

Abstract
Large-scale consortia including the Psychiatric Genomics Consortium, the Common Minds Consortium, BrainSeq and PsychENCODE, and many other studies taken together provide increasingly detailed insights into the genetic and epigenetic risk architectures of schizophrenia and offer vast amounts of molecular information, but with largely unexplored therapeutic potential. Here we discuss how epigenomic studies in human brain could guide animal work to test the impact of disease-associated alterations in chromatin structure and function on cognition and behavior. For example, transcription factors such as MYOCYTE-SPECIFIC ENHANCER FACTOR (MEF2C), or multiple regulators of the open chromatin mark, methyl-histone H3-lysine 4, are associated with the genetic risk architectures of common psychiatric disease and alterations in chromatin structure and function in diseased brain tissue. Importantly, these molecules also affect cognition and behavior in genetically engineered mice, including virus-mediated expression changes in prefrontal cortex and other key nodes in the circuitry underlying psychosis. Therefore, preclinical and small laboratory animal work could target genomic sequences affected by chromatin alterations in schizophrenia. To this end, in vivo editing of enhancer and other regulatory non-coding DNA by RNA-guided nucleases including CRISPR-Cas, and designer transcription factors, could be expected to deliver pipelines for novel therapeutic approaches aimed at improving cognitive dysfunction and other core symptoms of schizophrenia.
Introduction
Schizophrenia (SCZ) is a major psychiatric disorder often with onset in adolescence or young adulthood, with positive symptoms such as delusions and hallucinations, and negative symptoms such as social withdrawal and apathy. The lifespan of subjects diagnosed with SCZ is reduced on average by 15 years, in comparison with the general population, primarily due to cardiovascular disease and suicide [Hennekens and others 2005; Laursen and others 2012; Saha and others 2007]. Antipsychotic medications, while widely prescribed, mostly target dopaminergic and serotonergic receptor systems[Kim and Stahl 2010; Taly 2013], but many patients continue to suffer from debilitating symptoms [Lieberman and others 2005; Swartz and others 2007]. Cognitive symptoms in particular are severe, chronically disabling and often persistent during the course of illness[Ibrahim and Tamminga 2011], while ineffectively treated with antipsychotic medication[Green 2016]. Unfortunately, rationale drug development in schizophrenia is extremely challenging, given the lack of a unifying neuropathology[Catts and others 2013; Dorph-Petersen and Lewis 2011] in conjunction with highly heterogeneous genetic risk architectures[Andreassen and others 2014; Rodriguez-Murillo and others 2012]. On the other hand, owing recent conceptual and methodological advances in genetics and genomics, current sequencing technology is capable to process large numbers of samples ‘genome-wide’ at base pair resolution and within short time frames. As a result, the field is presently producing vast amounts of molecular information directly relevant for the study of schizophrenia, with yet largely untapped therapeutic potential. For example, in a study involving 150,000 subjects, the Psychiatric Genomics Consortium identified altogether 108 haplotypes that by individual small effect contribute to the heritable risk for schizophrenia[Schizophrenia Working Group of the Psychiatric Genomics 2014]. In addition, there are rapidly evolving databases for rare mutations and variants discovered by comprehensive sequencing of all protein coding genes (the ‘exome’, which comprises approximately 1% of total genome sequence) in >60,000 subjects[Ruderfer and others 2016] including thousands of subjects with schizophrenia[Genovese and others 2016]. In addition to these (still rapidly advancing) insights into the genetic risk architecture of the disease, we are predicting the emergence of an ‘epigenetic risk architecture’ for schizophrenia, which in the broadest terms of definitions may be described as any disease-relevant alteration in chromatin structure and function. For example, cytosine methylation as the predominant epigenetic DNA mark regulating gene expression, has been mapped in hundreds of post-mortem brain specimens from subjects diagnosed with schizophrenia and in controls across the lifespan[Hannon and others 2016; Jaffe and others 2016]. Similarly, there are ongoing efforts by the National Institutes of Health (NIMH)-sponsored PsychENCODE consortium[Psych and others 2015], and NIMH-Industry (Common Minds Consortium[Fromer and others 2016]), or Industry-sponsored Initiatives[BrainSeq 2015] to map chromatin including histone modifications, transcriptomes and nuclear proteomes in many hundreds of schizophrenia and control postmortem brain specimens. Thus, the field will soon be challenged with the task to ‘convert’ the molecular information provided by these various genetics and (epi)genomics resources into testable hypotheses aimed at gaining deeper insights into the neurobiology of schizophrenia. Perhaps more importantly, such newly gained information could be harnessed to develop novel therapies aimed at improving cognitive dysfunction. While there are already significant efforts to translate these evolving findings from schizophrenia genetics, genomics and epigenomics into drug discovery pipelines and clinical testing[Breen and others 2016], we predict that behavioral studies in genetically engineered mice and other small laboratory animals will serve as an critical preclinical intermediate towards this goal, in conjunction with molecular and cellular exploration of human cells in the culture dish[Brennand and others 2014]. Here, we discuss how epigenomic studies in human brain, combined with information on the genetic risk architecture of schizophrenia, could guide the design of preclinical studies in the mouse. Specifically, we will discuss cognitive and behavioural effects after targeting disease-relevant transcription factor binding sites in the mouse model, and the potential for novel discoveries resulting from genomic and epigenomic editing of sequence-specific regulatory elements important for gene expression.
DNA methylation and other epigenomic alterations in schizophrenia post-mortem brain are linked to the genetic risk architecture of the disorder
There is little doubt that dysregulation of neuronal gene expression in prefrontal cortex (PFC) and other areas of the cerebral cortex regions implicated in the neural circuitry of psychosis (reviewed in [Lewis and Sweet 2009]) contributes to the pathophysiology of schizophrenia, broadly affecting excitatory and inhibitory neurotransmission, metabolism, myelination and immune signaling [Arion and others 2015; Horvath and Mirnics 2015; Middleton and others 2002; Mirnics and others 2000; Vawter and others 2004; Volk and others 2015; Zhao and others 2015]. With the transcriptional process intimately connected to chromatin structure and function in human cells and model organisms alike[Brown and Celniker 2015; Lundberg and others 2016], one would therefore expect that epigenomic markers associated with open (‘active’, ‘loose’) chromatin permissive for gene expression, versus repressed and silenced chromatin, will show significant alterations in brain tissue from subjects diagnosed with schizophrenia. Such type of epigenomic explorations in schizophrenia postmortem brain initially focused on DNA methylation, one of the key epigenetic mechanisms involved in the regulation of gene expression [Klose and Bird 2006]. Methylation occurs at the position 5 of cytosine, primarily in the context of cytosine-guanine (CpG) dinucleotides. When located in gene promoters, the mark is often implicated in gene repression by directly impeding the binding of transcription factors, or by locally inducing repressive chromatin structure that is non-permissive to transcription[Bock and others 2012]. Early studies, examining DNA methylation status of candidate genes affected by dysregulated expression in brains of schizophrenia, reported differential DNA methylation profiles in diseased cerebral cortex for key regulators of neuronal connectivity such as REELIN(RELN) [Abdolmaleky and others 2005; Grayson and others 2005] and key transcription factors such a sex-determining region Y-box containing gene 10 (SOX10) [Iwamoto and others 2005], to mention just two examples out of many. These earlier candidate genes studies were superseeded by genome-wide mapping approaches [Mill and others 2008; Wockner and others 2014] reporting DNA methylation changes for various genes implicated in excitatory or inhibitory neurotransmission, among others. Recently, a study exploring the DNA methylome in the prefrontal cortex of 191 subjects with schizophrenia in comparison to 335 controls collected across the lifespan identified >2000 CpG sites with altered methylation levels in diseased tissue[Jaffe and others 2016]. This pool of epigenetically dysregulated sequences in [Jaffe and others 2016]showed significant overlap with sequences associated with common sequence polymorphisms in 108 haplotypes each associated with small but tractable genetic risk for schizophrenia[Schizophrenia Working Group of the Psychiatric Genomics 2014]. These included many CpGs that undergo robust DNA methylation changes during the transition from the pre- to the postnatal period, which speaks for a neurodevelopmental origin of the disorder[Hannon and others 2016; Jaffe and others 2016]. However, the functional implications of these overall extremely subtle methylation differences (on average, 1.3% difference between schizophrenia and control brains for significantly affected CpG sites in the aforementioned study by Jaffe and colleagues) remain unclear. Future studies, exploring the DNA methylome of specific brain cell populations in diseased vs. control brains[Jiang and others 2008; Siegmund and others 2007] as opposed to the aforementioned earlier studies which utilized tissue homogenate, or correlational analyses between the brain’s DNA methylomes and structural or functional defects in neurons[McKinney and others 2017; Ruzicka and others 2015], may provide deeper insight into the role of epigenetic dysregulation affecting the brain of subjects with schizophrenia.
In addition to DNA methylation, other epigenomic determinants of chromatin structure and function in fetal and adult human brain, including histone methylation and acetylation markings associated with cis-regulatory sequences such as gene promoters, enhancers and repressors, also show significant, up to 26-fold enrichment for single nucleotide polymorphisms associated with heritable risk for schizophrenia[Roussos and others 2014]. These effects were highly tissue-specific, because brain histone methylation landscapes showed no enrichment for polymorphisms associated with rheumatoid arthritis and other common disorders usually not affecting the central nervous system[Roussos and others 2014]. The fact that multiple epigenetic layers from brain cells have been linked to the genetic risk architecture of schizophrenia broadly implies that ‘epigenomic dysregulation’—or changes in chromatin structure and function that lead, further downstream, to alterations in the expression of specific genes and disruptions in the coordinated regulation of multiple transcriptional units in brain tissue from subjects with schizophrenia—could be key drivers in the pathophysiology of psychosis and are less likely to reflect epiphenomena.
Histone-based regulatory mechanisms associated with schizophrenia exert significant effects on cognition and behavior in the preclinical model
If, as we discussed in the preceding chapter, DNA variants and epigenetic alterations affecting promoter and enhancer functions are contributing to the pathophysiology of schizophrenia, then one would expect that ‘epigenomic interventions’ such as treatment with broadly acting chromatin modifying drugs, or alternatively, sequence specific genomic editing (defined as experimentally induced alterations in DNA sequence) or epigenomic editing (defined as experimentally induced alterations in local chromatin structure and function without altering DNA sequence), will exert therapeutic-like effects, including changes in cognition and behavior in the animal model. In the following, we will briefly discuss disease-relevant chemical modifications of the nucleosome histones, and early but exciting findings on behavioral and cognitive effects after neuron-specific ablations, or overexpression, of specific histone modifying enzymes in the preclinical model.
Chromatin regulation by virtue of chemical histone modifications is extremely complex, with far more than 100 amino acid residue-specific post-translational modifications (PTMs) in a typical vertebrate cell [Tan and others 2011]. These include, among several others, residue-specific mono (me1), di (me2)- and tri (me3) methylation, acetylation and crotonylation [Baumann 2015]. These site- and residue-specific PTMs are typically explored in the context of chromatin structure and function, with an epigenetic histone code (a combinatorial set of histone PTMs that differentiates between promoters, gene bodies, enhancer and other regulatory sequences, condensed heterochromatin, and so on [Zhou and others 2011]. The field is currently actively exploring the role of the histone modification machinery in the neurobiology of schizophrenia, including novel treatment options. Notably, clinically effective dopamine D2 antagonist and other antipsychotic drugs induce significant and, at least in the acute setting, massive increases in open chromatin-associated histone phosphoacetylation and acetylation in brain regions with rich dopaminergic innervation, including the striatum[Bertran-Gonzalez and others 2009; Li and others 2004]. Histone alterations after chronic exposure with atypical (beyond dopamine D2 antagonism) antipsychotic drugs could exert more subtle but still clinically very significant changes. These include reduced histone acetylation at a metabotropic glutamate receptor promoter, which might explain some of the drug’s side effects[Kurita and others 2012]. More broadly, these findings could imply that some of the mechanisms of action of antipsychotic drugs involve alterations in the activity of histone modifying enzymes, perhaps in locus- or gene-specific manner, thereby affecting only specific portions of chromatin. Interestingly, transcripts for a subset of histone deacetylase enzymes with broad activity against histones (and additional non-histone proteins) such HDAC1 and HDAC2 are reportedly expressed at altered levels in cortical tissue of some schizophrenia postmortem brain cohorts[Benes and others 2007; Schroeder and others 2016; Sharma and others 2008]. When overexpressed in neurons residing in adult mouse prefrontal cortex[Jakovcevski and others 2013], Hdac1 elicit significant impairments in working memory, a core executive function centered on transient holding and processing of information[Diamond 2013] which is impaired in many patients[Arnsten and others 2016]. Of note, compounds acting as histone deacetylase inhibitors broadly affect mood and affect[Covington and others 2009; Sandner and others 2011; Schroeder and others 2007; Whittle and others 2016], and learning and memory in animal models for neurodevelopmental and neuropsychiatric disorders[Benito and others 2015; Graff and Tsai 2013; Hasan and others 2013]. Perhaps unsurprisingly then, sodium butyrate as a prototype HDAC inhibitor drug is now entering a clinical trial for patients with schizophrenia (clinicalTrials.gov Identifier NCT03010865). In this context, one should mention sodium valproate, a short chain fatty acid derivative and FDA-approved mood stabilizer and anticonvulsant, while closely related to sodium butyrate and an effective HDAC inhibitor in cell culture. However, therapeutic levels in patients are most likely below the levels required to induce histone hyperacetylation (reviewed in [Hasan and others 2013]), and therefore not likely to be part of the clinically relevant mechanisms for action of this widely prescribed drug.
In addition to histone acetylation, mono-, di- and trimethyl-histone H3-lysine 4 (H3K4me1/2/3)—chromatin marks associated with promoters and active enhancers and gene expression[Zhou and others 2011]—are also of particular interest because multiple regulators for H3K4 methylation show by genome-wide association a surprisingly strong link to the genetic risk architecture of schizophrenia and related common psychiatric disorders [Pefanis and others 2015]. Furthermore, the H3K4me3 mark show neuron-specific regulation at genes involved in glutamatergic and dopaminergic signaling[Dincer and others 2015] with significant gene-specific alterations in cells and brain tissue from subjects with schizophrenia[Huang and others 2007; Kano and others 2013; Mitchell and others 2017]. In addition, mutations in various genes encoding H3K4-methyl regulators, including the histone H3-lysine 4 specific methyltransferase SETD1A/KMT2F have been linked to rare monogenic forms of neurodevelopmental disease and adult-onset schizophrenia [Singh and others 2016; Takata and others 2016; Takata and others 2014; Vallianatos and Iwase 2015]. Furthermore, neuron-specific ablation of the H3K4 methyltransferase Kmt2a/Mll1 in key nodes of the neural circuitry underlying psychosis, including prefrontal cortex and ventral striatum, was associated with defective synaptic plasticity, increased anxiety, altered response profiles to dopaminergic and impairments in working memory[Jakovcevski and others 2015; Shen and others 2016]. In addition, genetic ablation of H3K4 methytlransferase genes elicits robust defects in hippocampal learning and memory[Gupta and others 2010; Kerimoglu and others 2013]. However, it should be noted that these animal models cannot be viewed as specific for schizophrenia, given that alterations in anxiety, working memory, cognition and learning are broadly compromised in a wide range of psychotic and other psychiatric conditions[Lewandowski and others 2016; Lo and others 2016]. Therefore, it will be interesting to explore, in the preclinical model, changes in cognition and behavior after drug-induced interference with the H3K4-methyl-regulome.
Transcription factors associated with the genetic and epigenetic risk architectures of schizophrenia
Many aspects of epigenomic regulation, including DNA methylation and histone modification, are centered on facilitation, or inhibition of transcription factors and activator proteins binding to their designated target sequences in the genomic DNA. These mechanisms are critically important for promoters, commonly defined as cis-regulatory sequences within 1000 base pairs from the next gene transcription start site, and enhancers as cis-regulatory sequence positioned >1kb from the nearest transcription start site[Vernimmen and Bickmore 2015]. Promoters (but not enhancers) typically include a core promoter as docking site for general transcription factors (TFIIA/B/D/E/F/H) and parts of RNA polymerase II holoenzyme and preinitiation complex[Vernimmen and Bickmore 2015]. These core promoters drive low levels of basal transcription. However, gene expression is heavily stimulated by ‘activators’ or transcription factors that bind, in sequence-specific fashion, at the site of promoters and enhancers[Vernimmen and Bickmore 2015]. However, one cardinal challenge for the field is rooted in the fact that functional enhancers are comprised of clusters of transcription factor binding sites; typically cooperative binding of multiple TF in close proximity (<1 nucleosome length) is required for effective nucleosome eviction as pre-requisite for TF binding at the site of DNA motifs[Long and others 2016]. Currently, there are two main distinct models for enhancer architectures, the ‘enhanceosome’ defined by rigid motif organization and spacing, contrasting with the more flexible ‘billboard model’ which associates to each enhancer a set of TFs, with multiple permutational options of order, orientation, and cooperative binding of TF and TF coactivators such as the CBP/p300 histone acetyltransferase complex, the Mediator complex and many other multiprotein assemblies [Arnosti and Kulkarni 2005]. While comparative cross-species enhancer and synthetic enhancer-reporter studies point towards the flexible model[Smith and others 2013; Taher and others 2011], the majority of enhancers are likely to function neither as rigidly arranged ‘enhanceosomes’ nor as flexible ‘billboards’ but instead are somewhere in between these two extremes[Long and others 2016]. Complicating matters further, there is strong evidence that the majority of synergistic TF-TF interactions operate at novel consensus motifs that are very different from the composite motif (defined as complete motif for each of the two (or more) TF interspersed by spacing sequence)[Jolma and others 2015]. These findings, taken together, clearly highlight the extreme difficulties to link regulatory sequences within a schizophrenia risk haplotype to specific function by in silico analysis alone, and emphasize the critical importance of empirical ‘wet laboratory’-based approaches and animal model systems to explore genetic determinants of cognition and sequence-specific therapeutic strategies for cognitive disease.
To date, from the viewpoint of schizophrenia research, there are two not mutually exclusive categories of disease-relevant TF: The first category concerns TF genes associated with mutations or common polymorphisms directly contributing to genetic risk. This list includes, for example TCF4, encoding a basic helix-loop-helix (bHLH) transcription factor and well established neuropsychiatric risk gene with robust significance (P<10−12) in the most recent schizophrenia GWAS of the psychiatric genetics consortium[Schizophrenia Working Group of the Psychiatric Genomics 2014]. TCF4 expression in cerebral cortex, while highest in the pre- and perinatal period, is continuously expressed across all periods of the postnatal, adult and aging human and rodent cortex[Lein and others 2007; Rannals and others 2016], with disease-associated single nucleotide polymorphisms and haplotypes correlating with TCF4 gene expression in some cohorts of adult postmortem brain[Kim and others 2012]. Furthermore, epigenetic drugs, including histone deacetylase inhibitors, robustly ameliorate deficits in cognition and memory in adult Tcf4 haploinsufficient mice, in conjunction with normalized expression of Tcf4-sensitive hippocampal genes[Kennedy and others 2016]. Other examples include ZNF804A, encoding a zinc finger protein and contributing to the genetic risk architecture of schizophrenia and psychosis[Hess and others 2015; Xiao and others 2016]. ZNF804A is robustly expressed in brain across a wide age range, including adult cerebral cortex[Bernstein and others 2014; Hinna and others 2015].
Our second category of TF applies to transcriptional regulators with DNA binding motifs that match to sequence motifs with significant enrichment in the genetic and epigenetic risk architectures of schizophrenia. To this end, MEF2C encoding a member of the MEF (myocyte-specific enhancer factor) subfamily of MADS transcription factors[Adachi and others 2015; Leifer and others 1994], is of particular interest. Expression of MEF2C, a key molecule for neural plasticity and synapse formation[Barbosa and others 2008], is heavily regulated by FOXP2[Chen and others 2016], a TF essential for cortico-basal ganglia circuitry formation, and speech and language development in human[Konopka and Roberts 2016]. The MEF2C gene overlaps with one the 108 schizophrenia risk haplotypes by genome-wide association[Ohi and others 2016]. Furthermore, MEF2C harbors sequences that separate the human lineage from the rest of the primate tree[Doan and others 2016], and homozygous MEF2C mutations are associated with rare cases of neurodevelopmental disease[Doan and others 2016]. Thus MEF2C, in our classification system, certainly qualifies as ‘category 1’ TF. Interestingly, however, a recent study exploring motif enrichment within 2kb of sequence surrounding the lead polymorphisms in the set of 108 schizophrenia risk haplotypes[Schizophrenia Working Group of the Psychiatric Genomics 2014] identified 30 motifs, including at least two sets of sequences representing potential MEF2C target sites[Mitchell and others 2017]. In the same study, neuronal nucleosomes from prefrontal cortex of 17 subjects of schizophrenia, compared to 17 controls, were profiled by ChIP-seq for histone H3K4 trimethylation. Interestingly, among the (approximately 1000) nucleosomal sequences affected by H3K4 hypermethylation in the diseased cohort, MEF2C binding motifs were significantly overrepresented. Furthermore, MEF2C knock-down studies in the cell culture model resulted in excessive H3K4 methylation in the affected nucleosomes[Mitchell and others 2017], perhaps pointing to similar mechanisms in the diseased prefrontal neurons, including localized deficits in MEF2C chromatin occupancies and remodeling. Therefore, MEF2C is also a prototype for (in our classification) ‘category 2 ‘TF, given its very prominent footprints in the genetic and epigenetic risk architecture of schizophrenia. These insights then offer a very strong rationale to explore MEF2C mediated effects on schizophrenia-relevant cognition and behavior in the preclinical model (Figure 1A,B). Indeed, MEF2C plays a critical role during pre- and perinatal brain development[Adachi and others 2015; Barbosa and others 2008], with developmentally regulated expression in human prefrontal cortex due to a declining levels throughout childhood, adolescence and early adulthood[Mitchell and others 2017]. In addition, neuronal overexpression in adult prefrontal cortex improves working memory and object recognition memory in mice, in conjunction with spine remodeling in prefrontal projection neurons [Mitchell and others 2017] .Therefore, MEF2C provides an excellent example for the potential merit of multilayered integrative approaches to uncover the therapeutic potential of transcriptional regulators for schizophrenia and related disorders.
Figure 1.
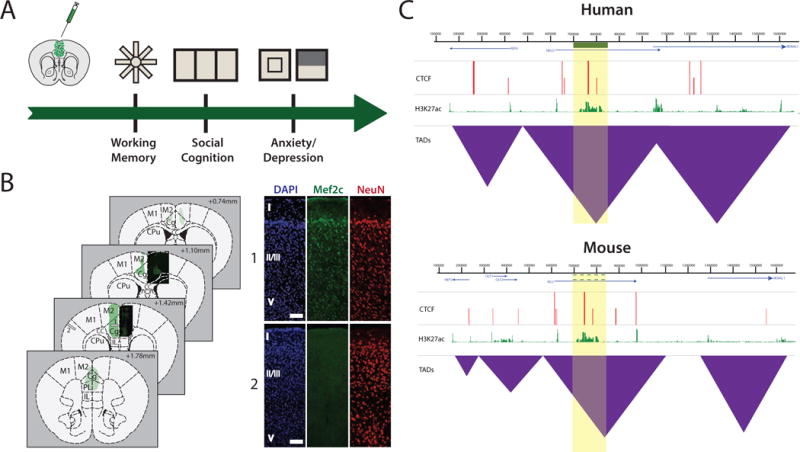
(A) Examples of schizophrenia-relevant behavioral assays in mice with genetically engineered (incl. virus vector-mediated transgene expression) PFC, including radial arm maze for working memory, three chamber social interaction choice, and the anxiety-related open field and light/dark box assays. (B) Representative serial sections of adult mouse brain tissue collected three weeks after infusion of human synapsin 1 promoter-driven AAV8-Mef2c-GFP as described[Mitchell and others 2017]. Areas shaded in green depict regions of viral expression in neurons, complemented by histological images near the injection site. Cortical distribution of Mef2c-GFP in neurons is shown in image set 1. GFP expression is not observed outside the PFC or neighboring regions (image set 2). Both sets correspond to gray bars 1 and 2 in the coronal diagram at +1.42mm. Scale bar = 50μm for both image sets. (C) Hypothetical 1.6 megabase-wide chromosomal locus with substantial epigenomic and genomic conservation in human and mouse cells with a hypothetical risk haplotype (green bar, yellow shade) associated with genetic risk for schizophrenia. There is conservation of mouse and human epigenomic landscapes, including similarities in linearly arranged gene order, and peak distributions for the CTCF chromosomal loop organizer and transcriptional regulator, and histone acetylation landscapes including histone H3-lysine 27 (H3K27ac) broadly associated with promoter and enhancer sequences[Zhou and others 2011]. There are similarities in organization of self-folding topologically associated chromatin domains (TAD)[Huang and others 2015]. Therefore, at this hypothetical locus, genomic and epigenomic editing in mouse prefrontal cortex and other anatomical structures associated with the circuitry of psychosis, may affect cognition and behavior.
Genomic and epigenomic editing of cis-regulatory sequences
In the paragraphs above, we provided specific examples of epigenomic and transcriptional regulators which are (epi)genetically associated with schizophrenia and with confirmed effects on cognition and behavior in the animal model. However, histone modification enzymes, transcription factors and many other chromatin regulatory proteins are likely to affect widespread areas of neuronal and glial genomes, thereby broadly affecting neuronal signaling and function. This could be associated with an increased likelihood for non-specific behavioral changes or even unwanted side effects. However, because non-coding DNA including many cis-regulatory elements such as promoters and enhancers play an important role in the genetic risk architecture of schizophrenia [Roussos and others 2014], one could argue that highly locus- and sequence-selective interventions, targeted against individual risk polymorphisms associated with schizophrenia, may bear greater promise to obtain specific therapeutic changes. In theory, the molecular toolboxes for such types of preclinical experiments already exist. Specifically, genome editing strategies via RNA-guided nucleases, including the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)-CRISPR-associated protein systems (CRISPR-Cas)(Figure 2A), introduced to the field only a few years ago[Doudna and Charpentier 2014], have now been widely adopted in all areas of genomic medicine, including the neurosciences[Heidenreich and Zhang 2016]. Mutations and disruption of enhancer sequences, even in instances where the enhancer was positioned on the linear genome many kilobase apart from the designated target promoter, has been accomplished for several neuropsychiatric risk genes, including the NMDA receptor gene GRIN2B [Bharadwaj and others 2014] and the FOXG1 transcription factor[Won and others 2016]. To mention one additional example, enhancer sequences, positioned 185kb from the transcription start site of the CACNA1C calcium channel gene, a gene robustly linked to schizophrenia by GWAS[Lencz and Malhotra 2015], when fused to a reporter gene expression, differentially drive expression, with the risk allele conveying decreased transcriptional activity in multiple cell lines[Roussos and others 2014]. While the precise molecular mechanisms underlying these phenomena, such as allele-specific binding of specific transcription factors, often remain incompletely understood, it is noteworthy that such types of experiments have been conducted to date primarily in cell culture systems. Typically transcriptional changes are explored after targeted mutations in the regulatory element, or by allele-specific comparisons of reporter gene expression activity. Therefore, the next phase of experiments should include in vivo genomic editing of risk-associated promoter and enhancer sequences, including the aforementioned CACNA1C and GRIN2B sites, and then test for changes in cognition and behavior in the animal. However, such types of genomic editing may carry drawbacks given that mutagenic interventions are likely to be irreversible. However, CRISPR-Cas and other RNA-guided nuclease systems can easily be converted into epigenomic editing tools (by using mutant protein with inactivated nuclease function, fused to a transcriptional activator such as VP64 or P300[Hilton and others 2015], or a repressor such as KRAB or DNA methyltransferase [Liu and others 2016; Thakore and others 2015], even with multi-locus manipulation[Stricker and others 2017] (Figure 2B,C). Recently, nuclease-deficient Cas9 DNA-binding protein was fused to a protein scaffold (‘SunTag’) to bind multiple copies of an antibody fusion to load 10 or even 24 copies of a specific transcriptional activator or repressor to a single sgRNA-Cas9 unit[Tanenbaum and others 2014]. With the underlying DNA sequence left intact, it will be interesting to explore whether simultaneous epigenomic targeting of enhancer and promoter sequences within multiple risk haplotypes could offer a promising approach to effectively alter cognition and behavior.
Figure 2.
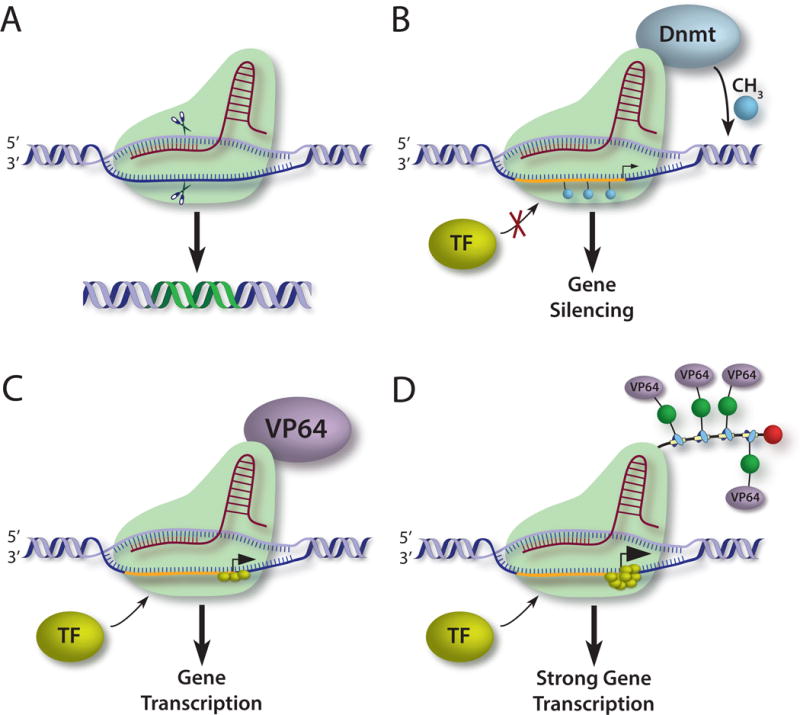
(A) sgRNA small guide RNA directs Cas9 nuclease in sequence-specific manner to mediate DNA mutations (affected DNA shown in green). (B) nuclease-deficient Cas9 is fused to a de novo DNA methyltransferase, targeting a gene promoter region. Promoter methylation silences transcription. (C) In constrast, Cas9-bound VP64 (or the p300 catalytic subunit) functions as transcriptional activator via mechanisms that include recruitment of basal transcription factors (TF). (D). Higher order systems, including SunTag protein scaffold (see text) are able to further amplify transcriptional activity by even more than one magnitude (see text for further details).
Obviously, preclinical assessment of cognitive and behavioral changes after such epigenomic editing of psychiatric risk haplotypes is only feasible for genomic sites that show at least some degree of conservation between human and mouse (or other laboratory animals) genomes. Such type of ‘epigenomic conservation’ could include similarities in sequential arrangements of genes and transcriptional units at the locus of interest, and conservation of histone modification landscapes, and similarities in chromosomal conformations including promoter-enhancer loopings important for transcriptional regulation. While a more detailed investigation on the epigenomic conservation for the genomic sites harboring schizophrenia risk haplotypes awaits further investigation, both genome-scale [Dincer and others 2015; Xiao and others 2012] and locus-specific [Bharadwaj and others 2013; Bharadwaj and others 2014] human-mouse comparative studies suggest that chromatin structure and function is conserved for a large number of regulatory non-coding sequences, and not necessarily accompanied by DNA sequence conservation (Figure 1C). With the recent accomplishment of region-specific multiplex gene editing in adult mouse brain in vivo [Zetsche and others 2017], we predict that the approaches proposed here will soon move center stage in preclinical schizophrenia research.
Conclusion
Chromatin structure and function, or from a genome-wide perspective, the ‘epigenome’ is an important study focus for neuropsychiatric disorders including schizophrenia. This is because exploration of DNA methylation and histone modification landscapes, the mapping of transcription factor occupancies, or the charting of promoter-enhancer loopings and other types of chromosomal conformations could inform about regulatory function of non-coding DNA associated with genetic risk. We hypothesize that such types of insights will also translate into novel therapeutic approaches for the preclinical model. Thus, we predict that targeted sequence-specific editing of the genome and the epigenome in specific brain cell types, guided by genetic schizophrenia studies, could affect cognition and behavior and offer potentially many advantages of pharmacological approaches that broadly target all brain regions with very little, if any, specificity for neural circuits of psychosis.
Acknowledgments
Work in the authors laboratory is supported by the grants from the National Institutes of Mental Health and the Brain Research Foundation (BRF). The authors report no financial conflicts of interest.
References
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2005;134B(1):60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Adachi M, Lin PY, Pranav H, Monteggia LM. Postnatal Loss of Mef2c Results in Dissociation of Effects on Synapse Number and Learning and Memory. Biological psychiatry. 2015 doi: 10.1016/j.biopsych.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Thompson WK, Dale AM. Boosting the power of schizophrenia genetics by leveraging new statistical tools. Schizophrenia bulletin. 2014;40(1):13–17. doi: 10.1093/schbul/sbt168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, Cacace AM, Zaczek R, Albright CF, Tseng G, Lewis DA. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry. 2015;20(11):1397–1405. doi: 10.1038/mp.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnosti DN, Kulkarni MM. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem. 2005;94(5):890–898. doi: 10.1002/jcb.20352. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Girgis RR, Gray DL, Mailman RB. Novel Dopamine Therapeutics for Cognitive Deficits in Schizophrenia. Biol Psychiatry. 2016 doi: 10.1016/j.biopsych.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, Olson EN. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A. 2008;105(27):9391–9396. doi: 10.1073/pnas.0802679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann K. Post-translational modifications: Crotonylation versus acetylation. Nat Rev Mol Cell Biol. 2015;16(5):265. doi: 10.1038/nrm3992. [DOI] [PubMed] [Google Scholar]
- Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104(24):10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, Awasthi A, Jain G, Capece V, Burkhardt S, Navarro-Sala M, Nagarajan S, Schutz AL, Johnsen SA, Bonn S, Luhrmann R, Dean C, Fischer A. HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest. 2015;125(9):3572–3584. doi: 10.1172/JCI79942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein HG, Steiner J, Dobrowolny H, Bogerts B. ZNF804A protein is widely expressed in human brain neurons: possible implications on normal brain structure and pathomorphologic changes in schizophrenia. Schizophr Bull. 2014;40(3):499–500. doi: 10.1093/schbul/sbt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Hakansson K, Borgkvist A, Irinopoulou T, Brami-Cherrier K, Usiello A, Greengard P, Herve D, Girault JA, Valjent E, Fisone G. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology. 2009;34(7):1710–1720. doi: 10.1038/npp.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj R, Jiang Y, Mao W, Jakovcevski M, Dincer A, Krueger W, Garbett K, Whittle C, Tushir JS, Liu J, Sequeira A, Vawter MP, Gardner PD, Casaccia P, Rasmussen T, Bunney WE, Jr, Mirnics K, Futai K, Akbarian S. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci. 2013;33(29):11839–11851. doi: 10.1523/JNEUROSCI.1252-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj R, Peter CJ, Jiang Y, Roussos P, Vogel-Ciernia A, Shen EY, Mitchell AC, Mao W, Whittle C, Dincer A, Jakovcevski M, Pothula V, Rasmussen TP, Giakoumaki SG, Bitsios P, Sherif A, Gardner PD, Ernst P, Ghose S, Sklar P, Haroutunian V, Tamminga C, Myers RH, Futai K, Wood MA, Akbarian S. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron. 2014;84(5):997–1008. doi: 10.1016/j.neuron.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell. 2012;47(4):633–647. doi: 10.1016/j.molcel.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BrainSeq AHBGC. BrainSeq: Neurogenomics to Drive Novel Target Discovery for Neuropsychiatric Disorders. Neuron. 2015;88(6):1078–1083. doi: 10.1016/j.neuron.2015.10.047. [DOI] [PubMed] [Google Scholar]
- Breen G, Li Q, Roth BL, O’Donnell P, Didriksen M, Dolmetsch R, O’Reilly PF, Gaspar HA, Manji H, Huebel C, Kelsoe JR, Malhotra D, Bertolino A, Posthuma D, Sklar P, Kapur S, Sullivan PF, Collier DA, Edenberg HJ. Translating genome-wide association findings into new therapeutics for psychiatry. Nat Neurosci. 2016;19(11):1392–1396. doi: 10.1038/nn.4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Landek-Salgado MA, Sawa A. Modeling heterogeneous patients with a clinical diagnosis of schizophrenia with induced pluripotent stem cells. Biol Psychiatry. 2014;75(12):936–944. doi: 10.1016/j.biopsych.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JB, Celniker SE. Lessons from modENCODE. Annu Rev Genomics Hum Genet. 2015;16:31–53. doi: 10.1146/annurev-genom-090413-025448. [DOI] [PubMed] [Google Scholar]
- Catts VS, Fung SJ, Long LE, Joshi D, Vercammen A, Allen KM, Fillman SG, Rothmond DA, Sinclair D, Tiwari Y, Tsai SY, Weickert TW, Shannon Weickert C. Rethinking schizophrenia in the context of normal neurodevelopment. Frontiers in cellular neuroscience. 2013;7:60. doi: 10.3389/fncel.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Kuo HY, Bornschein U, Takahashi H, Chen SY, Lu KM, Yang HY, Chen GM, Lin JR, Lee YH, Chou YC, Cheng SJ, Chien CT, Enard W, Hevers W, Paabo S, Graybiel AM, Liu FC. Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci. 2016;19(11):1513–1522. doi: 10.1038/nn.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29(37):11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A. Executive functions. Annu Rev Psychol. 2013;64:135–168. doi: 10.1146/annurev-psych-113011-143750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincer A, Gavin DP, Xu K, Zhang B, Dudley JT, Schadt EE, Akbarian S. Deciphering H3K4me3 broad domains associated with gene-regulatory networks and conserved epigenomic landscapes in the human brain. Transl Psychiatry. 2015;5:e679. doi: 10.1038/tp.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan RN, Bae BI, Cubelos B, Chang C, Hossain AA, Al-Saad S, Mukaddes NM, Oner O, Al-Saffar M, Balkhy S, Gascon GG, Homozygosity Mapping Consortium for A. Nieto M, Walsh CA. Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell. 2016;167(2):341–354 e312. doi: 10.1016/j.cell.2016.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Lewis DA. Stereological approaches to identifying neuropathology in psychosis. Biological psychiatry. 2011;69(2):113–126. doi: 10.1016/j.biopsych.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, Klei LL, Kramer R, Pinto D, Gumus ZH, Cicek AE, Dang KK, Browne A, Lu C, Xie L, Readhead B, Stahl EA, Xiao J, Parvizi M, Hamamsy T, Fullard JF, Wang YC, Mahajan MC, Derry JM, Dudley JT, Hemby SE, Logsdon BA, Talbot K, Raj T, Bennett DA, De Jager PL, Zhu J, Zhang B, Sullivan PF, Chess A, Purcell SM, Shinobu LA, Mangravite LM, Toyoshiba H, Gur RE, Hahn CG, Lewis DA, Haroutunian V, Peters MA, Lipska BK, Buxbaum JD, Schadt EE, Hirai K, Roeder K, Brennand KJ, Katsanis N, Domenici E, Devlin B, Sklar P. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19(11):1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landen M, Moran JL, Purcell SM, Sklar P, Sullivan PF, Hultman CM, McCarroll SA. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci. 2016;19(11):1433–1441. doi: 10.1038/nn.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Tsai LH. The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol. 2013;53:311–330. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(26):9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF. Impact of cognitive and social cognitive impairment on functional outcomes in patients with schizophrenia. J Clin Psychiatry. 2016;77(Suppl 2):8–11. doi: 10.4088/JCP.14074su1c.02. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30(10):3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, Troakes C, Turecki G, O’Donovan MC, Schalkwyk LC, Bray NJ, Mill J. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2016;19(1):48–54. doi: 10.1038/nn.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan A, Mitchell A, Schneider A, Halene T, Akbarian S. Epigenetic dysregulation in schizophrenia: molecular and clinical aspects of histone deacetylase inhibitors. Eur Arch Psychiatry Clin Neurosci. 2013;263(4):273–284. doi: 10.1007/s00406-013-0395-2. [DOI] [PubMed] [Google Scholar]
- Heidenreich M, Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat Rev Neurosci. 2016;17(1):36–44. doi: 10.1038/nrn.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekens CH, Hennekens AR, Hollar D, Casey DE. Schizophrenia and increased risks of cardiovascular disease. American heart journal. 2005;150(6):1115–1121. doi: 10.1016/j.ahj.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Hess JL, Quinn TP, Akbarian S, Glatt SJ. Bioinformatic analyses and conceptual synthesis of evidence linking ZNF804A to risk for schizophrenia and bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(1):14–35. doi: 10.1002/ajmg.b.32284. [DOI] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33(5):510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinna KH, Rich K, Fex-Svenningsen A, Benedikz E. The Rat Homolog of the Schizophrenia Susceptibility Gene ZNF804A Is Highly Expressed during Brain Development, Particularly in Growth Cones. PLoS One. 2015;10(7):e0132456. doi: 10.1371/journal.pone.0132456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Mirnics K. Schizophrenia as a disorder of molecular pathways. Biol Psychiatry. 2015;77(1):22–28. doi: 10.1016/j.biopsych.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27(42):11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Marco E, Pinello L, Yuan GC. Predicting chromatin organization using histone marks. Genome Biol. 2015;16:162. doi: 10.1186/s13059-015-0740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim HM, Tamminga CA. Schizophrenia: treatment targets beyond monoamine systems. Annu Rev Pharmacol Toxicol. 2011;51:189–209. doi: 10.1146/annurev.pharmtox.010909.105851. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Yamada K, Takao H, Iwayama-Shigeno Y, Yoshikawa T, Kato T. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. The Journal of Neuroscience. 2005;25(22):5376–5381. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19(1):40–47. doi: 10.1038/nn.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski M, Bharadwaj R, Straubhaar J, Gao G, Gavin DP, Jakovcevski I, Mitchell AC, Akbarian S. Prefrontal cortical dysfunction after overexpression of histone deacetylase 1. Biol Psychiatry. 2013;74(9):696–705. doi: 10.1016/j.biopsych.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski M, Ruan H, Shen EY, Dincer A, Javidfar B, Ma Q, Peter CJ, Cheung I, Mitchell AC, Jiang Y, Lin CL, Pothula V, Stewart AF, Ernst P, Yao WD, Akbarian S. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J Neurosci. 2015;35(13):5097–5108. doi: 10.1523/JNEUROSCI.3004-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, Taipale J. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015;527(7578):384–388. doi: 10.1038/nature15518. [DOI] [PubMed] [Google Scholar]
- Kano S, Colantuoni C, Han F, Zhou Z, Yuan Q, Wilson A, Takayanagi Y, Lee Y, Rapoport J, Eaton W, Cascella N, Ji H, Goldman D, Sawa A. Genome-wide profiling of multiple histone methylations in olfactory cells: further implications for cellular susceptibility to oxidative stress in schizophrenia. Mol Psychiatry. 2013;18(7):740–742. doi: 10.1038/mp.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AJ, Rahn EJ, Paulukaitis BS, Savell KE, Kordasiewicz HB, Wang J, Lewis JW, Posey J, Strange SK, Guzman-Karlsson MC, Phillips SE, Decker K, Motley ST, Swayze EE, Ecker DJ, Michael TP, Day JJ, Sweatt JD. Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep. 2016;16(10):2666–2685. doi: 10.1016/j.celrep.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimoglu C, Agis-Balboa RC, Kranz A, Stilling R, Bahari-Javan S, Benito-Garagorri E, Halder R, Burkhardt S, Stewart AF, Fischer A. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J Neurosci. 2013;33(8):3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Stahl SM. Antipsychotic drug development. Current topics in behavioral neurosciences. 2010;4:123–139. doi: 10.1007/7854_2010_47. [DOI] [PubMed] [Google Scholar]
- Kim S, Cho H, Lee D, Webster MJ. Association between SNPs and gene expression in multiple regions of the human brain. Transl Psychiatry. 2012;2:e113. doi: 10.1038/tp.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends in biochemical sciences. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Konopka G, Roberts TF. Insights into the Neural and Genetic Basis of Vocal Communication. Cell. 2016;164(6):1269–1276. doi: 10.1016/j.cell.2016.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, Heshmati M, Golden SA, Kennedy PJ, Takahashi N, Dietz DM, Mocci G, Gabilondo AM, Hanks J, Umali A, Callado LF, Gallitano AL, Neve RL, Shen L, Buxbaum JD, Han MH, Nestler EJ, Meana JJ, Russo SJ, Gonzalez-Maeso J. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15(9):1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Current opinion in psychiatry. 2012;25(2):83–88. doi: 10.1097/YCO.0b013e32835035ca. [DOI] [PubMed] [Google Scholar]
- Leifer D, Golden J, Kowall NW. Myocyte-specific enhancer binding factor 2C expression in human brain development. Neuroscience. 1994;63(4):1067–1079. doi: 10.1016/0306-4522(94)90573-8. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lencz T, Malhotra AK. Targeting the schizophrenia genome: a fast track strategy from GWAS to clinic. Mol Psychiatry. 2015;20(7):820–826. doi: 10.1038/mp.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski KE, Whitton AE, Pizzagalli DA, Norris LA, Ongur D, Hall MH. Reward Learning, Neurocognition, Social Cognition, and Symptomatology in Psychosis. Front Psychiatry. 2016;7:100. doi: 10.3389/fpsyt.2016.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest. 2009;119(4):706–716. doi: 10.1172/JCI37335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C, Konradi C, Akbarian S. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem. 2004;90(5):1117–1131. doi: 10.1111/j.1471-4159.2004.02569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. The New England journal of medicine. 2005;353(12):1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167(1):233–247 e217. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SB, Szuhany KL, Kredlow MA, Wolfe R, Mueser KT, McGurk SR. A confirmatory factor analysis of the MATRICS consensus cognitive battery in severe mental illness. Schizophr Res. 2016;175(1–3):79–84. doi: 10.1016/j.schres.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Long HK, Prescott SL, Wysocka J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell. 2016;167(5):1170–1187. doi: 10.1016/j.cell.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg SM, Tu WB, Raught B, Penn LZ, Hoffman MM, Lee SI. ChromNet: Learning the human chromatin network from all ENCODE ChIP-seq data. Genome Biol. 2016;17:82. doi: 10.1186/s13059-016-0925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney B, Ding Y, Lewis DA, Sweet RA. DNA methylation as a putative mechanism for reduced dendritic spine density in the superior temporal gyrus of subjects with schizophrenia. Transl Psychiatry. 2017;7(2):e1032. doi: 10.1038/tp.2016.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22(7):2718–2729. doi: 10.1523/JNEUROSCI.22-07-02718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, Jia P, Assadzadeh A, Flanagan J, Schumacher A, Wang SC, Petronis A. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. American journal of human genetics. 2008;82(3):696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28(1):53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Mitchell AC, Javidfar B, Pothula V, Ibi D, Shen EY, Peter CJ, Bicks LK, Fehr T, Jiang Y, Brennand KJ, Neve RL, Gonzalez-Maeso J, Akbarian S. MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice. Mol Psychiatry. 2017 doi: 10.1038/mp.2016.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi K, Shimada T, Nitta Y, Kihara H, Okubo H, Uehara T, Kawasaki Y. Specific gene expression patterns of 108 schizophrenia-associated loci in cortex. Schizophr Res. 2016;174(1–3):35–38. doi: 10.1016/j.schres.2016.03.032. [DOI] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP, Economides AN, Bradner JE, Rabadan R, Basu U. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161(4):774–789. doi: 10.1016/j.cell.2015.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, Geschwind DH, Mill J, Nairn AC, Abyzov A, Pochareddy S, Prabhakar S, Weissman S, Sullivan PF, State MW, Weng Z, Peters MA, White KP, Gerstein MB, Amiri A, Armoskus C, Ashley-Koch AE, Bae T, Beckel-Mitchener A, Berman BP, Coetzee GA, Coppola G, Francoeur N, Fromer M, Gao R, Grennan K, Herstein J, Kavanagh DH, Ivanov NA, Jiang Y, Kitchen RR, Kozlenkov A, Kundakovic M, Li M, Li Z, Liu S, Mangravite LM, Mattei E, Markenscoff-Papadimitriou E, Navarro FC, North N, Omberg L, Panchision D, Parikshak N, Poschmann J, Price AJ, Purcaro M, Reddy TE, Roussos P, Schreiner S, Scuderi S, Sebra R, Shibata M, Shieh AW, Skarica M, Sun W, Swarup V, Thomas A, Tsuji J, van Bakel H, Wang D, Wang Y, Wang K, Werling DM, Willsey AJ, Witt H, Won H, Wong CC, Wray GA, Wu EY, Xu X, Yao L, Senthil G, Lehner T, Sklar P, Sestan N. The PsychENCODE project. Nat Neurosci. 2015;18(12):1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannals MD, Hamersky GR, Page SC, Campbell MN, Briley A, Gallo RA, Phan BN, Hyde TM, Kleinman JE, Shin JH, Jaffe AE, Weinberger DR, Maher BJ. Psychiatric Risk Gene Transcription Factor 4 Regulates Intrinsic Excitability of Prefrontal Neurons via Repression of SCN10a and KCNQ1. Neuron. 2016;90(1):43–55. doi: 10.1016/j.neuron.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Murillo L, Gogos JA, Karayiorgou M. The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annual review of medicine. 2012;63:63–80. doi: 10.1146/annurev-med-072010-091100. [DOI] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, Stahl EA, Georgakopoulos A, Ruderfer DM, Charney A, Okada Y, Siminovitch KA, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Plenge RM, Raychaudhuri S, Fromer M, Purcell SM, Brennand KJ, Robakis NK, Schadt EE, Akbarian S, Sklar P. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9(4):1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Exome Aggregation C. Daly MJ, MacArthur DG, Fromer M, Purcell SM. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet. 2016;48(10):1107–1111. doi: 10.1038/ng.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka WB, Subburaju S, Benes FM. Circuit- and Diagnosis-Specific DNA Methylation Changes at gamma-Aminobutyric Acid-Related Genes in Postmortem Human Hippocampus in Schizophrenia and Bipolar Disorder. JAMA Psychiatry. 2015;72(6):541–551. doi: 10.1001/jamapsychiatry.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Archives of general psychiatry. 2007;64(10):1123–1131. doi: 10.1001/archpsyc.64.10.1123. [DOI] [PubMed] [Google Scholar]
- Sandner G, Host L, Angst MJ, Guiberteau T, Guignard B, Zwiller J. The HDAC Inhibitor Phenylbutyrate Reverses Effects of Neonatal Ventral Hippocampal Lesion in Rats. Front Psychiatry. 2011;1:153. doi: 10.3389/fpsyt.2010.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder FA, Gilbert TM, Feng N, Taillon BD, Volkow ND, Innis RB, Hooker JM, Lipska BK. Expression of HDAC2 but Not HDAC1 Transcript Is Reduced in Dorsolateral Prefrontal Cortex of Patients with Schizophrenia. ACS Chem Neurosci. 2016 doi: 10.1021/acschemneuro.6b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62(1):55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Sharma RP, Grayson DR, Gavin DP. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr Res. 2008;98(1–3):111–117. doi: 10.1016/j.schres.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen EY, Jiang Y, Javidfar B, Kassim B, Loh YE, Ma Q, Mitchell AC, Pothula V, Stewart AF, Ernst P, Yao WD, Martin G, Shen L, Jakovcevski M, Akbarian S. Neuronal Deletion of Kmt2a/Mll1 Histone Methyltransferase in Ventral Striatum is Associated with Defective Spike-Timing-Dependent Striatal Synaptic Plasticity, Altered Response to Dopaminergic Drugs, and Increased Anxiety. Neuropsychopharmacology. 2016;41(13):3103–3113. doi: 10.1038/npp.2016.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2(9):e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, Suvisaari J, Chheda H, Blackwood D, Breen G, Pietilainen O, Gerety SS, Ayub M, Blyth M, Cole T, Collier D, Coomber EL, Craddock N, Daly MJ, Danesh J, DiForti M, Foster A, Freimer NB, Geschwind D, Johnstone M, Joss S, Kirov G, Korkko J, Kuismin O, Holmans P, Hultman CM, Iyegbe C, Lonnqvist J, Mannikko M, McCarroll SA, McGuffin P, McIntosh AM, McQuillin A, Moilanen JS, Moore C, Murray RM, Newbury-Ecob R, Ouwehand W, Paunio T, Prigmore E, Rees E, Roberts D, Sambrook J, Sklar P, Clair DS, Veijola J, Walters JT, Williams H, Swedish Schizophrenia S. Study I. Study DDD. Consortium UK. Sullivan PF, Hurles ME, O’Donovan MC, Palotie A, Owen MJ, Barrett JC. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19(4):571–577. doi: 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, Ahituv N. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet. 2013;45(9):1021–1028. doi: 10.1038/ng.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18(1):51–66. doi: 10.1038/nrg.2016.138. [DOI] [PubMed] [Google Scholar]
- Swartz MS, Perkins DO, Stroup TS, Davis SM, Capuano G, Rosenheck RA, Reimherr F, McGee MF, Keefe RS, McEvoy JP, Hsiao JK, Lieberman JA. Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: findings from the NIMH CATIE study. The American journal of psychiatry. 2007;164(3):428–436. doi: 10.1176/ajp.2007.164.3.428. [DOI] [PubMed] [Google Scholar]
- Taher L, McGaughey DM, Maragh S, Aneas I, Bessling SL, Miller W, Nobrega MA, McCallion AS, Ovcharenko I. Genome-wide identification of conserved regulatory function in diverged sequences. Genome Res. 2011;21(7):1139–1149. doi: 10.1101/gr.119016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De Novo Synonymous Mutations in Regulatory Elements Contribute to the Genetic Etiology of Autism and Schizophrenia. Neuron. 2016;89(5):940–947. doi: 10.1016/j.neuron.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Xu B, Ionita-Laza I, Roos JL, Gogos JA, Karayiorgou M. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron. 2014;82(4):773–780. doi: 10.1016/j.neuron.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taly A. Novel approaches to drug design for the treatment of schizophrenia. Expert opinion on drug discovery. 2013;8(10):1285–1296. doi: 10.1517/17460441.2013.821108. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159(3):635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, Gersbach CA. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12(12):1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallianatos CN, Iwase S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics. 2015;7(3):503–519. doi: 10.2217/epi.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter MP, Shannon Weickert C, Ferran E, Matsumoto M, Overman K, Hyde TM, Weinberger DR, Bunney WE, Kleinman JE. Gene expression of metabolic enzymes and a protease inhibitor in the prefrontal cortex are decreased in schizophrenia. Neurochem Res. 2004;29(6):1245–1255. doi: 10.1023/b:nere.0000023611.99452.47. [DOI] [PubMed] [Google Scholar]
- Vernimmen D, Bickmore WA. The Hierarchy of Transcriptional Activation: From Enhancer to Promoter. Trends Genet. 2015;31(12):696–708. doi: 10.1016/j.tig.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA. Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry. 2015;172(11):1112–1121. doi: 10.1176/appi.ajp.2015.15010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle N, Maurer V, Murphy C, Rainer J, Bindreither D, Hauschild M, Scharinger A, Oberhauser M, Keil T, Brehm C, Valovka T, Striessnig J, Singewald N. Enhancing dopaminergic signaling and histone acetylation promotes long-term rescue of deficient fear extinction. Transl Psychiatry. 2016;6(12):e974. doi: 10.1038/tp.2016.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wockner LF, Noble EP, Lawford BR, Young RM, Morris CP, Whitehall VL, Voisey J. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational psychiatry. 2014;4:e339. doi: 10.1038/tp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, Lee C, Eskin E, Voineagu I, Ernst J, Geschwind DH. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538(7626):523–527. doi: 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Xie D, Cao X, Yu P, Xing X, Chen CC, Musselman M, Xie M, West FD, Lewin HA, Wang T, Zhong S. Comparative epigenomic annotation of regulatory DNA. Cell. 2012;149(6):1381–1392. doi: 10.1016/j.cell.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Luo XJ, Chang H, Liu Z, Li M. Evaluation of European Schizophrenia GWAS Loci in Asian Populations via Comprehensive Meta-Analyses. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-9990-3. [DOI] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35(1):31–34. doi: 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Xu J, Chen J, Kim S, Reimers M, Bacanu SA, Yu H, Liu C, Sun J, Wang Q, Jia P, Xu F, Zhang Y, Kendler KS, Peng Z, Chen X. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol Psychiatry. 2015;20(5):563–572. doi: 10.1038/mp.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]