]
- Ruggenenti P, Cravedi P, Remuzzi G (2010). The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol 6: 319–330. [DOI] [PubMed] [Google Scholar]
- Salem ES, Grobe N, Elased KM (2014). Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. Am J Physiol Renal Physiol 306: F629–F639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lo C‐S, Padda R, Abdo S, Chenier I, Filep JG et al (2015). Angiotensin‐(1–7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin‐converting enzyme 2 and MAS1 receptor expression in diabetic mice. Clin Sci 128: 649–663. [DOI] [PubMed] [Google Scholar]
- Soler M, Wysocki J, Sowers K (2006). Differential expression of ACE/ACE2 in renal tubules and glomerulus from db/db mice with established nephropathy. J Am Soc Nephrol 17: TH‐PO083. [Google Scholar]
- Sourris K, Morley A, Koitka A, Samuel P, Coughlan M, Penfold S et al (2010). Receptor for AGEs (RAGE) blockade may exert its renoprotective effects in patients with diabetic nephropathy via induction of the angiotensin II type 2 (AT2) receptor. Diabetologia 53: 2442–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman J, Benson H, Faccenda E, Pawson A, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TA S (2014). Diagnosis and classification of diabetes mellitus. Diabetes Care 37: S81. [DOI] [PubMed] [Google Scholar]
- Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C et al (2008). ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes 57: 1018–1025. [DOI] [PubMed] [Google Scholar]
- Tikellis C, Thomas M (2012). Angiotensin‐converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept 2012: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkoska E, Patel SK, Griggs K, Pickering RJ, Tikellis C, Burrell LM (2015). Short‐term treatment with diminazene aceturate ameliorates the reduction in kidney ACE2 activity in rats with subtotal nephrectomy. PLoS One 10: e0118758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE et al (2006). ACE and ACE2 activity in diabetic mice. Diabetes 55: 2132–2139. [DOI] [PubMed] [Google Scholar]
- Ye M, Wysocki J, Naaz P, Salabat MR, LaPointe MS, Batlle D (2004). Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice. Hypertension 43: 1120–1125. [DOI] [PubMed] [Google Scholar]
- Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D (2006). Glomerular localization and expression of angiotensin‐converting enzyme 2 and angiotensin‐converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 17: 3067–3075. [DOI] [PubMed] [Google Scholar]
- Zhang K, Meng X, Li D, Yang J, Kong J, Hao P et al (2015b). Angiotensin (1–7) attenuates the progression of streptozotocin‐induced diabetic renal injury better than angiotensin receptor blockade. Kidney Int 87: 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu J, Luo J‐Y, Tian XY, Cheang WS, Xu J et al (2015a). Upregulation of angiotensin (1‐7)‐mediated signaling preserves endothelial function through reducing oxidative stress in diabetes. Antioxid Redox Signal 23: 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study plan.
Figure S2 Negative staining for A. TGF, Collagen IV, AT1, AT2, Fibronectin and B. MAS1.
Table S1 Primers list for qRT‐PCR.