

Abstract
An important goal in the development of polo-like kinase 1 (Plk1) polo-box domain (PBD) binding inhibitors is selectivity for Plk1 relative to Plk2 and Plk3. In our current work we show that Plk1 PBD selectivity can be significantly enhanced by modulating interactions within a previously discovered “cryptic pocket” and a more recently identified proximal “auxiliary pocket.”
Keywords: Plk1, Selectivity, Polo-box domain, Cryptic binding pocket
Graphical Abstract
1. Introduction
Members of the polo family of serine/threonine kinases (polo-like kinases; Plks 1–5) play important roles in mammalian cellular physiology.1 Overexpression of the cell cycle regulatory Plk1 occurs in a number of cancers, where it is associated with a poor prognosis and Plk1 is recognized as a potentially promising anti-cancer target.2 In addition to their amino-terminal catalytic kinase domains (KD) (not present in Plk53), the Plks are characterized by C-terminal polo-box domains (PBDs), which mediate protein-protein interactions (PPIs) by recognizing and binding to phosphoserine (pSer) and phosphothreonine (pThr)-containing sequences.4 Development of Plk1 inhibitors is made complicated by a number of factors. Among these is the fact that Plks 2 and 3 may function as tumor suppressors, thereby making Plk1-selectivity an important objective.5 Issues of collateral cytotoxicity have arisen for KD-directed inhibitors, potentially due to similarities among the KDs of Plk1, 2 and 3 as well as high homology among protein kinases in general. Because both the KD and PBD are essential for proper Plk function,6 PBD-binding antagonists offer a potentially attractive alternative to KD-directed inhibitors for down-regulating Plk function.7 Such agents may offer advantages in terms of selectivity, since PBDs are unique to the Plks and the domains exhibit differences in their binding preferences for phosphoepitope sequences.8,9 Additionally, the results of blocking PBD-binding may not be the same as inhibiting kinase activity, opening the possibility of qualitatively different therapeutic profiles or synergism with KD inhibitors.10 Accordingly, significant effort has been devoted to develop Plk1 PBD-binding antagonists.2,7
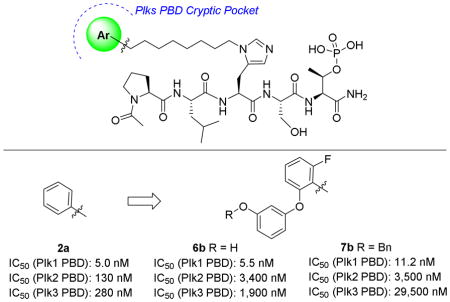
Competitive PBD-binding inhibitors have been approached using peptides or peptide mimetics, which are based on cognate recognition sequences. Starting from the polo-box interacting protein 1 (PBIP) pT78-derived peptide PLHSpT (1),11 we have previously reported that appending alkylphenyl groups from different positions on the peptide, including the His N(π)-position [the sequence having a (CH2)8Ph group appended is designated as PLH*SpT (2a)],12 the 4-position on the Pro residue (3)13 and an amino-terminal N-alkyl Gly residue (4),14 can result in up to one thousand-fold enhancement in Plk1 PBD-binding affinity (Figure 1). We found in each case that the alkylphenyl groups bind within a “cryptic binding pocket” on the PBD surface, which is defined by PBD residues Y417, Y421, Y481, L478, F482 and Y485 and revealed by rotation of the Y481 side chain (Figure 2). Abell has also shown that the same cryptic pocket can be accessed by the phenyl ring of the terminal Phe residue in the longer PBIP1 pT78-derived sequence FDPPLHSpTA15 and by the phenyl ring of 3-phenylpropylamide on the Pro residue of 1.16 This group has also shown that higher affinity can be realized by replacing the terminal Phe residue in the FDPPLHSpTA sequence with a residue that contains a more lipophilic 2-benzothiophene ring (5), which is consistent with the highly lipophilic nature of the cryptic pocket (Figure 1).17
Figure 1.

Structures of peptides discussed in the text.
Figure 2.

Plk1 PBD electrostatic surfaces (blue = positive; red = negative). (A) PLHSpT with bound 1(PDB accession code: 3HIK); (B) PLH*SpT with bound 2a (PDB accession code: 3RQ7) having the portion of the peptide accessing the cryptic pocket shown in yellow and the cryptic pocket overlain yellow mesh and a recently identified “auxiliary pocket” shown with overlain red mesh.
Recently, we examined access to the cryptic pocket from the His N(π)-position in peptide 1 using an oxime-based post-solid phase peptide diversification strategy18 and found that replacing the phenyl ring with motifs having two aryl rings arranged in an angular orientation, can result in several fold greater Plk1 PBD-binding affinity relative to the parent phenyl ring.19 We hypothesized that enhanced affinity may result from interactions within an auxiliary region that is contiguous with the canonical cryptic pocket. Yet, beyond the above, very little has been reported to exploit binding within the cryptic pocket to greater advantage. Therefore, as we report herein, we used the simple tethered phenyl ring of peptide 2a and our recently disclosed 2-fluoro-6-phenyloxylphenyl peptide 2b as starting points for the design of peptides 6a and 6b, which include hydroxyls at different locations. Our thought was that this added functionality could potentially afford interactions with hydroxyl or carbonyl groups of residues Y421 and Y481 or other residues proximal to the Plk1 PBD cryptic pocket. The benzyl protected precursors 7a and 7b also provided the opportunity to extend aromatic interactions in this region. Our ultimate goal was to more fully explore what could be achieved by altering known binding motifs, particularly effects related to Plk1, 2 and 3 selectivity.
2. Results and discussion
2.1. Chemistry
Peptides 6(a, b) and 7(a, b) were obtained by Fmoc-based solid-phase peptide synthesis using reagents 18(a-d), which were prepared as shown in Scheme 1.20 The synthesis began with the t-butyl and benzyl-protected alcohols 9(a, b), which were synthesized by protecting the free hydroxyl of the commercially available alcohol 2-(4-bomophenyl)ethan-1-ol (8). Coupling of the aryl bromides 9(a, b) and oct-7-yn-1-ol,21 followed by hydrogenation catalyzed by Pd·C (for 10a) or Wilkinson’s catalyst (for 10b) afforded alcohols 11(a, b) having t-butyl and benzyl protecting groups, respectively. Separately, SNAr reaction of 2,6-difluorbenzaldehyde (12) with 3-(tert-butoxy)phenol or 3-(benzyloxy)phenol gave the 2-fluoro-6-phenoxybenzaldehydes 13(a, b), respectively.22 Reduction of the aldehyde functionality in 13(a, b) and replacement of the hydroxyls in the resulting 14(a, b) with bromide gave the corresponding benzyl bromides 15(a, b). Coupling of 15(a, b) with hept-6-yn-1-ol or ((hept-6-yn-1-yloxy)methyl)benzene afforded the alkynes 16(a, b). Hydrogenation catalyzed by Pd·C or Wilkinson’s catalyst afforded the alcohols 17(a, b). Finally, reaction of alcohols 11(a, b) and 17(a, b) with either the 2,4-dimethoxylbenzyl ester or the benzyl ester of Nα-Fmoc-His using our previously reported methodology afforded the key orthogonally protected His N(π)-alkylated analogs 18(a-d). 19,20 Employing these reagents, we prepared peptides 19(a-d) on NovaSyn TGR resin. Cleavage of the peptides from the resin using a cocktail solution of TFA/H2O/TIS (95/2.5/2.5) and purification by HPLC gave the desired peptide products 6(a, b) and 7(a, b) (Scheme 1).
Scheme 1.

Preparation of peptides 6(a, b) and 7(a, b). Reagents and Conditions: i) Boc2O, Mg(ClO4)2; ii) Pd(PPh3)2Cl2, CuI, CH≡C(CH2)6OH, 70 °C; iii) H2, (Ph3P)3RhCl or Pd/C; iv) ArOH, K2CO3, DMA, 165 °C; v) NaBH4, MeOH; vi) CBr4, PPh3, CH3CN; vii) CuI, TBAI, Cs2CO3, CH≡C(CH2)5OH or nBuLi, CH≡C(CH2)5OBn; viii) CH≡CCH2OtBu, CuI, DIEA, Pd(PPh3)2Cl2; ix) Nα-Fmoc-His(Nτ-Trt)-OBn or Nα-Fmoc-His(Nτ-Trt)-ODMP, Tf2O; x) TFA/TIS or H2, Pd/C; xi) TFA/TIS/H2O.
2.3. Biological evaluation
2.3.1. PBD-binding activity in full-length Plk1
In order to evaluate PBD-binding affinities, we employed an ELISA assay, which measures the ability of the synthetic peptides to compete with an immobilized phosphopeptide “PMQSpTPLN” for binding to full-length Plk1.19 As we have recently reported, the parent peptide 1, which lacks an N(π)-alkyl group, shows almost no inhibitory potency in this assay (IC50 > 300 μM), while introduction of a -[CH2]8Ph at this position results in greater than an approximate 1000-fold enhancement in binding affinity (2a, IC50 = 180 nM, Table 1).19 Replacement of the phenyl ring in 2a with a 2-fluoro-6-phenoxybenzy group resulted in enhanced binding affinity (2b, IC50 = 127 nM).19 The focus of our current study was to examine the effects of introducing additional oxygen functionality into cryptic pocket-binding motifs. For this purpose, we prepared 6a as a 4-(2-hydroxyethyl)-containing analog of 2a and 6b as an analog of 2b, which included a 3-hydroxyl group at the 3-position of the phenyl ether moiety. Both 6a and 6b exhibited IC50 values (220 nM and 190 nM, respectively), which were approximately equal to 2a, indicating that the presence of the hydroxyl groups was compatible with maintenance of high affinity binding (Table 1). Given the large number of aromatic side chains bordering the cryptic pocket, we were also interested in observing the effects incurred by introducing additional aryl functionality. Peptides 7a and 7b, which are intermediates in the synthesis of 6a and 6b, respectively served this purpose. While the added benzyl group was well tolerated in 7a (IC50 = 150 nM), its presence in 7b caused an approximate four-fold loss of potency (IC50 = 860 nM) (Table 1).
Table 1.
Inhibitory binding potencies of peptides determined using full-length Plk1 and isolated PBDs of Plk1, Plk2 and Plk3.i
| |||||
|---|---|---|---|---|---|
| No. | Ar | IC50 (nM)ii | |||
| Plk1 ELISA | Plk1 PBD FP | Plk2 PBD FP | Plk3 PBD FP | ||
| 2a |
|
181 ± 13 | 5.0 ± 0.2 | 130 ± 10 (26 ×) iv | 280 ± 100 (56 ×) |
| 6a |
|
218 ± 11 | 5.5 ± 0.2 | 310 ± 25 (56 ×) | 960 ± 280 (175 ×) |
| 7a |

|
152 ± 4 | 9.7 ± 0.7 | 250 ± 20 (25 ×) | 920 ± 100 (95 ×) |
| 2biii |
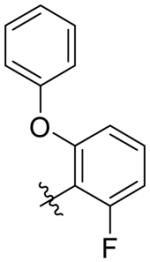
|
127 ± 21 | 6.6 ± 0.3 | 290 ± 45 (44 ×) | 1,450 ± 530 (220 ×) |
| 6b |
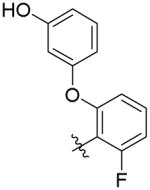
|
188 ± 22 | 5.5 ± 0.4 | 3,400 ± 60 (62 ×) | 1,900 ± 540 (345 ×) |
| 7b |
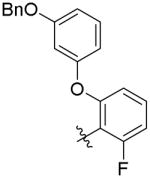
|
862 ± 113 | 11.2 ± 0.3 | 3,500 ± 800 (312 ×) | 29,500 ± 11,300 (2,634 ×) |
Assays were conducted as described in the Experiment Section.
Values represent the average ± SEM from three independent experiments and fit using nonlinear regression in GraphPad Prism 6.
See reference.19
Selectivity compared with Plk1.
2.3.2. Binding affinities measured using isolated PBDs of Plk1, 2 and 3
An important consideration in the development of PBD-binding inhibitors is selectivity for the PBD of Plk1 relative the PBDs of Plk2 and Plk3. While, there are no PBD-binding assays currently available that employ full-length Plk2 and Plk3, fluorescence polarization (FP) assays have been reported, which measure the abilities of inhibitors to compete with fluorescently labeled pThr-containing peptides for binding to the isolated PBDs of Plk1, 2 and 3.8,9 These assays employ fluorescently labeled peptides that have been optimized for binding to each individual PBD isoform.8,9 It is worth noting that the IC50 values measured against the isolated Plk1 PBD are more potent than those measured against full length Plk1.23,24 This difference can be attributed to inhibitory interactions between the PBD and KD, which affect equilibrium binding to phosphopeptides. We measured the abilities of 6(a, b) and 7(a, b) as well as 2(a, b) to compete with fluorescently-labeled phosphopeptide probes for binding to isolated PBDs of Plks 1–3 using FP assays (Table 1).
While the parent peptide 2a showed approximately 20-fold selectivity for the Plk1 PBD relative to the Plk2 and approximately 50-fold selectivity relative to the Plk3 PBD, introduction of functionality onto the cryptic pocket-binding phenyl ring of 2a was capable of further increasing selectivity for both domains (Table 1). Fold-selectivities for Plk1 relative to Plk2 and Plk3, respectively upon introduction of the indicated functionality were: 4-(2-hydroxylethyl) (6a) 50 and 175; 2-fluoro, 6-phenlyoxy (2b) 40 and 200; 2-fluoro, 6-(3-hydroxy)phenyloxy (6b) 60 and 350. Interestingly, introducing a benzyl group onto 6b to give 2-fluoro, 6-(3-benzyloxy)phenoxy-containing 7b, increased Plk1 PBD selectivity versus the Plk2 PBD to 300-fold and 2,500-fold relative to the Plk3 PBD. This data showed that modification of the phenyl group in 2a could result in significant improvements in selectivity for the Plk1 PBD relative to the Plk2 PBD and that near complete selectivity over the Plk3 PBD could be realized.
2.4 Comparison of the cryptic pocket of the Plk1 PBD with potential cryptic pockets in the PBDs of Plk2 and Plk3
A major focus of our current study was to examine the effects incurred by modifying interactions within the cryptic binding pocket. Although we prepared only a very limited number of analogs, we found that changes to the cryptic pocket-binding phenyl ring could have a profound impact on the selectivity for the Plk1 PBD relative to the Plk2 and Plk3 PBDs. We speculated whether there might be differences among the PBDs of Plk1, 2 and 3, which could potentially contribute to modulation of selectivity. The existence of “cryptic binding pockets” in the Plk2 and Plk3 PBDs have not been reported. Crystal structures of the Plk2 PBD are known,25,26 yet these structures do not contain bound ligands and to date, no crystal structures of the Plk3 PBD have been reported. Therefore, we set out to define putative interacting residues within potential cryptic pockets of Plk2 and Plk3 and to compare them with corresponding residues in the Plk1 PBD.
Lacking a crystal structure for the Plk3 PBD, we aligned the sequences of Plk1 with Plk3 and identified residues of Plk3 that corresponded to the Plk1 PBD. We then generated a homology model of the Plk3 PBD based on the Plk1 PBD (PDB accession code: 3FVH) through the online Robetta server.27 Using the crystal structure of the Plk1 PBD having ligand 2a bound (PDB accession code: 3RQ7) we superimposed the crystal structure of the apo Plk2 PBD (PDB accession code: 4RS6) and the Plk3 PBD homology model. Ligand 2a was oriented into the Plk2 and Plk3 PBDs as in the Plk1 PBD and residues proximal to the ligands were minimized (Figure 3). We found that all three PBDs were highly similar in the region of the cryptic pocket. Several of the residues from Plk1 (creme; Y417, Y421, Y481, F482 and Y485) and Plk2 (orange; Y510, Y514, Y574, F575 and Y578) are closely oriented. Although these residues were also found in the Plk3 PBD, certain side chains were rotated or slightly displaced in the latter domain (blue; Y470, F474, Y534, F535 and Y538) (Figure 3). Notable differences among the tree domains appeared to be a single residue at the terminus of the pocket, which consisted of L478, V571 and I531 for Plk1, Plk2 and Plk3, respectively. Additionally, while similar orientations were observed for K513 and K473 in Plk2 and Plk3, respectively, the corresponding K420 in Plk1 was considerably displaced. The most significant variations were in residues bordering a recently identified “auxiliary binding region”, which is contiguous with the canonical cryptic pocket.19 Here the differences for Plk1, Plk2 and Plk3 were found to be N484, H577 and S537; E488, E581 and Q541 and H489, N582 and H542, respectively. In spite of the general overall high similarity of residues proximal to the cryptic pocket among the three PBDs, it is possible that the minor differences noted as well as the significant variations in the auxiliary pocket could potentially contribute to significant modulation of ligand recognition. Although beyond the scope of the current work, replacing select residues identified above in the PBDs of Plk2 and Plk3 could help to resolve speculation regarding the roles that these may play in ligand selectivity.
Figure 3.

Superposition of residues judged to potentially impact interactions in the cryptic pocket as well as in the “auxiliary pocket” (encircled in red). Residue carbons and residue numbering are color keyed as shown in the legend.
3. Conclusions
Targeting the PBD offers a conceptually attractive alternative to KD-directed inhibitors for down-regulating Plk1 function.2,7 Achieving selectivity is an important consideration in the development of Plk1 PBD-binding inhibitors. In our current work we demonstrate that structural variation of functionality intended to interact within the cryptic pocket can have a significant impact on the relative affinities of peptides for the PBDs of Plk2 and Plk3 relative to Plk1. In the case of Plk3, specificity can exceed three orders-of-magnitude. We have found subtle differences in residues proximal to the putative cryptic pockets and particularly in residues forming a recently identified proximal auxiliary binding pocket. We suggest that these differences may contribute to ligand selectivity. Introducing extended functionality that permits access to the auxiliary pocket may be a valuable approach to increasing differences in binding affinity among the three domains.
A further significant challenge in developing clinically relevant PBD-binding inhibitors arises from the fact that PBD-binding peptides and peptide mimetics, even those showing extremely high affinities in extracellular assays, have frequently shown disappointing biological efficacies in cell-based systems.12 The discrepancies in potencies may result from poor cellular uptake. Accordingly, increasing cellular bioavailability of PBD-binding inhibitors represents an active area of investigation.28 Our current peptides are not clinically relevant, due in part to poor bioavailability. However, given the extreme enhancement in overall ligand affinity that can be achieved by accessing the cryptic pocket, we believe that optimizing interactions within this region may be highly relevant to developing more bioavailable agents. Accordingly, our current findings may facilitate the further development of clinical agents.
4. Experimental Section
4.1. General chemical methods
Proton (1H) and carbon (13C) NMR spectra were recorded on a Varian 400 MHz spectrometer or a Varian 500 MHz spectrometer and are reported in ppm relative to TMS and referenced to the solvent in which the spectra were collected. Solvent was removed by rotary evaporation under reduced pressure and anhydrous solvents were obtained commercially and used without further drying. Purification by silica gel chromatography was performed using Combiflash with EtOAc–hexanes or CH2Cl2-MeOH solvent systems. Electrospray ionization-mass spectrometra (ESI-MS) were acquired with an Agilent LC/MSD system equipped with a multimode ion source. High resolution mass spectrometric (HRMS) were acquired with a LTQ-Orbitrap-XL at 30 K resolution by LC/MS-ESI.
4.1.1. General procedure A for the synthesis of alkynes (10a, b) using Sonogashira reaction21
Appropriate terminal alkynes (0.45 mmol) were added to a mixture of bromides (9a, b) (0.3 mmol) and bis(triphenylphosphine)palladium (II) dichloride (9 μmol), DIEA (0.3 mmol) and copper (I) iodide (0.03 mmol) in DMF (1.5 mL). The reaction mixture was sealed and heated under argon (70 °C, 18 h). The crude mixture was purified by silica gel chromatography to provide alkynes (10a, b).
4.1.2. General procedure B for the synthesis of alcohols (11a, 17a) using Wilkinson’s catalyst29
A mixture of alkynes (10a, 16a) (1.0 mmol), tris(triphenylphosphine)rhodium(I) chloride (0.1 mmol) in THF (10 mL) and tBuOH (10 mL) was stirred under H2 (16 h). The crude mixture was purified by silica gel chromatography to provide alcohols (11a, 17a).
4.1.3. General procedure C for the synthesis of alcohols (11b, 17b) using Pd·C
To a solution of alkynes (10b, 16b) (10 mmol) in MeOH (10 mL) was added Pd·C (30 mg, 10%) and the mixture was hydrogenated under H2. When all starting material had been consumed (TLC), the reaction mixture was collected by filtration and concentrated. The crude residue was purified by silica gel chromatography to provide alcohols (11b, 17b).
4.1.4. General procedure D for the synthesis of aldehydes (13a, b)22
Potassium carbonate (100 mmol) was added to a solution of 2,6-difluorobenzaldehydes (12a) (102 mmol) and 3-(benzyloxy)phenol or 3-(tert-butoxy) phenol (100 mmol) in dimethylacetamide (DMA) (50 mL). The reaction mixture was stirred and refluxed (2 h). The crude mixture was partitioned between CH2Cl2 and H2O, dried (Na2SO4), concentrated and purified by silica gel chromatography to provide aldehydes (13a, b).
4.1.5. General procedure E for the synthesis of benzylalcohols (14a, b)
NaBH4 (20 mmol) was added portion-wise to the solution of aldehydes (13a, b) (20 mmol) in MeOH (100 mL) (0 °C). The reaction mixture was stirred (0 °C, 0.5 h) and concentrated. The crude mixture was partitioned between EtOAc and H2O, dried (Na2SO4), concentrated and purified by silica gel chromatography to provide benzylalcohols (14a, b).
4.1.6. General procedure F for the synthesis of benzylbromide (15a, b)
PPh3 (30 mmol) was added to a solution of benzylalcohols (14a, b) (20 mmol) in acetonitrile (100 mL). CBr4 (30 mmol) was added (0 °C). The formed suspension was stirred (30 min). The mixture was filtered and the filtrate was concentrated. The crude residue was purified by silica gel chromatography to provide benzylbromides (15a, b).
4.1.7. General procedure G for the synthesis of Nπ-alkylated His residues (18a–d)19,20
According to our previous reports,19,20 a solution of alcohols (11a, b and 17a, b) (2 mmol) and DIEA (2 mmol) in CH2Cl2 (1.5 mL) was added dropwise to a solution of trifluoromethanesulfonic anhydride (2 mmol, 1.0 M in CH2Cl2) in CH2Cl2 (2.5 mL) under argon (−78° C) and stirred (20 min). A solution of 2,4-dimethoxybenzyl Nα-Fmoc-Nτ-Trt-L-histidinate (2 mmol) or benzyl Nα-Fmoc-Nτ-Trt-L-histidinate (2 mmol) in CH2Cl2 (5 mL) was added (−78 °C). The mixture was stirred (16 h) and concentrated. The residue was deprotected using TFA:TIS (10:1, 10 mL) or hydrogenated using Pd·C (10%) in MeOH. The resultant crude residue was purified by silica gel chromatography to provide Nπ-alkylated His residues (18a–d).
4.1.8. 1-Bromo-4-(2-(tert-butoxy)ethyl)benzene (9a).30
Magnesium perchlorate (0.96 g, 4.3 mmol) and commercial available 2-(4-bromophenyl)ethanol 8 (8.6 g, 43 mmol) were dissolved in DCM (50 mL). (Boc)2O (21 g, 98 mmol) was added to the suspension. The mixture was stirred (35 °C, 16 h) and concentrated. The crude residue was purified by silica gel column chromatography to provide 9a as a colorless oil (7.6 g, 69% yield). 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.3 Hz, 2H), 3.54 (t, J = 7.2 Hz, 2H), 2.79 (t, J = 7.1 Hz, 2H), 1.18 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 138.57, 131.22 (2C), 130.79 (2C), 119.85, 72.90, 62.56, 36.78, 27.50 (3C).
4.1.9. 1-(2-(Benzyloxy)ethyl)-4-bromobenzene (9b)
2-(4-Bromophenyl)ethanol 8 (3.2 g, 16 mmol) and (bromomethyl)benzene (1.9 mL, 16 mmol) were dissolved in THF (50 mL). NaH (0.45 g, 18 mmol) was added (0 °C). The reaction mixture was stirred (from 0 °C to room temperature, 16 h), quenched by pouring into water, extracted by EtOAc, dried (Na2SO4) and concentrated. The crude residue was purified by silica gel chromatography to provide 9b as a colorless oil (1.9 g, 41% yield). 1H NMR (400 MHz, CDCl3) δ 7.45 - 7.43 (m, 2H), 7.39 - 7.31 (m, 5H), 7.15 - 7.13 (m, 2H), 4.55 (s, 2H), 3.70 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H).13C NMR (101 MHz, CDCl3) δ 138.26, 138.13, 131.38 (2C), 130.72 (2C), 128.40 (2C), 127.61 (3C), 120.03, 73.03, 70.77, 35.80.
4.1.10. 8-(4-(2-(tert-Butoxy)ethyl)phenyl)oct-7-yn-1-ol (10a)
Treatment of 9a and commercial available oct-7-yn-1-ol as outlined in general procedure A provided 10a as a yellow oil (41% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 3.66 (td, J = 6.6, 5.3 Hz, 2H), 3.53 (t, J = 7.3 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.42 (t, J = 7.0 Hz, 2H), 1.66 - 1.58 (m, 5H), 1.54 - 1.48 (m, 2H), 1.47 - 1.40 (m, 2H), 1.17 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 139.02, 131.36 (2C), 128.90 (2C), 121.67, 89.66, 80.67, 72.92, 62.86, 62.72, 37.24, 32.66, 28.73, 28.68, 27.49 (3C), 25.32, 19.36. ESI-MS m/z: 325.2 (MNa+).
4.1.11. 8-(4-(2-(Benzyloxy)ethyl)phenyl)oct-7-yn-1-ol (10b)
Treatment of 9b with commercial available oct-7-yn-1-ol as outlined in general procedure A provided 10b as a yellow oil (62% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 - 7.29 (m, 7H), 7.16 (d, J = 8.1 Hz, 2H), 4.53 (s, 2H), 3.68 (dt, J = 9.2, 6.8 Hz, 4H), 2.94 - 2.91 (m, 2H), 2.43 (t, J = 7.0 Hz, 2H), 1.74 (bs, 1H), 1.68 - 1.60 (m, 4H), 1.55 - 1.48 (m, 2H), 1.47 - 1.41 (m, 2H).
4.1.12. 8-(4-(2-(tert-Butoxy)ethyl)phenyl)octan-1-ol (11a)
Treatment of 10a as outlined in general procedure C provided 11a as a yellow solid (54% yield). 1H NMR (400 MHz, CDCl3) δ 7.17 - 7.11 (m, 4H), 3.65 (t, J = 6.6 Hz, 2H), 3.55 (t, J = 7.7 Hz, 2H), 2.82 (t, J = 7.7 Hz, 2H), 2.59 (t, J = 7.7 Hz, 2H), 11.63 - 1.56 (m, 4H), 1.40 - 1.30 (m, 8H), 1.21 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 140.60, 136.39, 128.85 (2C), 128.27 (2C), 72.90, 63.25, 63.03, 37.08, 35.56, 32.78, 31.53, 29.47, 29.36, 29.25, 27.55 (3C), 25.74. APCI-MS m/z: 307.2 (MNa+).
4.1.13. 8-(4-(2-(Benzyloxy)ethyl)phenyl)octan-1-ol (11b)
Treatment of 10b as outlined in general procedure B provided 11b as a colorless oil (29% yield). 1H NMR (400 MHz, CDCl3) δ 7.39 - 7.29 (m, 5H), 7.23 - 7.22 (m, 1H), 7.18 - 7.12 (m, 3H), 4.56 (s, 2H), 3.72 (q, J = 7.1 Hz, 2H), 3.65 (td, J = 6.6, 4.5 Hz, 2H), 2.94 (t, J = 7.2 Hz, 2H), 2.62 - 2.58 (m, 1H), 1.65 - 1.50 (m, 6H), 1.40 - 1.30 (m, 7H). 13C NMR (101 MHz, CDCl3) δ 140.72, 138.45, 136.04, 128.80 (2C), 128.38 (2C), 128.36 (2C), 127.64 (2C), 127.53 (2C), 72.95, 71.41, 63.05, 35.96, 35.57, 32.79, 31.54, 29.48, 29.37, 29.26, 25.75. DUIS-MS m/z: 341 (MH+).
4.1.14. 2-(3-(tert-Butoxy)phenoxy)-6-fluorobenzaldehyde (13a)
Treatment of commercial available 2,6-difluorobenzaldehyde 12 and 3-(tert-butoxy)phenol as outlined in general procedure D provided 13a as a yellow oil (78% yield). 1H NMR (400 MHz, CDCl3) δ 10.51 (s, 1H), 7.46 - 7.40 (m, 1H), 7.31 - 7.27 (m, 1H), 6.89 - 6.80 (m, 3H),6.73 (t, J = 2.2 Hz, 1H), 6.66 (d, J = 8.4 Hz, 1H), 1.37 (s, 9H).13C NMR (101 MHz, CDCl3) δ 186.78 (d, J = 2.4 Hz), 162.90 (d, J = 263.4 Hz), 160.43 (d, J = 5.2 Hz), 157.14, 155.93, 135.69 (d, J = 11.6 Hz), 129.90, 120.42, 115.95 (d, J = 9.4 Hz), 115.65, 114.64, 113.41 (d, J = 3.8 Hz), 110.77 (d, J = 21.2 Hz), 79.24, 28.84 (3C). ESI-MS m/z: 271.1 (MH+-OH), 311.1 (MNa+). [3-(tert-butoxy) phenol was prepared from commercial available 3-(benzyloxy)phenol through two steps below.30 Magnesium perchlorate (0.56 g, 2.5 mmol) and 3-(benzyloxy)phenol (5 g, 25 mmol) were dissolved in DCM (50 mL). (Boc)2O (12.5 g, 57 mmol) was added. The formed brown solution was stirred (35 °C, 16h) and concentrated. The crude residue was purified by silica gel chromatography to provide 1-(benzyloxy)-3-(tert-butoxy)benzene as a colorless oil (4.6 g, 72% yield). 1H NMR (400 MHz, CDCl3) δ 7.49 - 7.34 (m, 5H), 7.21 (t, J = 8.2 Hz, 1H), 6.77 (dd, J = 7.9, 1.9 Hz, 1H), 6.69 - 6.66 (m, 2H), 5.08 (s, 2H), 1.39 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 159.36, 156.62, 137.06, 129.15, 128.59 (2C), 127.96, 127.54 (2C), 116.73, 111.10, 109.87, 78.64, 70.08, 28.93 (2C). Hydrogenation of 1-(benzyloxy)-3-(tert-butoxy) benzene (4.0 g, 15.9 mmol) using Pd·C (10%, 60 mg) in MeOH (50 mL) (4 h) provided 3-(tert-butoxy)phenol a yellow solid (2.4 g, 93% yield). 1H NMR (400 MHz, CDCl3) δ 7.10 (t, J = 8.1 Hz, 1H), 7.05 (brs, 1H), 6.61 (ddd, J = 8.1, 2.4, 1.5 Hz, 2H), 6.55 (t, J = 2.3 Hz, 1H), 1.35 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.37, 155.67, 129.51, 116.52, 111.83, 111.31, 79.73, 28.79 (3C). ESI-MS m/z: 167.1 (MH+).]
4.1.15. 2-(3-(Benzyloxy)phenoxy)-6-fluorobenzaldehyde (13b)
Treatment of commercial available 2,6-difluorobenzaldehyde (12) with 3-(benzyloxy)phenol as outlined in general procedure D provided 13b as a brown oil (18% yield). 1H NMR (400 MHz, CDCl3) δ 10.40 (s, 1H), 7.37 - 7.18 (m, 7H), 6.82 - 6.77 (m, 1H), 6.75 (dd, J = 8.3, 2.3 Hz, 1H), 6.61 - 6.58 (m, 3H), 4.97 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 186.76, 162.90 (d, J = 263.5 Hz), 160.28, 160.08 (d, J = 5.2 Hz), 156.88, 136.42, 135.69 (d, J = 11.6 Hz), 130.61, 128.65 (2C), 128.14, 127.51 (2C), 116.21 (d, J = 9.5 Hz), 113.97 (d, J = 3.7 Hz), 111.97, 111.25, 111.05 (d, J = 21.2 Hz), 106.67, 70.26.
4.1.16. (2-(3-(tert-Butoxy)phenoxy)-6-fluorophenyl)methanol (14a)
Treatment of 13a as outlined in general procedure E provided 14a as a colorless oil (93% yield). 1H NMR (400 MHz, CDCl3) δ 7.24 (t, J = 8.1 Hz, 1H), 7.21 - 7.17 (m, 1H), 6.87 - 6.80 (m, 2H), 6.77 - 6.74 (m, 1H), 6.69 (t, J = 2.3 Hz, 1H), 6.65 (d, J = 8.5 Hz, 1H), 4.83 (d, J = 1.0 Hz, 2H), 2.32 (brs, 1H), 1.36 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 161.68 (d, J = 247.3 Hz), 156.97 (2C), 156.91 (d, J = 7.4 Hz), 129.67, 129.53 (d, J = 10.4 Hz), 119.59, 119.34 (d, J = 18.1 Hz), 114.93, 113.89, 113.71 (d, J = 3.3 Hz), 110.44 (d, J = 22.6 Hz), 79.06, 53.97 (d, J = 4.9 Hz), 28.84 (3C). ESI-MS m/z: 291.1 (MH+), 313.1 (MNa+).
4.1.17. (2-(3-(Benzyloxy)phenoxy)-6-fluorophenyl)methanol (14b)
Treatment of 13b as outlined in general procedure E provided 14b as a colorless oil (79% yield). 1H NMR (400 MHz, CDCl3) δ 7.46 - 7.34 (m, 5H), 7.30 - 7.26 (m, 1H), 7.25 - 7.20 (m, 1H), 6.92 - 6.87 (m, 1H), 6.80 (ddd, J = 8.4, 2.4, 0.9 Hz, 1H), 6.72 - 6.63 (m, 3H), 5.06 (s, 2H), 4.83 (d, J = 1.4 Hz, 2H), 2.18 (S, 1H). 13C NMR (101 MHz, CDCl3) δ 161.71 (d, J = 247.6 Hz), 160.20, 157.94, 156.52 (d, J = 7.3 Hz), 136.57, 130.44, 129.63 (d, J = 10.4 Hz), 128.64 (2C), 128.11, 127.54 (2C), 119.67 (d, J = 17.9 Hz), 114.34 (d, J = 3.3 Hz), 111.18, 110.75 (d, J = 22.5 Hz), 110.41, 105.81, 70.20, 54.04 (d, J = 5.1 Hz).
4.1.18. 2-(Bromomethyl)-1-(3-(tert-butoxy)phenoxy)-3-fluorobenzene (15a)
Treatment of 14a as outlined in general procedure F provided 15a as a colorless oil (90% yield). 1H NMR (400 MHz, CDCl3) δ 7.29 - 7.25 (m, 1H), 7.21 (td, J = 8.4, 6.5 Hz, 1H), 6.86 - 6.80 (m, 3H), 6.73 (t, J = 2.3 Hz, 1H), 6.63 (d, J = 8.5 Hz, 1H), 4.67 (s, 2H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 161.51 (d, J = 250.4 Hz), 156.95, 156.82 (d, J = 6.5 Hz), 156.73, 130.11 (d, J = 10.6 Hz), 129.65, 119.87, 117.08 (d, J = 17.2 Hz), 115.38, 114.44, 113.50 (d, J = 3.3 Hz), 110.11 (d, J = 21.6 Hz), 79.07, 28.87 (3C), 20.03 (d, J = 5.4 Hz).
4.1.19. 1-(3-(Benzyloxy)phenoxy)-2-(bromomethyl)-3-fluorobenzene (15b)
Treatment of 14b as outlined in general procedure F provided 15b as a colorless oil (87% yield). 1H NMR (400 MHz, CDCl3) δ 7.33 - 7.23 (m, 5H), 7.19 - 7.14 (m, 1H), 7.12 - 7.06 (m, 1H), 6.76 - 6.68 (m, 2H), 6.60 - 6.52 (m, 3H), 4.94 (s, 2H), 4.53 (d, J = 1.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 161.54 (d, J = 250.6 Hz), 160.18, 157.57, 156.51 (d, J = 6.5 Hz), 136.59, 130.40, 130.18 (d, J = 10.4 Hz), 128.65 (2C), 128.11, 127.57 (2C), 117.34 (d, J = 17.1 Hz), 113.93 (d, J = 3.3 Hz), 111.80, 110.74, 110.35 (d, J = 21.6 Hz), 106.37, 70.21, 20.04 (d, J = 5.4 Hz). APCI-MS m/z: 387.0, 389.0 (MH+).
4.1.20. 2-(8-(Benzyloxy)oct-2-yn-1-yl)-1-(3-(tert-butoxy)phenoxy)-3-fluorobenzene (16a).31
A solution of butyllithium in hexanes (10 mmol, 2.5 M) was added dropwise to a solution of ((hept-6-yn-1-yloxy)methyl)benzene (10 mmol) [which was prepared from commercial available hept-6-yn-1-ol using BnBr and NaH in THF (0°C) (78% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 - 7.35 (m, 4H), 7.33 - 7.29 (m, 1H), 4.53 (s, 2H), 3.50 (t, J = 6.5 Hz, 2H), 2.22 (td, J = 6.8, 2.6 Hz, 2H), 1.96 (t, J = 2.6 Hz, 1H), 1.71 - 1.61 (m, 2H), 1.60 - 1.47 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 138.65, 128.36 (2C), 127.62, 127.50 (2C), 84.53, 72.92, 70.23, 68.29, 29.29, 28.34, 25.42, 18.39. ESI-MS m/z: 203.1 (MH+).] in THF (20 mL) ( −78 °C). The mixture was stirred (−78 °C, 1 h). The solution of benzylbromides (15a) (3 mmol) in THF (10 mL) was added. The reaction mixture was stirred (−78 °C to room temperature, 16 h), quenched by pouring into ice, extracted by EtOAc and concentrated. The crude residue was purified by silica gel chromatography to provide benzylalkynes (16a) as a colorless oil (77% yield). 1H NMR (400 MHz, CDCl3) δ 7.38 - 7.3 (m, 4H), 7.33 - 7.27 (m, 1H), 7.21 (t, J = 8.2 Hz, 1H), 7.15 (td, J = 8.3, 6.5 Hz, 1H), 6.85 (t, J = 8.7 Hz, 1H), 6.79 - 6.72 (m, 2H), 6.67 (dd, J = 5.3, 3.0 Hz, 2H), 4.51 (s, 2H), 3.59 (brs, 2H), 3.46 (t, J = 6.6 Hz, 2H), 2.11 - 2.07 (m, 2H), 1.64 - 1.57 (m, 2H), 1.47 - 1.39 (m, 4H), 1.36 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 161.50 (d, J = 246.8 Hz), 157.63, 156.78, 155.95 (d, J = 7.3 Hz), 138.69, 129.36, 128.34 (2C), 127.95 (d, J = 10.2 Hz), 127.61 (2C), 127.46, 119.00, 117.51 (d, J = 18.2 Hz), 114.56, 114.46 (d, J = 3.3 Hz), 113.58, 110.52 (d, J = 22.3 Hz), 79.75, 78.84, 76.71, 72.86, 70.34, 29.28, 28.87 (3C), 28.70, 25.43, 18.74, 12.99 (d, J = 4.8 Hz).
4.1.21. 8-(2-(3-(tert-Butoxy)phenoxy)-6-fluorophenyl)octan-1-ol (17a)
Treatment of 16a as outlined in general procedure C provided 17a as a yellow solid (95% yield). 1H NMR (400 MHz, CDCl3) δ 7.21 (t, J = 8.1 Hz, 1H), 7.09 (td, J = 8.2, 6.5 Hz, 1H), 6.83 (t, J = 8.8 Hz, 1H), 6.77 - 6.74 (m, 1H), 6.70 - 6.66 (m, 2H), 6.61 (t, J = 2.3 Hz, 1H), 3.62 (td, J = 6.6, 1.3 Hz, 2H), 2.67 (td, J = 7.6, 1.6 Hz, 2H), 1.61 - 1.51 (m, 4H), 1.36 (s, 9H), 1.33 - 1.26 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 162.04 (d, J = 244.6 Hz), 158.02, 156.74, 155.84 (d, J = 8.3 Hz), 129.40, 126.85 (d, J = 10.4 Hz), 122.17 (d, J = 18.7 Hz), 118.68, 114.70 (d, J = 3.1 Hz), 114.02, 113.00, 110.50 (d, J = 23.1 Hz), 78.91, 63.00, 32.77, 29.43, 29.32, 29.25, 29.22, 28.84 (3C), 25.69, 22.98 (d, J = 2.7 Hz). ESI-MS m/z: 389.2 (MH+), 333.1 (MH+ - Bu).
4.1.22. 8-(2-(3-(Benzyloxy)phenoxy)-6-fluorophenyl)octan-1-ol (17b)
Treatment of 15b with hept-6-yn-1-ol using CuI, Cs2CO3 and TBAI in acetonitrile (70 °C, 20 h) as the reported method31 followed by using the synthesis as outlined in general procedure B provided 17b as a colorless oil (9% yield). 1H NMR (400 MHz, CDCl3) δ 7.45 - 7.35 (m, 5H), 7.28 - 7.22 (m, 1H), 7.14 - 7.08 (m, 1H), 6.88 - 6.83 (m, 1H), 6.76 - 6.70 (m, 2H), 6.61 - 6.56 (m, 2H), 5.05 (s, 2H), 3.64 (t, J = 6.6 Hz, 2H), 2.67 (td, J = 7.6, 1.6 Hz, 2H), 1.62 - 1.52 (m, 4H), 1.33 (qt, J = 8.3, 3.2 Hz, 8H). 13C NMR (101 MHz, CDCl3) δ 162.06 (d, J = 244.7 Hz), 160.10, 158.87, 155.56 (d, J = 8.3 Hz), 136.68, 130.18, 128.61 (2C), 128.06, 127.56 (2C), 126.93 (d, J = 10.3 Hz), 122.42 (d, J = 18.8 Hz), 115.12 (d, J = 3.1 Hz), 110.71 (d, J = 23.0 Hz), 110.47, 109.29, 105.03, 70.15, 63.06, 32.78, 29.44, 29.35, 29.29 (2C), 29.26, 25.71, 23.06, 23.04. ESI-MS m/z: 423.2 (MH+), 435.2 (MNa+).
4.1.23. Nα-(((9H-Fluoren-9-yl)methoxy)carbonyl)-Nπ-(8-(4-(2-(tertbutoxy) ethyl)phenyl)octyl)-L-histidine (18a)
Treatment of 11a as outlined in general procedure G provided 18a as a yellow oil (30% yield). DUIS-MS m/z: 667 (MH+).
4.1.24. Nα-(((9H-Fluoren-9-yl)methoxy)carbonyl)-Nπ-(8-(2-(3-(tertbutoxy) phenoxy)-6-fluorophenyl)octyl)-L-histidine (18b)
Treatment of 17a as outlined in general procedure G provided 18b as a yellow solid (30% yield). ESI-MS m/z: 748.3 (MH+).
4.1.25. Nα-(((9H-Fluoren-9-yl)methoxy)carbonyl)-Nπ-(8-(4-(2-(benzyloxy)ethyl)phenyl)octyl)-L-histidine (18c)
Treatment of 11b as outlined in general procedure G provided 18c as a colorless oil (64% yield). ESI-MS m/z: 700.3 (MH+).
4.1.26. Nα-(((9H-Fluoren-9-yl)methoxy)carbonyl)-Nπ-(8-(2-(3-(benzyloxy)phenoxy)-6-fluorophenyl)octyl)-L-histidine (18d)
Treatment of 11b as outlined in general procedure G provided 18d as a yellow oil (65% yield). ESI-MS m/z: 782.3 (MH+).
4.1.27. General solid-phase peptide synthesis (SPPS
Protected amino acids used were Fmoc-Thr(PO(OBzl)OH)-OH, Fmoc-Ser(OtBu)-OH, N(π)-alkylated Fmoc-His-OH (18a–d), Fmoc-Leu-OH, and Fmoc-Pro-OH (purchased from Novabiochem). Peptides were synthesized on a NovaSyn® TGR resin (Novabiochem Cat#. 855009) using standard Fmoc solid-phase protocols in N-methyl-2-pyrrolidone (NMP). 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (5.0 equivalents), and N,N-diisopropylethylamine (DIPEA) (10 equivalents) were used as coupling reagents. Unnatural amino acid residues were coupled using 2.5 equivalents. Deprotection was performed using 20% piperidine in DMF (15 min, twice). Amino-terminal acetylation was performed using 1-acetylimidazole. Finished resins were washed with NMP, MeOH, CH2Cl2, and Et2O, dried under vacuum, and then cleaved by treatment with a solution of TFA:H2O:TIS (95:2.5:2.5) (5 h). The resin was removed by filtration, and the filtrate was concentrated under vacuum and the resulting residue dissolved in 50% aqueous acetonitrile (5 mL) and purified by reverse-phase preparative high pressure liquid chromatography (HPLC) using a Waters 2535 system having photodiode array detection and Phenomenex C18 columns (Cat. No. 00G-4436-P0-AX, 250 mm × 21.2 mm 10 μm particle size, 110 A pore) at a flow rate of 10 mL/min. Binary solvent systems consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in acetonitrile were employed with gradients as indicated. Products (6a, b and 7a, b) were obtained as amorphous solids following lyophilization.
4.1.28. Peptides
Peptide 6a
Title peptide 6a was prepared using 18a as outlined in SPPS and purified by preparative HPLC (linear gradient of 0% B to 80% B over 30 min, retention time = 17.7 min). ESI-MS m/z: 907.4 (MH+). HRMS calcd for C42H68N8O12P(MH+) 907.4689, found 907.4676.
Peptide 6b
Title peptide 6b was prepared using 18b as outlined in SPPS and purified by preparative HPLC (linear gradient of 0% B to 80% B over 30 min, retention time = 19.7 min). ESI-MS m/z: 989.2 (MH+). HRMS calcd for C46H67FN8O13P(MH+) 989.4544, found 989.4516.
Peptide 7a
Title peptide 7a was prepared using 18c as outlined in SPPS and purified by preparative HPLC (linear gradient of 0% B to 80% B over 30 min, retention time = 21.6 min). ESI-MS m/z: 997.3 (MH+).
Peptide 7b
Title peptide 7b was prepared using 18d as outlined in SPPS and purified by preparative HPLC (linear gradient of 0% B to 80% B over 30 min, retention time = 22.3 min). ESI-MS m/z: 1079.2 (MH+). HRMS calcd for C53H73FN8O13P (MH+) 1079.5013, found 1079.4993.
4.2 Biological Experimental Procedures
Experimental procedures used to obtain the IC50 values shown in Table 1 have been previously reported24 and are described in detail in the Supplementary data.
4.3. Comparison of the PBDs of Plk1, Plk2 and Plk3 in the region corresponding to the cryptic binding pocket of Plk1
4.3.1. ICM Modeling protocols
Standard Molsoft parameters and procedures were used as defined in the unmodified ICM Chemist Pro version 3.8-5/MacOSXIL64bit/g4.2/Release/HardwareGL/Gui264199/WebKit/Video/FlexLM built Oct 17 2016 14:22. 4.8.7.
4.3.2. Sequence alignment and identification of cryptic pocket residues
ICM Modeling was preformed using Plk1 (PDB accession code: 3RQ7), Plk2 (PDB accession code: 4RS6) and Plk3 [(Robetta homology model based on PDB accession code: 3FVH (see Supplementary data)]. Sequences were aligned using 3RQ7 as the static reference. The 2a ligand from the 3RQ7 structure was placed in each structure based on the protein alignments, then all neighboring residues were minimized using the ICM standard protocol. Results are shown in Figure 3.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Detailed biological procedures used to prepare the IC50 values shown in Table 1 and the sequences and alignment for Plk1, Plk2 and Plk3 and assignment of residues corresponding their respective PBDs can be found in the online version at http://dx.doi.org/10.1016/j.bmc.2017.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Nat Rev Mol Cell Biol. 2014;15:433–452. doi: 10.1038/nrm3819. [DOI] [PubMed] [Google Scholar]
- 2.Lee KS, Burke TR, Park JE, Bang JK, Lee E. Trends Pharmacol Sci. 2015;36:858–877. doi: 10.1016/j.tips.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Carcer G, Escobar B, Higuero AM, Garcia L, Anson A, Perez G, Mollejo M, Manning G, Melendez B, Abad-Rodriguez J, Malumbres M. Mol Cell Biol. 2011;31:1225–1239. doi: 10.1128/MCB.00607-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elia AEH, Cantley LC, Yaffe MB. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 5.Strebhardt K. Nat Rev Drug Discov. 2010;9:643–659. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 6.Park JE, Erikson RL, Lee KS. Proc Natl Acad Sci U S A. 2011;108:8200–8205. doi: 10.1073/pnas.1102020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg A, Berg T. ChemBioChem. 2016;17:650–656. doi: 10.1002/cbic.201500580. [DOI] [PubMed] [Google Scholar]
- 8.Reindl W, Strebhardt K, Berg T. Anal Biochem. 2008;383:205–209. doi: 10.1016/j.ab.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Reindl W, Graeber M, Strebhardt K, Berg T. Anal Biochem. 2009;395:189–194. doi: 10.1016/j.ab.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 10.Archambault V, Lepine G, Kachaner D. Oncogene. 2015;34:4799–4807. doi: 10.1038/onc.2014.451. [DOI] [PubMed] [Google Scholar]
- 11.Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao C, Soung NK, Lee S, Yoon DY, Lim Y, Lee DH, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu F, Park JE, Qian WJ, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR., Jr Nat Chem Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, Park JE, Qian WJ, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. ACS Chem Biol. 2012;7:805–810. doi: 10.1021/cb200469a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu F, Park JE, Qian WJ, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. ChemBioChem. 2012;13:1291–1296. doi: 10.1002/cbic.201200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sledz P, Stubbs CJ, Lang S, Yang YQ, McKenzie GJ, Venkitaraman AR, Hyvoenen M, Abell C. Angew Chem, Int Ed. 2011;50:4003–4006. doi: 10.1002/anie.201008019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan YS, Sledz P, Lang S, Stubbs CJ, Spring DR, Abell C, Best RB. Angew Chem, Int Ed. 2012;51:10078–10081. doi: 10.1002/anie.201205676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sledz P, Lang S, Stubbs CJ, Abell C. Angew Chem Int Ed Engl. 2012;51:7680–7683. doi: 10.1002/anie.201202660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F, Stephen AG, Waheed AA, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr ChemBioChem. 2008;9:2000–2004. doi: 10.1002/cbic.200800281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao XZ, Hymel D, Burke TR., Jr Bioorg Med Chem Lett. 2016;26:5009–5012. doi: 10.1016/j.bmcl.2016.08.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian W, Liu F, Burke TR., Jr J Org Chem. 2011;76:8885–8890. doi: 10.1021/jo201599c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]
- 22.Fish PV, Ryckmans T, Stobie A, Wakenhut F. Bioorg Med Chem Lett. 2008;18:1795–1798. doi: 10.1016/j.bmcl.2008.02.036. [DOI] [PubMed] [Google Scholar]
- 23.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 24.Hymel D, Burke TR., Jr ChemMedChem. 2017;12:202–206. doi: 10.1002/cmdc.201600574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan HM, Wang T, Quan JM. Biochem Biophys Res Commun. 2015;456:780–784. doi: 10.1016/j.bbrc.2014.11.125. [DOI] [PubMed] [Google Scholar]
- 26.Kim JH, Ku B, Lee KS, Kim SJ. Proteins: Struct, Funct, Bioinf. 2015;83:1201–1208. doi: 10.1002/prot.24804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DE, Chivian D, Baker D. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian WJ, Park JE, Lim D, Lai CC, Kelley JA, Park SY, Lee KW, Yaffe MB, Lee KS, Burke TR. Biopolymers Pept Sci. 2014;102:444–455. doi: 10.1002/bip.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jourdant A, Gonzalez-Zamora E, Zhu J. J Org Chem. 2002;67:3163–3164. doi: 10.1021/jo025595q. [DOI] [PubMed] [Google Scholar]
- 30.Bartoli G, Bosco M, Locatelli M, Marcantoni E, Melchiorre P, Sambri L. Org Lett. 2005;7:427–430. doi: 10.1021/ol047704o. [DOI] [PubMed] [Google Scholar]
- 31.Kwon Y, Cho H, Kim S. Org Lett. 2013;15:920–923. doi: 10.1021/ol400073s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.