

Abstract
An important goal for improving vaccine and immunotherapy technologies is the ability to provide further control over the specific phenotypes of T cells arising from these agents. Along these lines, frequent administration of rapamycin (Rapa), a small molecule inhibitor of the mammalian target of rapamycin (mTOR), exhibits a striking ability to polarize T cells toward central memory phenotypes (TCM), or to suppress immune function, depending on the concentrations and other signals present during administration. TCM exhibit greater plasticity and proliferative capacity than effector memory T cells (TEFF), and therefore polarizing vaccine-induced T cells toward TCM is an intriguing strategy to enhance T cell expansion and function against pathogens or tumors. Here we combined biodegradable microparticles encapsulating Rapa (Rapa MPs) with vaccines composed of soluble peptide antigens and molecular adjuvants to test if this approach allows polarization of differentiating T cells toward TCM. We show Rapa MPs modulate DC function, enhancing secretion of inflammatory cytokines at very low doses, and suppressing function at high doses. While Rapa MP treatment reduced – but did not stop – T cell proliferation in both CD4+ and CD8+ transgenic T cell co-cultures, the expanding CD8+ T cells differentiated to higher frequencies of TCM at low doses of MP Rapa. Lastly, we show in mice that local delivery of Rapa MPs to lymph nodes during vaccination either suppresses or enhances T cell function in response to melanoma antigens, depending on the dose of drug in the depots. In particular, at low Rapa MP doses, vaccines increased antigen-specific TCM, resulting in enhanced T cell expansion measured during subsequent booster injections over at least 100 days.
Keywords: microparticle and nanoparticle, T cell, immunology, vaccine and immunotherapy, lymph node, rapamycin and mTOR inhibitor
Graphical abstract
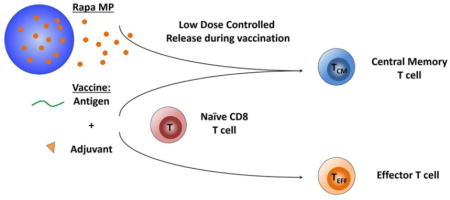
Controlled release of low doses of rapamycin during vaccination enhances T cell function by polarizing differentiating T cells toward central memory function and away from effector function.
INTRODUCTION
Most vaccines amplify immune responses elicited against foreign molecules (“antigens”) associated with a pathogen, exploiting the exquisite specificity the immune system harnesses to destroy pathogens without targeting host cells. Despite the importance of these technologies, one of the arising themes for new vaccines and immunotherapies is the need for approaches that not only generate large responses, but that also program specific functions and phenotypes of these responses.[1, 2] In this study we used peptide antigen and common molecular vaccine adjuvants to study how controlled release of low doses of modulatory drugs during T cell expansion alters immune cell differentiation and enhances immune function in cells and mice.
Our studies are motivated by fascinating recent work revealing that regular, low doses of immunosuppressive drugs can be used to enhance vaccination and immunotherapy by maintaining plasticity – that is, limiting the differentiation – of T cells. Rapamycin (Rapa), and analogs of this drug, are some of the most-well studied along these lines.[3, 4] Rapa has historically played a significant role as an immunosuppressant, but this drug class targets the mTOR pathway, which is intimately involved in metabolism, cell growth, and differentiation.[5, 6] Several seminal studies demonstrate regular, systemic administration of Rapa increases the durability of expanding antigen-specific CD8+ T cells, and in particular, that the phenotypes of these cells are polarized away from effector T cells (TEFF) and toward central memory T cells (TCM).[7, 8] While TEFF are needed to combat pathogenic cells, these cells become less proliferative as they differentiate from naïve T cells. In contrast, TCM are more plastic, exhibiting the highly proliferative capacity needed to rapidly generate large numbers of antigen-specific TEFF. Thus, expanding TCM against a target antigen might provide a route to generate large, durable populations of antigen-specific cells that mount potent responses by maintaining a highly proliferative characteristic.
Gaining control over the processes above to direct immune cell differentiation could have a transformative impact in combating infectious disease, immune dysfunction, and cancer. Cancer, for example, is difficult to treat due to the heterogeneity of disease and the ability of tumors to metastasize, evade and suppress the immune system, and cause relapse.[9] Over the past decade, adoptive cell therapy – in which tumor-primed T cells are transferred to animals or patients – have led to the revelation that TCM are important drivers of anti-tumor immunity.[10] The high proliferation rates of TCM support rapid response against established tumors and improved protection against relapse.[11–14] Unfortunately, as TCM expand, the differentiation to effector memory (TEM) or effector (TEFF) T cells results in reduced numbers of cells and cytokines (e.g., IL-2) to overcome the immunosuppressive tumor environment.[15, 16]
Adoptive cell therapy, cancer vaccination, and checkpoint blockade are some of the most studied emerging technologies in cancer research, and Rapa is also already being implemented clinically as a route to suppress the excess metabolic and proliferative function of cancer cells.[17–19] Newer pre-clinical studies are exploring modulation of low-doses of Rapa and other drugs to polarize T cell function during cancer vaccination in melanoma, Glioblastoma, and thymoma models.[20–23] However, inducing TCM in animals or people with Rapa or other drugs is hindered by short half-lives (t1/2=1–2 hrs), the need for frequent injections (1–5x/day), drug hydrophobicity, and the challenge of co-delivering drug cues with other vaccine components to target immune tissues such as lymph nodes (LNs), where tumor-specific TCM are primed by antigen-presenting cells (APCs).[24] Overcoming these hurdles might enable cancer vaccines that achieve adoptive transfer-like potency without need for isolation, expansion, and reinfusion of cells from a patient.
In LNs, APCs process and present antigens to activate T and B cells. Thus, vaccines must reach LNs to generate antigen-specific responses. Importantly, the specific type of response depends intimately on the soluble factors and surface molecules encountered by immune cells during antigen presentation. This characteristic has created enormous interest in harnessing biomaterials for vaccines against disease and cancer immunotherapies by targeting LNs, controlling delivery kinetics, and minimizing toxicity.[25–28] In this report, we combined common candidate vaccine formulations with polymer particles that slowly release minute doses of Rapa to test if this immunomodulator enhances immune response. We hypothesized that controlled release of low doses of Rapa during vaccination with peptide antigens and molecular adjuvants would moderate co-stimulation by APCs, alter cytokine secretion, and polarize differentiating T cells toward TCM that enhance the size and quality of vaccine response. We show in primary cell co-culture models and in mice that release of Rapa from polymer depots during vaccination preserves plasticity in differentiating T cells, allowing generation of antigen-specific T cells, but biasing these populations toward a central memory phenotype that enhances response in primary cell co-culture models and in mice.
MATERIALS AND METHODS
Peptides
Trp2 peptide (SVYDFFVWL) formulated as a disodium salt and MOG peptide (MEVGWYRSPFSRVVHLYRNGK) were synthesized by Genscript.
Particle synthesis and characterization
PLGA microparticles were synthesized using a double emulsion/solvent evaporation method previously described.[26] Briefly, an organic phase consisting of 80 mg of PLGA (Durect) dissolved in 5 mL dichloromethane was prepared. In samples encapsulating Rapa, polymer solution was added to a vial containing 2 mg of dried drug (LC Laboratories). A primary emulsion was formed by adding 500 μL of water with the organic phase followed by sonication for 30 s at 12 W. This primary emulsion was then added to 40 mL of 2% w/v polyvinyl alcohol (PVA, Sigma) and homogenized for 3 min at 16,000 rpm. Excess organic solvent was evaporated overnight under stirring. Particles formulations were passed through a 40 μm cell strainer and collected via centrifugation (5000 g, 5 min, 4° C). Particles were then washed three times in in 1 mL water, with centrifugation steps (5000 g, 5 min, 4° C) to collect particles in between, and resuspended in water. Particle diameter was determined using laser diffraction (Horiba LA-950). Yield was quantified by drying a known volume of particle suspensions under air, recording the weight, and back-calculating a total mass yield for each batch. To quantify Rapa loading, known masses of dried particles were dissolved in DMSO, and the absorbance at 278 nm was determined using UV/VIS spectrophotometry. Absorbance values were compared with a standard curve of known concentrations of Rapa to calculate drug loading per mass of particles.
Dendritic cell uptake, activation and cytokine secretion
CD11c+ cells were isolated from the spleens of 4–6 week old C57BL/6 mice (The Jackson Laboratory) using magnetic isolation according to the manufacturer’s protocol (Miltenyi). Cells were plated in 96 well plates at 105 cells/well for uptake and activation studies, or at 2.5 x 105 cells/well for studies to analyze cytokine secretion, in RPMI 1640 media (Lonza), supplemented with 10% fetal bovine serum (Corning), 2 mM L-glutamine (Gibco), 55 μM β-mercaptoethanol (Sigma-Aldrich), 1 × non-essential amino acids (Fisher Scientific), 10 mM HEPES (Fisher Scientific), and 1 × Penn/Strep (Gibco). With the exception of indicated control wells, cells were stimulated with lipopolysaccharide (LPS) (Invitrogen) at 1μg/mL; cells were treated with decreasing doses of soluble Rapa, matched-doses of encapsulated Rapa (Rapa MPs), or equivalent masses of empty MPs as a control. Soluble Rapa was dissolved in DMSO, and the final v/v % of DMSO in the well was 0.2%. After 4, 18, or 48 hrs of culture, cells were collected for analysis by flow cytometry and supernatants were collected for analysis of IL-6, IL-12p70 and IFNγ concentrations by ELISA (BD Biosciences). Briefly, cells were collected, washed in FACS buffer (1% bovine serum albumin in 1X PBS), and either resuspended in 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) for analysis of particle uptake by flow cytometry, or blocked with anti-CD16/CD32 (BD Biosciences). After blocking, cells were then stained with anti-CD40, anti-CD80, and anti-CD86 (BD Biosciences) for 20 min at room temperature, washed two more times as above, and resuspended in DAPI for analysis by flow cytometry. All flow cytometry data was collected on a Canto II (BD Biosciences) and analyzed using Flowjo software (Tree Star). In studies to analyze particle uptake by microscopy, CD11c + cells were isolated, as above, and 1 x 106 cells were plated in glass-bottom dishes with No. 1.5 thickness cover slips (MatTek). After 2 hrs of incubation of cells with fluorescent particles, cells were washed twice with PBS, fixed in 4% paraformaldehyde, and washed with PBS and additional three times. Fixed cells were stained with Wheat Germ Agglutinin, Texas Red-X Conjugate (Thermo Fisher) at 5 μg/mL for ten minutes, washed twice with PBS, and resuspended in Hoescht (Thermo Fisher) at 4 μg/mL for imaging.
Transgenic T cell co-culture studies
In order to characterize the effects of Rapa MPs on CD4 and CD8 T cell responses, co-cultures consisting of wild type dendritic cells (DCs) and T cells from transgenic mice were performed. T cells were from 2D2 mice C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J (The Jackson Laboratory) which have CD4+ T cell receptors specific for MOG, and from Trp2-clone 37 mice (National Cancer Institute, National Institutes of Health), which have CD8+ T cell receptors specific for Trp2. Splenic DCs from C57BL/6 mice were first isolated and plated at 105 cells/well, as above. With the exception of indicated controls, DCs were stimulated with 1 μg/mL LPS and either 1 μg/mL Trp2 for co-cultures with T cells from Trp2 mice or 1 μg/mL MOG for co-cultures with T cells from 2D2 mice. After culture for 24 hrs, splenic CD4+ T cells were isolated from 2D2 mice or CD8+ T cells were isolated from Trp2 mice using magnetic isolation according to the manufacturer’s protocol (STEMCELL Technologies). 3 × 105 Trp2 or 2D2 T cells were added to culture with DCs treated with Trp2 or MOG, respectively, and were cultured for an additional 48 hrs. For analysis of Trp2 T cell phenotype, cells from the Trp2 co-culture were collected and washed in FACS buffer, and blocked with anti-CD16/CD32 (BD Biosciences). Cells were then stained with anti-CD8a, anti-CD44 and anti-CD6L (BD Bioscience) for 20 min at room temperature, washed two more times as above, followed by analysis by flow cytometry. To analyze proliferation of T cells in co-cultures, identical experiments were performed as above, except T cells were labeled with carboxyfluorescein succinimidyl ester (CSFE) before addition to culture. T cells were labeled by resuspension in media at 50 × 106 cells/mL followed by incubation in CSFE at a final concentration of 5 μM for 5 min at room temperature and four washes in media. After 48 hrs of co-culture, cells were collected and proliferation was quantified by analysis of CSFE dilution using flow cytometry after staining with fluorescent anti-CD4 or anti-CD8 for 2D2 and Trp2 T cells, respectively.
i.LN. injection
i.LN. injection of C57BL/6 mice was performed as previously described.[26, 29–32] Briefly, the hair was removed from mice using a mild depilatory cream, and then mice were injected subcutaneously (s.c.) at the tail base with a tracer dye (Evan’s Blue). After allowing the tracer dye to drain to inguinal lymph nodes for 16 hr, both inguinal lymph nodes were identified and injected using a 31G insulin needle containing indicated treatments in a volume of 10 μL. Soluble vaccines consisted of two injections of 5 μg CpG with 25 μg Trp2, or 25 μg OVA in mode antigen studies. Soluble vaccines including rapamycin contained either 1 mg of Rapa MPs per injection (high dose) or 0.5 mg of Rapa MPs (low dose) per injection.
Tetramer and memory phenotype analysis
100 μL of peripheral blood was collected from mice by submandibular bleeding, followed by depletion of red blood cells by incubating with 750 μL of ACK lysis buffer. Cells were collected via centrifugation (800 g, 5 min, 4° C) and resuspended in 750 μL of ACK lysis buffer. Following the second round of ACK lysis, cells were blocked by incubation with anti-CD16/CD32, and then stained with a tetramer specific for Trp2 (MBL). Cells were then washed twice with 1% BSA in PBS, stained with fluorescent anti-CD8, anti-CD44 and anti-CD62L, followed by two washes in 1% BSA in PBS. After resuspension in DAPI, tetramer and memory marker expression was analyzed by flow cytometry.
Tumor challenge studies
After treating mice with the indicated vaccines, mice were shaved and injected s.c. at the hind flank with 3 × 105 B16-F10 (ATCC) cells in 100 μL of cold PBS. Mice were then weighed and monitored for tumor growth daily following inoculation. Tumor burden was calculated as the product of two orthogonal diameters. Mice were euthanized according to the IACUC-approved humane endpoints when aggregate tumor burden reached 150 mm2.
Statistical analysis
One-way ANOVA with a Tukey post-test was used to compare three or more groups during in vitro and in vivo studies. Significance for survival studies was carried out with a Log-rank test. T tests were used to compare the two groups for TCM:TEFF ratios. In all cases, analyses were carried out with Graphpad Prism (version 6.02). Error bars represent the mean ± SEM and p values were considered significant as defined by: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
RESULTS
Rapa is encapsulated in PLGA MPs and slowly released over time
To test our hypothesis that low levels of Rapa promote TCM during vaccine delivery, a well-established platform, PLGA MPs, was used to encapsulate and release Rapa. Rapa MPs were formed via double emulsion and exhibited Rapa loading levels of 17.3 ± 0.68 μg rapamycin/mg particle and average diameters of 2.45 ± 0.13 μm (Figure 1A,B). In order to quantify drug release from Rapa MPs, MPs were incubated in water at 37 °C using sink conditions. Rapa MPs released 65.2 ± 0.01% of drug over 14 days (Figure 1C).
Figure 1.

Rapa MPs gradually release rapamycin, are internalized by DCs without toxicity. (A) Table showing properties of Rapa MPs. (B) Histogram showing size distributions of Rapa MPs. (C) In vitro release kinetics of Rapa MPs. CD11c+ splenocytes were incubated with MPs encapsulating rapamycin and fluorescently labeled MOG peptide. Frequency of DCs internalizing MPs after 4 hrs was quantified by flow cytometry (D) and uptake was visualized by fluorescent microscopy at 2 hrs (E). (F) Viability of DCs was quantified with DAPI staining by flow cytometry after treatment of LPS stimulated DCs with decreasing doses of Rapa MPs.
MPs are internalized by primary DCs in vitro and do not cause toxicity
To test the ability of DCs to internalize MPs, MPs encapsulating fluorescent peptide and Rapa were synthesized and cultured with primary splenic DCs. After 4 hrs, a dose dependent uptake of MPs was measured using flow cytometry (Figure 1D); uptake was visualized by microscopy after 2 hrs of culture and indicated co-localization of MPs within DCs membranes (Figure 1E). To confirm MPs were non-toxic, primary DCs were stimulated with LPS and treated with decreasing doses of Rapa MPs. After 18 hrs no reduction in toxicity for any of the tested doses of Rapa MPs was observed by analysis with flow cytometry after DAPI staining (Figure 1F).
Rapa MPs transiently decrease DC activation and modulate inflammatory cytokine secretion in a dose dependent manner
In order to investigate the effects of Rapa dose during activation of DCs, splenic CD11c+ DCs were stimulated with LPS and treated with decreasing doses of soluble Rapa or Rapa MPs. DCs stimulated with LPS and treated with empty MPs at equivalent particle masses to the Rapa MP groups were included as controls in order to isolate the effect from encapsulated Rapa. After 18 hrs of culture, DCs treated with Rapa MPs exhibited modest decreases in expression of surface activation markers, CD40 (Figure 2A), CD80 (Figure 2B) and CD86 (Figure 2C) compared to empty MP controls. These observed effects were transient, as DCs treated with Rapa MPs did not exhibit decreased activation 48 hrs after treatment (Figure S1). The concentrations of IL-6, IL-12 and IFNγ in culture supernatants at 48 hrs were next measured to determine the effects of Rapa MPs on function, as indicated by cytokine profiles. A dosing effect was observed, where high Rapa MPs doses decreased IFNγ levels compared to untreated DCs stimulated with LPS. At these higher doses (i.e., 1, 0.1, 0.01 μg/mL), Rapa MPs significantly reduced IFNγ levels relative to empty particles; there were some non-specific effects from empty particles at high doses relative to LPS only treatment (Figure 3A). An intriguing dosing effect was observed with IL-12 levels (Figure 3B), and to a lesser extent, IL-6 (Figure 3C). For these cytokines, high levels of Rapa had no effect vs. LPS, whereas at intermediate Rapa MP concentrations, IL-12 and IL-6 levels peaked before returning at the lowest doses to the low cytokine levels observed for the LPS controls. No non-specific effects were observed with empty MP controls.
Figure 2.

Rapa MPs reduce DC activation. CD11c+ splenocytes were stimulated with LPS and treated with soluble rapamycin, Rapa MPs, or equivalent masses of empty MPs. After 18 hrs levels of expression of activation markers, CD40 (A), CD80 (B) and CD86 (C) were quantified by flow cytometry.
Figure 3.

Rapa MPs modulate DC cytokine secretion. CD11c+ splenocytes were stimulated with LPS and treated with soluble rapamycin, Rapa MPs, or equivalent masses of empty MPs. After 48 hrs concentrations of IFNγ (A), IL-12 (B), and IL-6 (C) in culture supernatants were quantified by ELISA.
Rapa MPs reduce CD4 and CD8 T cell proliferation and promote central memory phenotypes in vitro
To investigate if the differential dose effects of Rapa MPs on DC cytokine secretion alter T cell proliferation and phenotype, co-culture studies were performed using DC/transgenic CD4+ T cell co-culture to assess effects on antigens presented by the MHC-II pathway, and a DC/transgenic CD8+ T cell co-culture to investigate effects on antigens presented by the MHC-I pathway. In these studies, DCs from naïve C57/BL6 mice were treated with Rapa MPs with or without LPS stimulation in the presence of a CD4 epitope, MOG, or a CD8 epitope, Trp2. T cells were then isolated from transgenic 2D2 mice, which have CD4 T cells displaying T cell receptors specific for MOG presented in MHC-II, or Trp2 transgenic mice, where CD8 T cell receptors recognize Trp2 presented in MHC-I. Isolated T cells were then cultured with DCs in the presence of their respective cognate antigens. After 48 hrs of co-culture, Rapa MP treatment reduced 2D2 T cell proliferation compared to empty MP controls. These effects were seen in CD4 co-cultures performed both with or without LPS stimulation (Figure 4A,B). Concentrations of IL-17, an inflammatory cytokine characteristic of a TH17 polarized response were also measured in culture supernatants. In these studies, Rapa MPs decreased IL-17 secretion in 2D2 co-cultures with or without LPS stimulation (Figure 4C,D), indicating a suppressive function at each dose for MHC-II/CD4 co-culture.
Figure 4.

Rapa MPs decrease inflammatory cytokine secretion and proliferation in CD4+ T cell co-cultures in vitro. Wild type splenic DCs were cultured in the presence of MOG with or without LPS stimulation and were treated with soluble rapamycin, Rapa MPs or empty MPs. After 24 hours CSFE labeled splenic CD4+ T cells from transgenic 2D2 mice were added to culture. Levels of T cell proliferation in (A) LPS stimulated co-cultures and (B) unstimulated co-cultures were quantified by flow cytometry 48 hrs after the addition of T cells. IL-17 concentrations in LPS-stimulated co-cultures (C) or unstimulated co-cultures (D) were measured by ELISA.
In Trp2 co-cultures (CD8), Rapa MPs also decreased T cell proliferation relative to samples treated with Trp2 and empty MP controls, but still allowed an intermediate level of proliferation relative to that observed in untreated cells (Figure 5A,B). Representative flow cytometry plots from the mean CSFE dilution data shown in Figure 5A,B confirmed that despite a decreased percentage of proliferated cells, T cells treated with Rapa MPs proliferated for multiple generations compared to T cells in empty MP treated or untreated co-cultures stimulated with LPS and Trp2 (Figure S2). This trend was observed in both co-cultures stimulated with LPS or co-cultures left unstimulated. Interestingly, while the magnitude of reduction in proliferation from Rapa MP treatment was similar for LPS stimulated or unstimulated co-cultures, differences were seen in IFNγ levels in culture supernatants. While in the presence of LPS, Rapa MPs only significantly suppressed IFNγ at a dose of 1μg/mL Rapa MPs (Figure 5C), Rapa MPs completely suppressed IFNγ secretion in unstimulated co-cultures at all three doses (Figure 5D). In order to determine if Rapa MPs polarized T cells toward central memory phenotypes, CD44 and CD62L expression was quantified with flow cytometry. Rapa MPs increased the percentages of central memory phenotypes (CD8+/CD62L+CD44+) in both LPS-stimulated and unstimulated co-cultures, and interestingly, this effect peaked at an intermediate Rapa MPs doses (0.1μg/mL) (Figure 5E,F). In particular, while cultures stimulated with LPS and Trp2 and left untreated or treated with empty MPs exhibited low levels of CD8 T cells with central memory phenotypes (4.3±0.37 % and 5.83 ± 0.38 %, respectively), the percentage of central memory T cells was increased to 19.57 ± 1.42 % after treatment with 0.1 μg/mL Rapa MPs. Additionally, it was observed that Rapa MP treatment for both Trp2 co-cultures reduced the percentages of effector memory T cells (CD8+/CD62L− CD44+) (Figure S3), indicating a polarization of antigen-specific T cells during expansion.
Figure 5.

Rapa MPs decrease proliferation of CD8+ T cells and polarize central memory phenotypes in vitro. Wild type splenic DCs cultured in the presence of Trp2 with or without LPS stimulation were treated with soluble rapamycin, Rapa MPs or empty MPs. After 24 hours, CSFE labeled splenic CD8+ T cells from transgenic Trp2 mice were added to culture. Levels of T cell proliferation in LPS stimulated co-cultures (A) and unstimulated co-cultures (B) were quantified by flow cytometry 48 hrs after the addition of T cells. IFNγ levels in culture supernatants for LPS stimulated cultures (C) and unstimulated cultures (D) were quantified by ELISA. Frequencies of central memory T cells (CD8+/CD62L+ CD44+) in LPS stimulated (E) and unstimulated (F) co-cultures were quantified by flow cytometry.
Lymph node injection of soluble vaccines promotes efficient expansion of Trp2 specific T cells, and co-delivery of high doses Rapa MPs with vaccines suppresses T cell expansion
We recently developed a unique approach for controlling the combinations and concentrations of immune signals in LNs using intra-LN injection (i.LN.) of degradable, diffusion-limited microparticles.[26, 29–32] This idea builds on recent clinical trials exploring i.LN. injection of soluble antigens in the context of allergies and cancer.[33–36] Since LNs are one of the key sites where T cells develop, and in particular, pro-inflammatory T cells develop in tumor draining-LNs,[24] we reasoned this platform might allow direct investigation of local Rapa release during vaccine introduction and differentiation of T cells. We first compared the efficacy of LN injection to intra-muscular injection (i.m.), a common peripheral vaccination route. In these studies, mice were injected i.m. or i.LN. with a soluble vaccine consisting of soluble CpG mixed with soluble Trp2 peptide. The Trp2 peptide is a common, conserved human melanoma-associated antigen.[28] Fourteen days after vaccination, Trp2 specific T cells were quantified in peripheral blood using MHC-I tetramer staining. Vaccines delivered with i.LN. injection promoted significantly higher frequencies of Trp2 specific T cells compared to vaccines delivered i.m., which promoted no detectable T cell response (Figure 6A). Therefore, all further vaccine studies utilized i.LN. injection. To determine if localized co-delivery of Rapa MPs with soluble vaccines to LNs could enhance T cell response, mice were primed i.LN. with soluble vaccines (Trp2 + CpG) alone or soluble vaccines mixed with Rapa MPs (soluble CpG + soluble Trp2 + Rapa MP high). 15 days after the priming vaccine, mice were boosted s.c. at the tail base (Figure 6B). Increased Trp2 specific T cell frequencies in peripheral blood were observed one week after the prime in the mice treated with the soluble vaccine, while the inclusion of Rapa MPs at this (high) dose suppressed the expansion of Trp2 specific T cells to levels similar to those measured in untreated mice (Figure 6C,D). Tetramer levels for the soluble vaccine treated group then contracted to levels similar to untreated mice by 14 days after the prime (Figure 6E). However, a recall response was observed in mice treated with soluble vaccines while again no detectable response was detected in mice treated with soluble vaccines + Rapa MPs. One week after the boost (day 29 post prime) the soluble vaccine treated group had an average Trp2 tetramer frequency of 4.14 ± 1.44 % compared to 0.37 ± 0.04 % in mice treated with the soluble vaccine + Rapa MPs (Figure 6F). These results suggested that local LN delivery of Rapa MPs at this dose suppressed responses to of soluble vaccine components (i.e., peptide + adjuvant).
Figure 6.

i.LN. injection of soluble vaccines promotes Trp2 specific T cell expansion, and this effect is suppressed by co-delivery of high dose Rapa MPs. Mice were vaccinated i.LN. with soluble CpG + soluble Trp2 i.LN. or i.M, or were left untreated. (A) Fourteen days after vaccination frequency of Trp2 specific CD8+ T cells were quantified by tetramer staining. (B) Mice were primed i.LN. at day 0 with soluble vaccines, soluble vaccines mixed with Rapa MPs, or were left untreated. Fifteen days after the priming injection mice were boosted s.c with identical treatments. Frequencies of Trp2 specific T cells in peripheral blood were quantified at days 7 (C), 14 (D) and (21) (E). At day 28 post the prime mice were challenged with B16-F10 tumors, and survival was monitored (F).
i.LN. co-delivery of Rapa MPs with soluble vaccines enhances expansion of Trp2 specific T cells
In order to determine if a lower dose of Rapa MP could enhance T cell responses when combined with i.LN. vaccination, mice were vaccinated i.LN. with soluble vaccines or soluble vaccines mixed with Rapa MPs at half of the dose which suppressed T cell responses in the previous study (Figure 6). Further, these Rapa-loaded particles were administered only during the priming injection phase during which naïve T cells differentiate. Mice were primed on day 0 with the indicated treatments, then boosted with simple soluble vaccines (i.e., no Rapa MPs) s.c. at the tail base on day 15, 57 and 97 (Figure 7A). Interestingly, while both soluble vaccines and soluble vaccines + Rapa MPs promoted expansion of Trp2 specific T cells, tetramer frequencies were elevated throughout the duration of the study following the first boost in the soluble vaccine + Rapa MP treated group compared to mice receiving vaccine without Rapa MPs (Figure 7B). Soluble vaccines promoted a significant expansion of Trp2 specific T cells 7 days after the prime (Figure 7C), while at Day 7, soluble vaccines co-delivered with Rapa MPs promoted only a modest expansion. Tetramer frequencies contracted to similar levels observed in untreated mice by day 14 (Figure 7B). Strikingly, despite the lower levels observed during the primary response at day 7, an increased recall response was observed after the boost in mice primed with soluble vaccines combined with Rapa MPs. At day 21, an average tetramer frequency of 4.56 ± 1.14 % was observed in the soluble vaccine + Rapa MP group, compared to 3.19 ± 0.81 % in the soluble vaccine treated group (Figure 7C). Tetramer levels for soluble vaccine and soluble vaccine + Rapa MP groups contracted to similar levels by the second boost at day 57 (Figure 7B), and again a larger recall response was observed in the soluble vaccine + Rapa MP group compared to the soluble vaccine group after the second boost (Figure 7C). At this time point, the enhancement from inclusion of Rapa depots reached as high as 26.1% of CD8+ T cells in peripheral blood specific for Trp2, compared to a maximum frequency of 8.1% in mice receiving the same vaccine without Rapa (Figure 7D). Similar trends were observed at Day 104, one week after a final boost administered at Day 97. To begin testing if this approach and the larger number of Trp2-specific CD8+ T cells might enhance functional anti-tumor immunity, mice were then challenged with an aggressive infusion of B16-F10 tumor cells. Tumor growth was delayed in soluble vaccine + Rapa MP treated mice, with an average tumor burden at day 15 post challenge of 54.6 ± 18.4 mm2 compared to 80.8 ± 16.7 mm2 in mice receiving the vaccine only, and 110.64 ± mm2 in untreated mice (Figure 7E). These modest effects, however, were not statistically significant for tumor burden (p=0.111) or survival (p = 0.1884) comparing mice treated with soluble vaccines ± Rapa MPs (Figure 7F).
Figure 7.

i.LN. injection of low dose Rapa MPs with soluble vaccines promotes enhanced Trp2 T cell recall response. (A) Mice were primed i.LN. with soluble vaccines or soluble vaccines + Rapa MPs and boosted s.c with soluble vaccines at the indicated time points, followed by tumor challenge with B16-F10 tumors. (B) Trp2 tetramer frequencies were quantified weekly in peripheral blood after vaccination. (C) Average tetramer frequencies in each treatment group are shown at day 7, 21 and 64. (D) Flow cytometry plots of mice with the highest Trp2 tetramer frequency in each group are shown for day 64. (E) Scatter plots of tumor size at day 15, and (F) survival after tumor challenge are shown.
i.LN. delivery of Rapa MPs with soluble vaccines expands antigen-specific TCM
Since robust anti-tumor efficacy was not observed in mice treated with Trp2 vaccines combined with lose dose Rapa MPs, despite increased levels of cytotoxic T cells, we used a simpler regimen and a well-defined antigen to directly investigate polarization of central memory T cells. In these studies, mice were vaccinated i.LN. with soluble CpG and a model antigen, Ovalbumin (OVA), with or without low dose Rapa MPs. Booster injections were not administered to simplify the studies. Fourteen and twenty one days after vaccination, the frequency of CD8+ T cells specific for an immunodominant CD8 epitope of OVA—SIINFEKL (SIIN)— was assessed by MHC-I tetramer staining, along with the level of SIIN-specific CD8+ TCM (CD44+CD62L+) (Figure 8A). By fourteen days post vaccination, OVA vaccines combined with Rapa MPs increased SIIN-specific T cells, with 4.35 ± 0.37% of CD8+ T cells specific for SIIN, compared to 3.00 ± 0.22 % in mice immunized with OVA vaccines alone (Figure 8B). Further, phenotypic analysis revealed that inclusion of Rapa MPs in the vaccine led to a 60% increase (p<0.01) in the frequency of OVA-specific TCM cells by day 14 (Figure 8C). At day 21 after immunization, similar trends were observed for both tetramer frequencies and TCM levels (Figure 8B,C), but these differences were not significant.
Figure 8.

i.LN. injection of low dose Rapa MPs with soluble vaccines promotes enhanced antigen specific TCM. (A) Mice were primed i.LN. with soluble OVA or Trp2 vaccines with or without Rapa MPs, and tetramer and memory responses were analyzed in peripheral blood at the indicated time points. (B) SIIN tetramer frequencies in peripheral blood were quantified at day 14 and day 21 after vaccination with soluble OVA vaccines with or without Rapa MPs. (C) Frequency of SIIN tetramer specific TCM among CD8+ T cells. (D) Trp2 tetramer frequencies were quantified at day 14 and day 21 after vaccination in groups vaccinated with soluble Trp2 vaccines with or without Rapa MPs. (E) Frequency of Trp2 specific TCM among CD8+ T cells, and (F) ratio of TCM:TEFF frequencies among Trp2 specific CD8+ T cells.
Since Rapa MPs promoted antigen-specific central memory T cells when combined with a model antigen, we next determined if these effects occurred with vaccines integrating Trp2 antigen. Mice were vaccinated i.LN. using an identical regimen as just described for OVA (Figure 8A), but OVA was replaced with Trp2 peptide in each group. At day 14 after vaccination, Trp2 tetramer frequencies among CD8+ T cells increased similarly for groups vaccinated with or without Rapa MPs. By day 21, however, vaccines including Rapa MPs drove Trp2-specific T cell levels that were 40% higher compared to vaccines lacking the Rapa MPs (Figure 8D). Interestingly, at day 14, when Trp2 tetramer frequencies were similar for both treatment groups, TCM analysis revealed low dose Rapa MP treatment promoted a 36% increase in the frequency of Trp2-specific TCM compared with mice receiving only the vaccine. (Figure 8E). This trend was also observed at day 21 after vaccination, where the frequency of Trp2-specific TCM was significantly higher in the Rapa MP treated group compared to mice receiving only vaccines (Figure 8E). To investigate if Rapa MPs polarized differentiation toward TCM, the ratio of TCM to TEFF (CD44+CD62L−) among Trp2 specific T cells was analyzed. At both day 14 and day 21, the ratio of TCM to TEFF revealed a bias toward increased TCM:TEFF in the low dose Rapa MP group, although the level of significance was weaker for day 14 (p=0.051) and day 21 (p=0.057) (Figure 8F).
DISCUSSION
Regulating mTOR or other metabolic pathways offers new potential for tuning immune response by controlling plasticity to alter the phenotype and function of differentiating cells.[37, 38] Rapamycin, although, conventionally an immunosuppressant, is already used in cancer therapy, along with a number of structural analogs (“Rapalogs”) [17–19]. mTOR has two key signaling centers, mTORC1, and mTORC2, and many rapalogs seek to block both of these to limit the uncontrolled growth of tumor cells. With respect to vaccination, new studies are being carried to see if Rapa might serve the more subtle role we have investigated, as an immunoregulatory signal. The distinguishing features of this idea are that the doses are low, and that Rapa or other drugs are delivered in the presence of an adjuvant (compared with a traditional immunosuppressant application lacking pro-immune signals). For example, in infectious disease, several recent studies have used daily dosing of Rapa to promote T CM, [7, 8] and in particular, demonstrate that early exposure to Rapa during T cell priming drive increased IL-12 production that drives the improvements in durability and proliferative capacity (i.e., biasing toward TCM).[20] These effects also conferred a protective effect during live bacterial challenge with Listeria Monocytogenes.
The finding above is interesting, as in our studies Rapa could be used to drive IL-12 expression in a dose dependent manner when released from particles. Our in vitro studies revealed a suppressive effect on some aspects of DC function at high doses, while at decreasing doses this effect was reversed, promoting inflammatory function. In particular, the 1 μg/mL dose of Rapa MPs decreased IFNγ without impacting IL-12, but when the Rapa MP dose IL-12 (and IL-6 to a lesser level) spiked beyond above levels induced by LPS stimulation alone. IL-12 is a key cytokine that signals T cell differentiation and proliferation. Another interesting aspects of our studies is that doses where Rapa MPs didn’t affect DC activation or inflammatory cytokine secretion, these MPs still decreased T cell proliferation in culture. Additionally, at doses where Rapa MPs caused decreases in DC activation (Figure 2, S1), and increases in pro-immune cytokine secretion (Figure 3B, 3C), Rapa MPs still partially attenuated T cell proliferation in both CD4+ and CD8+ T cell co-cultures. This reduction in proliferation may potentially be attributed to effects of Rapa MPs directly – at least partially – on T cell function, independent from the modulatory effects on DCs. This explanation is consistent with findings from Araki et al. which demonstrate Rapa promotes central memory phenotypes in vivo when combined with LCMV challenge through a mechanism dependent on the inhibition of mTORC2 in T cells.[7] These differing effects of Rapa MPs on DCs and T cells observed in vitro may have important implications for the i.LN. vaccine platform we utilized in vivo. Rapa MPs are formulated at a size which is readily internalized by APCs (Figure 1E), providing a simple mechanism to target encapsulated drug to DCs. We have previously shown that MPs formulated similarly are retained in LNs over time, resulting in prolonged localized increases in concentrations of encapsulated cargo. Therefore this system may function as an efficient method to target DCs for enhanced immunostimulatory function, while Rapa MPs remain extracellularly to provide controlled release and locally exert additional immunomodulatory effects on T cells during priming in the LNs when co-delivered with vaccines.
In our cell culture studies, free Rapa was generally more potent in than Rapa MPs, however, these formulations clearly provide different drug availability and kinetics. In vivo, these considerations are significant since free drug is hydrophobic, exhibits a short half-life, and risks broad suppression if delivered in an uncontrolled or untargeted way. To address these problems, we used i.LN. delivery of depots loaded with Rapa as a means of directly testing the effect on T cell function. Several recent papers have explored regular administration of free Rapa during cancer vaccination, and one in particular has used i.LN. administration.[22] In this report free CpG and mRNA encoding antigen were administered to inguinal LNs, while Rapa was injected daily by i.p. injection. This approach improved survival, and interestingly – as with other reports [7] – the effect on T cell polarization depended on when the drug was delivered.
Our in vitro studies with CD8+ T cells revealed proliferation was reduced in Trp2 T cell co-cultures (Figure 5A, 5B), but their phenotype was polarized toward central memory (Figure 5E, 5F). These effects may allow for reduced T cell contraction and rapid proliferation after re-exposure to antigen, which could enhance T cell responses during vaccination in vivo. Our animal studies support this idea: low dose Rapa MPs combined with soluble vaccines initially (i.e., day 7) reduced T cell expansion after the prime compared to soluble vaccines alone (Figure 7C), but the recall response after subsequent boosts was larger and faster in the Rapa MP treated group. For example, Trp2 tetramer levels contracted to similar levels for soluble vaccine and soluble vaccine + Rapa MP treated mice at 14 days after the prime (Figure 7B) (1 day before the first boost), but expansion of Trp2 specific T cells was significantly higher on days 21 and 64 (one week after booster injections) for the soluble vaccines + Rapa MPs group (Figure 7B, 7C). The increased expansion after the boost supports the hypothesis that low dose Rapa MPs tune T cells to phenotypes with superior proliferative capacity.
During analysis of memory phenotype, we discovered that the addition of Rapa MPs enhanced the level and persistence of TCM. There are several implications from these studies. First, we observed similar or increased levels of antigen-specific T cells, as well as an increase in the frequency of circulating antigen-specific TCM cells (Figure 8). Both of these effects correlated with addition of the Rapa MP component to the vaccines. These two observations might indicate functionality of TCM that led to the increased expansion of Trp2-specific T cells measured in the boosting and recall studies (Figure 7). For example, the data in Figure 7B at Day 14 (just before the first boost) revealed similar levels of Trp2-specific T cells, but a large expansion the following week after boost when comparing groups receiving vaccine + Rapa MPs as the priming regimen to groups receiving only the vaccine. At this equivalent time in Figure 8B, although the tetramer levels were similar, the frequency of TCM and TCM:TEFF were significantly higher in the groups receiving the vaccine with Rapa MPs. Together this set of results supports the idea that local co-administration of low dose Rapa MPs with soluble vaccines polarizes TCM phenotypes to enhance T cell response. As discussed below, however, although Rapa MPs polarized TCM, these effects did not translate to potent anti-tumor immunity.
Our own results and the literature reports discussed above highlight an important role for both dose and kinetics in the proposed strategy. For example, past work reveals that repeated treatment during the priming T cell expansion phase leads to more durable T cell responses, while daily treatment during only the T cell contraction phase leads polarizes cells toward central memory.[7] Regular treatment during both phases provides both durability and improved memory function. At the same time, other studies in models such as human papilloma viruses, indicate Rapa does not provide protective effects and may be suppressive.[39] Thus, there is great potential for biomaterials in this area, both to enable new therapies, but also as tools to understand the impact of kinetics and dosing of drug delivery on mTOR signaling, and ultimately on T cell fate. For example, could tunable release kinetics be used to target only the expansion or only the contraction phase, and thus program durability/maintenance, polarization to TCM, or both?
In our studies, the expansion of T cells generated by incorporation of Rapa MPs provided only a modest impact during tumor challenge (Figure 7G, 7H), though we used an aggressive metastatic melanoma model. In the clinic, cancer vaccines are extremely unlikely to be used as monotherapies, but instead will be coupled with checkpoint blockage – where natural immune regulatory pathways are blocked, with pro-immune cytokines, or with other combination therapies.[9] Thus, while we measured greater expansion of tumor-specific T cells but limited efficacy, there are a variety of potential strategies which may function to enhance the anti-tumor function of T cells generated by vaccines combining metabolic modulators. In particular, checkpoint blockade is creating a renaissance for cancer vaccines using anti-CTLA4 or anti-PDL1 monoclonal anti-bodies that could be combined with vaccines and Rapa MP treatments to inhibit exhaustion of T cells driven by tumor evasion mechanisms. In addition it is possible that limited anti-tumor effectiveness we observed may be due in part to the use of a vaccine containing a single tumor antigen. Applying Rapa MPs to enhance T cell response toward broad sets of tumor-associated antigens, including multiple well-defined antigens or tumor cell lysates which contain highly immunogenic neoantigens, may serve as a potential strategy to significantly enhance anti-tumor immunity. As these strategies develop, there will also be important safety questions already facing some of the developing technologies. For example, how to balance the risk/reward profile of giving an immunosuppressant, or how to “turn-off” the effects of checkpoint blockage or greatly enhance proliferation during Rapa-containing vaccines therapies that may arise. Despite these hurdles, here we demonstrate an exciting new biomaterial-enabled strategy to program the local LN microenvironment for polarizing the phenotypes of developing T cells that could contribute to new options in for vaccines against infectious disease and for cancer immunotherapies.
CONCLUSION
In this study, we utilized a well-studied controlled release platform to investigate how the presence of low doses of a common regulatory drug alter the nature of immune response during vaccination with peptide antigens and molecular agonists. Rapa MPs reduced – but did not stop – DC activation and T cell proliferation. These effects were dose-dependent, and could be switched between suppression and enhancing functions, the latter resulting from polarization of antigen-specific T cells toward central memory function. Although only modest functional effects were observed during an aggressive tumor challenge, this report demonstrates the intriguing potential of using controlled release during vaccination to preserve T cells plasticity and enhance T cell function. In particular, this ability might enable new, simpler approaches to recapitulate the attractive features of adoptive cell therapies that currently require isolation from patients, expansion, and reinfusion to generate sufficient numbers of cells for efficacy.
Supplementary Material
Acknowledgments
This work was supported in part by the Damon Runyon Foundation (# DRR3415), the Melanoma Research Alliance (# 348963), NSF CAREER Award (# 1351688), Alex’s Lemonade Stand (# 27120), and the Alliance for Cancer Gene Therapy (# 15051543). J.M.G. is a grantee of the Pediatric Oncology Student Training Award (# 15082537) from Alex’s Lemonade Stand Foundation. L.H.T. is an NSF Graduate Fellow (# DGE1322106). Y.C.C. is a trainee on NIH Grant T32 CA154274. C.M.J. is a Damon-Runyon Rachleff Innovator supported by the Damon Runyon Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES CITED
- 1.Andorko JI, Hess KL, Jewell CM. Harnessing Biomaterials to Engineer the Lymph Node Microenvironment for Immunity or Tolerance. The AAPS journal. 2014 doi: 10.1208/s12248-014-9708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu TY, Singh M, Miller AT, De Gregorio E, Doro F, D’Oro U, Skibinski DA, Mbow ML, Bufali S, Herman AE, Cortez A, Li Y, Nayak BP, Tritto E, Filippi CM, Otten GR, Brito LA, Monaci E, Li C, Aprea S, Valentini S, Calabromicron S, Laera D, Brunelli B, Caproni E, Malyala P, Panchal RG, Warren TK, Bavari S, O’Hagan DT, Cooke MP, Valiante NM. Rational design of small molecules as vaccine adjuvants. Science translational medicine. 2014;6:263ra160. doi: 10.1126/scitranslmed.3009980. [DOI] [PubMed] [Google Scholar]
- 3.Waickman AT, Powell JD. Mammalian target of rapamycin integrates diverse inputs to guide the outcome of antigen recognition in T cells. Journal of immunology. 2012;188:4721–4729. doi: 10.4049/jimmunol.1103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–U117. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollizzi KN, Powell JD. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends in immunology. 2015;36:13–20. doi: 10.1016/j.it.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–U124. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi YW. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–U118. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gammon JM, Dold NM, Jewell CM. Improving the clinical impact of biomaterials in cancer immunotherapy. Oncotarget. 2016;7:15421–15443. doi: 10.18632/oncotarget.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews Immunology. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. The Journal of clinical investigation. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 13.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, Borman ZA, Kerkar SP, Scott CD, Finkelstein SE, Rosenberg SA, Restifo NP. Determinants of Successful CD8(+) T-Cell Adoptive Immunotherapy for Large Established Tumors in Mice. Clin Cancer Res. 2011;17:5343–5352. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker EB, Haley D, Petrausch U, Floyd K, Miller W, Sanjuan N, Alvord G, Fox BA, Urba WJ. Phenotype and functional characterization of long-term gp100-specific memory CD8+ T cells in disease-free melanoma patients before and after boosting immunization. Clin Cancer Res. 2008;14:5270–5283. doi: 10.1158/1078-0432.CCR-08-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunological reviews. 2014;257:264–276. doi: 10.1111/imr.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annual review of immunology. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 17.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Annals of oncology: official journal of the European Society for Medical Oncology. 2005;16:525–537. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 18.Nelson V, Altman JK, Platanias LC. Next generation of mammalian target of rapamycin inhibitors for the treatment of cancer. Expert opinion on investigational drugs. 2013;22:715–722. doi: 10.1517/13543784.2013.787066. [DOI] [PubMed] [Google Scholar]
- 19.Blagosklonny MV. Immunosuppressants in cancer prevention and therapy. Oncoimmunology. 2013;2:e26961. doi: 10.4161/onci.26961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Garcia K, Sun Z, Xiao Z. Temporal regulation of rapamycin on memory CTL programming by IL-12. PloS one. 2011;6:e25177. doi: 10.1371/journal.pone.0025177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Rao R, Vazzana J, Goedegebuure P, Odunsi K, Gillanders W, Shrikant PA. Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. Journal of immunology. 2012;188:3080–3087. doi: 10.4049/jimmunol.1103365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diken M, Kreiter S, Vascotto F, Selmi A, Attig S, Diekmann J, Huber C, Tureci O, Sahin U. mTOR inhibition improves antitumor effects of vaccination with antigen-encoding RNA. Cancer immunology research. 2013;1:386–392. doi: 10.1158/2326-6066.CIR-13-0046. [DOI] [PubMed] [Google Scholar]
- 23.Mineharu Y, Kamran N, Lowenstein PR, Castro MG. Blockade of mTOR signaling via rapamycin combined with immunotherapy augments antiglioma cytotoxic and memory T-cell functions. Molecular cancer therapeutics. 2014;13:3024–3036. doi: 10.1158/1535-7163.MCT-14-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiura T, Kagamu H, Miura S, Ishida A, Tanaka H, Tanaka J, Gejyo F, Yoshizawa H. Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. Journal of immunology. 2005;175:5058–5066. doi: 10.4049/jimmunol.175.8.5058. [DOI] [PubMed] [Google Scholar]
- 25.McHugh KJ, Guarecuco R, Langer R, Jaklenec A. Single-injection vaccines: Progress, challenges, and opportunities. Journal of controlled release: official journal of the Controlled Release Society. 2015;219:596–609. doi: 10.1016/j.jconrel.2015.07.029. [DOI] [PubMed] [Google Scholar]
- 26.Jewell CM, Lopez SC, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc Natl Acad Sci U S A. 2011;108:15745–15750. doi: 10.1073/pnas.1105200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials. 2011;32:5134–5147. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andorko JI, Tostanoski LH, Solano E, Mukhamedova M, Jewell CM. Intra-lymph node injection of biodegradable polymer particles. Journal of visualized experiments: JoVE. 2014:e50984. doi: 10.3791/50984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell reports. 2016;16:2940–2952. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andorko JI, Hess KL, Pineault KG, Jewell CM. Intrinsic immunogenicity of rapidly-degradable polymers evolves during degradation. Acta biomaterialia. 2016;32:24–34. doi: 10.1016/j.actbio.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andorko JI, Gammon JM, Tostanoski LH, Zeng Q, Jewell CM. Targeted Programming of the Lymph Node Environment Causes Evolution of Local and Systemic Immunity. Cellular and molecular bioengineering. 2016;9:418–432. doi: 10.1007/s12195-016-0455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Vries IJ, Lesterhuis WJ, Barentsz JO, Verdijk P, van Krieken JH, Boerman OC, Oyen WJ, Bonenkamp JJ, Boezeman JB, Adema GJ, Bulte JW, Scheenen TW, Punt CJ, Heerschap A, Figdor CG. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat Biotechnol. 2005;23:1407–1413. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- 34.Ribas A, Weber JS, Chmielowski B, Comin-Anduix B, Lu D, Douek M, Ragavendra N, Raman S, Seja E, Rosario D, Miles S, Diamond DC, Qiu Z, Obrocea M, Bot A. Intra-lymph node prime-boost vaccination against Melan A and tyrosinase for the treatment of metastatic melanoma: results of a phase 1 clinical trial. Clin Cancer Res. 2011;17:2987–2996. doi: 10.1158/1078-0432.CCR-10-3272. [DOI] [PubMed] [Google Scholar]
- 35.Akdis CA. Therapies for allergic inflammation: refining strategies to induce tolerance. Nat Med. 2012;18:736–749. doi: 10.1038/nm.2754. [DOI] [PubMed] [Google Scholar]
- 36.Witten M, Malling HJ, Blom L, Poulsen BC, Poulsen LK. Is intralymphatic immunotherapy ready for clinical use in patients with grass pollen allergy? J Allergy Clin Immun. 2013;132:1248–1252. doi: 10.1016/j.jaci.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, Kuchroo VK. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell. 2015;163:1413–1427. doi: 10.1016/j.cell.2015.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, Xia J, Kwon JY, Nevin J, Herbst RH, Yanai I, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell. 2016;166:1500–1511 e1509. doi: 10.1016/j.cell.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaoul N, Fayolle C, Desrues B, Oberkampf M, Tang A, Ladant D, Leclerc C. Rapamycin Impairs Antitumor CD8+ T-cell Responses and Vaccine-Induced Tumor Eradication. Cancer research. 2015;75:3279–3291. doi: 10.1158/0008-5472.CAN-15-0454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.