

Abstract
The history of the National Institute of General Medical Sciences (NIGMS) Research Centers in Peri-operative Sciences (RCIPS) is the history of clinical, translational and basic science research into the etiology and treatment of posttraumatic multiple organ failure (MOF). Born out of the activism of trauma and burn surgeons after the Viet Nam war, the P50 trauma research centers have been a nidus of research advances in the field, and the training of future academic physician-scientists in the fields of trauma, burns, sepsis and critical illness. For over 40 years, research conducted under the aegis of this funding program has led to numerous contributions at both the bedside and at the bench.
In fact, it has been this requirement for team science with a clinician scientist working closely with basic scientists from multiple disciplines which has led the RCIPS to its unrivaled success in the field. This review will briefly highlight some of the major accomplishments of the RCIPS program since its inception, how they have both led and evolved as the field moved steadily forward, and how they are responsible for much of our current understanding of the etiology and pathology of MOF. This review is not intended to be all encompassing nor a historical reference. Rather, it serves as recognition to the foresight and support of many past and present individuals at the NIGMS and at academic institutions who have understood the cost of critical illness and MOF to the individual and to society.
Keywords: team science, P50 grants, NIGMS, multiple organ failure
Introduction
In 1974, the National Institute of General Medical Sciences (NIGMS), under the leadership of Dr. Ruth Kirschstein, initiated a new funding program, entitled “P50 Research Centers in Injury and Peri-operative Sciences (P50 RCIPS)”. These large program project grants were to foster multidisciplinary translational research related to multiple system responses invoked by trauma, burns, and surgical injury. The history of these “team science” efforts have had unrivaled success in transforming knowledge and fundamentally changing the care for surgical intensive care unit (ICU) patients. Additionally, they have provided the research training environments for two generations of trauma surgeons that have allowed them to be active physician-scientists advancing basic knowledge related to their specialty. NIGMS has recently allowed the P50 RCIPS program to expire in its current form, and intends to replace it with a broader focused team science program. Given that the Coalition for National Trauma Research was recently formed to advocate for continued federal funding for trauma research, discussing the future of this type of trauma research funding is a timely topic (1). This manuscript will provide an overview of the P50 RCIPS granting mechanism, describe seminal contributions of the Trauma Research Centers (TRCs) in the context of the evolving epidemiology of multiple organ failure (MOF) secondary to trauma and sepsis, and summarize the lessons learned through these successful team science efforts.
Origins of the NIGMS P50 RCIPS
The Viet Nam War brought significant changes in the care of the severely injured soldier. Battlefield mortality of wounded soldiers decreased from 30% in World War II to 23% in the Viet Nam War (2). Trauma surgeon activists (including John Burke, William Curreri, C. James Carrico, William Altimeir, William Blaisdale, Don Trunkey, and others) argued that the progress made on the battlefield should be sustained and expanded for civilian populations, in which death from motor vehicle accidents peaked between 1966 and 1975 at rates 30% higher than the decade before and after (3) Senator Edward Kennedy (chairman of the Subcommittee on Health and Scientific Research) was lobbied extensively in the 1970’s to support the establishment of a National Institute of Trauma within the National Institutes of Health (NIH). While this effort failed, Senator Kennedy did use his influence to establish a research program dedicated to trauma, burn and perioperative injury. While this did not fall within the NIGMS’s primary charter of “research in the general and basic medical sciences”, the Institute also had the responsibility to assume research programs “outside the general area of responsibility of any other Institute”. Thus, despite the lack of legislative-mandated funding, Dr. Ruth Kirschstein, initially reluctant, championed the P50 RCIPS at the NIGMS. During its early formative years, the survival of the program benefitted from a number of strong advocates inside the Institute including Drs. Marvin Cassman, Lee Van Lenten, Rochelle Long, Yvonne Maddox, and Bruce Wetzel.
After more than 40 years, the P50 RCIPS program today continues its original goal of improving the basic understanding of the adaptive and maladaptive responses invoked by trauma or burns, and to directly translate this new knowledge into improved patient care. The program emphasizes hypothesis-based bi-directional bench to bedside research. It requires multiple projects led by successful scientists who synergistically promote team science that has value-added beyond a collection of independent R01 type projects. In general, these programs have focused on the greatest challenges of their time. The scope of research spans the time of injury through critical illness and subsequent recovery. Unlike other institutes under the NIH umbrella that focus on single disease, organ or system response, NIGMS and the RCIPS focus on multiple system responses. For example, traumatic brain injury and spinal cord injury are not within the specific mission of NIGMS, but would be appropriate for the National Institute of Neurologic Diseases and Stroke (NINDS). These P50 programs also have often included a component of mathematical and computational approaches to systems biology, using all the tools available to address the complex nature of illness. Some of the programs have included interventional studies, but only when they were consistent with the overall goal of improving the understanding of the underlying pathobiology. Figure 1 lists the NIGMS P50 RCIPS grants by first year funding since the program’s inception, their general focus, and their program directors and host institution.
Figure 1.

Depicts the P50 NIGMS RCIPS grants by first year of funding, general focus, program director(s) and host institution. Many were funding through multiple cycles and therefore had multiple program directors.
Many of the Centers have been refunded through several cycles with the longest program led initially by Dr. John Burke and subsequently by Dr. Ronald Tompkins at Massachusetts General Hospital having had continuous funding for over 40 years. Successfully refunded P50 programs have evolved over time, and by necessity the program directors and project program investigators have changed as the focus of the programs kept up with the advances in the field. Although any attempt to summarize the accomplishments of this 40 plus year program will be incomplete, progress in the field of trauma and burns has been due in considerable part to the success of the RCIPS Program and the support of NIGMS leadership even during periods of lean funding to the NIH.
Seminal Contributions Related to the Evolving Epidemiology of MOF
Despite tremendous advances in care over the last four decades, MOF following trauma and sepsis has remained the leading cause of prolonged ICU stays and late mortality (4). Therefore, it is not surprising that many of the trauma-related P50 programs focused on some aspect of MOF. MOF emerged in the early 1970s as a result of advances in ICU technology that allowed patients to survive single organ failure. Over the ensuing decades with continued advances in the management of the critically ill patient, the epidemiology of MOF has evolved (5). Figure 2 depicts the timeline of this evolution. The upper portion of this timeline depicts major advances in care and the lower portion depicts a series of predominant presentations (or phenotypes) that have been the focus of ongoing research efforts. The bottom of the figure lists various research topics that have been pursued by the P50 programs and will be included in the following summary discussion.
Figure 2.
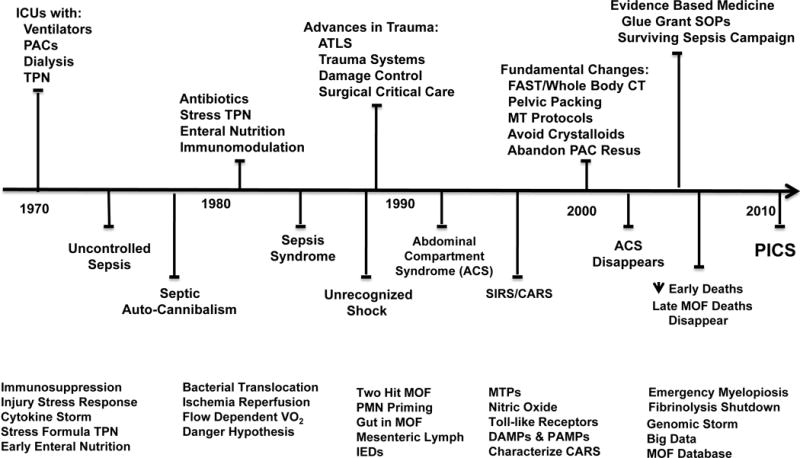
Depicts timeline of evolving epidemiology of multiple organ failure (MOF). ICU, intensive care unit; PACs, pulmonary artery catheters; TPN, total parenteral nutrition;VO2, oxygen consumption; ATLS, advanced trauma life support; PMN, neutrophil; SIRS, systemic inflammatory response syndrome; CARS, compensatory anti-inflammatory response syndrome; DAMP; damage associated molecular patterns; PAMPs, pathogen associated molecular patterns; IEDs, immune enhancing diets SOPs, standard operating procedures; PICS, persistent, inflammation, immunosuppression, catabolism syndrome. (Reprinted with permission of Wolters Kluwer Health, Inc, from Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012; volume 72, issue 6:1491–501).
Uncontrolled Sepsis
Initial case series in the 1970s concluded that MOF occurred due to uncontrolled sepsis (6). Research efforts were directed at the prevention and treatment of surgical infections. It was recognized that in large part these uncontrolled infections were due to failure in local and systemic host defenses (7,8). The early P50 RCIPS programs led by John Border, John Kinney, and Douglas Wilmore focused on the role of the injury stress response in MOF (9–11). The injury stress response is initially considered essential in mobilizing substrates, but when infections become uncontrolled, they induce “septic autocannibalism” (12). This term was coined to explain the tremendous losses of lean body mass that occur despite exogenous nutritional support. The associated hypermetabolism could be reproduced by the infusion of counter regulatory hormones (i.e, hydrocortisone, epinephrine and glucagon), but this did not fully recapitulate the ongoing catabolism, or the acute phase response. This could be induced by creating concurrent inflammation (by intramuscular injection of etiocholanolone) (11). At this time, expression cloning of new inflammation mediators was in its infancy, and what is now know as “cytokines” had only functional monikers such ‘lymphocyte activating factor’, ‘leukocyte endogenous mediator’, ‘cachectin’, among literally hundreds of individual mediators (13). However, with the recognition that these proteins are produced by every tissue of the body in massive quantities during sepsis, the term “cytokine storm” was popularized to describe uncontrolled sepsis. Importantly, studies from Dr. G. Thomas Shire’s program in New York City, initially showed that administration of key cytokines could reproduce bacteremic or endotoxemic shock (14) Additionally, it was demonstrated that the lethal administration of live bacteria or endotoxin produced a burst of TNFα followed by a more sustained rise in other proinflammatory cytokines, and that the resulting death could be prevented by blocking this endogenous TNFα response(15)
With the introduction of total parenteral nutrition (TPN) into clinical practice in the 1970’s, it is not surprising that the P50 investigators became interested in optimizing the nutrient composition of TPN to match the metabolic needs induced by injury stress response. Research indicated that the sequence of surgery/trauma-immunosuppression-late sepsis-MOF-death could be reversed by early TPN. (16,17). “Stress formula” TPNs enriched with L-glutamine. L-arginine, and branched chain amino acids were developed, widely utilized and subjected to clinical trials starting in the early 1980s. Unfortunately, their early use in critically ill surgical patients failed to improve outcome. Additionally, the TRC at Denver convincingly demonstrated in a series of clinical trials that early enteral nutrition (EEN) reduced nosocomial infections compared to TPN (18). The exact explanation for this benefit remained unclear, but it was presumed to be linked to beneficial effects on the gut. This sparked interest in role of the gut as the “motor” of MOF which remained a research interest in Denver and became a focus of subsequent P50 programs in both Houston and Newark (19, 20).
Sepsis Syndrome
In the mid-1980s, studies out of Europe reported that MOF frequently occurred after blunt trauma with no identifiable site of infection (21). The term “sepsis syndrome” was popularized to describe this phenomenon. (22) It became widely accepted that MOF could ensue after both infectious and non-infectious insults by a similar auto-destructive, systemic inflammatory response syndrome (SIRS). The research focus consequently shifted to determining the underlying mechanism(s) of noninfectious SIRS induced MOF (22). Multiple hypotheses have been put forward, including bacterial translocation (BT) (see figure 3) (23). Experimental work from Dr. Edward Dietch’s group provided persuasive evidence in support of BT and it became the accepted dogma (24). However, after the Denver group pursued the BT hypothesis in the laboratory and in clinical studies, they concluded that the BT hypothesis could not fully explain and was not likely a key a mechanism in early noninfectious MOF (25,26).
Figure 3.
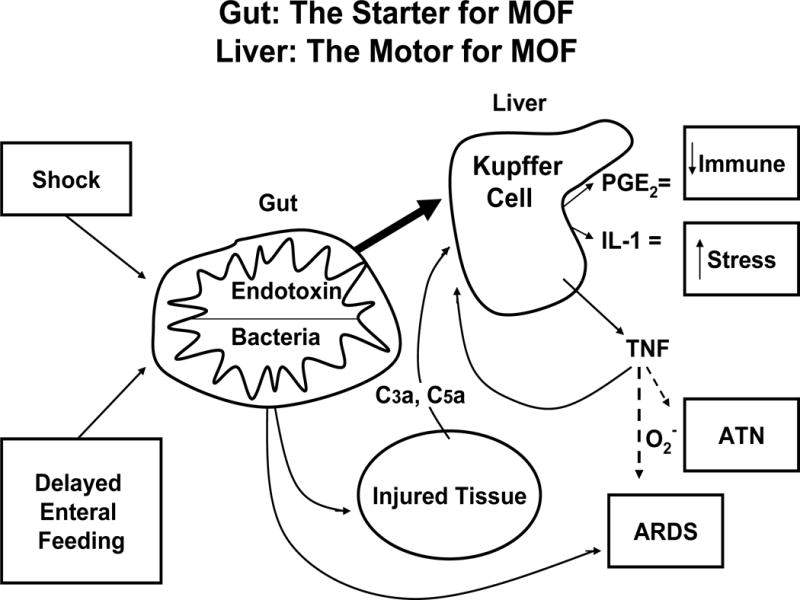
The gut is the starter and the liver is the motor of MOF. Complement factors C3a and C5a; PGE2, prostaglandin E2; IL-1, interleukin-1; O2−, superoxide radical; ATN, acute tubular necrosis; ARDS, acute respiratory distress syndrome. (Reprinted with permission of Wolters Kluwer Health, Inc, from Moore FA, Moore EE, Jones TN, McCroskey BL, Peterson VM. TEN versus TPN Following Major Abdominal Trauma – Reduced Septic Morbidity. J Trauma.1989; volume 29, issue 7:916–23).
Persistent unrecognized shock (as a result of impaired flow dependent oxygen consumption) was another popular alternative hypothesis for noninfectious MOF (22). Dr. William Shoemaker championed this concept and proposed that “supranormal oxygen delivery (DO2)” resuscitation was needed to eliminate it and thereby prevent MOF. As a result, in the late 1980s, it became standard of care to presumptively place pulmonary artery catheters (PACs) to guide early ICU resuscitation with the goal maximizing DO2. While this strategy was ultimately disproven, the resulting resuscitation protocols provided convincing evidence that the initial severity of shock plays a predominant role in the later development of MOF (27). At the same time, there was considerable interest in investigating the role of ischemia/reperfusion (IR) injury in noninfectious MOF. (22) Specifically, the Denver TRC focused on determining how gut IR activates circulating polymorphonuclear neutrophils (PMNs) to cause remote liver and lung injury (25). In the mid 1990’s, Dr. Polly Matzinger first proposed the ‘danger hypothesis’ based on the recognition that the release of endogenous compounds from dying, necrotic or pyrroptotic cells could act through the same signaling pathways responsible for the recognition of microbial products (28) This provided the mechanistic underpinnings for the role of damage associated molecular patterns (DAMPs) molecules [including mitochondrial DNA (mtDNA), HMGB1, S100A, and heat shock proteins] in eliciting inflammatory responses comparable to microbial DNA, endotoxin (LPS) and proteoglycans (29)
Two-Hit Model
This paradigm (depicted in figure 4) was based on clinical observation that when high risk trauma patients were subjected to supranormal DO2 resuscitation, nonresponders died early while the responders entered a quasi-stable state of systemic inflammation (22,27). However, some of these patients were prone to secondary inflammatory insults (e.g. aspiration, blood transfusion, femur fracture rodding) that precipitated MOF while others appeared to be protected when exposed to same insults. Based on this concept, hemorrhagic shock or gut IR models were modified to produce mild reversible MOF and then it was demonstrated that an appropriately timed delayed low dose of LPS could function as a “two-hit” in causing full blown MOF (30). PMNs were shown to play a key role in these two-hit models and this led to in-depth explorations of classic, G protein-coupled receptor mechanisms of PMN priming and activation (31). The clinical relevance of these laboratory observations was subsequently confirmed in high risk ICU patients (32).
Figure 4.
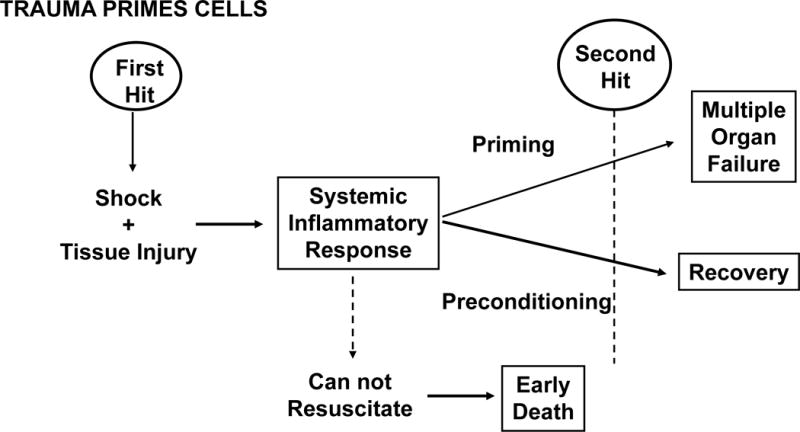
Depicts the “two hit” model of multiple organ failure (MOF). (Reprinted with permission of Elsevier from Moore FA. Presidential Address: Imagination Trumps Knowledge. Am J Surg. 2010, volume 200, issue 6:671–77).
Interest in the role of packed red blood cells (PRBC) transfusion as a second hit in this paradigm came from a series of MOF database analyses that showed > 6 units PRBC transfusion within the first 12 hours postinjury was a consistent, independent, and strong predictor for early MOF (33). This finding plus other compelling evidence suggested that PRBCs played a major role in the priming/activation of PMNs (34). It was found that during PRBC storage, cell wall degradation produced pro-inflammatory lipids (e.g. platelet activating factor) that were robust priming agents for PMN superoxide and elastase production. Although washing PRBCs was reasonably effective in attenuating their priming capacity, this was logistically problematic for patients requiring a massive transfusion (35). Recognizing that patients in hemorrhagic shock require an oxygen carrier led to the investigation of the potential role of a hemoglobin-based oxygen carrier for early resuscitation. It was documented in vivo that human polymerized hemoglobin avoided transfusion induced PMN priming (36). Ultimately this work lead to a large multi-institutional, FDA approved phase III clinical trial of PolyHeme™ for the early resuscitation of critically injured patients. Patients resuscitated with PolyHeme, without stored blood for up to 6 U in 12 hours postinjury, had outcomes comparable with those for the standard of care. Although there were more adverse events in the PolyHeme group, the benefit-to-risk ratio of PolyHeme is favorable when blood is needed but not available. (37)
Abdominal Compartment Syndrome (ACS)
During the 1980s, there were tremendous advances in trauma care including Advanced Trauma Life Support, Trauma Systems, damage control surgery and surgical critical care. As a result, early deaths from severe bleeding decreased substantially. Unfortunately, many of the survivors developed ACS in the first days after ICU admission and died from fulminant early MOF. By the early 1990s, this new MOF phenotype became a worldwide epidemic and was initially poorly understood. As part of the Denver and Houston P50 programs, high risk patients were managed by standard operating procedures (SOPs) and had their clinical trajectories tracked prospectively in their MOF data bases (38). A series of database analyses demonstrated that ACS was a surprisingly early event that could be predicted in the emergency department (ED) in a patient arriving with severe bleeding. Ongoing laboratory and clinical studies clarified underlying pathophysiology of ACS and its iatrogenic origins (i.e. excessive ED crystalloids, inadequate hemorrhage control, and supranormal DO2 ICU resuscitation). Concerted efforts were directed at refinement of the SOPs that lead to fundamental changes in early resuscitation [including a massive transfusion protocol (MTP) emphasizing early fresh frozen plasma (FFP), avoidance ED crystalloids and abandonment of PAC driven resuscitation) and the use of vacuum assisted wound closure in damage control surgery (39–41). These P50 program studies had tremendous impact on civilian and military trauma care. With these fundamental advances and more focus on initial hemorrhage control, the epidemic of ACS disappeared by the early 2000s and has now largely become an historic curiosity.
Nitric Oxide (NO)
As improved clinical management continued, the evolving field of molecular genetics was altering the research approaches to trauma and MOF. The Pittsburgh P50 TRC focused on the question of how injury activates inflammatory responses and why this progresses to immune dysfunction in some patients. One key inflammatory mechanism, the arginine → NO pathway discovered in the late 1980s, had major implications for the field of shock research. NO is a powerful endogenous vasorelaxant and can regulate many components of the immune system. The Pittsburgh TRC was the first to show that humans express the high output or inducible NO synthase (iNOS) and that iNOS is rapidly upregulated in severely injured humans (42,43). Studies in animals allowed them to exclude a major role for iNOS in decompensated hemorrhagic shock. They instead showed that iNOS contributes to the amplification of early SIRS and organ injury. (44) They provided evidence that targeting iNOS selectively or scavenging extracellular NO could protect animals subjected to hemorrhagic shock alone or hemorrhagic shock with trauma. In fact, NO scavenging prevented immune dysregulation manifested by SIRS, organ injury, or suppressed lymphocyte responses (45). The Pittsburgh TRC also showed that preservation of endothelial NOS (eNOS) function was an important consideration. This work would lead to patents targeted at iNOS-selective inhibitors and NO scavengers to prevent immune dysregulation.
Toll Like Receptors
Coming off their seminal observations regarding the importance of NO, the Pittsburgh TRC demonstrated that a series of pattern recognition receptors (PRR) known as toll-like receptors (TLR), originally discovered for their roles in microbial recognition, contribute to the activation of SIRS after hemorrhagic shock, tissue trauma, or a combination of hemorrhagic shock plus tissue trauma (46,47). The field of innate immunity had been revolutionized by the discovery of PRR that detect molecules expressed by invading microbes (i.e. pathogen associated molecular patterns [PAMPs]) or released by tissue injury or stress (i.e. DAMPs). Pittsburgh went on to show that TLR expression on both immune and non-immune cells is key to the host response to injury. TLR2, TLR4, and TLR9 appear to be especially relevant to the injury response and, in some cases, interact (i.e. TLR4-dependent upregulation of TLR2) (48). Importantly, they demonstrated that TLRs can be an appropriate therapeutic target to decrease organ damage and excessive inflammation. The Pittsburgh TRC, in collaboration with Drs. Kevin Tracey and Ping Wang at the Feinstein Institute, can be credited with showing that a prototypic DAMP, High Mobility Group Box-1 (HMGB1) is elevated early in humans after injury and that its concentration correlates with delayed complication (e.g. nosocomial infection)(49,50). Experimental studies confirmed that HMGB1 is required for SIRS after traumatic shock and is potentially targetable in preventing systemic injury.
SIRS/CARS
This paradigm (depicted in figure 5) was proposed in the mid-1990s to explain the dysregulated immune response related to the bimodal presentation of MOF (51). During successful resuscitation, patients develop SIRS (predominantly an innate immune response). If severe, it causes early fulminant MOF. Endogenous DAMPs or exogenous PAMPs signaling through PRRs activate inflammation signaling via transcription factors NF-kB, CREB1, C/EBP and IRFs to express proximal, pro-inflammatory mediators that activate overlapping cascades of other inflammatory mediators, effector cells and at the same time, activate endothelial cell dysfunction and prothrombotic events. The end-result is often inadequate tissue perfusion and organ injury. Fortunately, in the majority of patients SIRS can be successfully managed, and at the time it was proposed that survival to SIRS is followed by delayed immunosuppression, nosocomial infections and late onset MOF (52) Delayed immunosuppression after major trauma and surgical insult had been previously described, but its link to early SIRS was novel (53,54). In part this notion came from clinical studies showing an early vulnerable window of PMN priming/activation was followed by a period when circulating PMNs were functionally effete and bone marrow granulopoiesis was suppressed (32,53). This was also consistent with the observation that increase production of anti-inflammatory cytokines (e.g. interleukin (IL)-10) and cytokine antagonists (e.g. IL-1 receptor antagonist and soluble TNF receptor I) were delayed and prolonged compared to the early SIRS response meditators (54). Additionally, it was demonstrated that when nonhuman primates were administered live bacteria, inhibitors of SIRS also suppressed the later anti-inflammatory cytokine response (55). Thus, SIRS was viewed to be a pro-inflammatory “cytokine storm”, while CARS was a compensatory anti-inflammatory cytokine response to restore immunologic homeostasis.(56,57) However, the concept of CARS evolved to include depression in the adaptive immune response. Immunologists in late 1990s focused their effort on characterizing CARS to include increased lymphocyte and dendritic cell apoptosis, macrophage paralysis, elevation in regulatory T cells, decreased antigen presentation, suppressed T-cell proliferation, and a shift from type 1 helper T-cell (TH1) to TH2 lymphocyte phenotype (58–62).
Figure 5.
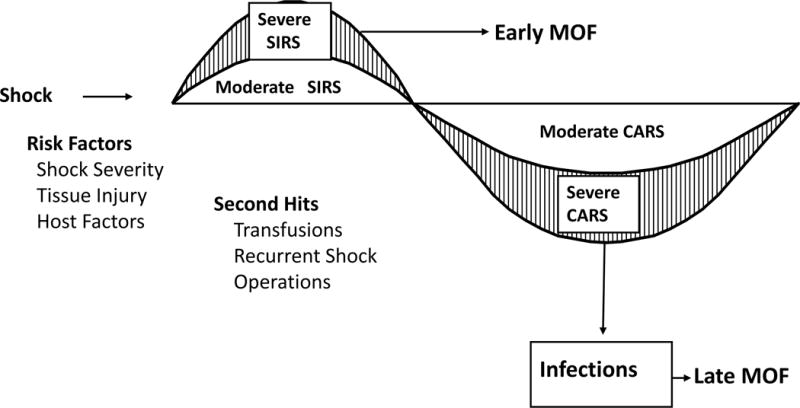
Depicts the SIRS/CARS paradigm for bimodal multiple organ failure (MOF). SIRS, systemic inflammatory response syndrome; CARS, compensatory anti-inflammatory response syndrome. (Reprinted with permission of Wolters Kluwer Health, Inc, from Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma. 1996, volume 40, issue 4, pp 501–12).
Role of The Gut in MOF
This was the focus of the Denver, Newark and Houston P50 programs (see figure 3).(19) Shock after trauma and sepsis followed by resuscitation results in gut IR. This activates gut phospholipase A2 with the release of pro-inflammatory lipid mediators which in turn, can prime/activate SIRS to cause early MOF (63). The mediators are released primarily via the mesenteric lymph rather than the portal vein (64). These early gut IR induced systemic pro-inflammatory events can be prevented by clinically relevant interventions (e.g. hypertonic saline and Poloxamer 188) (65–67). Gut IR injury also initiates a local pro-inflammatory response that results in a variety of gut dysfunctions (e.g. gastroparesis, gastric alkalinization, ileus, duodenogastric reflux, impaired mucosal blood flow, epithelial apoptosis, increased permeability, and impaired local gut immunity). Early crystalloid resuscitation can amplify gut inflammation, cause problematic edema and worsen ileus (68,69). Early laparotomy with bowel manipulation also promotes gut inflammation, mucosal injury and worsened ileus (70). Of note, standard ICU interventions worsen these gut dysfunctions, including the use of vasopressor agents (decrease mucosal perfusion), stress gastritis prophylaxis (worsens gastric alkalinization), narcotics (worsen ileus), antibiotics (promote bacterial overgrowth), and TPN (causes gut atrophy). Over a short period of time, the normally sterile upper GI tract becomes heavily colonized with drug resistant pathogens that are present in the ICU environment. Interestingly, stressful insults have recently been shown to stimulate a genomic response in quiescent gut bacteria such that they become more invasive and secrete more toxins (71). Additionally, the lack of enteral stimulation (i.e. use of TPN) causes a rapid and progressive decrease gut associated lymphoid tissue (GALT) which in turn adversely effects of the mucosal associated lymphoid tissue (MALT) (72). As a result of these events, the gut becomes a reservoir for virulent bacteria and toxic products which escape the gut via aspiration or translocation to cause late nosocomial infections and ongoing sepsis that characterize late MOF. This increased the susceptibility to the gut derived bacteria can be rapidly reserved by enteral feeding. This explains the reduced infections observed in the previously mention EEN vs TPN clinical trials as well as the more recent studies showing the added benefits using specialized immune enhancing enteral diets fortified with nutrients such as arginine, glutamine, and omega 3 fatty acids (73).
Post-Injury Fibrinolysis
In the 2000s, with renewed interest in optimizing MTPs to provide optimal blood products in initial ED resuscitation, trauma-induced coagulopathy (TIC) became a focus of research at the Denver TRC. They adopted whole blood viscoelastic assays [e.g. thrombelastography (TEG)] to guide blood component transfusion in severely injured patients and identified a surprisingly high incidence fibrinolysis (74). This was associated strongly with increased mortality in patients presenting with severe bleeding. The subsequent CRASH-2 study stimulated worldwide interest in the empiric administration of the antifibrinolytic agent tranexamic (TXA) to mitigate the contribution of fibrinolysis in the patient at risk for TIC. The unbridled use of TXA stimulated the Denver group to investigate the components of TIC. Principal Component Analysis (PCA) suggested the fundamental mechanisms driving inadequate clot formation were distinct from those responsible for enhanced clot degradation. Further investigation identified three distinct phenotypes of fibrinolysis in severely injured patients; i.e., hyperfibrinolysis (15%), physiologic fibrinolysis (20%), and fibrinolysis shutdown (65%) (75). While hyperfibrinolysis was a risk factor for early death due to uncontrolled bleeding, fibrinolysis shutdown was associated with late mortality due to MOF. (76) Parallel experimental work suggested hyperfibrinolysis was due to endothelial tissue plasminogen activator (tPA) release provoked by shock; whereas, tissue injury stimulated fibrinolysis shutdown due to elevated plasminogen activator-1 (PAI-1) from endothelium and platelet degranulation (77). To further refine the quantification of fibrinolysis phenotypes, a tPA challenge TEG assay was developed in which varying amounts of tPA were added to the patients’ whole blood to calibrate the responsiveness to endogenous tPA (78). Collectively, these clinical data and experimental findings prompted the recommendation for selective administration of TXA to injured patients (79). The CRASH-2 database was reassessed. Using markers of shock and tissue injury, subgroups of patients who likely benefit form TXA and those who may have adverse effects were identified. Additionally, a randomized trial in the surgical intensive care unit (NCT02901067) has been initiated, comparing heparin with or without the addition of aspirin and a statin, to attenuate fibrinolysis shutdown and reduce thromboembolic events.
Genomic Storm
Much of MOF research has been a reductionist approach of studying individual mediators. In the late 1990s, the Human Genome Program challenged this approach. Shortly thereafter, NIGMS (under Dr. Marvin Cassman) initiated the Glue Grant (GG) funding mechanism. This was envisioned to be a “P50 grant on steroids” with substantial funding (~ $5–8 million/year) to address questions of seminal importance. Ronald Tompkins from Boston developed a trauma/burn consortium GG called the “Inflammation and the Host Response to Injury”. The goal of the program was to describe the host response to trauma and burns using high-throughput genome-wide transcriptomics and proteomics (80) The most striking observation of this GG program was that severe trauma or burns caused a genome-wide expression in blood leukocyte populations of as many as 17,000 genes, representing greater than 75% of the human genome that are altered in response to these injurious signals (81). The term ‘ genomic storm’ was coined to reflect the broad diversity of the host response. Even more surprising was the high correlation in the initial rapid gene expression after trauma and burns. The principle difference was the duration of the response with resolution and return to homeostasis being notably longer in burns (>one year) compared to trauma (>one month) (82). The genomic and proteomic data has already resulted in early biomarkers for the development of MOF (83)
The other notable finding was the problematic observation that at the transcriptome level, the changes in gene expression in murine models of trauma and burn did not correlate well with their human orthologues (84) The results have been broadly misinterpreted to suggest that murine models have no value in burn and trauma research (85) In fact, the original report and subsequent reports have demonstrated that for many genes and gene pathways, there is good correlation between murine and human responses (86) What the report apparently failed to transmit was a warning that results from murine studies could not be universally accepted as equivalent to humans, and investigators should be required to validate their murine findings in patients.
Chronic Critical Illness (CCI) and the Emergence of the Persistent, Inflammation, Immunosupression, Catabolism Syndrome (PICS)
Starting in the late 1990’s, reports describing chronic critical illness (CCI) emerged under a variety of descriptive terms including the “neuropathy of critical illness”, “myopathy of critical illness”, “ICU acquired weakness” and most recently “post intensive care unit syndrome”. These reports largely originated from medical ICU’s, and included individuals with a wide variety of admission diagnoses, most commonly acute exacerbations of chronic diseases, who required prolonged mechanical ventilation and were often discharged to long-term care facilities. Given the clinical heterogeneity of this patient population, the underlying pathophysiology of CCI has remained ill defined. However, in more recent years as a result of improved implementation of evidence-based ICU care (e.g. Glue Grant SOPs and the Surviving Sepsis Campaign), the epidemiology of MOF has evolved. (87,88) Early in hospital mortality has decreased substantially and the incidence of late onset MOF deaths in the ICU has largely disappeared (89). As a result protracted low grade MOF has become a common cause of CCI. Based on substantial laboratory and clinical research data, the P50 program in Gainesville, Florida proposed the PICS paradigm (Figure 7) as a mechanistic framework in which to explain the increased incidence of CCI in surgical ICUs (5). Following an inflammatory insult, the host response can become overwhelming leading to an early fulminant MOF trajectory. Fortunately, earlier recognition and implementation of modern ICU care has reduced this trajectory’s fatal expression. If the severely insulted patients do not die of early MOF, there are two clinical outcomes. They can either rapidly recover, or their dysregulated immune response persists and many progress into this new predominant MOF phenotype that we have termed PICS. PICS is characterized by a persistent inflammatory and acute phase response with ongoing protein catabolism. Despite aggressive nutritional intervention, there is a tremendous loss of lean body mass and a proportional decrease in functional status and poor wound healing. Clinically, PICS patients are immunosuppressed and suffer from recurrent nosocomial infections, poor wound healing and are commonly discharged to long-term acute care facilities where they commonly experience sepsis recidivism requiring re-hospitalization, failure to rehabilitate and an indolent death. The value of the PICS paradigm is that it apportions CCI into a collection of testable hypotheses, and provides a framework for testing novel interventions. For example, recent laboratory work in chronic murine models of sepsis and trauma have identified the expansion of myeloid derived suppressor cells (MDSCs) to explain the persistent immune dysregulation observed in PICS patients. (90,91) A recent focused translational study of surgical patients with severe sepsis confirmed the clinical relevance of these laboratory observations. It showed that the numbers of MDSCs rapidly increase after sepsis and are persistently elevated out to 28 days (92). Importantly, these MDCSs were shown to suppress T lymphocyte proliferation and decrease the release of TH1 and TH2 cytokines. Moreover, MDSC expansion correlated with adverse outcomes including: a) early increased expansion was associated with early mortality, b) persistent expansion was associated with prolonged ICU stays and c) persistent expansion was a strong independent predictor of nosocomial infections and poor post discharge disposition. These data provide the theoretical basis for the use of immune-stimulants that modulate MDCSs similar to what has been successfully utilized in advanced malignancies to achieve durable response rates. However, monotherapies will likely be ineffective and multimodality intervention will be required to interrupt the inflammation, immunosuppression, and protein catabolism that characterizes PICS. Additionally early prediction models to identify appropriate candidates and novel biomarkers to assess responses will be needed to conduct these future interventional studies.
Figure 7.
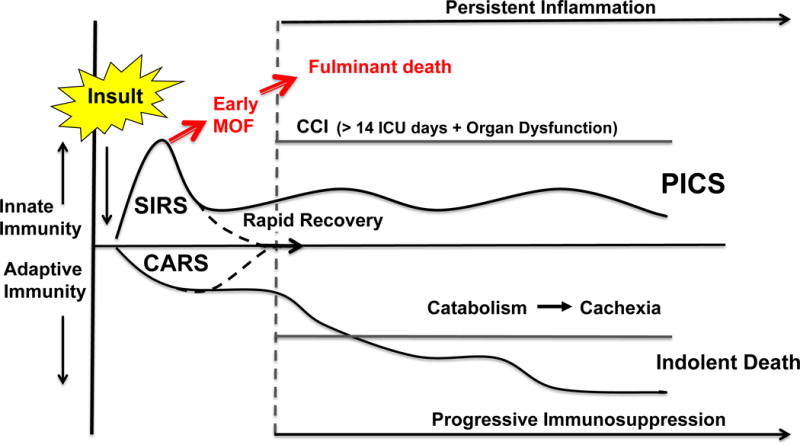
Depicts the persistent, inflammation, immunosuppression, catabolism syndrome (PICS). SIRS, systemic inflammatory response syndrome; CARS, compensatory anti-inflammatory response syndrome; CCI, chronic critical illness. (Reprinted with permission of Wolters Kluwer Health, Inc, from Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012; volume 72, issue 6:1491–501).
Big Data
Ongoing studies have revealed the complexity of the inflammatory response. Like many biological processes, inflammation is multi-dimensional. The advent of multiplexed platforms for gathering biological data, while providing an unprecedented level of detailed information, has paradoxically also flooded investigators with data they are often unable to utilize effectively. This problem is especially acute when the datasets involve time courses, since typical statistical analyses and data-driven modeling are geared towards single time points (93). The Pittsburgh TRC has addressed this complexity using dynamic approaches to data-driven and mechanistic computational modeling in order to decipher the dynamic patterns characteristic of inflammation and its attendant impact on immune dysregulation, organ dysfunction, and downstream clinical outcomes (94). Data-driven modeling describes a suite of tools that utilize the data themselves to define principal drivers and dynamic networks of a given biological system, and to infer putative central regulators of the underlying dynamic processes (95). The Pittsburgh group has focused gaining insights into trauma-induced inflammation using methods such as PCA, Dynamic Network Analysis (DyNA), and Dynamic Bayesian Network (DyBN) inference on human and animal data sets (96–99). Mechanistic modeling refers to an orthogonal, but synergistic, approach to understanding dynamic biological processes. In mechanistic modeling, one creates abstractions of the underlying biology at the molecular, cellular, tissue, organ, and/or whole-organism level using mathematical equations or computational rules. Mechanistic models can then be calibrated to data, and can allow for non-intuitive predictions and so-called “emergent phenomena”. They have used mechanistic modeling to carry out simulations of the host’s response to injury, both in pre-clinical settings and at the single-patient and population levels. These combined modeling studies have led to insights regarding dynamic networks, feedbacks, and regulatory switches; to in silico models of critically ill individuals and populations; and to methods for integrating data-driven and mechanistic modeling (100).
A large clinical translational database were initially developed in the late 1980s by the Denver group to prospectively characterize MOF and to provide patients for focused observational studies and interventional trials. This dataset was refined and expanded as it was incorporated into Houston P50 TRC and the Glue Grant databases. Most recently, data related to surgical sepsis has been incorporated into the dataset for the Gainesville TRC database and REDCap™ is now being utilized as a web based case report form to collect data for ongoing epidemiology studies and clinical trials. Over the years, this MOF dataset has produced a multitude of publications related to the evolving MOF epidemiology, MOF scoring, risk factors, biomarkers, prediction models, dysregulated immunity, cytokine patterns, genomics, acute kidney injury, ACS, resuscitation strategies, enteral nutrition, ICU monitoring, ARDS management, massive transfusions, coagulopathy, computerized clinical decision support, SOP performance improvement, sepsis screening, and sepsis management. This dataset has been an integral tool in promoting bi-directional bench to bedside research and multi-institutional collaborations.
Summary of Success
Over the past four decades, the NIGMS P50 RCIPS translational research program has had unrivaled success in transforming our understanding of the host response to trauma and burn, including MOF. This program has led directly to fundamental changes in the care of the critically ill surgical ICU patients with resultant substantial reductions in mortality. Successful programs have been led by clinician-scientists who have tackled an insurmountable clinical problem and in collaboration with basic scientists (who understand the complexity of the related science) developed a novel testable paradigm. This serves as the common theme around which a multidisciplinary team develops a program project that emphasizes bidirectional bench to bedside research. Understanding the requirement for a multidisciplinary approach, the P50 granting mechanism is unique because it has provided sufficient funding (beyond the financial scope of multiple R01s) to provide the core research infrastructure that facilitates synergy, economy and outreach. This involves a ‘teams of teams’ approach. An obligate Administrative Core assures meticulous financial oversight, ethical compliance and achievement of proposed research milestones. Additionally, this core organizes additional institutional support, fosters institutional collaboration, resolves academic or personnel issues and coordinates communication with the NIGMS. A requisite Clinical Core facilitates translational clinical studies central to the center’s success. Its key resource is a research nurse and laboratory technician capability, in many cases with 24/7 availability, to facilitate patient screening, consent and enrollment, prospectively collect clinical data and obtain/process biologic samples. Many of the centers have also employed an Animal Core which develops clinically relevant standardized laboratory models that are used in the different projects, but tailored to their specific needs. In addition, as the trend to larger more complex studies employing larger volumes of data, including -omics data, a Data Management and Biostatisical Core has become the de facto standard to assure correct/accurate data are being collected and ultimately made available to the scientific community. A computational modeling and systems biology expertise has also become essential to analyze diverse types of data to generate new alternative hypotheses and to understand the diversity of the human response for the development of personalized interventions and accurate patient stratification in clinical trials. What has been generally unappreciated by the broader scientific community, although not to NIGMS, has been the success of the program in developing young investigators who are often the catalyst for ongoing research, and in turn absorbing the cultural and scientific philosophies of team science which cannot be gained by working in a typical RO1 funded investigator lab. Not only are they mentored by older, more established physician-scientists, but they frequently have close interactions with basic health scientists, biomedical engineers, biostatisticians, computational biologists and medical ethicists. These trainees quickly become credible investigators who can independently pursue complementary lines of research which enhance the overall program evolution. One particular challenge to all of the centers has been the necessity to build and develop a successful infrastructure. The initial funding is generally viewed as an investment; successful centers continually grow and evolve, and require multiple funding cycle to achieve their ultimate success. Ongoing outreach is imperative to attract new investigators, novel investigative methods, interventions and additional funding. This outreach is also needed to attract divergent established investigators worldwide who through ongoing interactions add considerable insight and expertize into the complex problem being studied.
Figure 6.
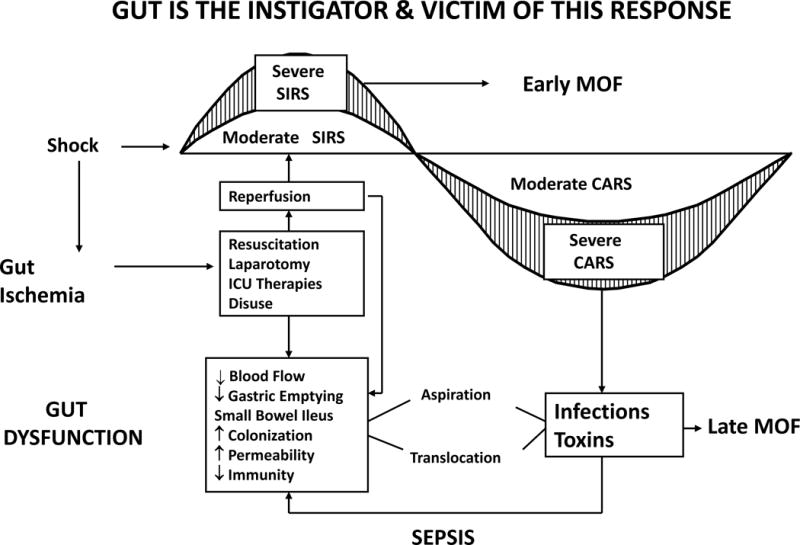
Depicts the role of the gut in multiple organ failure (MOF). SIRS, systemic inflammatory response syndrome; CARS, compensatory anti-inflammatory response syndrome. (Reprinted with permission of Elsevier from Moore FA. The Role of the Gastrointestinal Tract in Postinjury Multiple Organ Failure. Am J Surg. 1999, volume 78, issue 6:449–53).
Acknowledgments
All authors were principle investigators on NIGMS P50 grants whose work was highlighted in this review. All authors wrote sections related to their specific research and assisted in writing and editing of the entire manuscript.
Disclosures of funding: NIGMS P50 grants at the University of Colorado (P50 GM49222:1992-2020) University of Pittsburgh (P50-GM-53789:1997-2014) University of Texas Houston (P50 GM38529: 1988-2009) and University of Florida (P50 GM111152: 2014-2019)
Footnotes
Conflict of interest: we have no conflict of interests related to this publication
Meeting Presentation: Master Surgeon Lecture presented at the 75th annual meeting of the American Association for the Surgery of Trauma, September 14, 2016, in Waikola, Hawaii.
Disclaimer: The opinions contained in this review are solely those of the authors and do not reflect the opinions or positions of the National Institute of General Medical Sciences, the National Institutes of Health, the U.S. Public Health Service, and the Department of Health and Human Services.
Contributor Information
Frederick A. Moore, University of Florida.
Ernest E. Moore, University of Colorado.
Timothy R. Billiar, University of Pittsburg.
Yoram Vodovotz, University of Pittsburg.
Anirban Banerjee, University of Colorado.
Lyle L. Moldawer, University of Florida.
References
- 1.Coimbra R, Kozar RA, Smith JW, Hauser CJ, Moore FA, Bailey GA, Valadka A, Jurkovich GJ, Jenkins GH, Davis KA, et al. The Coalition for National Trauma Research (CNTR) Supports the Call for a National Trauma Research Action Plan. J Trauma Acute Care Surg. 2017;82(3):637–45. doi: 10.1097/TA.0000000000001353. [DOI] [PubMed] [Google Scholar]
- 2.Department of Veteran Affairs. America’s wars: fact sheets. 2016 Office of Public Affairs May. [Google Scholar]
- 3.National Highway Traffic Safety Administration. Traffic safety facts: research notes. DOT HS 812 318. 2016 [Google Scholar]
- 4.Sauaia A, Moore EE, Johnson JL, Chin TL, Banerjee A, Sperry JL, Maier RV, Burlew CC. Temporal trends of postinjury multiple-organ failure: still resource intensive, morbid, and lethal. J Trauma Acute Care Surg. 2014;76(3):582–92. doi: 10.1097/TA.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012;72:1491–501. doi: 10.1097/TA.0b013e318256e000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eiseman B, Beart R, Norton L. Multiple organ failure. Surg Gynecol Obstet. 1977;144(3):323–6. [PubMed] [Google Scholar]
- 7.Christou NV, McLean AP, Meakins JL. Host defense in blunt trauma: interrelationships of kinetics of anergy and depressed neutrophil function, nutritional status, and sepsis. J Trauma. 1980;20(10):833–41. doi: 10.1097/00005373-198010000-00003. [DOI] [PubMed] [Google Scholar]
- 8.McLoughlin GA, Wu AV, Saporoschetz I, Nimberg R, Mannick JA. Correlation between anergy and a circulating immunosuppressive factor following major surgical trauma. Ann Surg. 1979;190(3):297–304. doi: 10.1097/00000658-197909000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Border JR, Chenier R, McManamy RH, La Duca J, Seibel R, Birkhahn R, Yu L. Multiple systems organ failure: muscle fuel deficit with visceral protein malnutrition. Surg Clin North Am. 1976;56(5):1147–67. doi: 10.1016/s0039-6109(16)41035-2. [DOI] [PubMed] [Google Scholar]
- 10.Askanazi J, Carpentier YA, Elwyn DH, Nordenström J, Jeevanandam M, Rosenbaum SH, Gump FE, Kinney JM. Influence of total parenteral nutrition on fuel utilization in injury and sepsis. Ann Surg. 1980;191(1):40–6. doi: 10.1097/00000658-198001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watters JM, Bessey PQ, Dinarello CA, Wolff SM, Wilmore DW. Both inflammatory and endocrine mediators stimulate host responses to sepsis. Arch Surg. 1986;121(2):179–90. doi: 10.1001/archsurg.1986.01400020065008. [DOI] [PubMed] [Google Scholar]
- 12.Cerra FB, Siegel JH, Coleman B, Border JR, McMenamy RR. Septic autocannibalism. A failure of exogenous nutritional support. Ann Surg. 1980;192(4):570–80. doi: 10.1097/00000658-198010000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinarello CA. Historical insights into cytokines. Eur J Immunol. 2007 Nov;37(Suppl 1):S34–45. doi: 10.1002/eji.200737772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tracey KJ. Cachectin/tumor necrosis factor mediates changes of skeletal muscle plasma membrane potential. J Exp Med. 1986;164(4):1368–73. doi: 10.1084/jem.164.4.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. J Exp Med. 1987;330(6149):662–4. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 16.Daly JM, Dudrick SJ, Copeland EM. Effects of protein depletion and repletion on cell-mediated immunity in experimental animals. Ann Surg. 1978 Dec;188(6):791–6. doi: 10.1097/00000658-197812000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander JW, MacMillan BG, Stinett JD, Ogle CK, Bozian RC, Fischer JE, Oakes JB, Morris MJ, Krummel R. Beneficial effects of aggressive protein feeding in severely burned children. Ann Surg. 1980;192:505–7. doi: 10.1097/00000658-198010000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore FA, Feliciano DV, Andrassy RJ, McArdle AH, Booth FV, Morgenstein-Wagner TB, Kellum JM, Welling RE, Moore EE. Early Enteral Feeding, Compared with Parenteral, Reduces Postoperative Septic Complications:The Results of a Meta-analysis. Ann Surg. 1992;216(2):172–83. doi: 10.1097/00000658-199208000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrico CJ, Meakins JL, Marshall JC, Fry D, Maier RV. Multiple-organ-failure syndrome: the gastrointestinal tract. The “motor” of MOF. Arch Surg. 1986;121(2):196–208. doi: 10.1001/archsurg.1986.01400020082010. [DOI] [PubMed] [Google Scholar]
- 20.Hassoun HH, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Postinjury Multiple Organ Failure:The Role of the Gut. Shock. 2001;15(1):1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 21.Faist E, Baue AE, Dittmer H, Heberer G. Multiple organ failure in polytrauma patients. J Trauma. 1983;23(9):775–87. doi: 10.1097/00005373-198309000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995;75(2):257–77. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 23.Moore FA, Moore EE, Jones TN, McCroskey BL, Peterson VM. TEN versus TPN Following Major Abdominal Trauma – Reduced Septic Morbidity. J Trauma. 1989;29(7):916–23. doi: 10.1097/00005373-198907000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Deitch EA, Rutan R, Waymack JP. Trauma, shock, and gut translocation. New Horiz. 1996;4(2):289–99. [PubMed] [Google Scholar]
- 25.Moore EE, Moore FA, Franciose RJ, Kim FJ, Biffl WL, Banerjee A. The Postischemic Gut Serves As A Priming Bed for Circulating Neutrophils That Provoke Multiple Organ Failure. J Trauma. 1994;37(6):881–7. doi: 10.1097/00005373-199412000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Moore FA, Moore EE, Poggetti R, McAnena OJ, Peterson VM, Abernathy CM, Parsons PE. Gut bacterial translocation via the portal vein: a clinical perspective with major torso trauma. J Trauma. 1991;31(5):629–36. doi: 10.1097/00005373-199105000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Moore FA, Haenel JB, Moore EE, Whitehill TA. Incommensurate Oxygen Consumption in Response To Maximal Oxygen Availability Predicts Postinjury Multiple Organ Failure. J Trauma. 1992;33(1):58–66. doi: 10.1097/00005373-199207000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Matzinger P. An innate sense of danger. Semin Immunol. 1998 Oct;10(5):399–415. doi: 10.1006/smim.1998.0143. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007 Jan;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 30.Koike K, Moore FA, Moore EE, Poggetti RS, Tudor RM, Banerjee A. Endotoxin After Gut Ischemia/Reperfusion Causes Irreversible Lung Injury. J Surg Res. 1992;52:656–62. doi: 10.1016/0022-4804(92)90145-p. [DOI] [PubMed] [Google Scholar]
- 31.Botha AJ, Moore FA, Moore EE, Fontes B, Banerjee A, Peterson VM. Postinjury Neutrophil Priming and Activation States: Therapeutic Challenges. Shock. 1995;3(3):157–66. doi: 10.1097/00024382-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Botha AJ, Moore FA, Moore EE, Kim FJ, Banerjee A, Peterson VM. Postinjury Neutrophil Priming and Activation:An Early Vulnerable Window. Surgery. 1995;118:358–65. doi: 10.1016/s0039-6060(05)80345-9. [DOI] [PubMed] [Google Scholar]
- 33.Moore FA, Moore EE, Sauaia A. Blood Transfusion: An Independent Risk Factor for Postinjury Multiple Organ Failure. Arch Surg. 1997;132(6):620–5. [PubMed] [Google Scholar]
- 34.Silliman CS, Moore EE, Johnson JL, Gonzalez RJ, Biffl WL. Transfusion of the injured patient: Proceed with caution. Shock. 2004;21:291–9. doi: 10.1097/00024382-200404000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Biffl WL, Moore EE, Offner PJ, Ciesla DJ, Gonzalez RJ, Silliman CC. Plasma from aged stored red blood cells delays neutrophil apoptosis and primes for cytotoxicity: abrogation by poststorage washing but not prestorage leukoreduction. J Trauma. 2001;50(3):426–31. doi: 10.1097/00005373-200103000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Johnson JL, Moore EE, Gonzalez RJ, Fedel N, Partrick DA, Silliman CC. Alteration of the postinjury hyperinflammatory response by means of resuscitation with a red cell substitute. J Trauma. 2003;54(1):133–9. doi: 10.1097/00005373-200301000-00016. [DOI] [PubMed] [Google Scholar]
- 37.Moore EE, Moore FA, Fabian TC, Bernard AC, Fulda GJ, Hoyt DB, Duane TM, Weireter LJ, Jr, Gomez GA, Cipolle MD, et al. Human Polymerized Hemoglobin for the Treatment of Hemorrhagic Shock When Blood is Unavailable: The USA Multicenter Trial. J Am Coll Surg. 2009;208(1):1–13. doi: 10.1016/j.jamcollsurg.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 38.Balogh Z, McKinley BA, Cox CS, Allen SJ, Cocanour CS, Kozar RA, Moore EE, Miller CC, Weisbrodt NW, Moore FA. Abdominal Compartment Syndrome: The Cause or Effect of Postinjury Multiple Organ Failure. Shock. 2003;20(6):483–92. doi: 10.1097/01.shk.0000093346.68755.43. [DOI] [PubMed] [Google Scholar]
- 39.Balogh Z, McKinley BA, Cocanour CS, Kozar RA, Valdivia A, Sailors RM, Moore FA. Supra-normal Trauma Resuscitation Causes More Cases of Abdominal Compartment Syndrome. Arch Surg. 2003;138(6):637–43. doi: 10.1001/archsurg.138.6.637. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez EA, Moore FA, Holcomb JB, Miller CC, Kozar RA, Todd SR, Cocanour CS, Balldin BC, McKinley BA. Fresh Frozen Plasma Should Be Given Earlier to Patients Requiring Massive Transfusion. J Trauma. 2007;62(1):112–19. doi: 10.1097/01.ta.0000250497.08101.8b. [DOI] [PubMed] [Google Scholar]
- 41.Garner GB, Ware DN, Cocanour CS, Duke JH, McKinley BA, Kozar RA, Moore FA. Vacuum-Assisted Wound Closure Provides Early Fascial Reapproximation in Trauma Patients With Open Abdomens. Am J Surg. 2001;182(6):630–8. doi: 10.1016/s0002-9610(01)00786-3. [DOI] [PubMed] [Google Scholar]
- 42.Geller DA, Lowenstein CJ, Shapiro RA, Nussler AK, Di Silvio M, Wang SC, Nakayama DK, Simmons RL, Snyder SH, Billiar TR. Molecular cloning and expression of inducible nitric oxide synthase from human hepatocytes. Proc Natl Acad Sci USA. 1993;90(8):3491–5. doi: 10.1073/pnas.90.8.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly E, Shah NS, Morgan NN, Watkins SC, Peitzman AB, Billiar TR. Physiologic and molecular characterization of the role of nitric oxide in hemorrhagic shock: Evidence that type II nitric oxide synthase does not regulate vascular decompensation. Shock. 1997;7(3):157–63. doi: 10.1097/00024382-199703000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Hierholzer C, Harbrecht B, Menezes JM, Kane J, MacMicking J, Nathan CF, Peitzman AB, Billiar TR, Tweardy DJ. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med. 1998;187(6):917–28. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hierholzer C, Menezes JM, Ungeheuer A, Billiar TR, Tweardy DJ, Harbrecht BG. A nitric oxide scavenger protects against pulmonary inflammation following hemorrhagic shock. Shock. 2002;17(2):98–103. doi: 10.1097/00024382-200202000-00003. [DOI] [PubMed] [Google Scholar]
- 46.Levy RM1, Prince JM, Yang R, Mollen KP, Liao H, Watson GA, Fink MP, Vodovotz Y, Billiar TR. Systemic inflammation and remote organ damage following bilateral femur fracture requires toll-like receptor 4. Am J Physiol Regulatory Integrative Comp Physiol. 2006;291:970–6. doi: 10.1152/ajpregu.00793.2005. [DOI] [PubMed] [Google Scholar]
- 47.Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: Toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26(5):430–7. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- 48.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages: role in hemorrhage primed lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;290:738–46. doi: 10.1152/ajplung.00280.2005. [DOI] [PubMed] [Google Scholar]
- 49.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, Gallowitsch-Puerta M, Yang L, Yang H, Tracey KJ, Harbrecht BG, Billiar TR, Fink MP. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12(4):105–14. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, Mi Q, Barclay D, Zuckerbraun B, Peitzman AB, Sperry J, Billiar TR. Temporal Patterns of Circulating Inflammation Biomarker Networks Differentiate Susceptibility to Nosocomial Infection Following Blunt Trauma in Humans. Ann Surg. 2016;263(1):191–8. doi: 10.1097/SLA.0000000000001001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury Multiple Organ Failure: A Bimodal Phenomenon. J Trauma. 1996;40(4):501–12. doi: 10.1097/00005373-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 52.MAdib-Conquy M, Cavaillon JM. Compensatory anti-inflammatory response syndrome. Thromb Haemost. 2009 Jan;101(1):36–47. [PubMed] [Google Scholar]
- 53.Moore FA, Peterson VM, Jones TN, Moore EE. Inadequate granulopoiesis after major trauma: a hematopoietic regulatory paradox. Surgery. 1990;108:667–75. [PubMed] [Google Scholar]
- 54.Rogy MA, Coyle SM, Oldenburg HS, Rock CS, Barie PS, Van Zee KJ, Smith CG, Moldawer LL, Lowry SF. Persistently elevated soluble tumor necrosis factor receptor and interleukin-1 receptor antagonist levels in critically ill patients. J Am Coll Surg. 1994;178(2):132–8. [PubMed] [Google Scholar]
- 55.Fong Y, Tracey KJ, Moldawer LL, Hesse DG, Manogue KB, Kenney JS, Lee AT, Kuo GC, Allison AC, Lowry SF, et al. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170(5):1627–33. doi: 10.1084/jem.170.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS) Ann Intern Med. 1996;125:680–7. doi: 10.7326/0003-4819-125-8-199610150-00009. [DOI] [PubMed] [Google Scholar]
- 57.Mannick JA, Rodrick ML, Lederer JA. The immunologic response to injury. J Am Coll Surg. 2001;3:237–44. doi: 10.1016/s1072-7515(01)01011-0. [DOI] [PubMed] [Google Scholar]
- 58.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25(8):1298–307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 59.Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6(Suppl 1):S27–38. [PubMed] [Google Scholar]
- 60.Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, Lepape A. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. 2003;31(7):2068–71. doi: 10.1097/01.CCM.0000069345.78884.0F. [DOI] [PubMed] [Google Scholar]
- 61.De Waal Malefyt R, Haanen J, Spits H, Roncarolo MG, te Velde A, Figdor C, Johnson K, Kastelein R, Yssel H, de Vries JE. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174:915–24. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204(6):1463–74. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moore EE. Splanchnic hypoperfusion provokes acute lung injury via a 5-lipoxygenase-dependent mechanism. Am J Surg. 2010;200(6):681–9. doi: 10.1016/j.amjsurg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Senthil M, Brown M, Xu DZ, Lu Q, Feketeova E, Deitch EA. Gut-lymph hypothesis of systemic inflammatory response syndrome/multiple-organ dysfunction syndrome: validating studies in a porcine model. J Trauma. 2006 May;60(5):958–65. doi: 10.1097/01.ta.0000215500.00018.47. [DOI] [PubMed] [Google Scholar]
- 65.Gonzalez RJ, Moore EE, Ciesla DJ, Neto JR, Biffl WL, Silliman CC. Hyperosmolarity abrogates neutrophil cytotoxicity provoked by post-shock mesenteric lymph. Shock. 2002 Jul;18(1):29–32. doi: 10.1097/00024382-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 66.Gonzalez EA, Kozar RA, Suliburk JW, Weisbrodt NW, Mercer DW, Moore FA. Conventional Dose Hypertonic Saline Provides Optimal Gut Protection and Limits Remote Organ Injury After Gut Ischemia Reperfusion. J Trauma. 2006;61(1):66–74. doi: 10.1097/01.ta.0000224190.65542.e2. [DOI] [PubMed] [Google Scholar]
- 67.Hunter RL, Luo AZ, Zhang R, Kozar RA, Moore FA. Poloxamer 188 Inhibition of Ischemia/ Reperfusion Injury: Evidence for A Novel Anti-Adhesive Mechanism. Ann Clin Lab Sci. 2010;40(2):115–25. [PubMed] [Google Scholar]
- 68.Moore FA, McKinley BA, Moore EE. The next generation in shock resuscitation. Lancet. 2004;363(9425):1988–96. doi: 10.1016/S0140-6736(04)16415-5. [DOI] [PubMed] [Google Scholar]
- 69.Moore-Olufemi SD, Xue H, Allen SJ, Moore FA, Stewart RH, Laine GA, Cox CS., Jr Inhibition of Intestinal Transit by Resuscitation-Induced Gut Edema Is Reversed by L-NIL. J Surg Res. 2005;129:1–5. doi: 10.1016/j.jss.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 70.Türler A, Kalff JC, Moore BA, Hoffman RA, Billiar TR, Simmons RL, Bauer AJ. Leukocyte-derived inducible nitric oxide synthase mediates murine postoperative ileus. Ann Surg. 2006;244(2):220–9. doi: 10.1097/01.sla.0000229963.37544.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krezalek MA1, DeFazio J, Zaborina O, Zaborin A. The Shift of an Intestinal “Microbiome” to a “Pathobiome” Governs the Course and Outcome of Sepsis Following Surgical Injury. Shock. 2016;45(5):475–82. doi: 10.1097/SHK.0000000000000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang W, Gomez FE, Lan J, Sano Y, Ueno C, Kudsk KA. Parenteral nutrition impairs gut-associated lymphoid tissue and mucosal immunity by reducing lymphotoxin Beta receptor expression. Ann Surg. 2006;244:392–9. doi: 10.1097/01.sla.0000234797.42935.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moore FA. Effects of Immune Enhancing Diets on Infectious Morbidity and Multiple Organ Failure. JPEN. J Parenter Enteral Nutr. 2001;25(2Suppl):S36–42. doi: 10.1177/014860710102500209. [DOI] [PubMed] [Google Scholar]
- 74.Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, Barnett C, Biffl WL, Burlew CC, Johnson JL, Sauaia A. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252(3):434–44. doi: 10.1097/SLA.0b013e3181f09191. [DOI] [PubMed] [Google Scholar]
- 75.Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, Banerjee A, Sauaia A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014 Dec;77(6):811–7. doi: 10.1097/TA.0000000000000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, Sauaia A, Cotton BA. Acute Fibrinolysis Shutdown after Injury Occurs Frequently and Increases Mortality: A Multicenter Evaluation of 2,540 Severely Injured Patients. J Am Coll Surg. 2016 Apr;222(4):347–55. doi: 10.1016/j.jamcollsurg.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moore HB, Moore EE, Lawson PJ, Gonzalez E, Fragoso M, Morton AP, Gamboni F, Chapman MP, Sauaia A, Banerjee A, Silliman CC. Fibrinolysis shutdown phenotype masks changes in rodent coagulation in tissue injury versus hemorrhagic shock. Surgery. 2015;158(2):386–92. doi: 10.1016/j.surg.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moore HB, Moore EE, Chapman MP, Gonzalez E, Slaughter AL, Morton AP, D’Alessandro A, Hansen KC, Sauaia A, Banerjee A, Silliman CC. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. J Thromb Haemost. 2015;13(10):1878–87. doi: 10.1111/jth.13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moore EE, Moore HB, Gonzalez E, Sauaia A, Banerjee A, Silliman CC. Rationale for the selective administration of tranexamic acid to inhibit fibrinolysis in the severely injured patient. Transfusion. 2016;56(Suppl 2):S110–4. doi: 10.1111/trf.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tompkins RG. Genomics of injury: The Glue Grant experience. J Trauma Acute Care Surg. 2015;78(4):671–86. doi: 10.1097/TA.0000000000000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al. A genomic storm in critically injured humans. J Exp Med. 2011 Dec;208(13):19. 2581–90. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013 Feb;110(9):26. 3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cuenca AG, Gentile LF, Lopez MC, Ungaro R, Liu H, Xiao W, Seok J, Mindrinos MN, Ang D, Baslanti TO, Bihorac A, et al. Development of a genomic metric that can be rapidly used to predict clinical outcome in severely injured trauma patients. Crit Care Med. 2013 May;41(5):1175–85. doi: 10.1097/CCM.0b013e318277131c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013 Feb;110(9):26. 3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shay T, Lederer JA, Benoist C. Genomic responses to inflammation in mouse models mimic humans: we concur, apples to oranges comparisons won’t do. Proc Natl Acad Sci U S A. 2015 Jan 27;112(4) doi: 10.1073/pnas.1416629111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Cuenca AG, Ungaro R, Baslanti TO, McKinley BA, Bihorac A, et al. A better understanding of why murine models of trauma do not recapitulate the human syndrome. Crit Care Med. 2014;42(6):1406–13. doi: 10.1097/CCM.0000000000000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cuschieri J, Johnson JL, Sperry J, West MA, Moore EE, Minei JP, Bankey PE, Nathens AB, Cuenca AG, Efron PA, et al. Benchmarking Outcomes in the Critically Injured Trauma Patient and the Effect of Implementing Standard Operating Procedures. Ann Surg. 2012;255:993–9. doi: 10.1097/SLA.0b013e31824f1ebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McKinley BA, Moore LJ, Sucher JF, Todd SR, Turner KL, Valdivia A, Sailors RM, Moore FA. Computer Protocol Facilitates Evidence-Based Care of Sepsis in the Surgical Intensive Care Unit. J Trauma. 2011;70(5):1153–67. doi: 10.1097/TA.0b013e31821598e9. [DOI] [PubMed] [Google Scholar]
- 89.Sauaia A, Moore EE, Johnson JL, Chin TL, Banerjee A, Sperry JL, Maier RV, Cothren Burlew C. Temporal trends of postinjury multiple-organ failure: Still resource intensive, morbid, and lethal. J Trauma Acute Care Surg. 2014;76(3):582–93. doi: 10.1097/TA.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ochoa JB, Bernard AC, O’Brien WE, Griffen MM, Maley ME, Rockich AK, Tsuei BJ, Boulanger BR, Kearney PA, Morris SM., Jr Arginase I expression and activity in human mononuclear cells after injury. Ann Surg. 2001;233(3):393–9. doi: 10.1097/00000658-200103000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17:281–92. doi: 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mathias B, Delmas AL, Ozrazgat-Baslanti T, Szpila BE, Mohr AM, Moore FA, Brakenridge SC, Brumback BA, Moldawer LL, Efron PA. Human Myeloid-derived Suppressor Cells are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann Surg. 2016 May;:9. doi: 10.1097/SLA.0000000000001783. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.An GV Y. Translational Systems Biology: Concepts and Practice for the Future of Biomedical Research. New York, NY: Elsevier; 2014. [Google Scholar]
- 94.Namas RA, Mi Q, Namas R, Almahmoud K, Zaaqoq A, Abdul Malak O, Azhar N, Day J, Abboud A, Zamora R, et al. Insights into the role of chemokines, damage-associated molecular patterns, and lymphocyte-derived mediators from computational models of trauma-induced inflammation. Antiox Redox Signal. 2015;10:1370–87. doi: 10.1089/ars.2015.6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vodovotz Y, Billiar TR. In Silico Modeling: Methods and applications to trauma and sepsis. Crit Care Med. 2013;41:2008–14. doi: 10.1097/CCM.0b013e31829a6eb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mi Q, Constantine G, Ziraldo C, Solovyev A, Torres A, Namas R, Bentley T, Billiar TR, Zamora R, Puyana JC, et al. A dynamic view of trauma/hemorrhage-induced inflammation in mice: Principal drivers and networks. PloS One. 2011;6:e19424. doi: 10.1371/journal.pone.0019424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Namas R, Almahmoud K, Mi Q, Ghuma A, Namas R, Zaaqoq A, Sperry J, Zamora R, Billiar TR, Vodovotz Y. Individual-specific principal component analysis of circulating inflammatory mediators predicts early organ dysfunction in trauma patients. J Crit Care. 2016;36:146–53. doi: 10.1016/j.jcrc.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, Mi Q, Barclay D, Zuckerbraun B, Peitzman AB, et al. Temporal Patterns of Circulating Inflammation Biomarker Networks Differentiate Susceptibility to Nosocomial Infection Following Blunt Trauma in Humans. Ann Surg. 2016;263:191–8. doi: 10.1097/SLA.0000000000001001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Almahmoud K, Namas RA, Abdul-Malak O, Zaaqoq AM, Zamora R, Zuckerbraun BS, Sperry J, Peitzman AB, Billiar TR, Vodovotz Y. Impact of Injury Severity on Dynamic Inflammation Networks Following Blunt Trauma. Shock. 2015;44:105–9. doi: 10.1097/SHK.0000000000000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abboud A, Mi Q, Puccio A, Okonkwo D, Buliga M, Constantine G, Vodovotz Y. Inflammation Following Traumatic Brain Injury in Humans: Insights from Data-Driven and Mechanistic Models into Survival and Death. Front Pharmacol. 2016;7:342–8. doi: 10.3389/fphar.2016.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]