

Abstract
Pervasive transcription initiates from cryptic promoters and is observed in eukaryotes ranging from yeast to mammals. The Set2-Rpd3 regulatory system prevents cryptic promoter function within expressed genes. However, conserved systems that control pervasive transcription within intergenic regions have not been well established. Here we show that Mot1, Ino80 chromatin remodeling complex (Ino80C), and NC2 colocalize on chromatin and coordinately suppress pervasive transcription in S. cerevisiae and murine embryonic stem cells (mESCs). In yeast, all three proteins bind subtelomeric heterochromatin through a Sir3-stimulated mechanism and to euchromatin via a TBP-stimulated mechanism. In mESCs, the proteins bind to active and poised TBP-bound promoters along with promoters of polycomb-silenced genes apparently lacking TBP. Depletion of Mot1, Ino80C or NC2 by anchor away in yeast or RNAi in mESCs leads to near-identical transcriptome phenotypes, with new subtelomeric transcription in yeast, and greatly increased pervasive transcription in both yeast and mESCs.
Keywords: Ino80, Mot1, NC2, Promoter, Silencing, Polycomb, Sir3, Heterochromatin, Pervasive Transcription
Graphical Abstract
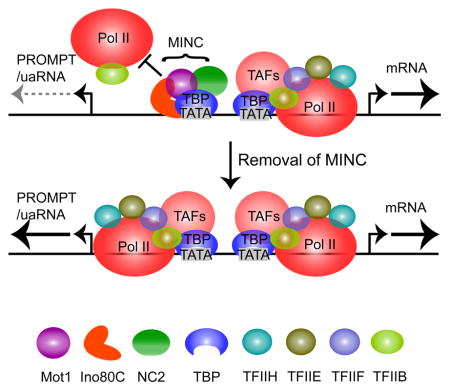
Introduction
Nucleosome depleted regions (NDRs) in both S. cerevisiae and mammalian promoters display closely situated divergent transcription by RNA polymerase II (Pol II). One RNA initiates in the forward direction into the gene. The other initiates in the antisense or reverse direction. In mammals, these RNAs are termed upstream antisense RNAs (uaRNAs) or promoter upstream transcripts (PROMPTs) (Chen et al., 2016). In yeast, three known classes of such transcripts exist including cryptic unstable transcripts (CUTs), stable unannotated transcripts (SUTs) and Xrn1-sensitive unstable transcripts (XUTs) (van Dijk et al., 2011; Xu et al., 2009). Collectively, these cryptic transcription units, mainly initiating within intergenic regions, lead to a phenomenon termed pervasive transcription. Pervasive transcription emerges from Pol II preinitiation complexes (PICs) detectable by ChIP-Exo (Pugh and Venters, 2016). In some cases, non-coding, pervasive transcription affects expression of nearby genes and can influence protein abundance, and thus it is probably regulated (Huber et al., 2016; Xu et al., 2011b). The magnitude of pervasive transcription relative to genic transcription has been difficult to assess because some of the RNAs are rapidly degraded by RNA surveillance systems like the exosome and Xrn1 but others generate stable RNAs. Additionally, some factors involved in Pol II elongation and RNA processing influence pervasive transcript levels (Almada et al., 2013; Colin et al., 2014; Roy et al., 2016; Yang et al., 2016). However, whether or how pervasive transcription is controlled at the transcriptional level has been largely unknown (Jensen et al., 2013; Kaplan, 2016).
A well-studied system that prevents initiation at cryptic promoters within a gene during transcription comprises Set2, Rpd3S, chromatin remodeling proteins (Chd1, Isw1), histone chaperones (i.e., FACT) and Pol II itself (Kaplan et al., 2003; Keogh et al., 2005; Li et al., 2007; Rando and Winston, 2012; Smolle and Workman, 2013; Venkatesh and Workman, 2013). Chromatin modification and remodeling disrupt nucleosomes to expose DNA for Pol II passage. However, chromatin must be reassembled to prevent cryptic promoters from initiating transcription. H3K36 methylation by Set2, which travels with elongating Pol II, prevents intragenic cryptic promoters from functioning. H3K36me3 recruits the Rpd3S histone deacetylase complex to create a repressive chromatin structure in the wake of Pol II (Lee et al., 2013; Smolle et al., 2012; Venkatesh et al., 2012). Set2-repressed antisense transcripts (SRATs) are a major class of the RNAs produced in Set2 mutants (Kim et al., 2016; Venkatesh et al., 2016). Like transcribed intragenic chromatin, the promoter NDR is generated by nucleosome remodeling and must likewise employ a mechanism to suppress transcription of uaRNAs/PROMPTs, CUTs, SUTs, XUTs and other ncRNAs. However, the identity of such systems has remained elusive.
Genic nucleosome organization in yeast is a complex process involving the coordinated action of numerous chromatin remodeling proteins including RSC, Chd1, Isw1, Isw2 and Ino80C (Krietenstein et al., 2016; Rando and Winston, 2012; Yen et al., 2012; Yen et al., 2013). RSC generates the NDR (Krietenstein et al., 2016; Lorch et al., 2014). However, Ino80C is recruited to AT-rich NDRs, possibly through its NHP10 subunit, where it positions the +1 and −1 nucleosomes around the promoter irrespective of gene activity (Yen et al., 2012; Yen et al., 2013). Ino80C can position the +1 and −1 nucleosomes in a purified system lacking other factors (Krietenstein et al., 2016). Ino80C is conserved from yeast to mammals (Conaway and Conaway, 2009). We have shown previously that deletion of Ino80C subunits in yeast leads to high levels of heterochromatin transcription and widespread intergenic non-coding transcription initiating upstream of the annotated TSS and also downstream of the TTS, in all cases accompanied by increased H3K79me3, a hallmark of active or recent transcription (Xue et al., 2015). This observation suggests that Ino80C is a component of a silencing system that acts outside of genes and in heterochromatin. However, several key questions remained unanswered. For example, how is Ino80C recruited to its sites of action, are there other potential proteins operating in combination with Ino80C, are these conserved, and finally, is the function of the system conserved across eukaryotes? Here we employed proteomics on heterochromatin and PICs in yeast to identify two known TBP binding proteins, NC2 and Mot1 (Auble, 2009; Sikorski and Buratowski, 2009), as additional components of this silencing system. Detailed in vivo analyses show that the binding and functions of NC2 and Mot1 directly overlap with Ino80C in yeast and mammalian cells, and in euchromatin and facultative heterochromatin, to suppress pervasive transcription and consequently, to ensure properly regulated gene expression.
Results
Mot1, Ino80C and NC2 Bind and Contribute to Silencing of Yeast Heterochromatin
Our previous studies determined that S. cerevisiae subtelomeric heterochromatin displayed a bi-stable state where embedded genes could become active even when bound by Sir proteins (Kitada et al., 2012). This observation suggested that other factors contribute to silencing. To identify these, we generated heterochromatin in vitro on a nucleosomal template bearing a GAL4-responsive promoter (Fig. 1A). This template was immobilized on paramagnetic beads, coated with purified Sir2, 3, and 4, and incubated with yeast nuclear extract. Bound proteins were eluted and subjected to MultiDimensional Protein Identification Technology (MuDPIT). The raw spectral data were analyzed using our in-house MS-SORT program, which generates a normalized spectral abundance factor for each protein complex identified (NSAFC). NSAFC is a semi-quantitative measure of a complex’s abundance in a mixture.
Figure 1.

MINC binds yeast heterochromatin. (A) Schematic of immobilized template procedure. (B) MuDPIT analysis of proteins binding to silent chromatin. Immobilized chromatin templates bearing or lacking Sir2,3,4 were incubated with yeast nuclear extract, washed and subjected to MuDPIT alongside purified Sir2,3,4. The chart shows a heat map depicting the Avg. NSAFC for proteins enriched by the presence of Sir2,3,4. Color scale bar in NSAFC is shown below. (C) Average binding profile of Sir3, Mot1, Arp5 (Ino80C) and Nc2β in subtelomeric regions of wild-type and sir3Δ strains by ChIP-seq. The moving averages of log2 MINC enrichment versus input and the Sir3 log2 ratio versus input (step size = 100 bp, window size = 20) were plotted by the distance from the telomere, from 0 to 15 kb (X-axis). (D) Browser plots of Sir3 and MINC in wild-type and sir3Δ strains by ChIP-seq on Chromosome III subtelomere. Y axis shows the normalized read counts of MINC and log2 ratio of Sir3 versus Input. See Table S1 and 2.
Figure 1B and Table S2 list NSAFc values for key proteins identified in our analysis. We detected >5-fold Sir2,3,4-dependent enrichment of several known transcriptional regulatory factors and chromatin remodeling proteins including Mot1, Ino80C and NC2 and lesser enrichments of other proteins. We will refer to Mot1, Ino80C and NC2 collectively as MINC with the caveat that although protein-protein interactions between the different components have been identified, we have no strong evidence for a free-standing complex. Upon purification, each of the components displayed a higher affinity for chromatin bound by Sir2,3,4 but did not bind cooperatively (data not shown). Two additional proteins, Rap1 and NHP6a, were also >5-fold enriched but subsequent analyses (below) show they play roles distinct from MINC.
ChIP-seq analysis of Mot1-Myc, Ino80C subunit Arp5, and Nc2β-Myc confirmed that the proteins localize within heterochromatin in vivo. Figure 1C shows the average binding profile within 15 kb of the telomere. Sir3 binding is highest in the region closest to the telomere, decreases throughout the subtelomeric region and reaches a minimum typically 5 to 15 kb from the telomere. This distribution is typical except for chromosome III bearing the telomere-proximal HML and HMR loci (Sperling and Grunstein, 2009). The bimodal shape of the average binding profile reflects the presence of Y′ repeat elements in about half of the subtelomeric regions. The data also reveal enrichment of MINC in subtelomeric regions with profiles similar to Sir3. The browser track in Figure 1D shows that MINC components co-localize in most instances. Deletion of SIR3 (sir3Δ) led to a decrease but not complete loss of binding of MINC (Fig. 1C and 1D). Using published datasets (Zentner and Henikoff, 2013), we could not consistently detect TBP in the regions of MINC binding within yeast heterochromatin. However, we cannot exclude the possibility that TBP is present but below the level of detection. We conclude that the Sir2,3,4 silencing complex contributes to MINC localization within subtelomeric heterochromatin in S. cerevisiae.
To understand the role of MINC in subtelomeric transcription, we employed the anchor away (AA) approach. Figure 2A shows that depletion of MINC components by AA led to an average increase in subtelomeric transcription versus a control strain (WT) with the differences becoming less evident as heterochromatin begins to merge with euchromatin. When comparing the ratio of transcription from the MINC-AA strains to the WT control (Fig. 2B), the combination of Ino80AA with either Mot1AA or NC2βAA led to subtelomeric transcription that exceeded the mild transcriptional increase observed in a sir3Δ strain. This point is illustrated by browser tracks plotting the increase in transcription in Ino80AA and Mot1AA strains upon rapamycin addition alongside that in sir3Δ. The increased transcription localized to precisely the same positions for each MINC component (Fig. 2C). We conclude that MINC controls subtelomeric transcription and likely functions together with the Sir2,3,4 complex to maintain subtelomeric gene silencing.
Figure 2.

Gene expression in heterochromatin after MINC depletion by anchor away (AA). (A) Average RNA levels in yeast subtelomeric domains after MINC depletion by AA. The moving average of log2 FPKM (fragments per kilobase of exon per million fragments mapped) (step size = 1, window size = 100) was plotted as a function of the distance from the telomere, up to 20 kb. (B) Same as A except using the Log2 ratio of RNA expression in MINC AA versus WT compared with sir3Δ. (C) Browser plot of RNA expression at chromosome X left side (Chr XL) and chromosome IX right side (Chr IXR) subtelomeric region comparing wild type to anchor away conditions for MINC components and sir3Δ.
MINC is Present Within PICs
During our proteomic analysis of yeast heterochromatin, we also employed MuDPIT to analyze PICs formed on immobilized chromatin templates incubated with yeast nuclear extract and GAL4-VP16. Figure 3A shows an MS-SORT chart demonstrating GAL4-VP16-mediated recruitment of select PIC components including the general transcription factors, and the co-activators TFIID, Mediator (MED) and SAGA. These data are consistent with a previous study (Sikorski et al., 2012). Notably, MINC was also recruited >4-fold. We conclude that recruitment of MINC is coincident with PIC assembly at a promoter in vitro.
Figure 3.

MINC binds to PICs and promoters. (A) PICs were captured from yeast nuclear extracts on chromatin bearing a GAL4-resposnsive promoter in the absence (first column) and presence (second column) of GAL4-VP16. NSAFc of listed complexes are shown in the chart with red denoting high abundance and green denoting low abundance proteins. Enrichment in presence of GAL4-VP16 is shown in the final column (Fold Enrich). Color scale bar in NSAFC is shown below. (B) Average metagene profile of MINC and TBP binding across the promoter and gene from 1 kb upstream of the TSS through 1 kb downstream of the TTS at all genes. Log2 ratios of immunoprecipitation (IP) versus input at significantly enriched windows were used. (C) Venn diagram showing the proportional overlap of MINC-bound genes, i.e., genes that show MINC binding +/− 0.5 Kb around the TSS were considered MINC bound genes. (D) Browser plot of a typical euchromatic region of chromosome XV demonstrating co-enrichment of MINC and TBP up and downstream of genes with RNA levels in wild type. Y axis shows normalized read counts. (E) Heat maps of TBP and MINC binding in WT and MINC in TBPAA from −1 kb from TSS to +1 kb from TTS. Genes were separated into 4 clusters (Clusters a-d: Ca 1003 genes, Cb 1387 genes, Cc 385 genes, Cd 2455 genes). Log2 ratios of IP versus input at significantly enriched windows were used. (F) Average metagene profile of MINC and TBP binding in WT and TBPAA in clusters a, b and c. See Figure S1.
We employed ChIP-seq to determine whether the MINC components co-localize at yeast promoters genome-wide using Mot1-Myc, Ino80-Myc, and Nc2β-Myc. A metagene analysis of all MINC components (Fig. 3B) revealed co-enrichment both upstream of the annotated TSS and downstream of the TTS. We found that MINC largely co-localizes with TBP within euchromatin using a published dataset (Zentner and Henikoff, 2013). Our data are consistent with previous ChIP-chip studies on Mot1 and NC2 (van Werven et al., 2008) and with many uncited studies. We conclude that MINC components co-localize at TBP binding sites in euchromatin. Note that Spearman correlation of replicates for the genomics data are in Table S3.
The enrichment of MINC and TBP is greater on the 5′ versus the 3′ end of genes. Figure 3C is a Venn diagram showing that the Mot1, Ino80 and NC2 co-localize at the majority of genes and Figure S1A shows the detailed number of overlapping genes. Figures S1B-E show that the upstream and downstream enrichment is not an informatics artifact due to the tight packing of genes in yeast causing misinterpretation of a TSS as a TTS. The average profile from the TSSs of divergent genes and the TTSs of convergent genes, positions where the signal cannot be misinterpreted, display MINC and TBP enrichment. These points are illustrated by the browser plot from a typical gene-dense region of the S. cerevisiae genome, where MINC and TBP co-localize, flanking most gene units (Fig. 3D, see dashed lines). Such binding may be independent or indicative of a gene loop.
To gain further insight into the determinants of MINC positioning, we employed TBP anchor away (TBPAA). Figure 3E shows heat maps, divided into 4 clusters (Ca-d) by k-means, comparing MINC binding before and after AA. The effects of AA were clearly evident in Ca-c but less so in Cd so we analyzed the first three clusters in more detail. Figures 3E and 3F reveal strong decreases in Mot1 binding at the TTS of all three clusters and at the TSS of all clusters except Cc. TBPAA had similar effects on Ino80 and NC2 leading to decreases in binding at the TSS and TTS in Ca, the TTS in Cb but interestingly, increased binding at the TSS in Cc. Indeed, in two replicates of this experiment, MINC binding around the TSS in Cc was the least changed upon TBPAA. Figure S1F enumerates the percentage of TFIID- versus SAGA-dependent promoters in each cluster, showing that Cc contains the highest percentage of SAGA-dependent promoters. These findings correlate well with the enrichment of TATA-containing promoters in Cc. This observation suggests TATA-containing promoters are more resistant to MINC depletion possibly because of a higher TBP affinity versus promoters of other clusters. This idea is supported by a previous study showing that TBP binding at TATA-containing promoters is more resistant to Mot1AA than at other promoters (Zentner and Henikoff, 2013). We conclude that TBP is mechanistically responsible for localizing MINC at a large number of genes. Although this should be predicted for Mot1 and NC2, the fact that Ino80 behaves similarly provides evidence for a functional link among the MINC components.
MINC is Responsible for Transcriptional Silencing in Intergenic Euchromatin
To determine the transcriptome consequences of depleting MINC, we performed strand-specific RNA sequencing in rapamycin-treated AA strains of MINC components and plotted the results as heat maps representing the fold change versus parental strains (blue is downregulated and red is upregulated by AA). It is known that depletion of Mot1, Ino80C and NC2 leads to increased levels of pervasive transcripts (Dasgupta et al., 2002; Gomez-Navarro et al., 2016; Xue et al., 2015). However, an important question was whether the Ino80AA transcriptome phenotype was distinct or quantitatively similar to the phenotypes of Mot1AA and NC2AA. Similar phenotypes would argue that Ino80C function is coordinated with that of Mot1 and NC2, whereas unique effects would suggest separate pathways. To address this distinction, the transcriptome changes were separated into antisense and sense heat maps, each of which was sorted into three clusters apiece (antisense C1–C3; sense C4–6) by k-means and then further analyzed by comparing individual browser tracks of affected genes.
Ino80AA led to near identical transcriptome phenotypes as those found in NC2α/βAA, and Mot1AA. Figure 4A shows that in C1, intergenic antisense transcript levels increased up and downstream of the gene upon AA. C2 revealed an increased level of antisense transcripts throughout the gene. C3 is a combination of weaker effects. All three clusters display similar TBP binding adjacent to the TSS. Combining Ino80AA with either NC2AA or Mot1AA revealed strengthening of antisense signals, including that of C3 (Fig. 4A). We also compared the gene expression level in each cluster and found that the median transcription level of C2 genes in untreated cells was lower than those of C1 and C3 (Fig. 4B).
Figure 4.

MINC depletion reveals similar transcriptome effects in S. cerevisiae. (A) Heatmaps indicating change in antisense transcription upon AA depletion of Mot1, Ino80, and NC2 α and β subunits, alongside NC2 and Mot1 in combination with Ino80 versus wild type from 1 kb upstream of TSS to 1 kb downstream of TTS at MINC bound genes. 2746 MINC overlap genes were separated into 3 clusters using k means (C1, 833 genes; C2, 843 genes; C3, 1070 genes). The change is log2 scale; red is upregulated and blue is downregulated RNAs. Heatmap of TBP enrichment (log2 scales of IP vs Input) within 1 kb upstream and downstream of TSS is shown to the right. (B) Boxplot of average WT genic mRNA levels of C1, C2 and C3 with p val significance indicated. (C) Same as A but for sense strand transcription (C4, 652 genes; C5, 1002 genes; C6, 1092 genes). (D) Same as B but for C4, C5 and C6 with p val significance indicated. (E). Browser track showing examples of different RNA effects upon MINC depletion from chromosome IV (upstream and genic antisense), chromosome XII (downstream and genic antisense), and chromosome XIII (upstream sense and genic sense, i.e., silent gene being expressed). TBP ChIP through affected region is shown (Top panel). Y axis shows the normalized read counts and the arrows indicate the direction of transcripts. Known 5′ Cap sites and directions are indicated as arrows in TSSs row. Known genic mRNAs are indicated as black boxed arrows and XUTs and SUTs are indicated as dark red boxed arrows. See Figure S2.
The data on the sense strand revealed additional insights into the silencing role of MINC. First, AA of MINC components led to a general increase in sense transcripts in intergenic regions mainly downstream of the TTS but also upstream of the TSS (Fig. 4C). These effects were amplified by combining Ino80AA with either Mot1AA or the NC2α/βAA. Second, sense transcripts in a large fraction of genes, represented by C5, increased substantially upon AA of MINC components. The median mRNA level of C5 genes in untreated cells was significantly lower than those of C4 and C6 (Fig. 4D). The different effects of AA strains on expression of non-coding RNAs could not be explained by differential binding of MINC or TBP with the possible exception of C5, which displayed less enrichment downstream (Fig. S2A–C), suggesting the possibility that intact PICs and MINC co-exist on some genes. Notably, the expression patterns for Rap1AA and Nhp6aΔ, two other proteins that were >5-fold enriched in the initial heterochromatin proteomic screen, were distinct suggesting the MINC effect is specific (Figs. S2D and S2E).
The browser plots of Figure 4E and Figure S2F (shaded regions) reveal the range of transcriptome phenotypes associated with MINC depletion. These RNAs shared 3 properties: i. they frequently co-localized with SUTs, XUTs and CUTs; ii. displayed proximal peaks of TBP distinct from the one of the adjacent gene; iii. and usually localized with capped RNAs (TSSs arrows). The plots show that the increase in antisense and sense transcripts occur in the same locations in the different MINC AA strains, and the effects are additive (Fig. S2G) or sometimes greater (Fig. 4E). The Chr XII example shows strong antisense transcription possibly originating from the TBP located at the 3′ end of YLR448W. Also, transcriptionally inactive genes normally bound by MINC such as YMR231W on chr XIII, become expressed upon individual and combinatorial AA of MINC components. In such cases, MINC appears to be silencing the entire gene unit. These observations support our model that Mot1, Ino80C and NC2 function together to suppress transcription from both cryptic promoters and silenced but physiological promoters. Finally, the enhanced levels of pervasive RNAs appear to be due to new transcription rather than stability as they are accompanied by increases in H3K79me3 upstream of the TSS and overlapping the TTS of the gene (Fig. S2H). H3K79 methylation is carried out by the histone methyltransferase Dot1, which associates with elongating Pol II.
Mammalian MINC Binds Within Promoters of Active and Poised Genes
The occurrence of pervasive transcription and conservation of MINC components between yeast and mammalian cells prompted us to investigate whether MINC was also responsible for suppressing pervasive transcription in murine embryonic stem cells (mESCs). Note that Mot1 is termed BTAF1 in mammalian cells (Tora, 2002) although we employ the yeast term here. To validate our ChIP-seq antibodies, we performed co-immunoprecipitation analysis from crude mESC nuclear extracts. Surprisingly, under native conditions, each MINC component displayed substantial co-association with its partners when compared to input (Fig. S3A).
We next determined the ChIP-seq binding profiles of Mot1, Ino80 and NC2 and compared them with a previously reported TBP dataset from mESCs (GSM555160). The metagene profile of MINC components in Figure 5A shows they largely co-localize with TBP upstream of the TSS. The Venn diagrams of Figure 5B and Figure S3B reveal considerable overlap of significant peaks among MINC components. This colocalization is clearly evident across a region of chromosome 9, where total RNA levels are shown (Fig. 5C). Unlike in yeast, we did not observe consistent binding of MINC to the regions downstream of the TTS.
Figure 5.

MINC binds active and poised promoters in ESCs. (A) Average metagene profile of MINC and TBP binding across the gene from 3 kb upstream of the TSS to 3 kb downstream from the TTS at all genes in mESCs. Log2 ratios of immunoprecipitation (IP) versus input at significantly enriched windows were used. (B) Venn diagram showing the proportional overlap of MINC bound genes. Genes which have MINC binding at +/− 1 Kb around the TSS were considered MINC bound genes. (C) Browser plot of Mot1, Ino80, Nc2β, TBP and Ring1B binding with RNA levels at a typical gene dense region of ESCs. Y axis shows the read counts. (D) Chromatin state chart of MINC binding showing enrichment of each MINC component in each chromatin state category. Scale bar is shown in the bottom. (E) Heat maps of Mot1, Ino80, Nc2β, TBP, Pol II, Med1, Pcgf2, Ring1B alongside genic RNA expression in Mock (log2 RPKM) separated into 3 clusters that were sorted first by Mot1 level, then TBP level using the average enrichment of +/−1 Kb around the TSS region. Log2 ratios of immunoprecipitation (IP) versus input at significantly enriched windows were used. The average of +/− 1 Kb around TSS region > 0.4 was used for sorting. See Figure S3.
To further understand MINC’s binding pattern, we employed a recent chromatin state model based on ChromHMM (Chronis et al., 2017). Chromatin states are assigned to specific loci based on the proportions of well-characterized histone modifications and variants. The location, size and enrichment of the assigned chromatin states in the genome are listed in Figure S3C. Figure 5D is a chart displaying the relative enrichment of each MINC component within each of the 18 chromatin states including active promoters (PromA), poised promoters (PromP), enhancers (EnhA, EnhM1, EnhM2, EnhW, EnhP), transcribed enhancers (TxEnhA, TxEnhB), various transcribed regions (Tx, Tx5′, Tx3′, TxWk3′, TxWk3′reg), coding regions bound by polycomb (ReprPC), H3K9me3 enriched heterochromatin (Het) and two regions where few modifications were detected (low signal). Strikingly, the data reveal that MINC is associated predominantly with chromatin states 1 and 2 comprising active (PromA) and poised (PromP) promoter regions, respectively. The slight bias for poised is due to the fact that ~25% more loci are represented in the PromP category versus PromA. A weaker association with different enhancer states is also observed.
Poised promoters bearing H3K27me3 are often bound by polycomb group proteins. We used published Polycomb repressive complex 1 (PRC1) ChIP-seq datasets (Kloet et al., 2016) along with our own Pol II and Med1 datasets to align with MINC and TBP. Ring1B is an H2AK119 ubiquitinase found in PRC1 and similar complexes [reviewed in (Di Croce and Helin, 2013)]. Pcgf2 is another complex member. Figure 5E shows heat maps sorted by either high (C_hi), low (C_low) or no (C_no) TBP binding at the TSS, compared with mRNA expression of the adjacent gene (RNA). The highly-expressed C_hi contains active promoters bearing MINC, TBP, Pol II, and Med1 centered near the TSS and low Ring1B and Pcgf2 binding. Conversely, C_low genes are less expressed, contain higher average levels of promoter-bound MINC, are enriched in Ring1B and Pcgf2 but contain low or undetectable TBP, Pol II, and Mediator. C_no genes are expressed poorly and contain little MINC, TBP, Pol II, Med1, Ring1B or Pcgf2. We conclude that MINC is present at active and polycomb-bound genes. As in yeast facultative heterochromatin, the C_low genes display low to undetectable TBP binding (Fig. 5E). This is also evident in the browser track of Figure 5C, where MINC is bound at promoters with Ring1B in either the presence or absence of TBP. MINC binding in the absence of detectable TBP appears to be a general characteristic of polycomb-rich promoter regions such as the Hoxd locus (Fig. S3D).
Depletion of MINC by RNAi Increases Pervasive Transcription in mESCs
To determine if the function of MINC in mESCs paralleled that in yeast, we performed total RNA-seq after RNAi to understand the effect of depleting each MINC component individually and in combination with Ino80. Figure S4A shows the knockdown efficiency of the various mRNAs for MINC components. The transcript changes upon knockdown were separated into antisense and sense heat maps, each of which was sorted into different clusters using k-means. The clusters are shown in Figure 6. Figure 6A illustrates heatmaps capturing the main trends for the upstream (Cluster 1, C1) and downstream antisense non-coding transcripts (C2) along with transcripts affected only mildly (C3). Both the upstream (C1) and downstream (C2) antisense transcripts are upregulated upon single or even greater in double knock down of MINC components. TBP occupancy of affected genes is not predictive of their sensitivity to depletion of MINC (Fig. 6A) but the new pervasive transcription is associated with slightly more active nearby genes (Fig. 6B). Analysis of increases in Sense transcripts revealed two clusters, C4 and C5. C4 displayed slightly enhanced levels within the gene and a stronger increase downstream of the TTS while there were mild to no effects of MINC depletion in C5 (Fig. 6C). The genes in C4 or C5 were expressed similarly (Fig. 6D). The average RNA profiles of C1–5 largely support the heatmap results (Fig. S4B).
Figure 6.

Depletion reveals similar transcriptome effects of MINC components in murine ESCs. (A) Heatmaps of change in antisense transcription upon depletion of Mot1, Ino80, and Nc2α individually and NC2α and Mot1 in combination with Ino80 versus control from 3 kb upstream of TSS to 3 kb downstream of TTS at MINC-bound genes. Clusters displaying similar transcription patterns under all conditions upstream of TSS and downstream of TTS are termed cluster 1 and 2 (C1, 1906 genes; C2, 1182 genes). All other clusters were merged and randomized as cluster 3 (C3, 9073 genes). Red is upregulated and blue is downregulated. Heatmap of TBP enrichment (log2 of IP vs Input) within 3 kb upstream and downstream of the TSS is shown to the right. (B) Boxplot of average mock genic RNA levels of C1, C2 and C3 with p val significance indicated. (C) Same as A but for sense strand transcription. The cluster with similar transcription patterns in the coding region and downstream of the TTS is termed cluster 4 (C4, 2502 genes). All other clusters were merged, randomized and included in cluster 5 (C5, 9659 genes). Red is upregulated and blue is downregulated RNAs. Heatmap of TBP enrichment is shown to the right. (D) Boxplot of average mock mRNA levels of C4 and C5 genes with p val significance indicated. (E) Browser tracks showing Pol II enrichment in the Mock and Ino80&Mot1 KD above TBP peaks. Representative examples of changes in Total RNA or Nascent RNA at the TSS (Suco) and TTS (Mef2d) upon MINC depletion. Y axis shows the normalized read counts and arrows indicate the direction of transcripts. Known 5′ Cap sites and direction of pervasive RNA are indicated in the TSSs row. Shaded areas represent pervasive transcripts in the antisense direction upstream of the TSS (Suco) and sense direction downstream of the TTS (Mef2d). See Figures S4 and S5.
We were unable to ascertain a root cause of the enhanced pervasive transcript levels based on comparison of TBP (Fig. S4C) and MINC enrichment (Fig. S4D and E). It was possible that we were observing effects of MINC depletion on RNA stability. However, similar results and clusters were obtained when we compared Total RNA with Nascent RNA-seq in the Mot1&Ino80 double knockdowns (Fig. S4F and G). Nascent RNA-seq captures chromatin- and Pol II-associated RNAs, arguing that the increased levels of pervasive transcripts are due to new transcription (Pandya-Jones and Black, 2009; Wuarin and Schibler, 1994)(Table S4). The increased transcription observed in the heatmaps of C1, C2 and C4 is supported by the average profile of the RNAs (Fig. S4H) and by the browser track examples representing C1 and C4 (Fig. 6E). However, whereas a mild increase in Pol II accompanied the increases in downstream transcription in C2 and C4 (Fig. S4F, G and H), the same was not observed for C1. One possible reason we did not observe increased Pol II levels at the promoter in C1 is because the increased transcription may be due to a change in Pol II distribution between the gene and divergent transcription units rather than total amount. More likely, the pervasive transcripts levels are very low compared to mRNAs and Pol II changes may not be easily detectable by ChIP.
Figure 6E shows browser plots representing a capped, antisense RNA initiating near the Suco TSS and a sense transcript downstream of the Mef2d TTS, where no capped RNAs were identified. The antisense upstream transcription shown in Figure 6A likely represents low-level existing divergent transcription increased by MINC knockdown. Divergent transcription has been described previously, corresponds with capped RNAs (Fort et al., 2014) and displays a TBP peak distinct from that of the nearby gene (Fig. 6E, Suco, compare TSS and TBP locations). Figure S5 illustrates several features of MINC-regulated pervasive transcription units. These include the sense transcription upstream of the Hat1 gene and the antisense transcription initiating downstream of the Cdk13 gene representing C2 (Fig. S5A). For C1, distinct peaks of TBP typically aligned with the gene TSS and with the known antisense TSS. However, in some examples (Suco), MINC was bound with TBP over the promoter of the gene but not with TBP at the promoter of the pervasive RNA (Fig. S5B). In other cases (Kpnb1), MINC bound both the genic and antisense promoters. Finally, we note that MINC downregulates pervasive transcription in facultative heterochromatin as illustrated by the Hoxa1 example (Fig. S5B). This example is curious because the nearby gene is active (not shown) reinforcing the possibility that gene activity contributes to MINC-regulated pervasive transcription. We conclude that the role of MINC in ESCs, like yeast, is to suppress pervasive transcription but its major effect is on upstream antisense RNA resulting from divergent transcription with a lesser effect on RNAs that extend beyond the normal TTS.
Discussion
MINC is reasonably well-conserved between S. cerevisiae and mammals unlike other major silencing systems including the Silencing Information Regulators (Sir 2,3,4 proteins) in S. cerevisiae, the Ring1B-associated polycomb group (PcG) in metazoans (Gartenberg and Smith, 2016; Piunti and Shilatifard, 2016), HP1 paralogs (Becker et al., 2016), and DNA methyltransferases (Klose and Bird, 2006). MINC coordinately downregulates thousands of pervasive transcription units in intergenic regions in euchromatin, while also playing a role in proper gene activation. We found that the binding and transcriptome phenotypes of all three MINC components overlap extensively in yeast and mESCs. We suggest that a major role of this conserved system is to suppress pervasive transcription initiating in intergenic regions, thus complementing the Set2-Rpd3S system for intragenic regions. However, our data show that MINC binding and regulation are superimposed on facultative heterochromatin in yeast, and binding is observed in mESC polycomb-bound regions, suggesting that this system may underlie mechanisms regulating heterochromatin function in two widely divergent eukaryotes.
We have not been able to obtain strong biochemical evidence that MINC exists as a free-standing complex although Mot1 and Ino80C have been shown to interact sub-stoichiometrically (Arnett et al., 2008), and NC2 and Mot1 interact in protein-protein interaction assays (Klejman et al., 2004) and in the co-crystal structure with TBP (Butryn et al., 2015). It should be emphasized that most components of the Set2-Rpd3 system do not interact in a larger complex but function coordinately to prevent intragenic cryptic promoter function (Rando and Winston, 2012). Nevertheless, we are able to observe co-immunoprecipitation of MINC components from mESC extracts under low stringency conditions but were unable to obtain similar results from yeast.
It is currently unclear how the MINC system functions. NC2α/β and Mot1 both bind simultaneously to the exposed surface of DNA-bound TBP in vitro (Butryn et al., 2015). NC2α/β specifically blocks recruitment of TFIIB (Inostroza et al., 1992), while Mot1 is an ATP-dependent SWI-like DNA translocase that evicts TBP from DNA (Auble et al., 1994; Auble et al., 1997). Ino80C subunit deletions lead to movement of the +1 nucleosome into the NDR in yeast (Yen et al., 2012; Yen et al., 2013) and depletion of Mot1 leads to redistribution of TBP (Zentner and Henikoff, 2013). In principle, the inward movement of nucleosomes could partially disrupt pre-existing PICs and redistribution of TBP could augment transcription from cryptic intergenic promoters.
TBP is a promiscuous DNA binding protein that recognizes many AT-rich sequences. Natural promoters employ several recognition sequences and multiple mechanisms, including evolutionary selection, to ensure that TBP, in the context of TFIID, recognizes physiological promoters with high affinity and a specific directionality (Kadonaga, 2012). If the roles of Mot1 and NC2 were to simply ensure that cryptic promoters were not utilized, then why do they position themselves within natural promoters and become recruited as part of PICs? It is plausible that by naturally downregulating TFIID function to a certain degree, most natural promoters, when activated, would overcome the inhibition via binding of the GTFs and co-activators, thereby displacing Mot1 and NC2. By contrast, most cryptic transcription units exposed by chromatin remodeling would not possess strong promoters because these would have been selected against evolutionarily. While TBP may bind to such promoters, the lack of recognition sites for TAFs along with the association of Mot1 and NC2 would restrict its ability to facilitate GTF and co-activator recruitment. In this scenario, the role of Ino80C would be to minimize exposure of cryptic promoters or disruption of the natural promoter via proper nucleosome organization.
Our data also reveal increases in RNA around the TTS in both yeast and mammalian genes after depletion of MINC. In yeast, MINC is enriched upstream of the TSS and downstream of the TTS, even on convergently transcribed genes. However, mESCs do not display consistent MINC or TBP binding near the TTS and our analyses of published Cap-seq data have not revealed significant downstream initiation sites. Although we observe increases in both sense and antisense RNA downstream of genes in mESCs, the sense signal is stronger and correlates with increased Pol II density. It is therefore plausible that the 3′ enrichment of RNA and Pol II is due to Downstream of Gene transcripts or DoGs (Vilborg and Steitz, 2016). DoGs are caused by stress, account for 20% of intergenic transcripts, and apparently result from failure by Pol II to terminate properly.
A key question is why MINC depletion leads to upregulation of pervasive transcription near only a subset of bound genes, i.e., C1 and C2 in mESCs represent 25% of mESC genes and C1 represents 30% of S. cerevisiae genes. Both convergently and divergently transcribed genes display MINC-responsive pervasive transcription initiating nearby. Neither Motif or Gene Ontogeny analyses revealed any distinguishing features not found in the less affected clusters. Moreover, there are thousands of unaffected pervasive transcripts. In mESCs there is a skew towards more highly expressed genes for MINC regulated uaRNAs. Indeed, little MINC regulated pervasive transcription occurs in Hox gene arrays, except near active genes (i.e., Hoxa1). However, other features such as the nucleosome organization, DNA replication and differentiation state of the cell might also affect MINC regulation. Additionally, many genes may have redundant mechanisms to control pervasive transcription or there may be additional unidentified components to the system, whose depletion might be necessary to observe an effect of MINC. MINC clearly plays a significant role in controlling thousands of pervasive transcription targets and an additional role in silencing inactive genes in yeast heterochromatin and murine and yeast euchromatin.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to Lead Contact Michael Carey (mcarey@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains
S. cerevisiae strains are derived from MJE7 (Matα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 tor1-1 fpr1::NAT rpl13A-2×FKBP12::TRP1 Δade2-1::ADE2). The full strain list is in Table S1.
Cell line
E14 murine embryonic stem cells E14TG2a were used here.
METHOD DETAILS
Preparation of Yeast Nuclear Extracts
Yeast nuclear extracts were prepared as described (Rani et al., 2004).
Immobilized Template Assay and MuDPIT Screen
Immobilized template assays were performed as described using G5E4T assembled into chromatin. Briefly, 5 micrograms of a 602-bp template encompassing G5E4T was assembled into chromatin as described (Kitada et al., 2012), immobilized onto paramagnetic beads and incubated with template-saturating amounts (~10 micrograms) of Sir3 and Sir2/4 in 2.5 ml of binding buffer (100 mM KOAc, 20 mM Hepes, pH 7.6, 1 mM EDTA and 10% glycerol) for 1h at 30°C. 12 mg of yeast nuclear extract were added and the incubation continued at 23°C for 45 min. The beads were washed three times with 1 ml of binding buffer. Proteins were eluted in 200 microliters of 50 mM Tris, pH 8.5 and 7 M urea. Alternately, the chromatin templates were incubated plus and minus ~2.5 micrograms of GAL4-VP16 prior to addition of nuclear extract after which the process was identical to above. MuDPIT analysis was performed as described (Chen et al., 2012). The raw NSAF files were analyzed by MS-SORT, a Python-based program that utilizes the manually curated list of protein complexes in the SGD database to arrange the identified proteins into complexes, and calculates the average NSAF of each complex with respect to its unique subunits. The data were imported into Excel, sorted by Avg. NSAF and a heat map was generated for both silent chromatin and PICs (More complete data in Table S2). The experiments were performed twice with similar results.
Murine Embryonic Stem Cell Culture
E14 murine embryonic stem cells (ESCs) were cultured on gelatin-coated plates without a MEF feeder layer under standard ESC conditions. Briefly, cells were cultured on 0.2% gelatinized (Sigma, G1890) tissue culture plates in ESC media containing DMEM-KO (Invitrogen, 10829-018) supplemented with 15% fetal bovine serum, 1000 U/ml LIF, 100 μM nonessential amino acids (Invitrogen, 11140-050), 2 mM L-glutamine (Invitrogen, 25030-081) and 8 nL/ml of 2- mercaptoethanol (Sigma, M7522).
siRNA Knockdown and Western blots
For the analysis of global levels of Ino80, Mot1 and NC2 upon knockdowns, mouse E14 ES cells were transfected with SMARTpool siRNA from Dharmacon. Knockdown was performed as per manufacturer’s protocol, and cells were harvested 72 hr following transfection. The efficiency was probed by RPKM analysis of the RNA-seq data. Mock transfection was performed with GFP siRNA. Cell lysates were prepared following the same protocol as for RNA-seq experiments.
ChIP-Seq of Yeast
All yeast cells were grown in YPD media at 30 °C in log phase for further treatment. For ChIP of MINC at the subtelomere, cells were collected and crosslinked with 1% formaldehyde for 45 minutes and quenched with 0.125 M glycine for 5 minutes. For ChIP of MINC in euchromatin, cells were synchronized in G1 phase with a-factor for 3 hours (4 hours for TBP anchor away), then treated with either DMSO or 8 μg/ml rapamycin with a factor for another two hours. Cells were harvested at O.D. ~1 and crosslinked with formaldehyde for 20 minutes. For ChIP of H3K79me3 changes, cells were treated with 8 μg/ml rapamycin for 24 hours and crosslinked with formaldehyde for 20 minutes. All samples were lysed with glass beads in lysis buffer (50 mM HEPES, 140 mM NaCl, 10 mM EDTA, 5 mM EGTA, 1% TritonX-100, 0.1% deoxycholate, 0.5% N-Lauroyl Sarcosine) with protease inhibitors. Chromatin was collected by centrifugation, resuspended in lysis buffer and subjected to sonication except for ChIP of MINC at the subtelomere, in which after lysis, chromatin was extensively washed using wash buffer (50 mM HEPES/KOH pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate), deoxycholate buffer (10 mM Tris/Cl pH 8.0, 0.25 M LiCl, 0.5% NP-40, 0.5% Na-Deoxycholate, 1 mM EDTA) at 4°C, and resusp end in lysis buffer before sonication. For MINC in euchromatin, chromatin was sonicated for 10 cycles at 15s each with 30s between cycles at 4°C with 20% power on a Qsonica Q800R2 sonicator. For H3K79Me3, chromatin was sonicated for 8 cycles at 30s each with 30s between cycles at 4°C with 60% power. For MINC at the subtelomere, chromatin was sonicated for 4 minutes with 30s on and 1 min off on a Biodisruptor. The supernatant from sonicated lysates were precleared with Protein A/G beads and ChIP was performed as described (Kitada et al., 2012) using commercial antibodies: anti-Myc (Abcam, ab32, GR255064-2), anti-Arp5 (Abcam, ab12099), anti-H3K79me3 (Grunstein lab, 644) and 0.5% BSA (w/v) pre-blocked Protein A/G beads. Libraries were prepared with a KAPA LTP kit and sequenced using the Illumina HiSeq 2000 or 4000 platform for 50 bp single end reads. All experiments were performed with biological replicates with similar results. Spearman correlation of most replicates is shown in Table S3.
ChIP-Seq of Mouse
ChIP was performed as in (Pradhan et al., 2016) with minor adaptations. E14 cells grown in 150 cm2 dishes were harvested at 70% confluency by trypsinization, followed by 1x DPBS wash. Cells were then formaldehyde-crosslinked to a final concentration of 0.75% for 10 min at room temperature, followed by 5 min quenching with 100 mM glycine. Cells were washed twice with cold PBS, the supernatant was aspirated and the cell pellet was flash frozen in liquid nitrogen. Frozen crosslinked cells were stored at −80°C. Crosslinked cells were resuspended and sonic ated in lysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA pH 8.0, 0.1% SDS, and 1% Triton X-100). Cells were sonicated for 10 cycles at 15s each with 30s between cycles at 4C on a Qsonica Q800R2 sonicator. Sonicated lysates were cleared and incubated overnight at 4°C with 5–10 micrograms of antibody: Ino80: Proteintech, 18810-1-AP; NC2 beta: Abcam, ab50783; murine Mot1: Abcam, ab72285, Med1: Santa Cruz, sc-5334, Pol II: QED Bioscience, 70101. DNA/Protein-antibody conjugates were captured using Protein A/G beads blocked with 0.5% BSA (w/v) in PBS. Beads were washed twice each and sequentially with wash buffer A (50 mM HEPES-KOH pH7.9, 140 mM NaCl, 1 mM EDTA pH 8.0, 0.1% Na-Deoxycholate, 1% Triton X-100, 0.1% SDS), buffer B (50 mM HEPES-KOH pH7.9, 500 mM NaCl, 1 mM EDTA pH 8.0, 0.1% Na-Deoxycholate, 1% Triton X-100, 0.1% SDS), buffer C (20 mM Tris-HCl pH8.0, 250 mM LiCl, 1 mM EDTA pH 8.0, 0.5% NaDeoxycholate, 0.5% IGEPAL C-630, 0.1% SDS) and buffer D (TE with 50 mM NaCl). DNA was eluted in elution buffer (50 mM Tris-HCL pH 8.0, 10 mM EDTA, 1% SDS). Crosslinks were reversed overnight. RNA and protein were digested using RNase A and Proteinase K, respectively, and DNA was purified with phenol chloroform extraction and ethanol precipitation. Libraries were prepared with a KAPA LTP kit and sequenced using the Illumina HiSeq 2000 or 4000 platform for 50 bp single end reads. All experiments were performed with biological replicates. Spearman correlation of replicates is shown in Table S3.
Data Analysis of ChIP-Seq
All sequenced reads were mapped to yeast genome version sacCer3 (SacCer_Apr2011) or mouse genome version mm9 using bowtie 0.12.9 and default settings (Langmead et al., 2009). After filtering out the clonal reads, the IP and Input samples were shuffled to similar reads, IP samples were normalized to input using a custom script. The S. cerevisiae and Mus musculus genome was divided into 50-bp windows, and significant windows with a p value lower than 0.001 were selected as described (Ferrari et al., 2012). The log2 ratio of ChIP vs. input at significant windows was used to generate metagene profiles around the TSS and TTS or profiles centered at the TSS or TTS. The annotation of yeast TSS and TTS have been described (Malabat et al., 2015). The gene list of SAGA or TFIID dominated TSSs was described (Zentner and Henikoff, 2013). The gene list of TATA-containing genes was obtained from (Basehoar et al., 2004). The annotation of mouse TSS and TTS were from UCSC and gene_ID beginning with NM were used during analysis. Unless specified, plots with log2 ratios of H3K79me3 and Pol II in anchor away vs wild type or knockdown versus mock were generated using normalized raw reads from all windows. The Sir3 ChIP-chip data were described (Sperling and Grunstein, 2009). The Ring1B and Pcgf2 ChIP-seq data were downloaded from GEO (Kloet et al., 2016) and TBP ChIP-seq data were download from GSM555160. We analyzed both of the biological duplicates and showed one dataset in the Figures. Another dataset showed similar results and the data are deposited in GEO. Spearman correlation of replicates is shown in Table S3.
Co-IP
Mouse embryonic stem cells were harvested at 70% confluency by trypsinization without formaldehyde crosslinking. Cell pellets were resuspended in lysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% SDS, and 1% Triton X-100 supplemented with protease inhibitors (1 mM PMSF, 1 g/ml pepstatin, 1 g/ml aprotinin, 1 g/ml leupeptin). Cell lysates were further homogenized with 20 strokes of pestle B Kontes glass homogenizer. Lysates were clarified by centrifugation at 16,000 g for 20 min at 4°C. Clarified supernatants were precleared with Protein A dynabeads (Invitrogen) for 1h at 4°C. 20 mg of precleared lysates were used for immunoprecipitation. Immunoprecipitation was performed with 5 μg of antibody-proteinA/G conjugates, in the presence of 7.5 units of RNase A/T1 mixture for 3h at 4°C. The beads were wa shed twice each with lysis buffer, lysis buffer containing 250 mM NaCl and then TE (10 mM Tris, pH 8.0, 1 mM EDTA). All the washes were performed at 4°C for 5 min, and the buffers were supplemented with fresh protease inhibitors. Proteins were extracted by boiling the beads at 95°C with SDS-PAGE loading buffer, loaded on SDS gels, immunoblotted and quantified a using Li-Cor Odyssey imaging system using dilutions of the input samples in the linear range.
RNA extraction
For yeast MINC anchor away, yeast cells were first grown in YPD to log phase and treated with DMSO or rapamycin (8 μg/ml) for 24 hours except for RAP1-FRB, which is an 8-hour treatment. During treatment, cells were sustained in log phase by dilution into the same media. For nhp6a/bΔ, mutant and WT cells were grown in YPD to log phase and collected. Total RNA was extracted using the hot acid phenol extraction method. The extracted RNA samples were treated with DNase I (Ambion TURBO DNA-free Kit) and further purified with Trizol regents (Ambion). For mouse total RNA extraction, total RNA was extracted using Trizol (Invitrogen) according to manufacturer’s instruction. RNAs were treated with DNase before library preparation. For mouse nascent RNA extraction, mESCs were harvested, washed with DPBS and lysed in 200 μl of ice-cold lysis buffer (10 mM Tris-HCl [pH 7.5], 0.1% NP40, 150 mM NaCl). The cell lysate was gently layered over 500 μl of chilled sucrose cushion (24% RNAse-free sucrose in lysis buffer) in a new Eppendorf tube and centrifuged for 10 min at 4°C, 10,000xg. The supernatant (cytoplasmic fraction) was removed and the pellet (nuclei) was washed once with 200 ml of ice-cold 1× PBS/1 mM EDTA. The nuclear pellet was resuspended in 100 μl of prechilled glycerol buffer (20 mM Tris-HCl [pH 7.9], 75 mM NaCl, 0.5 mM EDTA, 0.85 mM DTT, 0.125 mM PMSF, 50% glycerol) by gentle flicking of the tube. An equal volume (100 μl) of cold nuclei lysis buffer (10 mM HEPES [pH 7.6], 1 mM DTT, 7.5 mM MgCl2, 0.2 mM EDTA, 0.3 M NaCl, 1 M urea, 1% NP-40) was added. The mix was vortexed vigorously for 2 seconds. The sample was incubated for 2 min on ice, and then centrifuged for 2 min at 4°C and 10,000xg. The supe rnatant (nuclear fraction/nucleoplasm) was removed and the pellet (chromatin) was gently rinsed with ice-cold 1× PBS/1 mM EDTA. 1 ml of Trizol reagent was added to the chromatin and incubated for 30 min at 50 °C to dissolve it. Nasce nt RNA was extracted following the manufacturer’s manual. Fractionation and sample purity was monitored by immunoblotting using a-tubulin for cytoplasm, U1 70K for nuclear extract and histone H3 for chromatin. Two biological duplicates were prepared and sequencing data were similar and both were uploaded to GEO. The validity of the chromatin or Nascent RNA-seq is established by 6-fold higher intron/exon ratio of read counts as summarized in Table S4.
RNA libraries preparation, sequencing and data analysis
For yeast, libraries of mRNA were prepared with a KAPA stranded mRNA-Seq kit. For mouse total and nascent RNA, libraries were prepared with KAPA stranded RNA-Seq with RiboErase kit. Libraries were sequenced on the Illumina HiSeq 2500 for 50 bases in paired end read mode. Sequenced reads were aligned as above using tophat2.2.1 with option –g 1 –N 2 --no-mixed --no-discordant --library-type=fr-firststrand (Langmead et al., 2009). For yeast telomere, reads were aligned with option –g 1 –N 2. Yeast gene transcription levels were normalized to FPKM using cuffdiff 2.0.2 (Trapnell et al., 2010). Mouse gene transcription levels were normalized to RPKM using SAMMate (Xu et al., 2011a). For log2 ratio and log2 FPKM/RPKM calculations, all transcripts lower than 0.1 FPKM/RPKM were replaced with 0.1. Mapped reads in the mutants were also normalized to reads in the wild type or mock using a custom script. Because we were mapping changes in transcription, windows without any overlapping reads in the wild type or mutant cells were replaced with 0.1 for further treatment of the mutant transcription patterns. The log2 ratio of mutants vs. wild type at all windows were plotted against the distance from telomere ends or metagene with a custom script. For strand specific reads, the mapped plus or minus strand transcripts were separately plotted against genes on the minus or plus strand for metagene analysis. Yeast antisense or sense transcription on MINC-bound genes was separated into three clusters using k means (cluster 3.0). The mouse antisense transcription on MINC-bound genes was first separated into 6 clusters using k means. Clusters displaying similar transcription patterns under all conditions upstream of TSS and downstream of TTS are termed cluster 1 and 2. All other clusters were merged and randomized as cluster 3. The mouse sense transcription on MINC-bound genes were first separated into 4 clusters using k means and clusters with similar transcription pattern under all KD conditions at coding and downstream of TTS is included as cluster 4. All other clusters were merged and randomized as cluster 5. The RNA-seq data of WT (BY4741) and sir3Δ were obtained from GSE52000 (Xue et al., 2015). The yeast data for anchor away (except Nc2β) and mouse total RNA-Seq are a combination of two repeats. We showed one dataset of yeast Nc2β anchor away and mouse nascent RNA-seq in the Figures and we also have the biological duplicates, which yielded similar results. All the datasets are deposited in GEO. Spearman correlation of replicates is shown in Table S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical significance of gene expression between cluster results from Figure 4 and 6 was assessed using the unpaired Student’s t test.
DATA AND SOFTWARE AVAILABLITY
The accession number for the sequencing data reported in this paper is GEO: GSE95633.
MENDELEY DATA LINK
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 GM074701 to M.F.C., GM54096 to C.L.P. and CA178415 to S.K.K. C.C. was supported by P01 GM099134 to Kathrin Plath. We thank Gabe Zentner and Sebastian Grunberg for helpful advice on yeast promoter analysis. We thank Maria Koster for construction of certain yeast strains. The authors declare no conflicts.
Footnotes
Author Contributions
Y.X. performed yeast and S.K.P and F.S. performed mESC ChIP- and RNA-seq. N.T. performed immobilized template experiments, Y.X. and T.S. performed data analysis and C.C. performed chromatin state analysis. A.V. and J.W. performed MuDPIT experiments and data analysis. J.W. M.F.C. and S.K.K. directed all research and assisted in data interpretation. H.T.M.T., C.V. and C.L.P provided reagents, yeast strains and experimental guidance.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett DR, Jennings JL, Tabb DL, Link AJ, Weil PA. A proteomics analysis of yeast Mot1p protein-protein associations: insights into mechanism. Mol Cell Proteomics. 2008;7:2090–2106. doi: 10.1074/mcp.M800221-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auble DT. The dynamic personality of TATA-binding protein. Trends Biochem Sci. 2009;34:49–52. doi: 10.1016/j.tibs.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auble DT, Hansen KE, Mueller CG, Lane WS, Thorner J, Hahn S. Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev. 1994;8:1920–1934. doi: 10.1101/gad.8.16.1920. [DOI] [PubMed] [Google Scholar]
- Auble DT, Wang D, Post KW, Hahn S. Molecular analysis of the SNF2/SWI2 protein family member MOT1, an ATP-driven enzyme that dissociates TATA-binding protein from DNA. Mol Cell Biol. 1997;17:4842–4851. doi: 10.1128/mcb.17.8.4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- Becker JS, Nicetto D, Zaret KS. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016;32:29–41. doi: 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butryn A, Schuller JM, Stoehr G, Runge-Wollmann P, Forster F, Auble DT, Hopfner KP. Structural basis for recognition and remodeling of the TBP:DNA:NC2 complex by Mot1. Elife. 2015;4 doi: 10.7554/eLife.07432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XF, Lehmann L, Lin JJ, Vashisht A, Schmidt R, Ferrari R, Huang C, McKee R, Mosley A, Plath K, et al. Mediator and SAGA have distinct roles in Pol II preinitiation complex assembly and function. Cell Rep. 2012;2:1061–1067. doi: 10.1016/j.celrep.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Pai AA, Herudek J, Lubas M, Meola N, Jarvelin AI, Andersson R, Pelechano V, Steinmetz LM, Jensen TH, et al. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat Genet. 2016;48:984–994. doi: 10.1038/ng.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. 2017;168:442–459. e420. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin J, Candelli T, Porrua O, Boulay J, Zhu C, Lacroute F, Steinmetz LM, Libri D. Roadblock termination by reb1p restricts cryptic and readthrough transcription. Mol Cell. 2014;56:667–680. doi: 10.1016/j.molcel.2014.10.026. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci. 2009;34:71–77. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Darst RP, Martin KJ, Afshari CA, Auble DT. Mot1 activates and represses transcription by direct, ATPase-dependent mechanisms. Proc Natl Acad Sci U S A. 2002;99:2666–2671. doi: 10.1073/pnas.052397899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Su T, Li B, Bonora G, Oberai A, Chan Y, Sasidharan R, Berk AJ, Pellegrini M, Kurdistani SK. Reorganization of the host epigenome by a viral oncogene. Genome research. 2012;22:1212–1221. doi: 10.1101/gr.132308.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort A, Hashimoto K, Yamada D, Salimullah M, Keya CA, Saxena A, Bonetti A, Voineagu I, Bertin N, Kratz A, et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet. 2014;46:558–566. doi: 10.1038/ng.2965. [DOI] [PubMed] [Google Scholar]
- Gartenberg MR, Smith JS. The Nuts and Bolts of Transcriptionally Silent Chromatin in Saccharomyces cerevisiae. Genetics. 2016;203:1563–1599. doi: 10.1534/genetics.112.145243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Navarro N, Jordan-Pla A, Estruch F, JEPO Defects in the NC2 repressor affect both canonical and non-coding RNA polymerase II transcription initiation in yeast. BMC Genomics. 2016;17:183. doi: 10.1186/s12864-016-2536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber F, Bunina D, Gupta I, Khmelinskii A, Meurer M, Theer P, Steinmetz LM, Knop M. Protein Abundance Control by Non-coding Antisense Transcription. Cell Rep. 2016;15:2625–2636. doi: 10.1016/j.celrep.2016.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inostroza JA, Mermelstein FH, Ha I, Lane WS, Reinberg D. Dr1, a TATA-binding protein-associated phosphoprotein and inhibitor of class II gene transcription. Cell. 1992;70:477–489. doi: 10.1016/0092-8674(92)90172-9. [DOI] [PubMed] [Google Scholar]
- Jensen TH, Jacquier A, Libri D. Dealing with pervasive transcription. Mol Cell. 2013;52:473–484. doi: 10.1016/j.molcel.2013.10.032. [DOI] [PubMed] [Google Scholar]
- Kadonaga JT. Perspectives on the RNA polymerase II core promoter. Wiley Interdiscip Rev Dev Biol. 2012;1:40–51. doi: 10.1002/wdev.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD. Pairs of promoter pairs in a web of transcription. Nat Genet. 2016;48:975–976. doi: 10.1038/ng.3649. [DOI] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, Winston F. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003;301:1096–1099. doi: 10.1126/science.1087374. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee BB, Oh YM, Zhu C, Steinmetz LM, Lee Y, Kim WK, Lee SB, Buratowski S, Kim T. Modulation of mRNA and lncRNA expression dynamics by the Set2-Rpd3S pathway. Nature communications. 2016;7:13534. doi: 10.1038/ncomms13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Kuryan BG, Tran NN, Song C, Xue Y, Carey M, Grunstein M. Mechanism for epigenetic variegation of gene expression at yeast telomeric heterochromatin. Genes Dev. 2012;26:2443–2455. doi: 10.1101/gad.201095.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klejman MP, Pereira LA, van Zeeburg HJ, Gilfillan S, Meisterernst M, Timmers HT. NC2alpha interacts with BTAF1 and stimulates its ATP-dependent association with TATA-binding protein. Mol Cell Biol. 2004;24:10072–10082. doi: 10.1128/MCB.24.22.10072-10082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloet SL, Makowski MM, Baymaz HI, van Voorthuijsen L, Karemaker ID, Santanach A, Jansen PW, Di Croce L, Vermeulen M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol. 2016;23:682–690. doi: 10.1038/nsmb.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Krietenstein N, Wal M, Watanabe S, Park B, Peterson CL, Pugh BF, Korber P. Genomic Nucleosome Organization Reconstituted with Pure Proteins. Cell. 2016;167:709–721. e712. doi: 10.1016/j.cell.2016.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Wu J, Li B. Chromatin remodelers fine-tune H3K36me-directed deacetylation of neighbor nucleosomes by Rpd3S. Mol Cell. 2013;52:255–263. doi: 10.1016/j.molcel.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Lorch Y, Maier-Davis B, Kornberg RD. Role of DNA sequence in chromatin remodeling and the formation of nucleosome-free regions. Genes Dev. 2014;28:2492–2497. doi: 10.1101/gad.250704.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabat C, Feuerbach F, Ma L, Saveanu C, Jacquier A. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. Elife. 2015;4 doi: 10.7554/eLife.06722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Black DL. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009;15:1896–1908. doi: 10.1261/rna.1714509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 2016;352:aad9780. doi: 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- Pradhan SK, Su T, Yen L, Jacquet K, Huang C, Cote J, Kurdistani SK, Carey MF. EP400 Deposits H3.3 into Promoters and Enhancers during Gene Activation. Mol Cell. 2016;61:27–38. doi: 10.1016/j.molcel.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh BF, Venters BJ. Genomic Organization of Human Transcription Initiation Complexes. PLoS One. 2016;11:e0149339. doi: 10.1371/journal.pone.0149339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Winston F. Chromatin and transcription in yeast. Genetics. 2012;190:351–387. doi: 10.1534/genetics.111.132266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani PG, Ranish JA, Hahn S. RNA polymerase II (Pol II)-TFIIF and Pol II-mediator complexes: the major stable Pol II complexes and their activity in transcription initiation and reinitiation. Mol Cell Biol. 2004;24:1709–1720. doi: 10.1128/MCB.24.4.1709-1720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K, Gabunilas J, Gillespie A, Ngo D, Chanfreau GF. Common genomic elements promote transcriptional and DNA replication roadblocks. Genome Res. 2016;26:1363–1375. doi: 10.1101/gr.204776.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski TW, Buratowski S. The basal initiation machinery: beyond the general transcription factors. Curr Opin Cell Biol. 2009;21:344–351. doi: 10.1016/j.ceb.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski TW, Joo YJ, Ficarro SB, Askenazi M, Buratowski S, Marto JA. Proteomic analysis demonstrates activator- and chromatin-specific recruitment to promoters. J Biol Chem. 2012;287:35397–35408. doi: 10.1074/jbc.M112.391581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol. 2012;19:884–892. doi: 10.1038/nsmb.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta. 2013;1829:84–97. doi: 10.1016/j.bbagrm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling AS, Grunstein M. Histone H3 N-terminus regulates higher order structure of yeast heterochromatin. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13153–13159. doi: 10.1073/pnas.0906866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tora L. A unified nomenclature for TATA box binding protein (TBP)-associated factors (TAFs) involved in RNA polymerase II transcription. Genes Dev. 2002;16:673–675. doi: 10.1101/gad.976402. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk EL, Chen CL, d’Aubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix-Ne P, Loeillet S, et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–117. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- van Werven FJ, van Bakel H, van Teeffelen HA, Altelaar AF, Koerkamp MG, Heck AJ, Holstege FC, Timmers HT. Cooperative action of NC2 and Mot1p to regulate TATA-binding protein function across the genome. Genes Dev. 2008;22:2359–2369. doi: 10.1101/gad.1682308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Li H, Gogol MM, Workman JL. Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nature communications. 2016;7:13610. doi: 10.1038/ncomms13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455. doi: 10.1038/nature11326. [DOI] [PubMed] [Google Scholar]
- Venkatesh S, Workman JL. Set2 mediated H3 lysine 36 methylation: regulation of transcription elongation and implications in organismal development. Wiley Interdiscip Rev Dev Biol. 2013;2:685–700. doi: 10.1002/wdev.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Steitz JA. Readthrough transcription: How are DoGs made and what do they do? RNA Biol. 2016:1–5. doi: 10.1080/15476286.2016.1149680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin J, Schibler U. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol Cell Biol. 1994;14:7219–7225. doi: 10.1128/mcb.14.11.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Deng N, Zhao Z, Judeh T, Flemington E, Zhu D. SAMMate: a GUI tool for processing short read alignments in SAM/BAM format. Source Code Biol Med. 2011a;6:2. doi: 10.1186/1751-0473-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Clauder-Munster S, Smolik M, Huber W, Steinmetz LM. Antisense expression increases gene expression variability and locus interdependency. Mol Syst Biol. 2011b;7:468. doi: 10.1038/msb.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Van C, Pradhan SK, Su T, Gehrke J, Kuryan BG, Kitada T, Vashisht A, Tran N, Wohlschlegel J, et al. The Ino80 complex prevents invasion of euchromatin into silent chromatin. Genes Dev. 2015;29:350–355. doi: 10.1101/gad.256255.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li W, Hoque M, Hou L, Shen S, Tian B, Dynlacht BD. PAF Complex Plays Novel Subunit-Specific Roles in Alternative Cleavage and Polyadenylation. PLoS Genet. 2016;12:e1005794. doi: 10.1371/journal.pgen.1005794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. 2012;149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Pugh BF. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013;154:1246–1256. doi: 10.1016/j.cell.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S. Mot1 redistributes TBP from TATA-containing to TATA-less promoters. Mol Cell Biol. 2013;33:4996–5004. doi: 10.1128/MCB.01218-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.