

Abstract
Protein ubiquitination plays a role in essentially every process in eukaryotic cells. The attachment of ubiquitin (Ub) or Ub-like (UBL) proteins to target proteins is achieved by parallel but distinct cascades of enzymatic reactions involving three enzymes: E1, E2, and E3. The E1 enzyme functions at the apex of this pathway and plays a critical role in activating the C-terminus of ubiquitin or UBL, which is an essential step that triggers subsequent downstream transfer to their cognate E2s resulting in the fidelity of the Ub/UBL conjugation machinery. Despite the central role of the E1 enzyme in protein modification, a quantitative method to measure Ub/UBL activation by E1 is lacking. Here, we present a mass spectrometry-based assay to accurately measure the activation of Ub/UBL by E1 independent of the E2/E3 enzymes. Our method does not require radiolabeling of any components and therefore can be used in any biochemical laboratory having access to a mass spectrometer. This method allowed us to dissect the concerted process of E1-E2-catalyzed Ub conjugation in order to separately characterize the process of Ub activation and how it is affected by select mutations and other factors. We found that the hydrophobic patch of Ub is important for the optimal activation of Ub by E1. We further show that the blockers of the Ub-proteasome system such as ubistatin and fullerenol inhibit Ub activation by E1. Interestingly, our data indicate that the phosphorylation of Ub at the S65 position augments its activation by the E1 enzyme.
Introduction
Post-translational modification by a 76 amino acid protein ubiquitin (Ub) is an essential step to regulate the functions of myriad of proteins in eukaryotic cells1. While the best characterized role of ubiquitination is to target the modified proteins for proteasomal degradation2, other well-defined outcomes of such modifications include receptor endocytosis3, transcription4, subcellular localization5, and regulation of several kinases6,7. Defective regulation of ubiquitination is the main cause for various pathological conditions ranging from developmental abnormalities8 and autoimmune diseases9 to cancer10 and neurodegenerative diseases11.
In addition to Ub, there are other Ub-like proteins (UBLs), NEDD8 and SUMO12, whose covalent attachment alters the functions of several target proteins, and in most cases this leads to different biological consequences. For example, the modification of cullins by NEDD8 regulates the activity of the SCF complex which is important for development, cytokinesis, and signal transduction13. On the other hand, SUMO modification results in regulation of proteins involved in nuclear transport, signal transduction, stress response, and cell division14.
The attachment of Ub or UBLs to their target proteins is achieved by parallel but distinct cascades of enzymatic reactions involving sequential actions of Ub-activating (E1), Ub-conjugating (E2), and Ub-protein ligase (E3) enzymes15,16. The activation of Ub and UBLs by their cognate E1 enzymes is an essential step that triggers subsequent downstream transfer of Ub/UBL to their cognate E2s resulting in the fidelity of the Ub/UBL conjugation pathways17,18. In order to attach Ub/UBL to a target protein, firstly, the E1 in the presence of Mg2+ and ATP forms a high energy E1∼Ub/UBL thioester bond between the catalytic cysteine of E1 and the C-terminus of Ub/UBL19,20. Secondly, an E2 binds to E1∼Ub/UBL and transfers the activated Ub/UBL to its own catalytic cysteine by a process termed E1-E2 thioester transfer21. Finally, ‘charged’ E2 (E2∼Ub/UBL) interacts with the corresponding E3 ligases to promote the formation of a covalent and stable isopeptide bond between the C-terminal glycine of Ub/UBL and the ε-amine of a lysine residue of the target protein22. The E3 ligases in the pathway provide the substrate specificity.
The E1 enzyme is either a single polypeptide (110-120 kDa), such as the E1 for Ub23, or a heterodimeric complex, such as the E1s for NEDD824 and SUMO25. The activation of Ub/UBL by their respective E1s is a three-step process. First, Ub/UBL interacts with the adenylation domain (AD) of E1 in the presence of Mg2+-ATP and gets adenylated26. Second, a series of conformational changes allows the active cysteine of E1 to attack the acyl adenylate, resulting in the formation of E1∼Ub/UBL thioester linkage26. This presumably pulls the Ub/UBL away from the AD as soon as the active site cysteine occupies its original position. Finally, a second Ub/UBL binds to the AD and gets adenylated such that, under a steady-state condition, two molecules of Ub/UBL are bound to the same E1 molecule: one noncovalently at the AD site and the other covalently to the catalytic cysteine via a thioester bond19,27. Now, E1 is ready to interact with the E2 enzyme to transfer the thioester-linked Ub to the E2's active-site cysteine.
Despite the central role of the E1 enzyme in protein ubiquitination, a quantitative method to accurately measure the catalytic cysteine-mediated (complete) activation of Ub by E1 independently of the E2/E3 enzymes is lacking. Previous methods have either indirectly measured the partial activation of Ub by monitoring the ATP-PPi exchange during the adenylation step or detected the E1∼Ub or E1∼NEDD8 thioester formation using gel electrophoresis24,28,29. To address this deficiency, here we present a mass spectrometry-based assay to detect and accurately quantitate the activation of Ub by E1. Using this method, we examine how mutations in the hydrophobic patch as well as several other residues of Ub affect its activation by the E1 enzyme. Other factors, such as ubistatin A (C92), fullerenol, and phosphorylation of Ub at S65 also affect the E1-mediated Ub activation. Finally, we demonstrate that this method can be extended to quantitate the activation of UBLs, such as NEDD8 and SUMO, by their respective E1s.
Experimental Section
Mass Spectrometry-based method to measure the activation of Ub by E1
Here we devised a method (see Figure 1) that detects and quantitates the activation of Ub by its cognate E1 enzyme. In this method, an isotopically-labeled Ub (here we used 15N enrichment) is incubated with the E1 enzyme in the presence of ATP, MgCl2, and MESNa. MESNa attacks the Ub covalently attached at the active site cysteine of E1 to produce Ub with a thioester at its C- terminus (Ub-COSR) which ensures that the reaction moves forward. In fact, as we demonstrated elsewhere, Ub-COSR generated using this procedure is chemically active and can be conjugated to a lysine residue on a protein without E230,31. Equal amounts of samples are aliquoted out at different time points. The reactions are stopped by adding molar excess of EDTA to chelate Mg2+, followed by addition of an equimolar amount of unlabeled Ub as an internal standard. Then the samples are exchanged into water containing 0.006% TFA (pH 6.0) and injected (in triplicate) into an ESI-mass spectrometer (ESI-MS) to detect the conversion of 15N Ub into 15N Ub-COSR. The use of an internal standard, inspired by the Ub-AQUA method32,33, helps negate pipetting errors as well as possible errors in measuring the Ub concentration using UV-spectrometry (there may be up to 10% error due to the low extinction coefficient of Ub at A280). Ideally, the internal standard and the protein whose activation by E1 is analyzed (we term it reactant protein) should be identical but differentially isotope-labeled proteins such that they have the same physical characteristics except for their masses. In addition, care should be taken to ensure that the internal standard and the reactant proteins are well resolved in MS spectrum, and at the same time the masses of these two proteins should not coincide with the mass of the thioesterified protein. For example, if an isotope-labeled (such as 15N-enriched) Ub is used as a reactant protein, the internal standard should be either unlabeled Ub (as in Figure 1B) or differently isotope-labeled Ub (e.g. 13C-labeled) or vice versa (as in Figure S1). In most of our experiments, we used unlabeled Ub as the standard and 15N-labeled Ub as a reactant protein. We have also successfully used unlabeled Ub as the reactant protein and 15N-labeled Ub as the standard marker (Figure S1).
Figure 1.
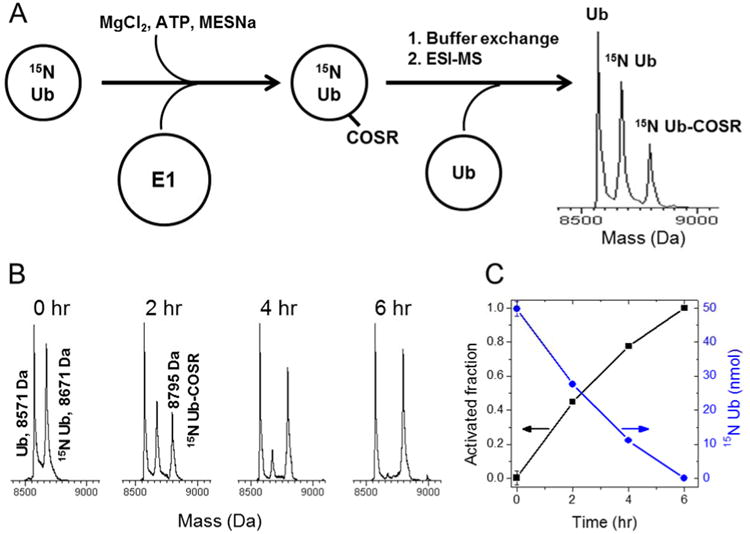
Mass spectrometry-based method for measuring the activation of ubiquitin by the E1 enzyme. (A) Schematic representation of the method. 15N-enriched Ub is incubated for a given time period with the E1 enzyme, MgCl2, ATP, and MESNa in a 20 mM sodium phosphate buffer at 30 °C. The reaction is stopped by adding EDTA followed by addition of equimolar amount of unlabeled Ub. The mixture is buffer exchanged into water and then subjected to ESI-MS analysis. (B,C) Activation of 15N-enriched Ub by the E1 enzyme. Unlabeled Ub was used as internal standard. Shown are representative spectral regions for each time point. (C) The activated fraction of Ub (black squares) and the remaining amount of unmodified Ub (blue circles) as a function of time. The arrows point to the corresponding axes.
Since the reaction product (15N-Ub-COSR) has different physical properties than the internal standard (such as ionization efficiency, water solubility etc), the amount of 15N-Ub-COSR was quantified indirectly by measuring the normalized signal intensity of the unactivated reactant protein (15N-Ub) at different time points and subtracting it from the normalized intensity of 15N-Ub at time zero. Since the E1 concentration in the reaction is negligible compared to Ub, the amount of E1-associated (catalytic Cys bonded and/or adenylated) Ub is also negligible; that is why the amount of Ub-COSR can be accurately assessed by subtracting the remaining unactivated Ub from the total Ub. The fraction of 15N-Ub converted to 15N-Ub-COSR (further referred to as the ‘activated fraction’) was quantified as (I0-It)/I0, where I0 and It are the normalized MS signal intensities of 15N-Ub at 0 hr and at a given time point t, respectively (Figure 1C).
All ESI-MS spectra were acquired on either JEOL AccuTOF-CS mass spectrometer or LTQ Orbitrap mass spectrometer in a positive electrospray mode. To ensure that the MS signal intensity corresponds to the amount of protein added, unlabeled Ub was mixed with 15N-Ub in various molar ratios prior to injection into the mass spectrometer, and the intensities of Ub and 15N-Ub MS signals were quantified and compared. The results (Figure S2) confirm that the intensities measured by MS are in good agreement (within up to 10% error) with the amounts of protein present in the sample.
To establish that the method described in this manuscript is a quantitation of the activated Ub at the catalytic cysteine of E1, we mutated that cysteine to a serine (C632S) and tested the ability of E1C632S to form Ub-COSR. A lack of Ub-COSR formation by the E1 mutant (Figure S3) indicates that this adduct forms as a result of MESNa reaction with the Ub∼E1 thioester rather than of MESNa attacking the Ub adenylate intermediate. The mutation also dramatically impaired formation of Ub conjugates although it did not affect the enzyme's ability to bind Ub.
Using our method we assessed the kinetic parameters for E1 activation of wild-type Ub assuming Michaelis-Menten kinetics. The rate of Ub activation increased linearly with E1 concentration (Figure S4) but did not depend on Ub concentration in the 25 μM-200 μM range, consistent with the published Km values (0.2-2 μM).34 The initial rates of Ub activation reaction are shown in Table S1. Since at saturation the initial rate equals kcat [E1]0, we determined the turnover rate for Ub to be kcat = 4.28 ± 0.90 min-1, in good agreement with the published value (4.3 ± 1.2 min-1).34
Results and Discussion
Hydrophobic patch of Ub is required for optimal activation of its C-terminus
Crystal structures of Ub in complex with the E1 enzyme reveal that both Ub molecules, at the adenylation site as well as covalently attached to the active site cysteine, interact with the E1 mainly through the Ub surface hydrophobic patch (comprising residues L8, I44, and V70)23,35,36. This functionally important hydrophobic patch is involved in Ub interactions with numerous receptors and is necessary for Ub and polyUb recognition in several signaling pathways37-39. However, the role of the hydrophobic patch in Ub activation has not been tested previously, mainly due to the lack of a method to directly measure the catalytic cysteine-mediated activation of Ub by E1.
To determine whether the surface hydrophobic patch of Ub is required for its C-terminal activation by E1, we first purified two mutants of Ub: (i) L8A& I44A and (ii) V70A and assessed their activation along with wild type Ub (UbWT). As expected, the intensity of 15N-UbWT signal decreased over time consistent with the activation of 15N-Ub and totally disappeared at the 6 hr time point (Figure 1B,C), indicating that 15N-UbWT was totally activated by E1 at a time point between 4 – 6 hr. At the same time, a signal corresponding to 15N-Ub-COSR appeared at 2 hr and its intensity increased concomitantly with the decreased intensity of 15N-Ub over time. Parallel reactions involving UbL8A&I44A or UbV70A (Figures 2A and S5) showed that both mutants were activated by E1. However, the rate of activation of UbL8A&I44A was slower than for UbWT as even after 6 hours a measurable fraction of the mutant Ub remained unactivated (compare different time points in Figure S5A and B). The activation of UbV70A was more similar to UbWT (Figure S5C). Indeed, the quantitation of the activated fraction of Ub over time confirmed that mutating L8 and I44 to alanines in Ub slowed down significantly its activation by E1, while the V70A variant behaved more or less like UbWT (Figure 2A). This suggests that Ub residues L8 and I44 play significant role in the activation of Ub by E1. Interestingly, mutating V70 of Ub did not slow down its activation by E1; instead, the activation of the C-terminus of UbV70A was even slightly accelerated (Figure 2A, Table S1). To assess the effect of simultaneous alanine replacement of all three hydrophobic-patch residues (L8A+I44A+V70A) in Ub, we constructed UbL8A&I44A&V70A and tested its activation by E1. We confirmed by NMR that the triple mutant was well folded, and the chemical shifts for the majority of the amide signals resembled those of UbWT (Figure S6). Surprisingly, the V70A mutation in combination of L8A+I44A, but not alone, significantly slowed down the activation of Ub by E1 (Figures 2A and S5D, Table S1). This suggests that the hydrophobic patch residues in Ub are important, though not critical, for its activation by the E1 enzyme. This observation is corroborated by the published structures where L8 and I44 of Ub make hydrophobic contacts with the AD and the catalytic cysteine half-domain of the E1 enzyme23,35. This suggests a possibility that mutations of some of the hydrophobic-patch residues, singly or in combination, may significantly weaken Ub's interactions with the AD and/or the catalytic cysteine half-domain of E1 (see Fig S13), resulting in slower Ub activation.
Figure 2.
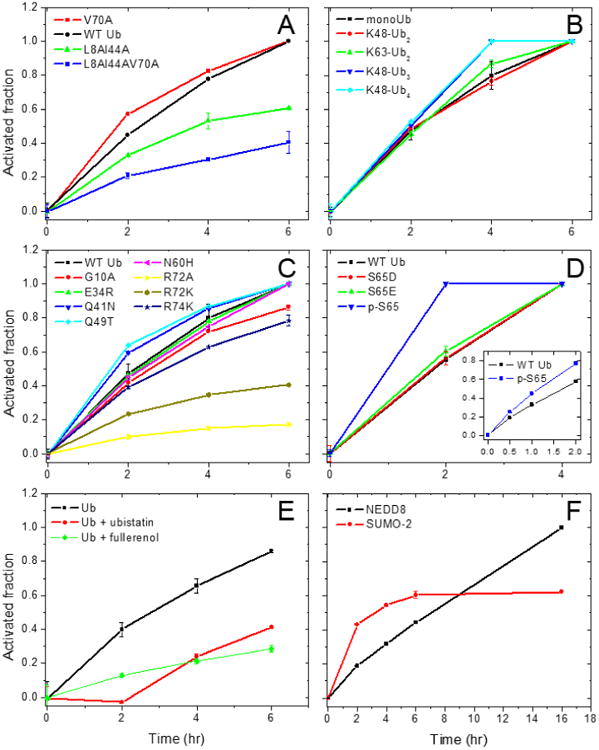
Comparison of the time course of activation of Ub, its mutants, and the UBLs by cognate E1 enzymes. Shown is the activated fraction of each protein as a function of time for (A) hydrophobic patch mutants of Ub, (B) mono- and polyUb, (C) other Ub mutants studied here, (D) p-S65 Ub and its phosphomimics, (E) Ub in the presence of ubistatin A or fullerenol, and (F) NEDD8 and SUMO-2. The inset in D shows the activation of p-S65-Ub and UbWT measured at earlier time points (0, 0.5, 1, and 2 hr). See also Figures S11-S12.
To further investigate whether the hydrophobic patch of Ub is required for E2-catalyzed Ub chain formation, we used E2-25K (also known as UBE2K in human and a homolog of yeast UBC1) which exclusively assembles K48-linked Ub chains40. We set up and ran in vitro ubiquitination reactions with all three hydrophobic patch mutants of Ub and compared the results with UbWT. The reactions involved Ub, E1, E2, ATP, and MgCl2; the products were separated using SDS-PAGE and visualized by coomassie staining (see Supporting Information for further details). Surprisingly, although the UBA domain of E2-25K interacts with the hydrophobic patch of Ub, and an intact UBA domain is essential for Ub chain formation41; all three variants UbV70A, UbL8A&I44A, and UbL8A&I44A&V70A were assembled into polymeric chains comparably to UbWT (Figure 3A-B). In fact, UbV70A was even faster than UbWT to form polymeric chains (compare the time points in Figure 3A-B), possibly due to its accelerated activation by the E1 enzyme. The observed lack of reduction of the assembly rate of polymeric chains when using the hydrophobic patch mutants of Ub suggests that the surface hydrophobic patch residues (L8, I44, V70) of Ub are not required for E2-25K catalyzed Ub-chain formation. This is surprising because the UBA domain of E2-25K, important for E2-25K's ability to synthesize free polyUb chains41, interacts with the hydrophobic patch of Ub both in solution41 and in crystals42. On the other hand, a study on the yeast homolog of E2-25K, UBC1, found that the UBA domain of UBC1 is not required for its ability to assemble free K48-linked Ub chains; rather the catalytic UBC domain is sufficient to assemble free Ub chains as well as to provide the specificity for K48-linkage43. This is intriguing, in particular, with regard to the role of the hydrophobic patch of Ub in E2-25K catalyzed Ub-chain formation, and warrants further investigation of the interactions between Ub and E2-25K.
Figure 3.
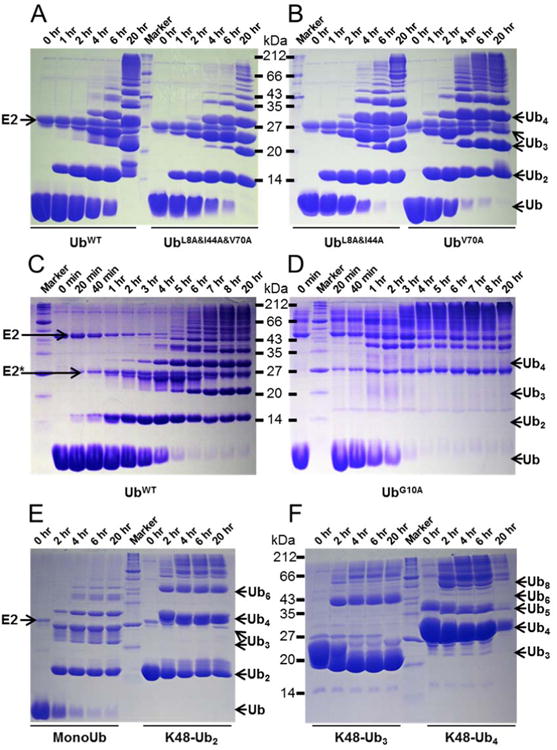
E1 and E2-25K-catalyzed polymerization of hydrophobic patch mutants of Ub and of K48-linked polyUbs. SDS-PAGE gels of in vitro E1 and E2-25K-catalyzed polymerization reactions for UbWT and its mutants (A-D) or preassembled polyUbs (E,F), as indicated. The reactions were performed and analyzed as detailed in Supporting Information. The running positions of the E2 and select Ub polymers are indicated by arrows.
R72 and G10 of Ub are crucial residues required for E1 and E2 activity, respectively
Besides the hydrophobic patch residues, several other surface residues in Ub are important for survival of the cells. A recent study revealed the effect of all possible point mutations in Ub on the growth of budding yeast44. We therefore selected few Ub mutations that render cells either sensitive, G10A, E34R, R72A, R72K, Q49T, or tolerant, Q41N, R74K, N60H. First, we tested these mutants for their ability to get activated by E1. All mutants, except for R72 substitutions, were activated by E1 with a rate comparable to UbWT (Figure 2C, Table S1). As expected, UbR72A was significantly impaired for its activation by E1 which is concordant with both the crystal structures and the biochemical data indicating that arginine at position 72 is critical for Ub interaction with E123,28,29,35,36. Even an R to K replacement at this position significantly affected the activation of Ub by E1 (Figure 2C, Table S1). Note also that UbR72A mimics NEDD845; thus, our results are consistent with the suggestion that residue 72 plays a key role in discrimination between Ub and NEDD8 by Ub E129. These results emphasize that residue 72 of Ub has to be an arginine for proper Ub activation and subsequent ubiquitination and degradation of cellular proteins, which agrees with our earlier report44 that the substitution of arginine at position 72 with any other amino acid is lethal to yeast cells.
To examine whether the other mutants, G10A, E34R, and Q49T, are impaired in their ability to form Ub chains, we set up in vitro ubiquitination reactions using E1 and E2-25K. UbE34R and UbQ49T formed polymeric chains with a rate similar to UbWT (Figure S7). However, the ability of the G10A mutant to form polymeric chains was significantly abrogated (Figure 3C-D). The dimer of UbG10A was hardly visible on the SDS-PAGE with no apparent appearance of trimer, tetramer, or higher chains. The appearance of slower running bands (>35 kDa) in the UbG10A ubiquitination reaction (Figure 3D) is likely due to the attachment of UbG10A to lysine residues of E2-25K as most of these bands are above the band corresponding to E2-25K (25 kDa) and GST-E2-25K (50 kDa). Combining the Ub activation and E2-25K ubiquitination reactions, it is clear that G10 is required for the E2-catalyzed conjugation reaction while R72 is required for Ub activation by E1. To further investigate whether the mutation of G10 affects the E2-catalyzed conjugation reactions broadly, we tested the ability of UbG10A to polymerize into K63-linked (assembled by UBC13/MMS246) or K11-linked (assembled by UBE2S47) Ub chains (Figure S8). Remarkably, in both cases the G10A mutant was significantly impaired to form higher polymeric chains. Since the G10A mutant was activated by E1 with a rate comparable to UbWT, our findings suggest that the impaired ability of UbG10A to polymerize into free chains is mainly due to the inability of the E2 enzymes to recognize UbG10A as an “acceptor” of the Ub linked to the active site cysteine of the E2. Notably, the charging of the E2 by UbG10A appeared “normal” as we noticed the presence of ubiquitinated E2s in the reaction (slower running bands in Figure 3D, above 35 kDa), which indicates that UbG10A attached to the active site cysteine of the E2 enzymes does act as a “donor”.
These results indicate that G10 is required for Ub's ability to form polymeric chains, in agreement with our published data44 that yeast cells are sensitive to mutations of the G10 residue in Ub and, in particular, that G10A mutation is not tolerated.
K48-linked Ub chains are activated by E1
There is an abundance of unanchored polyUb chains in cells, and recently their physiological roles have begun to emerge; the examples range from protein kinase activity48, innate immune signaling49 to the aggresome response50. To understand whether Ub chains can be activated by E1, we tested the ability of E1 to activate preassembled K48-linked dimer, trimer, and tetramer of Ub. We found that all three chains were activated by E1 (Figure S9). The K48-linked dimers of Ub were activated with a rate similar to monoUb. Since K48-linked dimer of Ub adopts a “closed” conformation (in which the hydrophobic patches are sequestered at the Ub:Ub interface) at physiological pH51 and the hydrophobic patch residues were important for optimal activation of Ub, we assessed the rate of activation of K48-linked Ub dimer in comparison with a K63-linked Ub dimer, which adopts an extended, “open” conformation with no hydrophobic Ub:Ub interface 52. Interestingly, the formation of the hydrophobic interface in K48-linked dimer of Ub did not affect its activation by E1, as both K48-linked and K63-linked dimers were activated with the rates comparable to each other and to monoUb (Figure 2A). This is consistent with the observation that K48-linked Ub dimer is not locked in the closed state but is in fast dynamic equilibrium between the closed and open conformations51,53. Intriguingly, the K48-linked trimers and tetramers were activated at a slightly faster rate (Figures 2A and S11B, Table S1), indicating that longer K48-linked chains also are conformationally flexible. Since the polyUb chains were activated by E1, we further tested the ability of these preassembled chains to form longer polyUb chains using E2-25K. Interestingly, all of them assembled into longer polyUb chains, albeit the efficiency decreased with the increasing length of polyUb chains (Figure 3E-F). This indicates that it is mainly the E2 enzyme that differentially recognizes polyUb chains compared to monoUb, at least at the conditions tested here. This report expands on the earlier findings for Ub dimers40,51 and demonstrates that longer polyUb chains are not only activated by the E1 enzyme but also these activated polyUbs are transferred to E2s and form longer chains.
Ubistatin and fullerenol block the activation of Ub by E1
Ubistatins are a class of small molecule compounds that inhibit proteasomal degradation of ubiquitinated cargos by binding to the hydrophobic patch on Ub and blocking it from recognition by proteasomal receptors54. Recently we reported that another compound, fullerenol (polyhydroxylated [60] fullerene), also binds to the hydrophobic surface patch on Ub and inhibits the E2-25K-catalyzed Ub chain formation55. Since both these compounds bind to Ub, we tested whether they can interfere with Ub activation by E1. Remarkably, the presence of equimolar (to Ub) amount of ubistatin A (compound C92 in ref. 54) or of fullerenol in the reaction significantly slowed down the activation of Ub (Figures 2E and S10, Table S1). Since both compounds bind to Ub through its hydrophobic patch and the residue R72 of Ub is very close to this patch, an attractive but purely speculative explanation of the significantly slower activation of Ub in the presence of these molecules would be that ubistatin as well as fullerenol also blocks/binds the R72 residue of Ub such that it has limited access to the E1 enzyme. This would lead to limited recognition of the C-terminus as well as the hydrophobic patch of Ub by the E1 enzyme thus resulting in a slower activation of Ub.
Ubistatins have been used in experiments to block the degradation of ubiquitinated proteins54 as well as to inhibit Ub's protein-protein interactions56. However, their role as a blocker of the activity of the E1 and/or E2 enzymes has not been investigated. Here we report, for the first time, that ubistatin A as well as fullerenol inhibit the activation of Ub by E1. This means that ubistatin not only blocks the recognition of ubiquitinated proteins by their downstream receptors but also inhibits the activation of Ub by the E1 enzyme and therefore can have an overall effect on Ub-mediated pathways. Indeed, our in vitro ubiquitination assays showed that the presence of ubistatin A slowed down the assembly of free Ub chains by E2-25K (Figure 4A-B).
Figure 4.
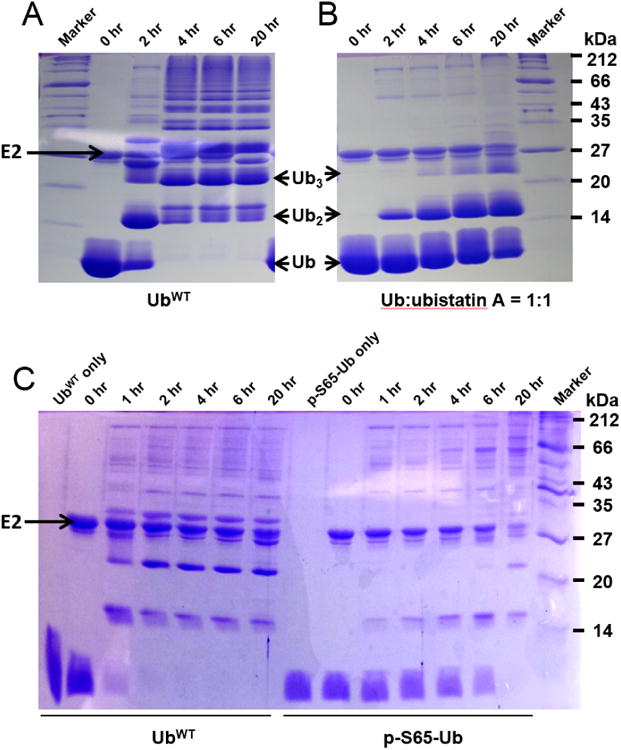
The effect of ubistatin or phosphorylation of Ub at S65 on E1 and E2-25K catalyzed Ub chain formation. SDS-PAGE gels of in vitro E1 and E2-25K-catalyzed polymerization reactions for UbWT (A and left panel in C) or equimolar mixture of UbWT with ubistatin A (B) or p-S65-Ub (right panel in C).
Phosphorylation at S65 of Ub enhances its C-terminal activation
Ub can be phosphorylated on several residues57,58. Among them, phosphorylation of Ub at S6559 has recently generated significant interest due to its implications in mitophagy (autophagosomal degradation of mitochondria) as well as in Parkinson's disease60,61. The biological research has shown that upon mitochondrial damage, such as loss of membrane potential, a mitochondrial protein kinase PINK1 accumulates on damaged mitochondria62 and this leads to rapid accumulation of an E3-ligase parkin on the mitochondrial outer membrane63. Next, catalytically active PINK1 phosphorylates Ub60 as well as the N-terminal UBL domain of parkin64. This sequence of events leads to a striking increase in ubiquitination65 and degradation of the mitochondria66.
Using the method devised here, we tested the activation of phosphorylated (at S65) Ub in comparison with the non-phosphorylated Ub. Surprisingly, phosphorylated Ub (p-S65-Ub) was activated faster than the non-modified Ub (Figures 2D and S10, Table S1). Since S65D and S65E have been used in several studies to mimic p-S65-Ub60,61,67, we also tested the activation of these phospho-mimics by E1 and found that the rate of activation of both S65D and S65E Ub was comparable with that of UbWT (Figure 2D). These results indicate that the phosphorylation of Ub at residue S65 accelerates its activation by E1. However, mutating S65 in Ub to either D or E does not adequately reproduce this feature of phosphorylated Ub, indicating that E1 differentiates p-S65-Ub from the phosphomimics. To further assess the ability of p-S65-Ub to be discharged from E1/E2 and assemble into polymeric chains, we set up an in vitro ubiquitination reaction of p-S65-Ub catalyzed by E2-25K. Consistent with the published data for other E2s68,69, phosphorylated Ub was significantly inhibited in its ability to assemble into polymeric chains (Figure 4C), suggesting that E2-25K discriminates between p-S65-Ub and UbWT.
The extension of the MS-based method to measure the activation of the UBLs
Inspired by the ability to determine the amount of E1-activated Ub, we have endeavored to extend our method to quantify the activation of other UBLs, NEDD8 and SUMO, by their cognate E1s. Similar to Ub, 15N-labeled NEDD8 or SUMO-2 were incubated with their respective E1s (APPBP1/UBA3 for NEDD8 and SAE1/SAE2 for SUMO-2) in the presence of ATP, MgCl2, and MESNa. The reactions were aliquoted and stopped at different time points by adding EDTA. An internal standard was added, and then the sample was subjected to ESI-MS. The appearance of NEDD8-COSR and SUMO-2-COSR MS signals confirmed that both NEDD8 and SUMO-2 were activated by their respective E1s (Figure 5A-B). A concomitant decrease in the unmodified NEDD8 and SUMO-2 compared with the internal standards clearly indicates that the UBLs get activated. Similar to Ub, the fraction of activated Nedd8 or SUMO-2 was calculated and plotted over time (Figure 2F, Table S1). These results demonstrate that our method for measuring the activation by E1 is equally applicable to the UBLs.
Figure 5.
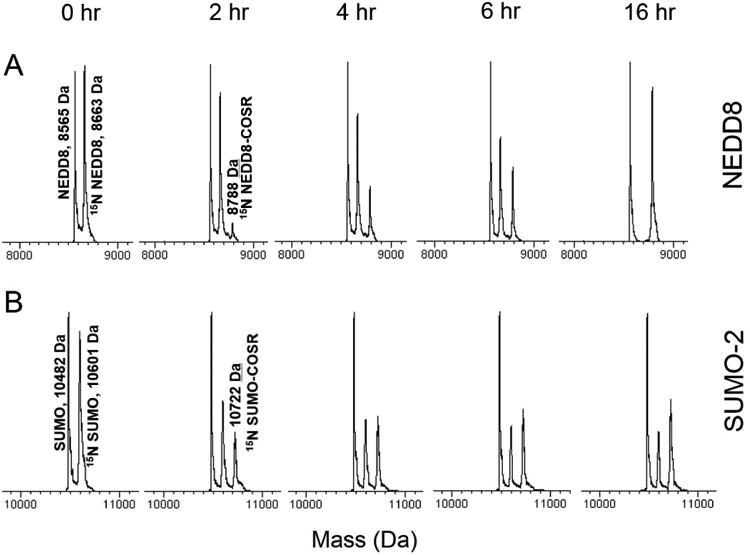
Activation of NEDD8 and SUMO-2 by their cognate E1 enzymes. 15N-enriched NEDD8 (A) or SUMO-2 (B) were incubated with their respective E1 enzymes (APPBP1/UBA3 for NEDD8 and SAE1/SAE2 for SUMO-2) in the presence of MgCl2, ATP, and MESNa. The reactions were aliquoted and stopped at different time points by adding EDTA. An internal (unlabeled) standard was added, and the samples were subjected to ESI-MS. The representative spectral regions are shown.
Conclusions
The MS-based method presented here enables comparative study of the structure-function relationship of any E1 enzymes with their respective UBLs. Unlike previously described methods that relied on the use of radio-labeled ATP (32P, 3H) or/and radio-labeled Ub (125I, 32P)21,24,27-29,70, our method does not require radio-labeling of any of the components and therefore can be readily available in any biochemical laboratory having access to a mass spectrometer. Moreover, in contrast to the previous approaches our method quantifies Ub activation through direct measurement of the remaining amount of unactivated Ub and uses internal standards for absolute quantification. In principle, using separately prepared thioesterified protein (Ub/UBL-COSR) as an internal standard it should be possible to directly measure the amount of E1-activated Ub or UBL by our MS-based method. Furthermore, by separating Ub activation by E1 from the downstream transfer of the activated Ub to a conjugating enzyme E2, we were able to dissect the complex process of E1-E2-catalyzed Ub conjugation. This allowed us to characterize the process of Ub activation independently of E2 and how it is affected by mutations of several key surface residues of Ub and other factors. Our results revealed that the rates of activation and conjugation are affected differently and emphasize the need for characterization of E1 activity separately from E2.
Recently, the E1s of Ub and UBLs have come into focus as attractive targets for the development of therapeutic approaches against cancers71-73, and a number of inhibitors have been developed which specifically target the E1 activity74,75. As the method described in this paper is quantitative and capable of deciphering the structure-function relationship of the E1 enzyme, it will provide a valuable tool to strengthen our understanding of the E1 activity relevant to drug design as well as for future research on novel strategies for treating cancers and other diseases.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM065334, GM095755, and GM021248 to D.F. and HHMI undergraduate research fellowship to Y.K., and utilized NMR instrumentation supported in part by NSF grant DBI1040158. We thank Catherine Fenselau and Yan Wang for useful discussions during the materialization of this study. We are indebted to Christopher Lima for kindly providing plasmids for SUMO-2 and E1 of SUMO, Brenda Schulman for the E1 of NEDD8 plasmid, Miratul Muqit for PINK1 plasmid, Randall King for ubistatin A, and Mariapina D'Onofrio for fullerenol. We also thank Adithya Sundar for repeating several ubiquitination reactions in this work.
Footnotes
Supporting Information. Experimental procedures for protein purification and analysis, abbreviations, tabulated rates of E1 activation of the proteins studied, MS spectra, SDS PAGE gels of polymerization reactions, comparison of the kinetics of protein activation, and NMR data.
References
- 1.Pickart CM. Trends Biochem Sci. 2000;25:544–548. doi: 10.1016/s0968-0004(00)01681-9. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Haglund K, Dikic I. J Cell Sci. 2012;125:265–275. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- 4.Geng F, Wenzel S, Tansey WP. Annu Rev Biochem. 2012;81:177–201. doi: 10.1146/annurev-biochem-052110-120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shan J, Brooks C, Kon N, Li M, Gu W. Ernst Schering Found Symp Proc. 2008:127–136. doi: 10.1007/2789_2008_105. [DOI] [PubMed] [Google Scholar]
- 6.Chen ZJ. Immunol Rev. 2012;246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Z, Hunter T. Annu Rev Biochem. 2009;78:435–475. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehman NL. Acta Neuropathol. 2009;118:329–347. doi: 10.1007/s00401-009-0560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Maldonado MA. Cell Mol Immunol. 2006;3:255–261. [PubMed] [Google Scholar]
- 10.Shi D, Grossman SR. Cancer Biol Ther. 2010;10:737–747. doi: 10.4161/cbt.10.8.13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng C, Geetha T, Babu JR. Neurodegener Dis. 2014;14:161–175. doi: 10.1159/000367694. [DOI] [PubMed] [Google Scholar]
- 12.van der Veen AG, Ploegh HL. Annu Rev Biochem. 2012;81:323–357. doi: 10.1146/annurev-biochem-093010-153308. [DOI] [PubMed] [Google Scholar]
- 13.Lee EK, Diehl JA. Oncogene. 2014;33:2011–2018. doi: 10.1038/onc.2013.144. [DOI] [PubMed] [Google Scholar]
- 14.Gareau JR, Lima CD. Nat Rev Mol Cell Biol. 2010;11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickart CM. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 16.Streich FC, Lima CD. Annu Rev Biophys. 2014;43:357–379. doi: 10.1146/annurev-biophys-051013-022958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL, Holton JM, Schulman BA. Mol Cell. 2003;12:1427–1437. doi: 10.1016/s1097-2765(03)00452-0. [DOI] [PubMed] [Google Scholar]
- 18.Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, Schulman BA. Nat Struct Mol Biol. 2004;11:927–935. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haas AL, Rose IA. J Biol Chem. 1982;257:10329–10337. [PubMed] [Google Scholar]
- 20.Haas AL, Warms JV, Rose IA. Biochemistry. 1983;22:4388–4394. doi: 10.1021/bi00288a007. [DOI] [PubMed] [Google Scholar]
- 21.Haas AL, Warms JV, Hershko A, Rose IA. J Biol Chem. 1982;257:2543–2548. [PubMed] [Google Scholar]
- 22.Metzger MB, Hristova VA, Weissman AM. J Cell Sci. 2012;125:531–537. doi: 10.1242/jcs.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee I, Schindelin H. Cell. 2008;134:268–278. doi: 10.1016/j.cell.2008.05.046. [DOI] [PubMed] [Google Scholar]
- 24.Walden H, Podgorski MS, Schulman BA. Nature. 2003;422:330–334. doi: 10.1038/nature01456. [DOI] [PubMed] [Google Scholar]
- 25.Lois LM, Lima CD. EMBO J. 2005;24:439–451. doi: 10.1038/sj.emboj.7600552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsen S, Capili A, Lu X, Tan D, Lima C. Nature. 2010;463:906–912. doi: 10.1038/nature08765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciechanover A, Heller H, Katz-Etzion R, Hershko A. Proc Natl Acad Sci U S A. 1981;78:761–765. doi: 10.1073/pnas.78.2.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burch TJ, Haas AL. Biochemistry. 1994;33:7300–7308. doi: 10.1021/bi00189a035. [DOI] [PubMed] [Google Scholar]
- 29.Whitby FG, Xia G, Pickart CM, Hill CP. J Biol Chem. 1998;273:34983–34991. doi: 10.1074/jbc.273.52.34983. [DOI] [PubMed] [Google Scholar]
- 30.Castaneda C, Liu J, Chaturvedi A, Nowicka U, Cropp TA, Fushman D. J Am Chem Soc. 2011;133:17855–17868. doi: 10.1021/ja207220g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh RK, Sundar A, Fushman D. Angew Chem Int Ed Engl. 2014;53:6120–6125. doi: 10.1002/anie.201402642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung JW, Bae SJ, Kang GY, Kim KH, Yeo WS, Park SH, Seol JH, Yi EC, Kim KP. Rapid Commun Mass Spectrom. 2013;27:339–346. doi: 10.1002/rcm.6447. [DOI] [PubMed] [Google Scholar]
- 33.Phu L, Izrael-Tomasevic A, Matsumoto ML, Bustos D, Dynek JN, Fedorova AV, Bakalarski CE, Arnott D, Deshayes K, Dixit VM, Kelley RF, Vucic D, Kirkpatrick DS. Mol Cell Proteomics. 2011;10:M110.003756. doi: 10.1074/mcp.M110.003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wee KE, Lai Z, Auger KR, Ma J, Horiuchi KY, Dowling RL, Dougherty CS, Corman JI, Wynn R, Copeland RA. J Protein Chem. 2000;19:489–498. doi: 10.1023/a:1026501515450. [DOI] [PubMed] [Google Scholar]
- 35.Schäfer A, Kuhn M, Schindelin H. Acta Crystallogr D Biol Crystallogr. 2014;70:1311–1320. doi: 10.1107/S1399004714002910. [DOI] [PubMed] [Google Scholar]
- 36.Olsen SK, Lima CD. Mol Cell. 2013;49:884–896. doi: 10.1016/j.molcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C. Proc Natl Acad Sci USA. 1996;93:861–866. doi: 10.1073/pnas.93.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fushman D, Wilkinson K. F1000 Biology Reports. 2011;3:1–10. doi: 10.3410/B3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pickart CM, Fushman D. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z, Pickart CM. J Biol Chem. 1990;265:21835–21842. [PubMed] [Google Scholar]
- 41.Wilson RC, Edmondson SP, Flatt JW, Helms K, Twigg PD. Biochem Biophys Res Commun. 2011;405:662–666. doi: 10.1016/j.bbrc.2011.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko S, Kang GB, Song SM, Lee JG, Shin DY, Yun JH, Sheng Y, Cheong C, Jeon YH, Jung YK, Arrowsmith CH, Avvakumov GV, Dhe-Paganon S, Yoo YJ, Eom SH, Lee W. J Biol Chem. 2010;285:36070–36080. doi: 10.1074/jbc.M110.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodrigo-Brenni MC, Foster SA, Morgan DO. Mol Cell. 2010;39:548–559. doi: 10.1016/j.molcel.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN. J Mol Biol. 2013;425:1363–1377. doi: 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh RK, Zerath S, Kleifeld O, Scheffner M, Glickman MH, Fushman D. Mol Cell Proteomics. 2012;11:1595–1611. doi: 10.1074/mcp.M112.022467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann RM, Pickart CM. J Biol Chem. 2001;276:27936–27943. doi: 10.1074/jbc.M103378200. [DOI] [PubMed] [Google Scholar]
- 47.Wu T, Merbl Y, Huo Y, Gallop JL, Tzur A, Kirschner MW. Proc Natl Acad Sci U S A. 2010;107:1355–1360. doi: 10.1073/pnas.0912802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, Yao TP. Mol Cell. 2013;51:819–828. doi: 10.1016/j.molcel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varadan R, Walker O, Pickart C, Fushman D. J Mol Biol. 2002;324:637–647. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 52.Varadan R, Assfalg M, Haririnia A, Raasi S, Pickart C, Fushman D. J Biol Chem. 2004;279:7055–7063. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- 53.Ryabov Y, Fushman D. Proteins. 2006;63:787–796. doi: 10.1002/prot.20917. [DOI] [PubMed] [Google Scholar]
- 54.Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. Science. 2004;306:117–120. doi: 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- 55.Zanzoni S, Ceccon A, Assfalg M, Singh RK, Fushman D, D'Onofrio M. Nanoscale. 2015;7:7197–7205. doi: 10.1039/c5nr00539f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ. Nat Struct Mol Biol. 2008;15:444–451. doi: 10.1038/nsmb.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 58.Swatek KN, Komander D. Cell Res. 2016;26:399–422. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 60.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N. J Cell Biol. 2015;209:111–128. doi: 10.1083/jcb.201410050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Narendra D, Tanaka A, Suen DF, Youle RJ. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo S, Burnette R, Zhao L, Vanderford NL, Poitout V, Hagman DK, Henderson E, Ozcan S, Wadzinski BE, Stein R. J Biol Chem. 2009;284:759–765. doi: 10.1074/jbc.M806314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D. EMBO J. 2015;34:307–325. doi: 10.15252/embj.201489847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Swaney DL, Rodríguez-Mias RA, Villén J. EMBO Rep. 2015;16:1131–1144. doi: 10.15252/embr.201540298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jonnalagadda S, Ecker DJ, Sternberg EJ, Butt TR, Crooke ST. J Biol Chem. 1988;263:5016–5019. [PubMed] [Google Scholar]
- 71.Soucy T, Smith P, Milhollen M, Berger A, Gavin J, Adhikari S, Brownell J, Burke K, Cardin D, Critchley S, Cullis C, Doucette A, Garnsey J, Gaulin J, Gershman R, Lublinsky A, McDonald A, Mizutani H, Narayanan U, Olhava E, Peluso S, Rezaei M, Sintchak M, Talreja T, Thomas M, Traore T, Vyskocil S, Weatherhead G, Yu J, Zhang J, Dick L, Claiborne C, Rolfe M, Bolen J, Langston S. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 72.Liu J, Shaik S, Dai X, Wu Q, Zhou X, Wang Z, Wei W. Biochim Biophys Acta. 2015;1855:50–60. doi: 10.1016/j.bbcan.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mattoscio D, Chiocca S. Future Oncol. 2015;11:1599–1610. doi: 10.2217/fon.15.41. [DOI] [PubMed] [Google Scholar]
- 74.Leung CH, Chan DS, Yang H, Abagyan R, Lee SM, Zhu GY, Fong WF, Ma DL. Chem Commun (Camb) 2011;47:2511–2513. doi: 10.1039/c0cc04927a. [DOI] [PubMed] [Google Scholar]
- 75.Zhao L, Yue P, Lonial S, Khuri FR, Sun SY. Mol Cancer Ther. 2011;10:2415–2425. doi: 10.1158/1535-7163.MCT-11-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.