

Abstract
Background & Aim
Primary sclerosing cholangitis (PSC) typically develops in middle-age adults. Little is known about phenotypic differences when PSC is diagnosed at various ages. Therefore, we sought to compare the clinical characteristics of a large PSC cohort based on the age when PSC was diagnosed.
Methods
We performed a multicenter retrospective review to compare the features of PSC among those diagnosed between 1–19 years (yrs) (n=95), 20–59 yrs (n=662), and 60–79 yrs (n=102).
Results
Those with an early diagnosis (ED) PSC were more likely to have small duct PSC (13%) versus those with a middle age diagnosis (MD) (5%) and late diagnosis (LD) groups (2%), p<0.01 and appeared to have a decrease risk of hepatobiliary malignancies: ED vs. MD: HR, 0.25; 95% CI 0.06–1.03 and ED vs LD: HR, 0.07; 95% CI 0.01–0.62. Cholangiocarcinoma (CCA) was diagnosed in 78 subjects (ED n=0; MD n=66, LD n=12) and was more likely to be diagnosed within a year after the PSC diagnosis among those found to have PSC late in life: ED 0% (0/95), MD 2% (14/662), LD 6% (6/102), p=0.02. Similarly, hepatic decompensation was more common among those with LD PSC versus younger individuals: LD vs. MD: HR,1.64; 95% CI 0.98–2.70 and LD vs ED: HR, 2.26; 95% CI 1.02–5.05.
Conclusions
Those diagnosed with PSC early in life are more likely to have small duct PSC and less likely to have disease related complications. Clinicians should be vigilant for underlying CCA among those with PSC diagnosed late in life.
Keywords: Primary sclerosing cholangitis, Cholangiocarcinoma, Liver transplantation, Inflammatory bowel disease
Introduction
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterized by inflammation of the intra- and or extra-hepatic bile ducts. Population based cohorts from Europe and North America report that 68–73% of PSC patients have concurrent inflammatory bowel disease (IBD), typically ulcerative colitis (UC). PSC can be associated with progressive liver fibrosis and complications stemming from portal hypertension and PSC associated malignancies.1
While PSC is typically diagnosed in middle-aged adults, smaller studies have suggested that the clinical features of PSC may vary depending upon the age when PSC is diagnosed. For example, among pediatric patients with PSC several studies have suggested that autoimmune hepatitis and small duct PSC is more prevalent while cholangiocarcinoma (CCA) is relatively rare when compared to their older counterparts.2–5
Overall there is a dearth of information concerning the features of PSC and risk of disease related complications when it is detected at the extremes of the age spectrum and few studies have been large enough to compare key phenotypic differences across age groups. Furthering our understanding of the heterogeneity of PSC can shed additional light on the natural history of the disorder, which can influence clinical practice. Consequently, we sought to compare the characteristics of a large cohort of PSC patients on the basis of the age when they were diagnosed with the disorder.
Methods
Subjects
Patients with PSC were identified and recruited between 2004–2013 from the PROGRESS (PSC Resource Of Genetic Risk, Environment and Synergy Studies) consortium and seen at one of 3 study sites (Mayo Clinic Rochester, MN, Mayo Clinic Jacksonville, FL or Mayo Clinic Scottsdale, AZ). Patients diagnosed with PSC by either cholangiography or liver biopsy were included and those suspected of having a secondary cause of sclerosing cholangitis were excluded. When PSC was suspected a magnetic resonance cholangiogram was performed. If this was negative and the suspicion for PSC did persist we typically performed a liver biopsy. Individuals who identified themselves as having PSC but did not have medical records available to confirm the diagnosis were excluded. This study was approved by the institutional review board for each participating center and conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
Data Collection & Key Definitions
The lead study center requested and reviewed the medical records of all study participants. This included medical records from all sites where the subjects received medical care either before or after being seen at one of the study sites. In order to determine the optimum cut-off for age groups, the lower and upper decile for the age of diagnosis was used for the early diagnosis (ED) and late diagnosis (LD) group, respectively. The median interquartile (IQR) age at the time of PSC diagnosis was 40 (27–51) years (yrs). The lower and upper decile for the age distribution for the entire cohort was 19 yrs and 60 yrs, correspondingly. Consequently, the age groups utilized in the study were ≤19 yrs, 20–59 yrs and ≥60 yrs for ED, middle-age diagnosis (MD) and LD-PSC, respectively.
Small duct PSC and overlap with concomitant autoimmune hepatitis were diagnosed using standard definitions.6, 7 Hepatic decompensation was defined as the development of ascites, variceal hemorrhage or encephalopathy. The development of hepatobiliary malignancies, CCA, hepatocellular carcinoma (HCC) and gallbladder cancer, was used as a composite endpoint. CCA, HCC and gallbladder cancer were diagnosed in the setting of a positive cytology or biopsy or definitive imaging features of cancer. The fibrosis stage was determined by applying the Batts-Ludwig criteria among patients who underwent a liver biopsy within1 year of their PSC diagnosis.8 Sequential laboratory tests were abstracted beginning from the first available laboratory tests after the diagnosis of PSC up to the time of their last clinical encounter. The model for end-stage liver disease (MELD) score at the time of diagnosis was also abstracted. Serum alkaline phosphatase (SAP) values were divided by the upper limit of normal (ULN) for that laboratory tests as the reference ranges for this assay varies between institutions and have changed over time. A SAP cutoff of 1.5 x the ULN was selected for additional analysis since this cutoff has been shown to have prognostic implications.9
Statistical Analysis
For the unadjusted results, categorical data are reported as percentages and compared using the chi-square test. Continuous data are reported as medians (IQR) and compared using the Kruskal-Wallis rank sum test. Cox proportional hazards regression analysis was performed for several outcomes of interest including colectomy, colorectal cancer (CRC) or high grade dysplasia (HGD), hepatobiliary malignancy, hepatic decompensation, liver transplantation and PSC related death. The baseline was time of PSC diagnosis for hepatobiliary outcomes of interest and time of IBD diagnosis for IBD-related events. For those who did not develop an endpoint, follow-up was censored at the time of their last follow up or liver transplant. However, in the Cox regression analysis for the development of colon cancer or HGD (whichever was earlier), subjects who underwent a colectomy for a non-malignant indication were censored at that time. Cumulative incidence for these endpoints was calculated treating death (and where appropriate, liver transplantation) as competing risks. The analysis was performed using SAS 9.4 (SAS Institute; Cary, NC) and R 3.2 (R Foundation for Statistical Computing; Vienna, Austria).
Results
Primary Sclerosing Cholangitis Features
Eight-hundred and fifty-nine patients with PSC were included (ED n=95, MD n=662, LD n=102) from Mayo Clinic in Rochester, MN (n=710), Jacksonville, FL (n=83) or Scottsdale, AZ (n=66). The patients were followed for a median (IQR) of 9.18 (4.31–16.10) yrs. Those with LD-PSC were followed for a shorter duration of time than the younger age groups (Table 1). Demographic features and basic characteristics are shown in Table 1. Small duct PSC was more prevalent in the ED group, 13% vs. 2–5% (p<0.01). Similarly, those with ED-PSC had a higher prevalence of autoimmune hepatitis when compared to the MD group (7% vs. 3%, p=0.03) but not the LD cohort (7% vs. 5%, p=0.56). When subjects with autoimmune hepatitis were excluded the prevalence of small duct PSC continued to be higher in the ED group (12%) versus those in the MD (4%) and LD PSC cohorts (2%), p<0.01. The fibrosis stage was similar across age groups among the 292 individuals who had a liver biopsy within 1 year of their PSC diagnosis (Table 1).
Table 1.
Selected Features of Primary Sclerosing Cholangitis
| % (n) or Median (IQR)
|
P-value
|
|||||
|---|---|---|---|---|---|---|
| Early (n=95) | Middle-Age (n=662) | Late (n=102) | Early vs. Middle | Late vs. Middle | Early vs. Late | |
| Age (yrs) at Diagnosis | 16.6 (14.1–18.2) | 39.5 (30.2–48.2) | 65.2 (62.0–69.1) | <0.001 | <0.001 | <0.001 |
|
|
||||||
| Female | 36% (34/95) | 40% (262/662) | 30% (31/102) | 0.50 | 0.08 | 0.45 |
|
|
||||||
| Small Duct PSC | 13% (11/88) | 5% (29/624) | 2% (2/98) | <0.01 | 0.42 | <0.01 |
|
|
||||||
| Autoimmune Hepatitis | 7% (7/95) | 3% (18/662) | 5% (5/102) | 0.03 | 0.22 | 0.56 |
|
|
||||||
| SAP x ULN at Diagnosis | 0.95 (0.5–1.8) | 2.2 (1.3–3.8) | 2.2 (1.2–3.1) | <0.001 | 0.29 | <0.001 |
|
|
||||||
| SAP <1.5 x ULN at Diagnosis | 68% (27/40) | 34% (70/207) | 38% (18/47) | <0.001 | 0.61 | 0.01 |
|
|
||||||
| MELD score | 7.45 | 8.61 | 9.17 | 0.05 | 0.26 | 0.02 |
|
|
||||||
| Symptoms at Diagnosis | 34% (25/74) | 44% (214/485) | 47% (42/90) | 0.10 | 0.73 | 0.11 |
|
|
||||||
| Fibrosis Stage on Biopsy | 0.72 | 0.86 | 0.79 | |||
|
|
||||||
| • Stage 0 | 4% (2/52) | 4% (8/211) | 7% (2/29) | |||
|
|
||||||
| • Stage 1 | 29% (15/52) | 30% (64/211) | 24% (7/29) | |||
|
|
||||||
| • Stage 2 | 46% (24/52) | 36% (76/211) | 38% (11/29) | |||
|
|
||||||
| • Stage 3 | 13% (7/52) | 19% (40/211) | 21% (6/29) | |||
|
|
||||||
| • Stage 4 | 8% (4/52) | 11% (23/211) | 10% (3/29) | |||
|
|
||||||
| Inflammatory Bowel Disease | 85% (80/94) | 80% (522/554) | 67% (67/100) | 0.27 | <0.01 | <0.01 |
|
|
||||||
| • Ulcerative Colitis | 72% (68/94) | 66% (434/554) | 60% (60/100) | 0.29 | 0.22 | 0.10 |
|
|
||||||
| • Crohn’s Disease | 5% (5/94) | 6% (39/554) | 2% (2/100) | 1.00 | 0.15 | 0.27 |
|
|
||||||
| • Indeterminate Colitis | 7% (7/94) | 8% (49/554) | 5% (5/100) | 1.00 | 0.53 | 0.60 |
|
|
||||||
| Follow up time (yrs) | 7.6 (3.8–12.3) | 10.5 (4.9–18.0) | 5.8 (2.5–8.6) | - | - | - |
|
|
||||||
Continuous variables expressed as medians (interquartile ranges)
Abbreviations: vs (versus); PSC (primary sclerosing cholangitis); yrs (years); SAP (serum alkaline phosphatase); ULN (upper limit of normal); MELD (model for end-stage liver disease).
The ED group also had a lower MELD score and SAP at the time of diagnosis (Table 1). Indeed, among the 326 individuals (ED n=37, MD n=237, LD n=52) with serial SAP measurements beyond 2 years the SAP was more likely to remain <1.5 x ULN over the course of 2 years in the ED group (60%) when compared to the MD (24%) and LD (21%) PSC subjects (p<0.001). These results remained similar after individuals with either concurrent autoimmune hepatitis or small duct PSC were excluded (data not shown). The presence or absence of symptoms when PSC was diagnosed was determined among 649 subjects: 368 were asymptomatic (ED n=49, MD n=271, LD n=48) and 281 were symptomatic (ED n=25, MD n=214, LD n=42). Thirty-six asymptomatic individuals also had a normal SAP at the time of the diagnosis (ED n=13, MD n=19, LD n=4) and had imaging for unrelated reasons that ultimately led to a diagnosis of PSC. However, the majority (90%) of asymptomatic individuals had an elevated SAP (n=332), which prompted additional testing to confirm the presence of PSC (ED n=36, MD n=252, LD n=44). Individuals with symptoms at diagnosis who also had a normal alkaline phosphatase (n=16) was uncommon (ED n=5, MD n=7, LD n=4).
Hepatobiliary Complications
Hepatobiliary malignancies were uncommon in ED PSC (4%) (CCA n=0; HCC n=4; gallbladder cancer n=0). The 4 individuals with HCC had cirrhosis when malignancy was detected and all had either radiographic or histologic evidence of classic HCC. In contrast, 81(12%) individuals in the MD group developed a hepatobiliary cancer (CCA n=66; HCC n=12; gallbladder cancer n=4) and 18 (18%) of subjects in the LD group had a hepatobiliary malignancy detected (CCA n=12; HCC n=2; gallbladder cancer n=4). Indeed, hepatobiliary malignancies were relatively rare in ED-PSC when compared to MD PSC (HR, 0.25; 95% CI 0.06–1.03, p=0.06) and LD PSC (HR, 0.07; 95% CI 0.01–0.62, p=0.02) (Figure 1). Among all patients, CCA was more likely to be diagnosed within a year after the PSC diagnosis among those found to have PSC late in life: ED 0% (0/95), MD 2% (14/662), LD 6% (6/102), p=0.02. In other words, when CCA did occur (n=78), it was more than twice as likely to be detected within 1 year of the initial PSC diagnosis in the LD patients (50%, 6/12) when compared to their middle-age counterparts (21%, 14/66) (Figure 2). Notably, the prevalence of IBD was similar between individuals in the MD and LD groups (71% vs 67% respectively) who had CCA detected within 1 year of a PSC diagnosis.
Figure 1.
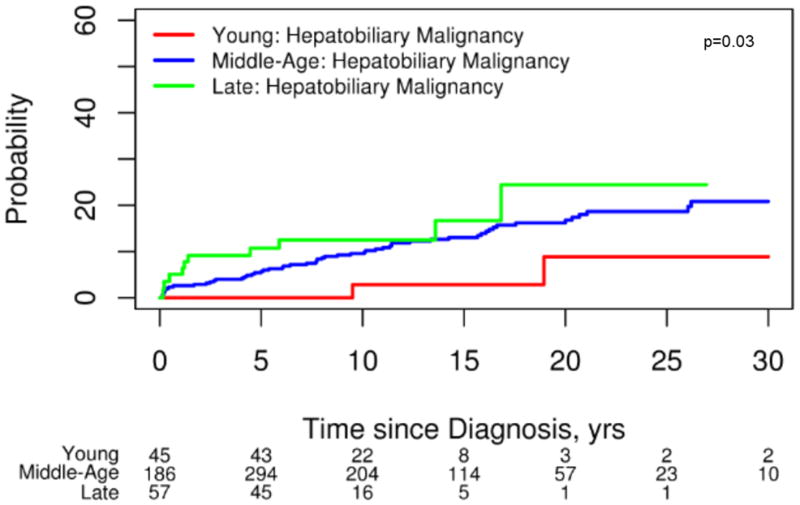
Development of Hepatobiliary Malignancies
Figure 2.

Proportion of Cholangiocarcinomas Detected within First Year of PSC Diagnosis
Hepatic decompensation was more common among those with LD-PSC (ascites n=17, variceal hemorrhage n=1, encephalopathy n=12) compared to ED-PSC (ascites n=2, variceal hemorrhage n=1, encephalopathy n=1), HR, 2.26; 95% CI 1.02–5.05, p=0.05. While not statistically significant, those with LD-PSC also appeared to be at a higher risk of hepatic decompensation when compared to the MD group (ascites n=35, variceal hemorrhage n=14, encephalopathy n=17), HR, 1.64; 95% CI 0.98–2.70, p=0.06 (Figure 3).
Figure 3.
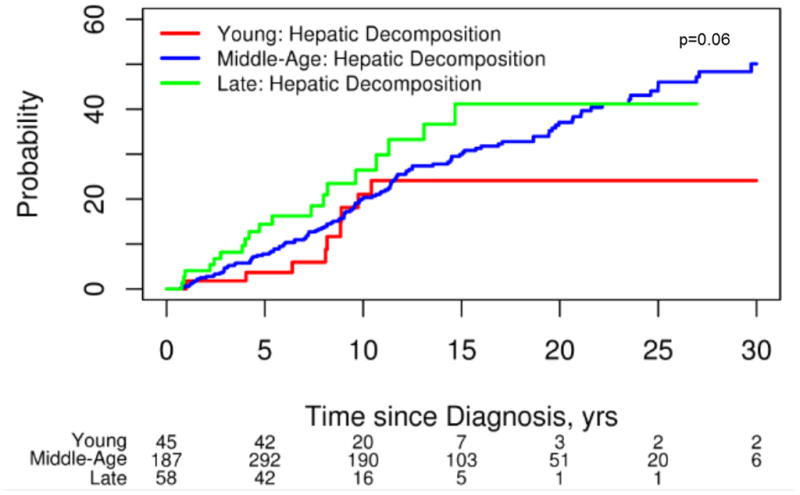
Development of Hepatic Decompensation
Two-hundred and twenty-nine (27%) subjects underwent a liver transplant (ED n=23, MD n=191, LD n=15). Hepatic decompensation (53%) was the leading indication for transplant followed by malignancy (19%), PSC related symptoms (17%) or some other indication (11%). There was no difference in liver transplantation among the 3 groups: (ED vs MD) HR, 1.04; 95% CI 0.67–1.63, p=0.86; (MD vs LD) HR, 1.41; 95% CI 0.80–2.50, p=0.23; (LD vs ED) HR, 0.71; 95% CI 0.35–1.44, p=0.34. Death secondary to a PSC-related cause occurred in 44 (5%) individuals (ED n=2, MD n=31, LD n=11). The majority of deaths (66%) were secondary to malignancy (CCA n=25, HCC n=2, colorectal cancer n=2) followed by complications stemming from advanced liver disease (18%), cholangitis (7%), other infection (7%), intraoperative death at time of transplant (2%). PSC-related mortality did not differ between the 3 groups: (ED vs MD) HR, 1.00; 95% CI 0.96–1.04, p=1.00; (MD vs LD) HR, 0.99; 95% CI 0.81–1.20, p=1.00; (LD vs ED) HR, 1.00; 95% CI 0.96–1.04, p=0.89.
Inflammatory Bowel Disease & Related Complications
A diagnosis of IBD was present in 669 patients (78%) and was less prevalent in the LD subgroup (Table 1). The majority of individuals with IBD (76%) had an established diagnosis of IBD before PSC was detected (ED n=59, MD n=365, LD n=56). IBD was often clinically apparent for years when PSC was detected later in life. For example, the median (IQR) duration of IBD at the time of PSC diagnosis among the ED, MD and LD groups was 1.00 (0.01–1.71), 8.50 (1.28–17.70), 22.50 (11.0–31.30) yrs, p<0.001.
CRC developed in 41 patients (ED n=0, MD n=31, LD n=10) while HGD occurred in 25 individuals (ED n=2, MD n=21, LD n=2). The development of either CRC or HGD did not differ between groups: (ED vs MD) HR, 0.75; 95% CI 0.17–3.37, p=0.70; (MD vs LD) HR, 1.18; 95% CI 0.38–3.70, p=0.77; (LD vs ED) HR, 3.63; 95% CI 0.28–47.44, p=0.33. One-hundred and eighty-one patients underwent a colectomy (ED n=14, MD n=143, LD n=24) for either neoplasia (ED n=3, MD n=74, LD n=14), refractory IBD (ED n=10, MD n=56, LD n=7) or an unknown indication (ED n=1, MD n=13, LD n=3). Undergoing a colectomy (for any indication) did not differ between groups: (ED vs MD) HR, 1.36; 95% CI 0.67–2.77, p=0.40; (MD vs LD) HR, 1.13; 95% CI 0.52–2.47, p=0.76; (LD vs ED) HR, 0.83; 95% CI 0.24–2.90, p=0.77.
Discussion
To date, this is the largest study to examine differences among individuals diagnosed with PSC at various age groups. Our results illustrate that phenotypic differences exist among those with PSC diagnosed at the extremes of the age spectrum. These findings have clinical implications and further illuminate our understanding of the natural history of PSC.
Overall, individuals with ED PSC had several prognostic factors associated with less aggressive phenotype when compared to their older counterparts. For example, SAP was lower in those with EO PSC and tended to remain < 1.5 x the ULN years after the initial diagnosis. A lower SAP has been associated with an improved prognosis.9–12 In addition, their MELD score was significantly lower at the time of diagnosis. In contrast to large duct PSC, small duct PSC is a sub-phenotype associated with an improved prognosis.13 It has been postulated that small duct PSC may represent an earlier, more mild phenotype when compared to large duct PSC. Indeed, small duct PSC associated with IBD has a similar human leukocyte antigen profile when compared to those with large duct PSC.14 The prevalence of small duct PSC was higher in the ED group (13%) when compared to their older counterparts (Table 1). This observation appears to be consistent with other series that have reported a prevalence of small duct PSC in children that is higher than the prevalence of small duct PSC within predominately adult cohorts.2, 15
In addition to improved prognostic markers, ED PSC appeared to have a decreased risk of hepatobiliary malignancies during this study interval (Figure 1). Notably, there were no cases of CCA detected among those with ED PSC despite having nearly 8 years of follow up. This observation is consistent with other reports that suggest pediatric onset CCA among children with PSC is rare.4, 5 Hence, CCA screening in asymptomatic pediatric PSC patients is likely unnecessary. However, the true risk of CCA development in adulthood years after a pediatric PSC diagnosis is unknown. The decreased risk of hepatobiliary complications among those with ED-PSC did not translate into a diminished risk of colorectal neoplasia or a lower likelihood of undergoing a colectomy when compared to other age groups. Our observations between prognostic variations between age groups did not change after excluding individuals with concurrent autoimmune hepatitis (data not shown).
Persons diagnosed with PSC late in life were more likely to have malignant hepatobiliary complications. Our findings suggest that hepatobiliary malignancies, particularly CCA, are more common when PSC is detected at a later age (Figure 1). An older age at the time of a PSC diagnosis and an increased risk of CCA has been reported in several studies.16, 17 Population-based studies have shown that approximately one-third of all biliary cancers are detected within a year of a diagnosis of PSC.15, 17 This indirectly suggests that the development of CCA can bring previously asymptomatic patients with PSC to the attention of clinicians. Importantly, this phenomenon appears to be more common in those with LD-PSC (Figure 2). Therefore, it is important for clinicians to have a high index of suspicion for an occult biliary cancer in this age group shortly after a diagnosis of PSC is established. Notably, the prevalence of IBD was similar among those with CCA detected within the first year across age groups. Because of this and the cholangiographic features of these subjects it is highly unlikely that this observation can be explained by de novo CCA and secondary sclerosing cholangitis among those with CCA detected late in life. While survival free of liver transplant and PSC related mortality was similar across age groups, those with LD-PSC were more likely to have hepatic decompensation particularly when compared to those with ED-PSC (Figure 3). The increased prevalence of malignant and non-malignant complications in the LD-PSC group may be related to a longer subclinical PSC disease course. For example, biliary cancer in the setting of PSC is a prototypical malignancy that occurs in the context of chronic inflammation and it is possible that the risk of malignant degeneration increases as the duration of inflammation and biliary epithelial damage increases.18
Our study has several limitations. First, it was a retrospective study conducted at 3 academic medical centers and may not be representative of the entire PSC population. A second limitation is we were only able to determine the time of PSC diagnosis rather than the actual time when the disease started. PSC may have a subclinical disease course without symptoms or biochemical abnormalities.19 A key strength of the study is both the size and phenotypic detail we were able to collect. This was achieved by us collecting medical records from medical centers across the United States when participants were seen at facilities other than the Mayo Clinic.
In conclusion, our study reinforces that PSC is a heterogeneous disorder that also varies across the age spectrum. Individuals diagnosed with PSC at an earlier age do not appear to have a more aggressive phenotype whereas PSC detected late in life was more likely to be associated with hepatobiliary malignancies and hepatic decompensation. Clinicians should have a high index of suspicion for CCA shortly after PSC is detected in older individuals.
Acknowledgments
Grant Support: The project was supported by the following NIH grants: DK 84960 and the A. J. and Sigismunda Palumbo Charitable Trust.
Abbreviations
- PSC
Primary sclerosing cholangitis
- IBD
inflammatory bowel disease
- UC
ulcerative colitis
- CCA
cholangiocarcinoma
- ED
early diagnosis
- LD
late diagnosis
- MD
middle-age diagnosis
- IQR
median interquartile
- HCC
hepatocellular carcinoma
- SAP
serum alkaline phosphatase
- ULN
upper limit of normal
- CRC
colorectal cancer
- HGD
high grade dysplasia
- MELD
model for end-stage liver disease
Footnotes
Disclosures: None
Transcript Profile: None.
Writing Assistance: Not utilized.
Author Contributions:
John E. Eaton. Study concept and design; data collection; interpretation of data; drafting of the manuscript; critical revision of the manuscript.
Brian D. Juran. Study concept and design; data collection; interpretation of data; critical revision of the manuscript.
Elizabeth J. Atkinson. Study design; analysis and interpretation of data; critical revision of the manuscript.
Erik M. Schlicht. Data collection; critical revision of the manuscript.
Bryan M. McCauley. Study design; analysis and interpretation of data; critical revision of the manuscript.
Mariza de Andrade. Study design; analysis and interpretation of data; critical revision of the manuscript.
Konstantinos N. Lazaridis. Guarantor of the article; study concept and design; critical revision of the manuscript.
References
- 1.Eaton JE, Talwalkar JA, Lazaridis KN, Gores GJ, Lindor KD. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology. 2013;145:521–36. doi: 10.1053/j.gastro.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A retrospective single-center review of primary sclerosing cholangitis in children. Clin Gastroenterol Hepatol. 2009;7:239–45. doi: 10.1016/j.cgh.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 3.Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary sclerosing cholangitis in children: a long-term follow-up study. Hepatology. 2003;38:210–7. doi: 10.1053/jhep.2003.50289. [DOI] [PubMed] [Google Scholar]
- 4.Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary sclerosing cholangitis, autoimmune hepatitis, and overlap in Utah children: epidemiology and natural history. Hepatology. 2013;58:1392–400. doi: 10.1002/hep.26454. [DOI] [PubMed] [Google Scholar]
- 5.Bjornsson E, Angulo P. Cholangiocarcinoma in young individuals with and without primary sclerosing cholangitis. Am J Gastroenterol. 2007;102:1677–82. doi: 10.1111/j.1572-0241.2007.01220.x. [DOI] [PubMed] [Google Scholar]
- 6.Manns MP, Czaja AJ, Gorham JD, et al. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:2193–213. doi: 10.1002/hep.23584. [DOI] [PubMed] [Google Scholar]
- 7.Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–78. doi: 10.1002/hep.23294. [DOI] [PubMed] [Google Scholar]
- 8.Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. 2007;47:598–607. doi: 10.1016/j.jhep.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2012 doi: 10.1016/j.jhep.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 10.de Vries EM, Wang J, Leeflang MM, et al. Alkaline phosphatase at diagnosis of primary sclerosing cholangitis and 1 year later: evaluation of prognostic value. Liver Int. 2016;36:1867–75. doi: 10.1111/liv.13110. [DOI] [PubMed] [Google Scholar]
- 11.Lindstrom L, Hultcrantz R, Boberg KM, Friis-Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2013;11:841–6. doi: 10.1016/j.cgh.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 12.Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis. 2011;43:309–13. doi: 10.1016/j.dld.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjornsson E, Olsson R, Bergquist A, et al. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology. 2008;134:975–80. doi: 10.1053/j.gastro.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 14.Naess S, Bjornsson E, Anmarkrud JA, et al. Small duct primary sclerosing cholangitis without inflammatory bowel disease is genetically different from large duct disease. Liver Int. 2014;34:1488–95. doi: 10.1111/liv.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boonstra K, Weersma RK, van Erpecum KJ, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58:2045–55. doi: 10.1002/hep.26565. [DOI] [PubMed] [Google Scholar]
- 16.de Valle MB, Bjornsson E, Lindkvist B. Mortality and cancer risk related to primary sclerosing cholangitis in a Swedish population-based cohort. Liver Int. 2012;32:441–8. doi: 10.1111/j.1478-3231.2011.02614.x. [DOI] [PubMed] [Google Scholar]
- 17.Bergquist A, Ekbom A, Olsson R, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002;36:321–7. doi: 10.1016/s0168-8278(01)00288-4. [DOI] [PubMed] [Google Scholar]
- 18.Rizvi S, Eaton JE, Gores GJ. Primary Sclerosing Cholangitis as a Premalignant Biliary Tract Disease: Surveillance and Management. Clin Gastroenterol Hepatol. 2015;13:2152–65. doi: 10.1016/j.cgh.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunder AK, Hov JR, Borthne A, et al. Prevalence of Sclerosing Cholangitis Detected by Magnetic Resonance Cholangiography in Patients With Long-term Inflammatory Bowel Disease. Gastroenterology. 2016;151:660–69e4. doi: 10.1053/j.gastro.2016.06.021. [DOI] [PubMed] [Google Scholar]