

Abstract
Background and Purpose
The phosphatase SHIP1 negatively regulates the PI3K pathway, and its predominant expression within cells of the haematopoietic compartment makes SHIP1 activation a novel strategy to limit inflammatory signalling generated through PI3K. AQX‐1125 is the only clinical‐stage, orally administered, SHIP1 activator. Here, we demonstrate the prophylactic and therapeutic effects of AQX‐1125, in a mouse model of bleomycin‐induced lung fibrosis.
Experimental Approach
For prophylactic evaluation, AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or dexamethasone (1 mg·kg−1·d−1, i.p.) were given to CD‐1 mice starting 3 days before intratracheal administration of bleomycin (0.1 IU per mouse) and continued daily for 7 or 21 days. Therapeutic potentials of AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or pirfenidone (90 mg·kg−1·d−1, p.o.) were assessed by initiating treatment 13 days after bleomycin instillation and continuing until day 28.
Key Results
Given prophylactically, AQX‐1125 (10 and 30 mg·kg−1) reduced histopathological changes in lungs, 7 and 21 days following bleomycin‐induced injury. At the same doses, AQX‐1125 reduced the number of total leukocytes, neutrophil activity, TGF‐β immunoreactivity and soluble collagen in lungs. Administered therapeutically, AQX‐1125 (10 and 30 mg·kg−1) improved lung histopathology, cellular infiltration and reduced lung collagen content. At 30 mg·kg−1, the effects of AQX‐1125 were similar to those of pirfenidone (90 mg·kg−1) with corresponding improvements in disease severity.
Conclusions and Implications
AQX‐1125 prevented bleomycin‐induced lung injury during the inflammatory and fibrotic phases. AQX‐1125, given therapeutically, modified disease progression and improved survival, as effectively as pirfenidone.
Abbreviations
- BAL
bronchoalveolar lavage
- IPF
idiopathic pulmonary fibrosis
- MPO
myeloperoxidase
- SHIP1
Src‐homology 2 domain‐containing inositol‐5‐phosphatase
Introduction
Fibrosis occurs in the end stage of many pathological, inflammatory conditions as the result of an excessive accumulation of collagen‐rich, extracellular matrix in inflamed or damaged tissues leading to organ failure and death (Wynn and Ramalingam, 2012). Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive lung disease with a median survival time of 2.5 years from diagnosis (Mura et al., 2012). While idiopathic in nature, the prevailing hypotheses regarding the pathogenesis of this condition are focused on dysfunctional interactions between the epithelium and mesenchyme, promoting continued epithelial injury and fibroblast activation. The disease is likely to be multi‐factorial in nature with dysregulation occurring within multiple biological pathways affecting inflammation, chemotaxis, wound healing and tissue remodelling (Todd et al., 2012).
The phosphatase Src‐homology 2 domain‐containing inositol‐5‐phosphatase 1 (SHIP1; INPP5D) is a 145 kDa intracellular enzyme predominantly expressed in cells of the haematopoietic lineage. The enzymic function of SHIP1 is to degrade phosphatidylinositol (3,4,5)‐trisphosphate, the product of PI3K and a key signalling molecule that promotes cellular survival and growth via recruitment and activation of downstream effector kinases such as Akt and Btk. By opposing PI3K signalling in immune cells, SHIP1 can dampen processes contributing to inflammation and cell migration (Krystal et al., 1999). Mice deficient in SHIP1 are prone to autoimmune reactions and develop a pulmonary inflammatory disease leading to pulmonary fibrosis (Oh et al., 2007).
Small molecule compounds originally isolated from marine natural extracts have been found to act as activators of SHIP1, possessing anti‐inflammatory activity both in vitro and in vivo (Yang et al., 2005; Ong et al., 2007; Stenton et al., 2013a,b). Thus, in recent years, pharmacological activation of SHIP1 has emerged as a novel approach for the experimental therapy of various inflammatory diseases. AQX‐1125 ((1S,3S,4R)‐4‐[(3aS,4R,5S,7aS)‐4‐(aminomethyl)‐7a–methyl‐1‐methylidene‐octahydro‐1H‐inden‐5‐yl]‐3‐(hydroxymethyl)‐4‐methylcyclohexan‐1‐ol; acetic acid salt) is a novel, clinical‐stage, low MW activator of SHIP1 that has shown promising anti‐inflammatory effects in a number of animal models of pulmonary inflammation (Stenton et al., 2013a) and reduced the late response to allergen challenge, with a trend towards reducing sputum inflammatory cell numbers, in asthmatic human subjects (Leaker et al., 2014).
The model of pulmonary injury induced by bleomycin has been extensively used as a model for evaluating the anti‐fibrogenic potential of compounds (Moeller et al., 2007). It is characterized by an initial acute inflammatory phase marked by extensive leukocyte infiltration and the production of pro‐inflammatory cytokines including interleukin‐1, interleukin‐6, interferon‐γ and tumour necrosis factor‐α. The resolution of the inflammatory phase is followed by an increase in the levels of pro‐fibrotic markers, such as transforming growth factor‐β1, fibronectin and procollagen‐1. The timing of compound administration can be used to distinguish therapeutic agents with anti‐inflammatory as distinct from anti‐fibrotic, potential (Chaudhary et al., 2006). The current study was designed to characterize the effects of AQX‐1125 on pulmonary inflammation and fibrosis in comparison to either a standard anti‐inflammatory drug, dexamethasone, or pirfenidone, a clinically relevant compound with anti‐fibrotic activity approved for treatment of IPF.
Methods
Animal studies
All animal care and experimental procedures were in compliance with guidelines approved by the Institutional Animal Care and Use Committee (IACUC) following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 400 adult (6–8 weeks old), male CD‐1 mice weighing between 27 and 32 g (Harlan Nossan, Milan, Italy) were used in the studies.Mice were housed in groups of three in polycarbonate cages (300 × 180 mm × 150 mm) with corn cob bedding, within laminar flow rooms, and were allowed free access to a diet of standard laboratory chow and water. A 12 h light/dark cycle was maintained, with controlled temperature (20–26°C) and humidity (40–70%).
Bleomycin induces a severe pulmonary injury with high morbidity or mortality, similar to IPF in humans and is the most widely acceptable model for this disease. The bleomycin‐induced model of lung injury and fibrosis in rodents has been extensively characterized and is commonly used to test the anti‐fibrotic potential of novel drugs to treat IPF (Moeller et al., 2007), justifying the choice of species for these studies. The CD‐1 strain used in the study are commonly used in research for efficacy and safety testing and are susceptible to bleomycin‐induced pulmonary fibrosis (Raisfeld, 1979). All efforts were made to minimize animal suffering and distress and humane endpoints were used in compliance with IACUC guidelines. Welfare checks occurred daily with assessments of body weight, general appearance and behaviour. Any animal exhibiting progressive deterioration or in distress prior to the study end was humanely euthanized, and the study day where this occurred was recorded.
Bleomycin ‐induced pulmonary injury and treatment
Mice were randomly allocated to treatment groups, and bleomycin (Santa Cruz, Italy) was administered once by a single intra‐tracheal instillation at 0.1 IU per mouse in 0.9% saline in a volume of 100 μL (n = 15 or 40 for prophylactic or therapeutic models respectively), whereas the sham control groups (n = 5 or 10 for prophylactic or therapeutic models respectively) received an instillation of 0.9% saline only, without bleomycin. In the prophylactic model, animals were orally dosed, once daily, with vehicle (saline, n = 15) or AQX‐1125 (3, 10 or 30 mg·kg−1 in saline, n = 15 per group) starting 3 days prior to administration of bleomycin and continuing to the end of the study (Day 7 or 21). The reference control drug, dexamethasone (dissolved in saline), was given by daily intraperitoneal injection at 1 mg·kg−1 (n = 15). For the therapeutic model, mice were dosed orally, once per day, with AQX‐1125 (3, 10 or 30 mg·kg−1 in saline, n = 40), or saline vehicle (n = 40) starting 13 days after the bleomycin challenge. The reference control compound, pirfenidone (n = 40), was given three times per day (in saline) to achieve a total daily dose of 90 mg·kg−1 up until the end of the study (Day 28). At the end of the study, animals were killed by pentobarbitone overdose. Lung tissue was collected and used for assessment of oedema, myeloperoxidase, histology, soluble collagen and/or hydroxyproline content, depending on the study.
Bronchoalveolar lavage (BAL)
At the end of the studies, tracheas were immediately cannulated with an i.v. polyethylene catheter (Neo Delta Ven 2, Delta Med, Viadana, Italy) equipped with a 24‐gauge needle on a 1 mL syringe. Lungs were lavaged once with 0.5 mL D‐PBS (GIBCO, Paisley, UK). In >95% of the mice, the recovery volumes were over 0.4 mL. The BAL fluid was centrifuged at 500× g, the supernatants were stored and the pelleted cells were collected. Total BAL cells were enumerated by counting on a haemocytometer in the presence of Trypan blue. Cytospins were prepared from resuspended BAL cells. Cytospins of BAL cells were made by centrifuging 50 000 cells onto microscope slides using a Shandon Cytospin 3 (Shandon, Astmoore, UK). Slides were allowed to air dry and were then stained with Diff‐Quick Stain Set (Diff‐Quick; Baxter Scientific, Miami, FL, USA). A total of 400 cells were counted from randomly chosen high power microscope fields for each sample.
Measurement of lung oedema
The wet lung tissue weights were measured by careful excision of the lung from other adjacent extraneous tissues. The tissues were exposed for 48 h at 180°C, and the dry weight was subsequently measured. Water content of the lungs was subsequently calculated as the ratio of wet : dry weight of the tissue.
Histological examination
Lung biopsies were fixed at room temperature in buffered formaldehyde solution (10% in phosphate buffered saline), and histological sections were prepared and stained with van Giessen trichrome stain or by haematoxylin–eosin and evaluated using an Axiovision Zeiss (Milan, Italy) microscope. The severity of lung fibrosis was scored on a scale from 0 to 8 by examining randomly chosen sections, with 5 fields per sample at a magnification of 100×. Criteria for grading lung fibrosis were as follows: grade 0, normal lung; grade 1, minimal fibrous thickening of alveolar or bronchiolar walls; grade 3, moderate thickening of walls without obvious damage to lung architecture; grade 5, increased fibrosis with definite damage to lung structure and formation of fibrous bands or small fibrous masses; grade 7, severe distortion of structure and large fibrous areas; grade 8, total fibrous obliteration of fields (Ashcroft et al., 1988).
Myeloperoxidase (MPO) assay
Lung tissue was removed, weighed, homogenized in a solution containing 0.5% hexa‐decyl‐trimethyl‐ammonium bromide dissolved in 10 mM potassium phosphate buffer (pH 7) and centrifuged for 30 min at 20 000× g at 4°C. An aliquot of the supernatant was then allowed to react with a solution of tetra‐methyl‐benzidine (1.6 mM) and 0.1 mM H2O2. The rate of change in absorbance was measured spectrophotometrically at 650 nm. MPO activity was defined as the quantity of enzyme degrading 1 μmol of peroxide per min at 37°C and was expressed in units per gram weight of wet tissue (Mullane et al., 1985).
Soluble collagen assay
The total lung collagen content was measured by performing the Sircol Soluble Collagen Assay (Biocolor, Newtownabbey, Northern Ireland), based on a modification of the Sirius Red method, following the manufacturer's instructions. Explanted lung tissue was homogenized, and samples were then incubated at 4°C for 2 h and centrifuged at 15 000× g. Supernatants (20 mL) were diluted five times in lysis buffer, added to 1 mL of Sircol Dye Reagent and then mixed for 30 min at room temperature in a mechanical shaker. The collagen–dye complex was precipitated by centrifugation at 10 000× g for 10 min. The unbound dye solution was then carefully removed. The precipitated complex was resuspended in 1 mL of alkali reagent. The obtained solution was finally placed in a 96‐well flat‐bottomed plate and evaluated in a plate reader (absorbance = 540 nm). Absolute collagen content was calculated by comparing sample values to a standard curve.
Immunohistochemical localization of TGF‐β and collagen‐1
Lung tissues were fixed in 10% (w/v) PBS‐buffered formaldehyde, and 8 μm sections were prepared from paraffin embedded tissues. After deparaffinization, endogenous peroxidase was quenched with 0.3% (v/v) hydrogen peroxide in 60% (v/v) methanol for 30 min. The sections were permeabilized with 0.1% (w/v) Triton X‐100 in PBS for 20 min. Non‐specific adsorption was minimized by incubating the section in 2% (v/v) normal goat serum in PBS for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with biotin and avidin (DBA, Milan, Italy) respectively. Sections were incubated overnight with anti‐TGF‐β antibody (1:500 v/v in PBS, Santa Cruz Biotechnology, CA, USA) or anti‐collagen‐1 (1:400 v/v in PBS, Santa Cruz Biotechnology, CA), washed with PBS and incubated with secondary antibody. Specific labelling was detected with a biotin‐conjugated goat anti‐rabbit or anti‐mouse IgG and avidin‐biotin peroxidase complex (DBA, Milan, Italy).
The counter stain was developed with DAB (brown colour) and nuclear fast red (red background). A positive staining (brown colour) was found in the sections, indicating that the immunoreactions were positive. To verify the binding specificity for TGF‐β, some sections were also incubated with primary antibody only (no secondary antibody) or with secondary antibody only (no primary antibody). In these situations, no positive staining was found in the sections indicating that the immunoreactions were positive in all the experiments carried out. No positive staining (pink colour) was observed in the sections indicating that the immunoreactions were negative.
Immunohistochemical images were collected using a Zeiss microscope and Axio Vision software. For graphic display of densitometric analyses, the intensity of positive staining (brown staining) was measured by computer‐assisted colour image analysis (Leica QWin V3, UK). The percentage area of immunoreactivity (determined by the number of positive pixels) was expressed as percent of total tissue area (red staining).
Hydroxyproline quantification
Twenty‐eight days after intratracheal instillation of bleomycin or saline, mice were killed. Tissue from the left lung of each animal was used for hydroxyproline quantification as described previously (Christensen et al., 1999). Briefly, the explanted tissue was washed in PBS, weighed, minced and diluted in 10 mL PBS per mg lung. Samples (100L) were hydrolyzed in 12 M HCl in duplicate. Samples were analysed for hydroxyproline content, expressed as μg hydroxyproline per mg of lung tissue.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are expressed as the mean ± SEM with n representing the number of individual animals per experimental cohort. Statistical analysis was performed by ANOVA followed by Dunnett's post hoc test. Survival was compared using a log‐rank test on the Kaplan–Meier curves. All statistical analyses were performed using GraphPad Prism® Version 6.07 software. Probability values of P ≤ 0.05 were considered statistically significant.
Materials
All materials, unless otherwise indicated, were obtained from Sigma‐Aldrich (Poole, UK). AQX‐1125 was prepared from commercially available 5‐dehydroepiandrosterone as described (Raymond et al., 2014).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to the corresponding entries in http://guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Prophylactic effects of AQX‐1125 on bleomycin‐induced pulmonary inflammation and fibrosis
A single intratracheal administration of bleomycin induces acute lung inflammation leading to fibrosis (Izbicki et al., 2002). In rats, the initial inflammation is characterized by an influx of leukocytes, particularly neutrophils, into the airways and an elevation in pro‐inflammatory cytokines and gene expression of pro‐fibrotic markers. After Day 9, the inflammatory cytokine levels begin to resolve and markers associated with fibrosis, such as TGF‐β, and collagen synthesis and deposition increase (Chaudhary et al., 2006). In mice, similar phases have been identified by gene signatures with an active inflammation phase (Days 1–2), an active fibrosis phase (Days 7–14) and a late fibrosis phase (Days 21–35) (Peng et al., 2013). Accordingly, in this model, the anti‐inflammatory potential of AQX‐1125, given before the bleomycin challenge, was tested in the acute inflammatory/initial fibrotic phase (7 days), into the late fibrotic phase (21 days).
Bleomycin‐instillation resulted in substantial histological tissue damage in the bleomycin‐treated animals, characterized by consolidation of the parenchyma and the presence of an extensive inflammatory infiltrate at both 7 and 21 days (Figure 1A, B respectively). In contrast, mice treated with AQX‐1125 at 10 and 30 mg·kg−1, starting 3 days before challenge, appeared to show less tissue damage and a greater preservation of pulmonary architecture (Figure 1A, B respectively). Fibrosis, graded histologically according to the Ashcroft criteria (Ashcroft et al., 1988), was also increased with bleomycin instillation as compared to the sham‐treated group. The bleomycin group had an average score of 6.4 on Day 7 and 6.8 and Day 21, indicative of severe damage to the lung structure and the presence of large fibrous areas even during the early inflammatory and fibrotic stage. At both 7 and 21 days (Figures 1C, D respectively), these scores were significantly reduced to 3–4 by the highest doses of AQX‐1125, with only moderate thickening of the alveolar or bronchiolar walls without obvious damage to the lung architecture.
Figure 1.
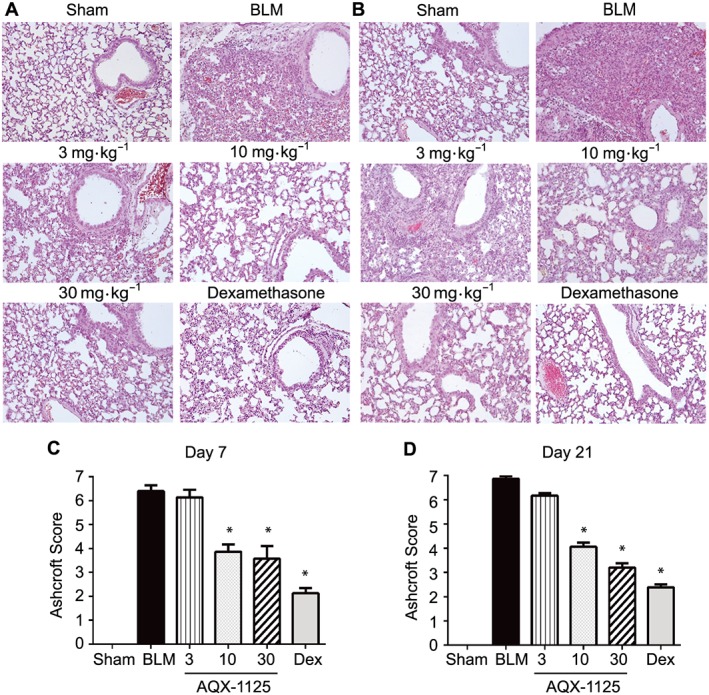
Effects of prophylactic AQX‐1125 on bleomycin‐induced changes in lung histology and fibrosis. Mice were treated prophylactically with either saline vehicle (sal, p.o., daily), AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or dexamethasone (dex, 1 mg·kg−1·d−1, i.p.) for 3 days prior to receiving a single intratracheal administration of bleomycin (BLM). The sham‐treated group received intratracheal saline. Histology and Ashcroft scores were performed on Day 7 (A, C) and Day 21 (B, D) on haematoxylin and eosin stained sections. *P < 0.05, significantly different from vehicle. Results are given as mean ± SEM from n = 5–15 (surviving animals from bleomycin‐treated groups) or n = 5 (sham‐treated group) (C, D).
Extensive increases in BAL cellularity (Figure 2A, B) and neutrophil activity, as measured by myeloperoxidase (MPO) levels (Figure 2C, D), occurred at Days 7 and 21, in the bleomycin‐treated mice as compared to sham controls. Even at Day 21, the bleomycin‐treated group had an increase in BAL leukocytes as compared to the sham control (Figure 2B), similar to previous studies in this strain of mice (Kakugawa et al., 2004; Lee et al., 2010). AQX‐1125 at 10 and 30 mg·kg−1, significantly reduced the total number of BAL leukocytes in bleomycin‐challenged mice, up to a maximum of 60% at 7 days (Figure 2A) and 63% at 21 days at 30 mg·kg−1 (Figure 2B). In contrast, dexamethasone at 1 mg·kg−1 reduced BAL leukocytes by 83 and 82%, at Days 7 and 21 respectively. The maximal reduction was similar across all cell types, the majority of which were macrophages and neutrophils (Table 1). At 30 mg·kg−1 AQX‐1125 reduced MPO activity by 54% at Day 7 (Figure 2C) and by 74% at Day 21 (Figure 2D) where the corresponding reductions by dexamethasone were 87% and 94%. In addition to the influx of leukocytes, the inflammatory phase of BLM is also characterized by substantial lung oedema, as measured by the ratio of wet : dry lung weight, which was significantly diminished by AQX‐1125 (10 and 30 mg·kg−1), and dexamethasone, at both 7 and 21 days (Figure 2E, F respectively).
Figure 2.

Effects of prophylactic AQX‐1125 on bleomycin‐induced cell influx, myeloperoxidase activity (MPO) and pulmonary oedema. Mice were treated prophylactically with either saline vehicle (sal, p.o., daily), AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or dexamethasone (dex, 1 mg·kg−1·d−1, i.p.) for 3 days prior to receiving a single intratracheal administration of bleomycin (BLM). The sham‐treated group received intratracheal saline. Cells recovered from the bronchoalveolar lavage fluid on Day 7 (A) and Day 21 (B) were counted by haemocytometry. MPO activity was measured in homogenized lung tissue on Day 7 (C) and Day 21 (D). Lung oedema at Day 7 (E) and Day 21 (F) was measured as a ratio of wet to dry weight of excised lung tissue. *P < 0.05, significantly different from vehicle. Results are given as mean ± SEM from n = 5–15 (surviving animals from bleomycin‐treated groups) or n = 5 (sham‐treated group).
Table 1.
Differential leukocyte counts in the BALF of mice at 7 and 21 days in prophylactic model
| Neutrophils (×106) | Macrophages (×106) | Lymphocytes (×106) | ||||
|---|---|---|---|---|---|---|
| Day 7 | Day 21 | Day 7 | Day 21 | Day 7 | Day 21 | |
| Sham | 0.00 ± 0.00* | 0.00 ± 0.00* | 0.22 ± 0.02* | 0.21 ± 0.01* | 0.00 ± 0.00* | 0.00 ± 0.00* |
| Bleomycin | 0.29 ± 0.03 | 0.12 ± 0.02 | 0.60 ± 0.03 | 0.66 ± 0.05 | 0.11 ± 0.01 | 0.23 ± 0.02 |
| AQX‐1125 3 mg·kg−1 | 0.27 ± 0.02 | 0.11 ± 0.01 | 0.58 ± 0.04 | 0.57 ± 0.05 | 0.10 ± 0.01 | 0.23 ± 0.02 |
| AQX‐1125 10 mg·kg−1 | 0.10 ± 0.02* | 0.05 ± 0.01* | 0.40 ± 0.07* | 0.38 ± 0.01* | 0.05 ± 0.01* | 0.10 ± 0.01* |
| AQX‐1125 30 mg·kg−1 | 0.09 ± 0.01* | 0.05 ± 0.01* | 0.40 ± 0.02* | 0.35 ± 0.02* | 0.04 ± 0.01* | 0.10 ± 0.01* |
| Dexamethasone 1 mg·kg−1 | 0.04 ± 0.01* | 0.03 ± 0.00* | 0.28 ± 0.01* | 0.28 ± 0.01* | 0.03 ± 0.00* | 0.04 ± 0.01* |
Data are the mean ± SEM of the absolute leukocyte counts (×106 cells) (n = 5–15).
P < 0.05 shows a significant effect of AQX‐1125 or dexamethasone, compared to the vehicle‐treated bleomycin group.
TGF‐β is an important mediator of fibrosis and is thought to contribute to the pathogenesis of IPF (Fernandez and Eickelberg, 2012). Instillation of bleomycin dramatically increased TGF‐β immunoreactivity in the lungs of treated mice at Day 21, during the fibrotic phase of the disease. In accordance with the observed attenuation of inflammation and the reduced Ashcroft fibrotic score, AQX‐1125 (10 and 30 mg·kg−1) also significantly reduced TGF‐β detected in the lung (Figure 3A, B) by up to 97%, similar to dexamethasone. The anti‐fibrotic effect of AQX‐1125 was further supported by a decrease in soluble lung collagen, by up to 60%, at 30 mg·kg−1 (Figure 3C). We did not measure lung mechanical changes in these studies; however, the collagen content has been shown to correlate with the dynamic parameters of resistance and elastance in this model (Dolhnikoff and Mauad, 1999).
Figure 3.
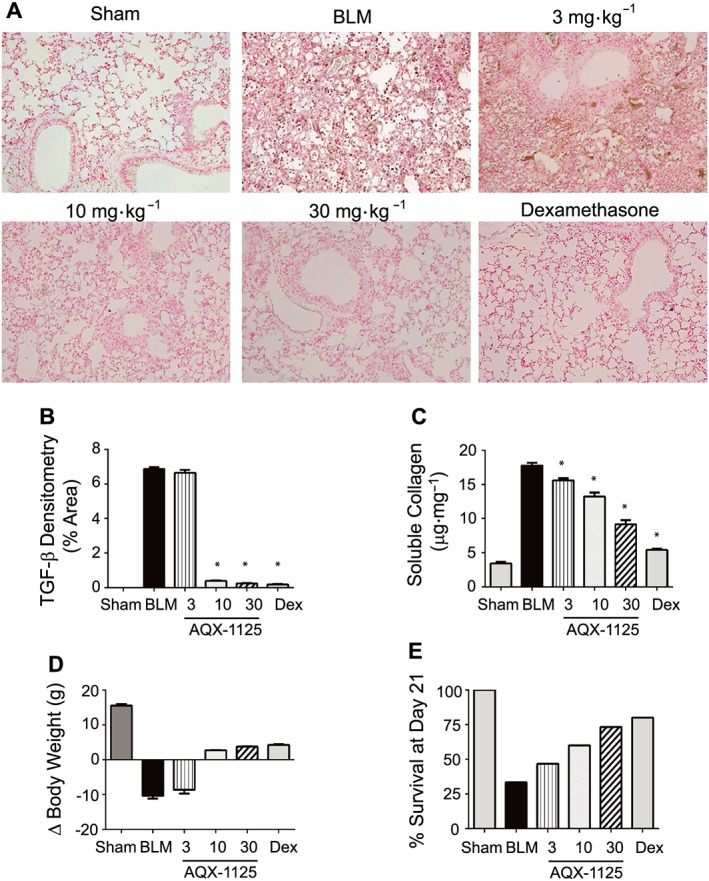
Effects of prophylactic AQX‐1125 on bleomycin ‐induced up‐regulation of TGF‐β, soluble collagen and body weight loss. Mice were treated prophylactically with either saline vehicle (sal, p.o., daily), AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or dexamethasone (dex, 1 mg·kg−1·d−1, i.p.) for 3 days prior to receiving a single intratracheal administration of bleomycin (BLM). The sham‐treated group received intratracheal saline. At Day 21, lung tissues were processed for immunohistochemical detection of TGF‐β (A), and the total area positive for TGF‐β was calculated by densitometry (B). Total soluble lung collagen (C) was measured using the Sircol assay. Body weight loss (D) was calculated by taking the difference in mouse weights on Day 0 and Day 21. The percent of each group remaining at Day 21 were also calculated (E). *P < 0.05, significantly different from vehicle. Results are given as mean ± SEM from n = 5–15 (surviving animals from bleomycin‐treated groups) or n = 5 (sham‐treated group).
The severe lung injury induced by bleomycin results in a loss of body weight and decreased survival. AQX‐1125‐treated animals were partly protected from bleomycin‐induced body weight loss, and at 10 and 30 mg·kg−1, a small weight gain was observed in these groups at Day 21 (Figure 3D). Similarly, these same treatment groups had an increase in the percent of surviving animals from 33% in the bleomycin‐treated group to 60 and 73% at 10 and 30 mg·kg−1 groups respectively. The dexamethasone‐treated group had a survival of 73%, the same as the 30 mg·kg−1 AQX‐1125‐treated group.
Effect of AQX‐1125 administered therapeutically following bleomycin exposure
Many anti‐inflammatory drugs have shown benefit in the bleomycin model when given prophylactically, potentially by interfering with the induction of the response (Chaudhary et al., 2006). In order to separate the anti‐inflammatory activity of AQX‐1125 from its anti‐fibrotic potential, we initiated daily treatment, therapeutically, beginning 13 days following BLM exposure. Corticosteroids do not prevent fibrosis when given therapeutically (Chaudhary et al., 2006; Cortijo et al., 2009), and therefore, pirfenidone, a clinically relevant anti‐fibrotic compound approved for treatment of IPF (Noble et al., 2015), was selected as a reference control.
As observed after prophylactic treatment, therapeutic administration of AQX‐1125 (10 and 30 mg·kg−1) was still able to attenuate the bleomycin‐induced changes to the lung (Figure 4A). A significant reduction in the Ashcrift fibrosis score (Figure 4B), pulmonary neutrophil activity (Figure 4C) and BAL cell counts (Table 2) were still observed at both 10 and 30 mg·kg−1. At 30 mg·kg−1, AQX‐1125 reduced the Ashcroft score from 7, indicative of large fibrotic areas and severe distortion of pulmonary architecture, to a 2.5, reflective of thickening of walls but without damage to lung structure. These results were equal to those obtained with the 90 mg·kg−1 daily dose of pirfenidone.
Figure 4.
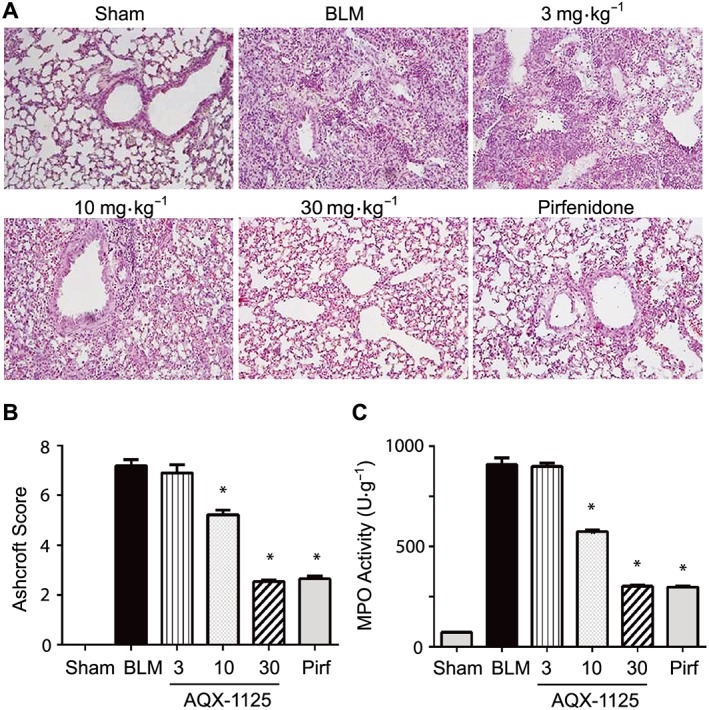
Therapeutic effects of AQX‐1125 on bleomycin‐induced changes in lung histology and fibrosis. Mice were treated with either saline vehicle (sal, p.o., daily), AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or pirfenidone (pirf, 90 mg·kg−1·d−1, p.o., three times daily) starting on Day 13 after receiving a single intratracheal administration of bleomycin (BLM). The sham‐treated group received intratracheal saline. Lung tissue was sectioned and stained with haematoxylin and eosin (A), and Ashcroft scores were calculated (B) at Day 28 as described in the Methods. MPO activity (C) was measured in homogenized lung tissue Day 28. *P < 0.05, significantly different from vehicle. Results are given as mean ± SEM from n = 11–29 (animals surviving to Day 28 in each of the bleomycin‐treated groups) or n = 10 (sham‐treated group) (B, C).
Table 2.
Differential leukocyte counts in the BALF of mice at 28 days in therapeutic model
| Neutrophils (×106) | Macrophages (×106) | Lymphocytes (×106) | |
|---|---|---|---|
| Sham | 0.00 ± 0.00* | 0.18 ± 0.01* | 0.00 ± 0.00* |
| Bleomycin | 0.30 ± 0.02 | 0.60 ± 0.02 | 0.10 ± 0.00 |
| AQX‐1125 3 mg·kg−1 | 0.28 ± 0.02 | 0.54 ± 0.02* | 0.09 ± 0.00* |
| AQX‐1125 10 mg·kg−1 | 0.13 ± 0.01* | 0.39 ± 0.01* | 0.05 ± 0.00* |
| AQX‐1125 30 mg·kg−1 | 0.05 ± 0.00* | 0.26 ± 0.01* | 0.03 ± 0.00* |
| Pirfenidone 90 mg·kg−1 | 0.05 ± 0.00* | 0.25 ± 0.01* | 0.03 ± 0.00* |
Data are the mean ± SEM of the absolute leukocyte counts (×106 cells) (n = 10–29).
P < 0.05 shows a significant effect of AQX‐1125 or pirfenidone, compared to the vehicle‐treated bleomycin group.
At 28 days post‐challenge, extensive TGF‐β (Figure 5A, C) and collagen I immunoreactivity (Figure 5B, D, respectively) were detectable in lung sections from the bleomycin animals. AQX‐1125, administered therapeutically at 10 and 30 mg·kg−1, retained its ability to significantly reduce the tissue area positive for TGF‐β (Figure 5A, C) and collagen I (Figure 5B, D). These doses also resulted in significant decreases in both soluble collagen (Figure 5E), as measured using the Sircol assay, and this effect was mirrored in the hydroxyproline content, which was included as a measure of total collagen (Figure 5F). The 30 mg·kg−1 dose of AQX‐1125 was nearly equivalent to pirfenidone, effecting reductions of 82 and 78% for soluble collagen and hydroxyproline content, respectively, as compared to decreases of 92 and 84% for these markers in the pirfenidone group.
Figure 5.
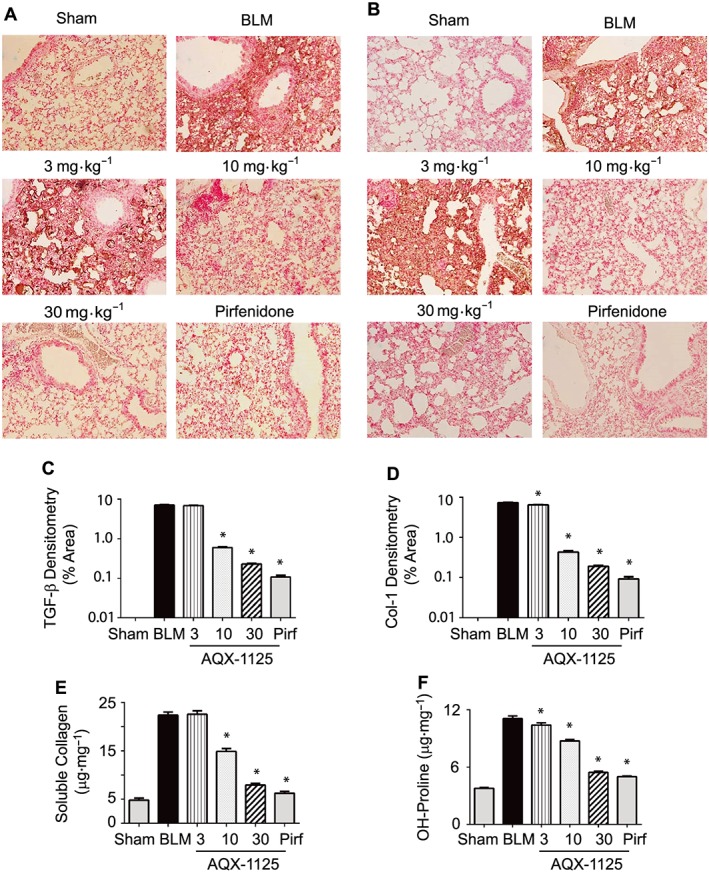
Therapeutic effects of AQX‐1125 on bleomycin‐induced parameters of lung fibrosis. Mice were treated with either saline vehicle (sal, p.o., daily), AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or pirfenidone (pirf, 90 mg·kg−1·d−1, p.o.) starting on Day 13 after receiving a single intratracheal administration of bleomycin (BLM). The sham‐treated group received intratracheal saline. Lung tissue collected at Day 28 was sectioned and stained with antibodies to either TGF‐β (A) or collagen‐1 (B), and the relative positive immunoreactive areas for TGF‐β (C) and collagen‐1 (D) were quantified by densitometry. Soluble collagen was quantified using the Sircol assay (E), and the hydroxyproline content (F) was assessed. *P < 0.05, significantly different from vehicle. Results are given as mean ± SEM from n = 11–29 (animals surviving to Day 28 in each of the bleomycin‐treated groups) or n = 10 (sham‐treated group).
Prior to therapeutic treatment with AQX‐1125 on Study Day 13, the body weight decreases observed in the pretreatment phase was either partly (3 mg·kg−1) or fully reversed (10 and 30 mg·kg−1) during Study Days 14–28 in surviving animals. The saline control group continued to lose body weight during the same period (Figure 6A). Kaplan–Meier survival plots show treatment with AQX‐1125 had a dose‐dependent effect on survival across all dose levels with significant results at 10 and 30 mg·kg−1 , compared with saline vehicle control (Figure 6B). The efficacy observed with AQX‐1125 at 30 mg·kg−1 was considered equivalent to the pirfenidone treatment. The survival rates from Day 0 to 13 were similar between all groups, but following treatment initiation on Day 13, the number of surviving animals was stabilized in all AQX‐1125‐treated groups compared to the saline vehicle, and no additional deaths occurred in the 30 mg·kg−1 AQX‐1125 and pirfenidone groups (Figure 6C).
Figure 6.
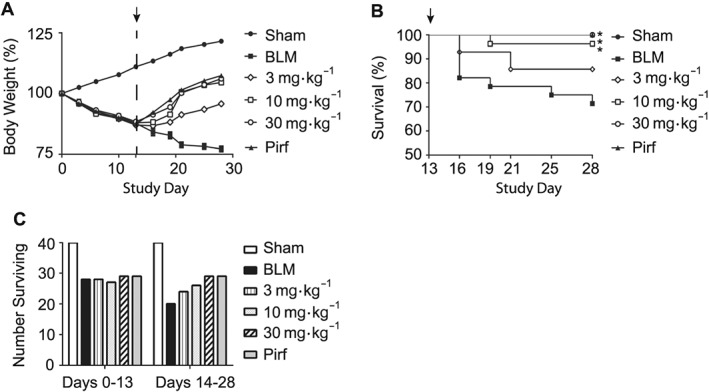
Effect of therapeutically administered AQX‐1125 on bleomycin‐induced body weight loss and survival. Mice were treated with either saline vehicle (sal, p.o., daily), AQX‐1125 (3, 10 or 30 mg·kg−1·d−1, p.o.) or pirfenidone (pirf, 90 mg·kg−1·d−1, p.o.) starting on Day 13 (indicated by ↑) after receiving a single intratracheal administration of bleomycin (BLM). The sham‐treated group received intratracheal saline. Body weights were measured throughout the study and normalized to the starting weight at Day 0 (100%), in order to visualize the percent change over 28 days (A). Mean survival was also plotted for each group following the initiation of treatment (B) with the number of remaining animals shown for each group in the period prior to compound treatment and following initiation of dosing (C). *P < 0.05, significantly different from vehicle using a log‐rank test. All bleomycin‐treated groups had n = 40 at Day 0.
Discussion
AQX‐1125 is a novel, clinical‐stage compound that activates the SHIP1 lipid phosphatase. In vitro, AQX‐1125 suppressed chemotaxis in human neutrophils and lymphocytes and reduced pro‐inflammatory cytokine release from murine splenocytes (Stenton et al., 2013b). Across several in vivo rodent models of pulmonary inflammation, induced by LPS, ovalbumin or cigarette smoke, AQX‐1125 consistently reduced BAL leukocyte influx (Stenton et al., 2013a). Similarly, we have now shown that AQX‐1125 is able to reduce cellular influx and neutrophil activity in bleomycin‐induced lung injury, and this resulted in improved outcomes through the fibrotic phase (up to Day 21) including a reduction in the pro‐fibrotic mediator, TGF‐β.
Many anti‐inflammatory drugs, when administered in a preventative regime, have shown promising anti‐fibrotic results in the bleomycin model, but have failed to show substantial clinical benefit in IPF, including prednisone and etanercept (Veeraraghavan, 2015). In studies where glucocorticoids have been administered 9 days following bleomycin, after the acute inflammatory phase, they have failed to demonstrate any effect on fibrosis (Chaudhary et al., 2006; Cortijo et al., 2009), and the evidence supporting the clinical benefits of steroid use in IPF patients is correspondingly weak (Veeraraghavan, 2015). The advantages conferred by prophylactic administration of these compounds in the bleomycin model are most likely a result of the reduction in the acute inflammation. Dexamethasone treatment initiated 3 days after bleomycin reduced excess collagen deposition but did not significantly affect BAL infiltration or BAL levels of TGF‐β (Dik, 2003), consistent with other work using corticosteroids, showing that inhibition of total lung TGF‐β in the bleomycin model was a result of the reduction in BAL leukocytes (Khalil et al., 1993) and not a direct effect on mediator release. In contrast to steroids, pirfenidone, a clinically approved compound for the treatment of IPF, inhibits induction of TGF‐β by bleomycin, at the transcriptional level (Iyer et al., 1999), and this may be one of the mechanisms by which pirfenidone mediates its anti‐inflammatory and anti‐fibrotic effects.
One of the major criticisms of the bleomycin model is that it does not reproduce many aspects of the human condition in a clinically meaningful way. Unlike bleomycin, the initial inflammatory insult leading to IPF in patients is unknown, and, at the time of clinical presentation, IPF manifests as chronic, progressive fibrosis. This disparity between the animal model and IPF may be a cause of the lack of clinical translation of many anti‐inflammatory drugs, and it has been suggested that compounds that are able to show a benefit when administered therapeutically in the bleomycin model are more likely to result in clinical benefit (Chaudhary et al., 2006; Moeller et al., 2007). Therefore, we sought to test if the activity of AQX‐1125 was due strictly to its anti‐inflammatory properties by giving it at Day 13, following the initial inflammatory burst. At this time, AQX‐1125 maintained the ability to suppress indicators of fibrosis, such as TGF‐β and the increased lung collagen, and reversed the body weight loss while stabilizing mortality levels. At the highest dose tested (30 mg·kg−1), the anti‐fibrotic, anti‐inflammatory effects and survival rates were similar to that of pirfenidone at 90 mg·kg−1.
More recently, gene expression signatures from the bleomycin model show that there are three distinct temporal phases of the response, an initial inflammatory phase (Days 1–2), an active fibrosis phase (Days 7–14) and a late fibrosis phase (Days 21–35). The gene signature obtained during the inflammatory phase was not the same as that in lung tissue from IPF patients, further supporting the notion that drugs that strictly target the inflammation are unlikely to translate into clinical benefit. However, there was a high correlation between the gene signature obtained from the lung tissue of bleomycin‐challenged mice collected during the active fibrotic phase and that obtained from IPF fibroblasts collected from patients with a rapidly progressing disease (Peng et al., 2013). These data indicate that the bleomycin model used here is in fact relevant to IPF, with similarities to the subset of patients with a faster course of disease, and they underscore the potential for clinical translation of AQX‐1125.
The anti‐fibrotic actions of AQX‐1125 are supported by recent literature highlighting a role for the PI3K–SHIP1 pathway in fibrosis. As a negative regulator of the PI3K pathway, SHIP1 can modulate immune cell activation, phagocytosis (Kerr, 2010) and neutrophil chemotaxis (Nishio et al., 2006; Mondal et al., 2012). The role of SHIP1 in pulmonary inflammation and fibrosis was first established in the SHIP1−/− mice. These mice show a progressive pulmonary infiltration of leukocytes where the severity of the phenotype is influenced by the genetic background (Helgason et al., 1998; Liu et al., 1998; Maxwell et al., 2011), and they also develop spontaneous intestinal inflammation leading to fibrosis (McLarren et al., 2011). Clinically, pathological conditions like scleroderma (Yan et al., 2016) and the progression of lung fibrosis in patients with systemic sclerosis (Christmann et al., 2016) are associated with high levels of mIR‐155, a negative regulator of SHIP1 expression. A decrease in SHIP1 levels in whole blood has been shown to inversely correlate with the histological stage of liver fibrosis (Katsounas et al., 2011), further linking immune regulation by SHIP1 to fibrotic outcomes.
One avenue by which SHIP1 could affect the development of fibrosis is via negative regulation of cytokine and chemokine production. Bone marrow‐derived macrophages from SHIP1−/− mice secrete enhanced levels of cytokines and chemokines upon stimulation, including the chemokine CCL2 (Gabhann et al., 2010), which plays an important role in the recruitment of immune cells and fibrocytes. CCL2 deficiency is protective in the bleomycin model (Gharaee‐Kermani et al., 2003), and its levels are significantly elevated in the BAL fluid of IPF patients (Suga et al., 1999). Although CCL2 was not measured specifically in this study, AQX‐1125 has previously been shown to be able to decrease levels of CCL2 in the BAL fluid of LPS‐mediated pulmonary inflammation in rats (Stenton et al., 2013a). Therefore, one possible mechanism by which SHIP1 activators like AQX‐1125 could limit fibrosis is by limiting the production of profibrotic mediators that drive the recruitment of leukocytes and fibrocytes.
In vitro, bleomycin‐induced growth and collagen production in fibroblasts is dependent on PI3K/Akt activation (Lu et al., 2009). In vivo, PI3Kϒ−/− mice have attenuated pulmonary fibrosis and mortality following bleomycin challenge (Russo et al., 2010), while sustained activation of the PI3K pathway exacerbates bleomycin‐induced pulmonary fibrosis (Kral et al., 2016). Prophylactic administration of the PI3Kϒ inhibitor, AS605240, can attenuate bleomycin‐induced lung inflammation and collagen deposition in rats (Wei et al., 2010). Beyond bleomycin, the PI3Kϒ isoform has uniquely been shown to be up‐regulated in IPF lung tissues, and the use of either siRNA or low MW inhibitors can reduce proliferation and fibrogenic activities in IPF‐derived fibroblasts (Conte et al., 2013).
These studies highlight the critical role of the PI3K/Akt pathways, especially PI3Kϒ, in pulmonary fibrosis and have spurred great interest in designing isoform specific inhibitors of this isoform as therapeutic agents. However, the clinical advancement of PI3Kϒ‐specific inhibitors for inflammatory and fibrotic diseases has been limited, possibly in part due to the difficulty in achieving the degree of selectivity required to circumvent toxicities such as disturbed glucose/insulin homeostasis, which occurs when the p110α isoform is inhibited (Lamb et al., 2013). Activation of SHIP1 represents a novel mechanism to suppress PI3K signalling, and AQX‐1125 has been generally well‐tolerated across three Phase II trials, with adverse event rates similar to placebo (Nickel et al., 2016; Vestbo et al., 2016). Therefore, suppression of the PI3K pathway, via SHIP1 activation, may provide an alternative therapeutic strategy in the treatment of fibrosis with a more favourable safety profile.
In summary, the current report describes the in vivo anti‐inflammatory and anti‐fibrotic effects of the SHIP1 activator AQX‐1125, in a murine model of bleomycin‐mediated pulmonary inflammation and fibrosis. The effects of AQX‐1125 were demonstrated both prophylactically and therapeutically. AQX‐1125 has good oral bioavailability, a terminal half‐life suited to once per day dosing and distributes to the lung at high concentrations (Stenton et al., 2013b). AQX‐1125 also has a novel mechanism of action that is distinct from anti‐inflammatory compounds, such as steroids, and functions to down‐regulate the PI3K/Akt pathway without side effects of hyperglycaemia or neutropenia, as seen with PI3K inhibitors (Akinleye et al., 2013). Therefore, the current anti‐fibrotic data, coupled with previously published data demonstrating the efficacy of this compound in rodent models of inflammation (Stenton et al., 2013a), support the view that SHIP1 activators, such as AQX‐1125, may have significant therapeutic potential for amelioration of inflammatory and fibrotic disease.
Author contributions
J.C. wrote the manuscript and participated in review and analysis of the data. C.H. oversaw synthesis of the compound and was involved in preparation of the manuscript and review of the data. G.S., C.S. and L.M. conceived of the project and worked with SC to design the experiments and in review of the manuscript. T.G., R.D.P. and E.E. performed the experiments, data collection and analysis.
Conflict of interest
All work performed in this article was financially supported by Aquinox Pharmaceuticals (Canada) Inc., a for‐profit organization involved in the development of low MW activators of SHIP1, as potential human therapeutic agents. Authors are employees, former employees, consultants, contractors or stockholders of Aquinox Pharmaceuticals (Canada) Inc.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Cross, J. , Stenton, G. R. , Harwig, C. , Szabo, C. , Genovese, T. , Di Paola, R. , Esposito, E. , Cuzzocrea, S. , and Mackenzie, L. F. (2017) AQX‐1125, small molecule SHIP1 activator inhibits bleomycin‐induced pulmonary fibrosis. British Journal of Pharmacology, 174: 3045–3057. doi: 10.1111/bph.13934.
References
- Akinleye A, Avvaru P, Furqan M, Song Y, Liu D (2013). Phosphatidylinositol 3‐kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol 6: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft T, Simpson JM, Timbrell V (1988). Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 41: 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary NI, Schnapp A, Park JE (2006). Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med 173: 769–776. [DOI] [PubMed] [Google Scholar]
- Christensen PJ, Goodman RE, Pastoriza L, Moore B, Toews GB (1999). Induction of Lung Fibrosis in the Mouse by Intratracheal Instillation of Fluorescein Isothiocyanate Is Not T‐Cell‐Dependent. Am J Pathol 155: 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann RB, Wooten A, Sampaio‐Barros P, Borges CL, Carvalho CRR, Kairalla RA et al. (2016). miR‐155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res Ther 18: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte E, Gili E, Fruciano M, Korfei M, Fagone E, Iemmolo M et al. (2013). PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: in vitro effects of its inhibition. Lab Invest 93: 566–576. [DOI] [PubMed] [Google Scholar]
- Cortijo J, Iranzo A, Milara X, Mata M, Cerdá‐Nicolás M, Ruiz‐Saurí A et al. (2009). Roflumilast, a phosphodiesterase 4 inhibitor, alleviates bleomycin‐induced lung injury. Br J Pharmacol 156: 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolhnikoff M, Mauad T (1999). Extracellular Matrix and Oscillatory Mechanics of Rat Lung Parenchyma in Bleomycin‐induced Fibrosis. Am J Respir Crit Care Med 160: 1750. [DOI] [PubMed] [Google Scholar]
- Dik WA (2003). Short course dexamethasone treatment following injury inhibits bleomycin induced fibrosis in rats. Thorax 58: 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez IE, Eickelberg O (2012). The impact of TGF‐β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc 9: 111–116. [DOI] [PubMed] [Google Scholar]
- Gabhann JN, Higgs R, Brennan K, Thomas W, Damen JE, Larbi NB et al. (2010). Absence of SHIP‐1 Results in Constitutive Phosphorylation of Tank‐Binding Kinase 1 and Enhanced TLR3‐Dependent IFN‐ Production. J Immunol 184: 2314–2320. [DOI] [PubMed] [Google Scholar]
- Gharaee‐Kermani M, McCullumsmith RE, Charo IF, Kunkel SL, Phan SH (2003). CC‐chemokine receptor 2 required for bleomycin‐induced pulmonary fibrosis. Cytokine 24: 266–276. [DOI] [PubMed] [Google Scholar]
- Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM et al. (1998). Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes &Amp. Development 12: 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SN, Gurujeyalakshmi G, Giri SN (1999). Effects of pirfenidone on transforming growth factor‐beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther 291: 367–373. [PubMed] [Google Scholar]
- Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R (2002). Time course of bleomycin‐induced lung fibrosis. Int J Exp Pathol 83: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakugawa T, Mukae H, Hayashi T, Ishii H, Abe K, Fujii T et al. (2004). Pirfenidone attenuates expression of HSP47 in murine bleomycin‐induced pulmonary fibrosis. Eur Respir J 24: 57–65. [DOI] [PubMed] [Google Scholar]
- Katsounas A, Trippler M, Kottilil S, Lempicki RA, Gerken G, Schlaak JF (2011). Altered expression of SHIP, a Toll‐like receptor pathway inhibitor, is associated with the severity of liver fibrosis in chronic hepatitis C virus infection. J Infect Dis 204: 1181–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr WG (2010). Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann N Y Acad Sci 1217: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil N, Whitman C, Zuo L, Danielpour D, Greenberg A (1993). Regulation of alveolar macrophage transforming growth factor‐beta secretion by corticosteroids in bleomycin‐induced pulmonary inflammation in the rat. J Clin Investig 92: 1812–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kral JB, Kuttke M, Schrottmaier WC, Birnecker B, Warszawska J, Wernig C et al. (2016). Sustained PI3K activation exacerbates BLM‐induced lung fibrosis via activation of pro‐inflammatory and pro‐fibrotic pathways. Sci Rep 6: 23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal G, Damen JE, Helgason CD, Huber M, Hughes MR, Kalesnikoff J et al. (1999). SHIPs ahoy. Int J Biochem Cell Biol 31: 1007–1010. [DOI] [PubMed] [Google Scholar]
- Lamb D, Lunn G, O'Reilly M, Butler C, Kilty I (2013). In vitro and In vivo Assessment of Pi3Kg inhibitors for anti‐inflammatory indications: challenges of selectivity over Pi3Ka. J Pulm Respir Med 3: 157. [Google Scholar]
- Leaker BR, Barnes PJ, O'Connor BJ, Ali FY, Tam P, Neville J et al. (2014). The effects of the novel SHIP1 activator AQX‐1125 on allergen‐induced responses in mild‐to‐moderate asthma. Clin Exp Allergy 44: 1146–1153. [DOI] [PubMed] [Google Scholar]
- Lee J, Yang H‐S, Cho J‐W, Kwon S, Kim Y‐B, Her J‐D et al. (2010). Dose‐response effects of bleomycin on inflammation and pulmonary fibrosis in mice. Toxicol Res 26: 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Mariathasan S, Bouchard D, Jones J, Sarao R, Kozieradzki I (1998). The Inositol polyphosphate 5‐phosphatase ship is a crucial negative regulator of B cell antigen receptor signaling. J Exp Med 188: 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Azad N, Wang L, Iyer AKV, Castranova V, Jiang B‐H et al. (2009). Phosphatidylinositol‐3‐kinase/akt regulates bleomycin‐induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol 42: 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell MJ, Duan M, Armes JE, Anderson GP, Tarlinton DM, Hibbs ML (2011). Genetic segregation of inflammatory lung disease and autoimmune disease severity in SHIP‐1−/− mice. J Immunol 186: 7164–7175. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarren KW, Cole AE, Weisser SB, Voglmaier NS, Conlin VS, Jacobson K et al. (2011). SHIP‐deficient mice develop spontaneous intestinal inflammation and arginase‐dependent fibrosis. Am J Pathol 179: 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Ask K, Warburton D, Gauldie J, Kolb M (2007). The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol 40: 362–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, Subramanian KK, Sakai J, Bajrami B, Luo HR (2012). Phosphoinositide lipid phosphatase SHIP1 and PTEN coordinate to regulate cell migration and adhesion. Mol Biol Cell 23: 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullane KM, Kraemer R, Smith B (1985). Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods 14: 157–167. [DOI] [PubMed] [Google Scholar]
- Mura M, Bargagli E, Sergiacomi G, Zompatori M, Sverzellati N, Taglieri A et al. (2012). Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3‐year prospective study. Eur Respir J 40: 101–109. [DOI] [PubMed] [Google Scholar]
- Nickel JC, Egerdie B, Davis E, Evans R, Mackenzie L, Shrewsbury SB (2016). A phase II study of the efficacy and safety of the novel oral SHIP1 activator AQX‐1125 in subjects with moderate to severe interstitial cystitis/bladder pain syndrome. J Urol 196: 747–754. [DOI] [PubMed] [Google Scholar]
- Nishio M, Watanabe K‐I, Sasaki J, Taya C, Takasuga S, Iizuka R et al. (2006). Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol 9: 36–44. [DOI] [PubMed] [Google Scholar]
- Noble PW, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA et al. (2015). Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 47: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S‐Y, Zheng T, Kim Y‐K, Zhu Z (2007). Src homology 2 domain–containing inositol 5‐phosphatase 1 deficiency leads to a spontaneous allergic inflammation in the murine lung. J Allergy Clin Immunol 119: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CJ, Ming‐Lum A, Nodwell M, Ghanipour A, Yang L, Williams DE et al. (2007). Small‐molecule agonists of SHIP1 inhibit the phosphoinositide 3‐kinase pathway in hematopoietic cells. Blood 110: 1942–1949. [DOI] [PubMed] [Google Scholar]
- Peng R, Sridhar S, Tyagi G, Phillips JE, Garrido R, Harris P et al. (2013). Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for ‘active’ disease. PLoS One 8: e59348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisfeld IH (1979). Bleomycin‐Induced Pulmonary Toxicity: A Model for the Study of Pulmonary Fibrosis. Springer‐Verlag: New York. [Google Scholar]
- Raymond JR, Han K, Zhou Y, He Y, Noren B (2014). Indene derivatives as pharmaceutical agents. US Patent 8,673,975 B2.
- Russo RC, Garcia CC, Barcelos LS, Rachid MA, Guabiraba R, Roffê E et al. (2010). Phosphoinositide 3‐kinase γ plays a critical role in bleomycin‐induced pulmonary inflammation and fibrosis in mice. J Leukoc Biol 89: 269–282. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J et al. (2013a). Characterization of AQX‐1125, a small‐molecule SHIP1 activator Part 2. Br J Pharmacol 168: 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J et al. (2013b). Characterization of AQX‐1125, a small‐molecule SHIP1 activator. Br J Pharmacol 168: 1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M (1999). Clinical significance of MCP‐1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J 14: 376. [DOI] [PubMed] [Google Scholar]
- Todd NW, Luzina IG, Atamas SP (2012). Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan S (2015). Pharmacologic therapies for idiopathic pulmonary fibrosis, past and future. Ann Med 47: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestbo J, Tam P, Mackenzie L, Harwig C, Lamb D, Lunn G et al. (2016). Safety and Efficacy of AQX‐1125 in chronic obstructive pulmonary disease (COPD)–results of the phase 2 FLAGSHIP study. Am J Respir Crit Care Med 193: A6842. [Google Scholar]
- Wei X, Han J, Chen Z‐Z, Qi B‐W, Wang G‐C, Ma Y‐H et al. (2010). A phosphoinositide 3‐kinase‐gamma inhibitor, AS605240 prevents bleomycin‐induced pulmonary fibrosis in rats. Biochem Biophys Res Commun 397: 311–317. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR (2012). Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Chen J, Li W, Bao C, Fu Q, Del Bianco E et al. (2016). Targeting miR‐155 to treat experimental scleroderma. Sci Rep 6: 20314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Mui A, Ong C, Krystal G, van Soest R (2005). Synthesis of pelorol and analogues: activators of the inositol 5‐phosphatase SHIP. Org Lett 7: 1073. [DOI] [PubMed] [Google Scholar]