

Abstract
In centralized immune monitoring for a multi-center allergen immunotherapy trial, we observed frequent loss of CD4+ T cell integrity following staining of cultured PBMCs with our regulatory T cell flow cytometry panel. Samples were marked by a loss of total cellular events, altered scatter properties, and reduced CD3+CD4+ events. This occurred only in samples that were stained with Foxp3 and were therefore treated with Foxp3 fixation-permeabilization buffer. We identified granulocyte contamination in samples associated with a loss of integrity, and went on to test the impact of granulocyte depletion on day-old blood samples. Granulocyte depletion prevented loss of cell integrity and CD3+CD4+ events, and reduced variability in detection of Foxp3+ cells. Addition of purified neutrophils back to PBMCs altered scatter properties and detection of CD4+ T cells. Implementation of a granulocyte depletion step in our standard operating protocols has reduced assay failure due to loss of sample integrity from 31% to 0%. Routine incorporation of a granulocyte depletion step during PBMC isolation is recommended prior to downstream immune monitoring in blood with next-day processing.
Introduction
Immune monitoring in recent multi-center intervention trials has focused on the role of regulatory T cells in immunotherapy-induced tolerance. A central laboratory for immune monitoring has numerous advantages, primarily related to reducing assay variability by removing operator and equipment variability (Butterfield et al., 2011). The main disadvantage is the requirement to uniformly ship blood samples from geographically dispersed clinical trial sites.
In the Consortium of Food Allergy Research (CoFAR), we have implemented a temperature-controlled GreenBox™ shipping system that maintains temperature between 20–30 °C, as confirmed by in-shipment temperature monitoring. Yield of PBMCs and functional responses to allergen stimulation were well maintained after shipping. In two multi-center trials conducted through CoFAR, we performed short-term stimulation of PBMCs with allergen for 6 or 18h, followed by flow cytometric identification of allergen-responsive T cells through detection of upregulation of the activation marker CD154 (CD40L). In samples analyzed as part of the primary analysis for one trial (Jones et al., 2017), we observed an overall failure rate of 31% in our regulatory T cell panel. Assay failure was due to low event count during acquisition, and these samples were characterized by altered scatter properties, and a low frequency of CD3+CD4+ T cells. In contrast, the effector cell panel, which did not incorporate Foxp3 staining, had a failure rate of 11%, and loss of sample integrity was not a factor in assay failure. Cells used for the two panels were derived from the same samples, were in culture for the same length of time, and used the same antibodies for detection of CD3+CD4+ T cells. Samples differed in the use of a commercial Foxp3 Fixation/Permeabilization Buffer in the regulatory T cell panel, which is necessary for Foxp3 staining. Here we outline the steps we took to identify and address contaminating granulocytes as the cause of assay failure in our regulatory panel.
Methods
Human subjects
Informed consent was obtained from all subjects or parents/guardians. Blood samples were obtained from a clinical trial conducted by the Consortium of Food Allergy Research (CoFAR) investigating epicutaneous immunotherapy for peanut allergy (COFAR6, ClinicalTrials.gov identifier NCT01904604). All procedures were approved by the Institutional Review Boards of 5 participating CoFAR institutions. Blood from healthy adult volunteers was obtained from the Icahn School of Medicine at Mount Sinai through an IRB-approved protocol.
Shipping
Blood was collected in sodium heparin green-top vacutainer tubes at the clinical sites, and shipped for next-morning delivery in temperature-controlled GreenBox™ shipping system (ThermoSafe, Arlington Heights, IL), with temperature logger included and checked with each shipment. PBMCs were isolated immediately on shipment arrival.
PBMC Isolation
Conventional PBMC Isolation
Plasma was collected by centrifugation (450 × g, 15 minutes, acceleration and brakes off, at room temperature). The remaining blood cells were diluted with 3 × volume of phosphate buffered saline (PBS) (Gibco, Grand Island, NY), underlaid with Ficoll-Paque PLUS (GE Healthcare Bio-Sciences, Pittsburgh, PA) and centrifuged at 400 × g, 30 minutes, acceleration and brakes off, at room temperature. Centrifugation conditions were according to Ficoll-Paque PLUS manufacturer’s recommendations. Cells were collected from the interface and washed with PBS. Cells were resuspended in Aim V media (ThermoFisher, Grand Island, NY), and washed once in Aim V.
Granulocyte Depletion
Plasma collection was performed as above. Plasma volume was replaced by PBS + 2% fetal bovine serum (Gibco, Grand Island, NY). RosetteSep Human Granulocyte Depletion cocktail (StemCell, Vancouver, Canada) was added at 5uL/mL of whole blood, and incubated for 20 min at room temperature. After incubation, blood was diluted with 3 × volume of PBS + 2% FBS and underlaid with Ficoll-Paque PLUS. Blood was centrifuged at 1200 × g, 20 minutes, acceleration and brakes off, at room temperature. Centrifugation conditions were according to manufacturer’s recommendations for the granulocyte depletion kit. Interface cells were collected and diluted with PBS+2%FBS to wash cells. Cells were resuspended in AimV media (ThermoFisher), and washed once in Aim V.
Neutrophil purification
Neutrophils were purified from whole blood by negative selection (StemCell), and co-cultured with granulocyte-depleted PBMCs at a ratio of 1:10. Purity of neutrophils and PBMCs was verified by staining as described below.
Cell subset staining to identify contaminating populations
To determine if there was a different cell composition in PBMC samples that passed or failed quality control measures, we stained an aliquot of PBMCs from 11 sequential CoFAR6 samples immediately after PBMC isolation and prior to knowledge of assay results for assessment of cell subset composition by flow cytometry prior to culture. PBMCs were stained with a cocktail of 12 antibodies, shown in Table 1 to identify cell subsets in samples that were deemed to pass or fail quality control. Supplementary Figure 1 shows gating strategy to identify cell subsets, which was based on a publication by Hensley-McBain et al (Hensley-McBain et al., 2014).
Table 1.
Staining panel for determination of PBMC cell subsets
| Target | Fluorochrome | Clone |
|---|---|---|
| CD45 | Pacific Orange | H130 |
| CD3 | AF700 | UCHT1 |
| CD4 | PE-CF594 | rpa-t4 |
| CD8 | APC | SK1 |
| CD19 | PE | HIB19 |
| CD56 | BV605 | HCD56 |
| CD14 | PE-Cy7 | HCD14 |
| CD16 | APC-H7 | 3G8 |
| HLADR | AF488 | LN3 |
| CD123 | BV650 | 6H6 |
| CD11c | PacBlue | 3.9 |
| BDCA2 | PerCP/Cy5.5 | 201A |
To quantify neutrophils following granulocyte depletion, cells were stained with CD45 Pacific Orange (Invitrogen, Carlsbad, CA) and CD16 APC-H7 (Biolegend, San Diego, CA) and cells identified by scatter and CD16 staining.
PBMC culture and stimulation
PBMCs were plated with Aim V + 2.5% autologous plasma at a concentration of 4 million cells per ml per well of a 24-well cell culture plate. PBMCs were stimulated with 5 μL/mL anti-CD3/CD28 beads (Invitrogen) for 18 hours. GolgiPlug (BD Biosciences, San Jose, CA) was added 4 hours prior to harvesting.
Staining and Flow Cytometry
Surface Staining: Harvested PBMCs were stained with fixable Live/Dead stain (Invitrogen) in PBS for 30 min on ice. Cells were washed and stained with surface markers in FACS buffer (PBS/1%BSA /2mM EDTA) for 30 min on ice.
Fixation and staining of intracellular markers: For the effector panel, cells were washed prior to fixation in 4% PFA for 5 min, 37 C. Cells were washed in FACS buffer, and resuspended in permeabilization buffer (eBioscience, San Diego, CA) for 20 mins at room temperature. Antibodies against intracellular targets were added and cells were incubated for 45 mins, on ice. For the regulatory panel, cells were washed with FACS buffer, and fixed with Foxp3 Fix/Perm buffer (eBioscience, San Diego, CA) for 30 mins, on ice. After washing in permeabilization buffer, cells were stained with antibodies detecting intracellular markers in permeabilization buffer for 45 mins, on ice.
Stained and permeabilized cells were washed twice in permeabilization buffer before resuspension in FACS buffer, and subsequently acquired on an LSR Fortessa (BD Biosciences). Antibodies used for the regulatory panel are shown in Table 2.
Table 2.
Regulatory T cell staining panel
| Target | Fluorochrome | Clone |
|---|---|---|
| Live/Dead | Aqua | |
| CD3 | APC-eFluor780 | SK7 |
| CD4 | BV605 | OKT4 |
| CD25 | BV650 | BC96 |
| CD127 | BV785 | A019D5 |
| CCR4 | PerCP-Cy5.5 | 1G1 |
| CCR6 | PE-Cy7 | 11A9 |
| CCR9 | AlexaFluor488 | 112509 |
| CD154 | PE | 24-31 |
| IFNg | AlexaFluor700 | B27 |
| IL-10 | PE-CF594 | JES3-19F1 |
| FoxP3 | AlexaFluor647 | 206D |
Results and Discussion
Figure 1 shows representative flow cytometry of trial samples stained with our effector and regulatory panels, showing an example of samples that passed and failed quality control (QC). Samples that failed QC were characterized by a marked shift in FSC, and a reduction in cell yield that was particularly marked in the CD3+CD4+ compartment. This phenomenon of altered FSC and reduced yield was only observed in the regulatory panel, that differed from the effector panel primarily in the method of fixation. The quality control cut-off was a frequency of less than 20% CD3+CD4+ T cells. To assess if the cell composition of PBMCs was altered in samples resulting in assay failure, we quantified 14 different blood subsets, and compared the composition among samples that passed or failed QC using multiple T tests with FDR adjustment (5%) for multiple comparisons. Four of 11 samples were classified as QC failures, and these samples were characterized by a marked increase in frequency of neutrophils but not other granulocytes such as eosinophils or basophils (Figure 2) compared to the 7 samples that were classified as QC pass.
Figure 1.
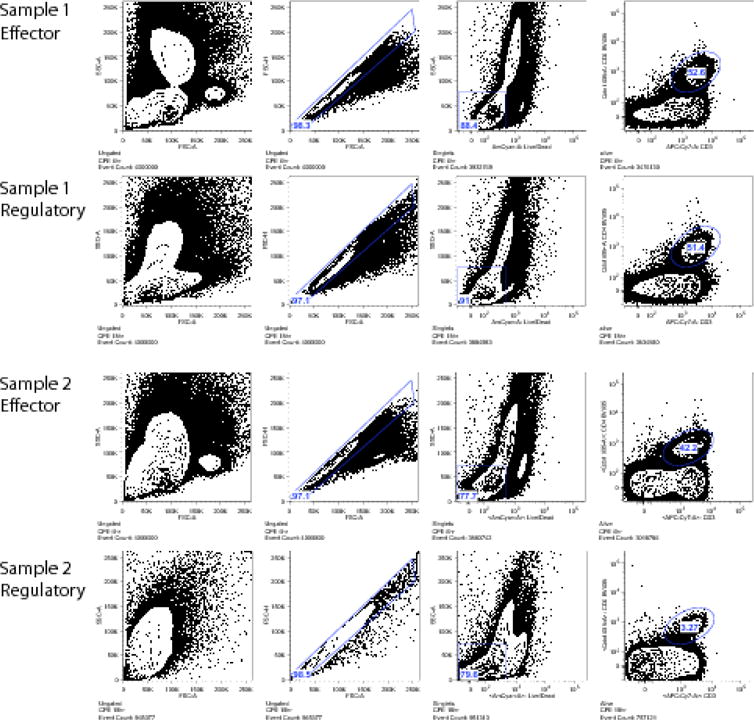
Flow cytometry plots showing representative examples of regulatory panels that passed (Sample 1) or failed (Sample 2) quality control. From left to right: Scatter properties, singlets, live/dead staining, and CD3/CD4 staining. In the regulatory panel of Sample 2, note the leftward shift of forward scatter properties, and the markedly reduced CD3+CD4+ cells as compared to the effector panel, which includes cells isolated and cultured in the same conditions and time frame from the same subject.
Figure 2.
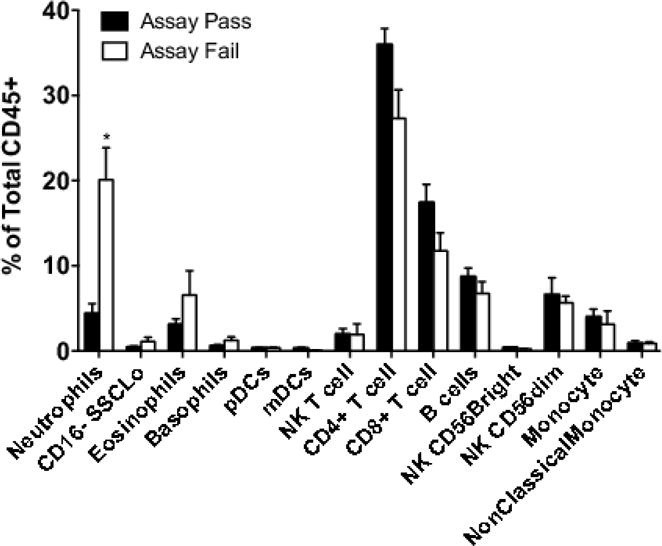
Proportion of cell subsets measured immediately after PBMC isolation in 11 sequential study samples. 4 of 11 samples failed quality control measures at the end of the experimental procedure (“assay fail”) while the remaining 7 passed quality control (“assay pass”).*p=0.0007, T test with FDR correction.
Neutrophil contamination is known to increase in PBMCs isolated from blood that is not immediately used for PBMC isolation (Nicholson et al., 1984). To determine if this granulocyte contamination influenced sample integrity in our regulatory T cell panel, we performed depletion of granulocytes using a CD66b-based depletion system. Blood was obtained from 4 healthy volunteers, and PBMCs isolated 24 hours after blood draw. Following the manufacturer’s instructions, we observed a greater than 99% depletion of contaminating CD16+ neutrophils (Figure 3A). There was a difference in centrifugation speed between our previous protocol and that used for granulocyte depletion (based on manufacturer’s recommendations for Ficoll-Paque Plus vs. Granulocyte Depletion Kit). Higher centrifugation speed alone reduced some granulocyte contamination but was not sufficient to eliminate granulocyte contamination. PBMCs were stimulated for 18h with anti-CD3/CD28 stimulator beads or left unstimulated prior to staining using our regulatory panel protocol. Samples were divided, and an aliquot was acquired immediately after staining and the second aliquot the following day (according to our SOP procedures). We observed a marked loss of CD3+CD4+ T cells without granulocyte depletion, particularly with next-day acquisition (Figure 3B). Frequency of Foxp3+ cells was also significantly reduced in the absence of granulocyte depletion (Figure 3C). In contrast, samples depleted of granulocytes demonstrated stable CD3+CD4+ and Foxp3+ cell frequencies, whether samples were acquired immediately or the next day. We then added back neutrophils to granulocyte-depleted PBMCs (Figure 3D–F). Addition of neutrophils at a ratio of 1:10 (similar to the frequency observed in contaminated PBMC preparations) resulted in a mild shift in forward scatter of the lymphocyte population, and a reduction in detection of CD3+CD4+ cells and Foxp3+ cells. We did not observe complete loss of cell integrity of samples as in our study samples or in PBMCs without granulocyte depletion, suggesting that there may be contribution from additional factors.
Figure 3.
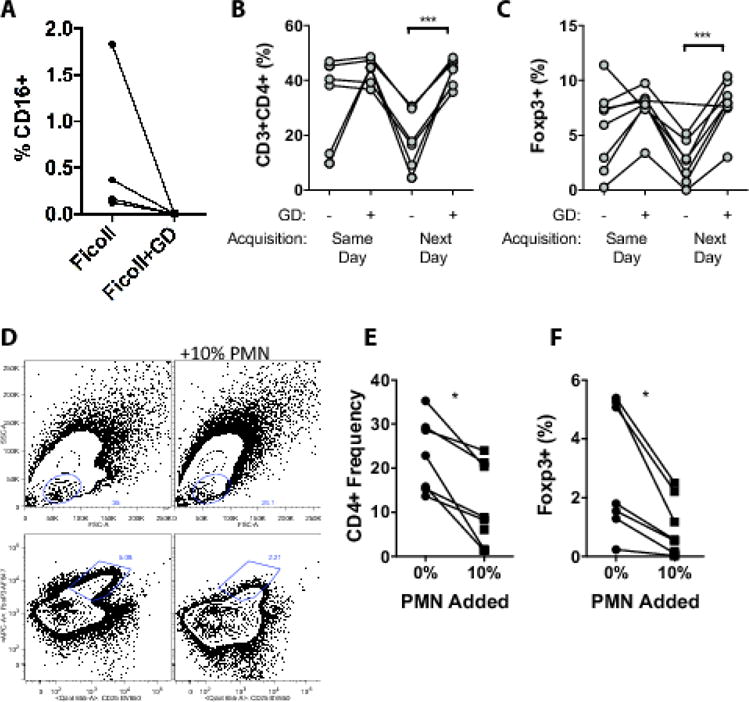
(A) CD16+ granulocyte frequency in PBMCs purified by conventional Ficoll, or with a granulocyte depletion (GD) step. Frequency of CD3+CD4+ T cells (B), or Foxp3+ cells/CD4+ cells (C), in PBMCs acquired immediately after stimulation and staining (day 0) or the following day (day 1), in PBMCs isolated by conventional Ficoll or with granulocyte depletion. (D) Representative plots showing impact of addition of 10% purified PMNs on scatter properties of PBMCs (top row) and Foxp3+CD25+ cells (bottom row) of PBMCs after stimulation. Impact of adding 10% PMNs to PBMCs on CD3+CD4+ frequency (E), and Foxp3 frequency (F). *p<0.05.
Based on these findings, we incorporated a granulocyte depletion step into our standard operating procedures. We performed titrations to determine that we could use 10% of the recommended antibody cocktail to effectively deplete CD16+ granulocytes from our PBMCs (data not shown). Implementation of a granulocyte depletion step reduced our assay failure due to loss of sample integrity from 31% to 0% (Figure 4), a highly significant reduction.
Figure 4.
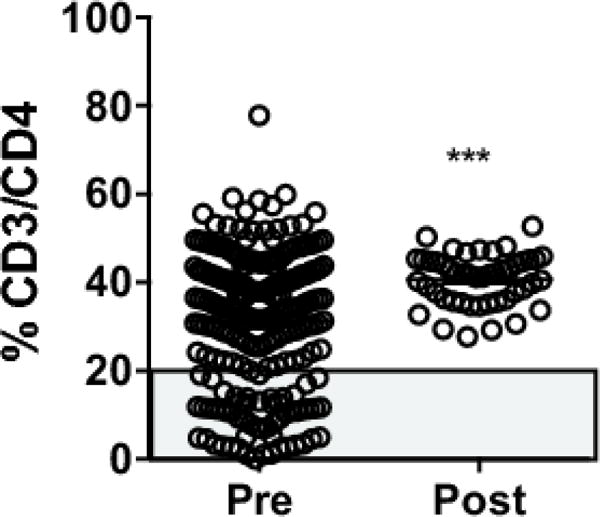
CD3/CD4 frequency in study samples completed before (pre) or after (post) implementation of a granulocyte depletion step in the standard operating protocol. *** p < 0.001 by unpaired T test. Grey box indicates QC failures.
The mechanism by which contaminating granulocytes interfere with sample integrity, and only in the presence of Foxp3 Fixation/Permeabilization buffer, is not clear. The more extensive permeabilization of this buffer, which allows for nuclear staining, is clearly detrimental to sample integrity only in the presence of granulocytes. Limitations of the study include a lack of analysis of the functional impact of neutrophil contamination on cytokine production or activation parameters. However, it has been previously reported that neutrophils can influence T cell activation (Kalyan and Kabelitz, 2014). We have not examined the impact of this contamination on viability or function after cryopreservation, which may also be of interest. However, based on our results we have incorporated granulocyte depletion as a routine standard operating protocol when working with blood samples from clinical studies that are processed the next day. Other approaches, such as the use of CPT tubes to separate PBMCs from granulocytes and red blood cells prior to shipping may be beneficial in eliminating this granulocyte contamination when whole blood is not required for assays.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Butterfield LH, Palucka AK, Britten CM, Dhodapkar MV, Hakansson L, Janetzki S, Kawakami Y, Kleen TO, Lee PP, Maccalli C, Maecker HT, Maino VC, Maio M, Malyguine A, Masucci G, Pawelec G, Potter DM, Rivoltini L, Salazar LG, Schendel DJ, Slingluff CL, Jr, Song W, Stroncek DF, Tahara H, Thurin M, Trinchieri G, van Der Burg SH, Whiteside TL, Wigginton JM, Marincola F, Khleif S, Fox BA, Disis ML. Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers. Clin Cancer Res. 2011;17:3064–76. doi: 10.1158/1078-0432.CCR-10-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley-McBain T, Heit A, De Rosa SC, McElrath MJ, Andersen-Nissen E. Optimization of a whole blood phenotyping assay for enumeration of peripheral blood leukocyte populations in multicenter clinical trials. J Immunol Methods. 2014;411:23–36. doi: 10.1016/j.jim.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Sicherer SH, Burks AW, Leung DY, Lindblad RW, Dawson P, Henning AK, Berin MC, Chiang D, Vickery BP, Pesek RD, Cho CB, Davidson WF, Plaut M, Sampson HA, Wood RA, Consortium of Food Allergy, R Epicutaneous immunotherapy for the treatment of peanut allergy in children and young adults. The Journal of allergy and clinical immunology. 2017;139:1242–1252 e9. doi: 10.1016/j.jaci.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyan S, Kabelitz D. When neutrophils meet T cells: beginnings of a tumultuous relationship with underappreciated potential. Eur J Immunol. 2014;44:627–33. doi: 10.1002/eji.201344195. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Jones BM, Cross GD, McDougal JS. Comparison of T and B cell analyses on fresh and aged blood. J Immunol Methods. 1984;73:29–40. doi: 10.1016/0022-1759(84)90028-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.