

Summary
Osteoarthritis (OA) is a common chronic disorder that affects an increasing number of the ageing population. Despite the prevalence, there are currently no therapies. Defining new therapies that target specific pathogenic phases of disease development relies on the effective separation of the different stages of OA. This manuscript reviews the tissues and models that are being used to separate these stages of disease, in particular initiation and early and late progression. These models include human tissues with known initiating factors, the use of anatomical locations with defined relationships to the primary cartilage lesion area, timing of OA development in well‐described animal models and the versatility of a non‐invasive model of murine knee joint trauma.
Keywords: cartilage, mouse models, osteoarthritis
Introduction
Osteoarthritis (OA) is the most common degenerative disease of synovial joints affecting more than 6.6 million people in England alone. Despite this high prevalence, there are currently no effective therapies for patients with osteoarthritis. Determining new targets for therapy, for both prevention and slowing of disease, relies on defining the different stages occurring during osteoarthritis development. These involve initiation, early and late stages of progression, each of which can conceivably be targeted selectively to delay the need for joint replacement surgery (Figure 1).
Figure 1.
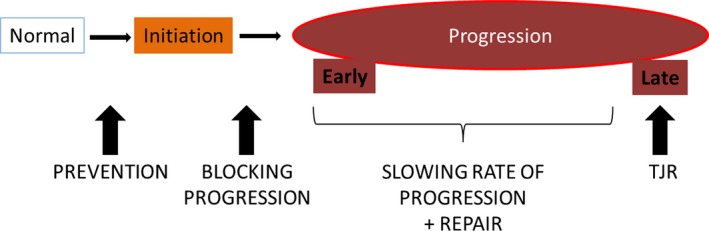
Stages of osteoarthritis and potential target points for therapy. Prevention therapy can be used before disease initiation in normal joints; blocking progression from taking place may be targeted through events that make joint become progressively worse with time. During progression, early and late stages should be separated as slowing of the rate of progression and repair potentials will be more successful in the earlier stages of disease, whereas total joint replacement (TJR) may be the only option left for late stages of disease.
A number of risk factors are known to be involved in OA, including genetics, mechanical instability and joint injuries, ageing, obesity. OA development often relies on the interactions between various factors, making it difficult to pinpoint which mechanism should be altered to slow disease progression, or to prevent disease initiation in specific populations. In addition, the idea that a treatment should be linked to initiating factor is plausible: some studies in mouse models have now shown that specific treatment/mechanisms can modify ageing‐associated and trauma/mechanical‐induced OA differently (Little & Zaki 2012; Rowe et al. 2017; Usmani et al. 2016). Hallmarks of OA include articular cartilage (AC) degradation, with loss of proteoglycan and collagen type II, two of the most prevalent components of cartilage, subchondral bone sclerosis, osteophyte formation, and synovial hyperplasia and activation. The sequence of these changes (i.e. do changes start in the cartilage or in the bone?) is still disputed (Brandt et al. 2006; Little & Fosang 2010) and will be dependent on the patients and models used for research. Indeed, it has been shown that overexpression of EphB4 specifically to bone protects from OA development and AC degradation (Valverde‐Franco et al. 2012). In contrast, global and cartilage‐specific (Col2a1) deletions of MMP13 both protect against post‐traumatic OA (Little et al. 2009)(Wang et al. 2013)), suggesting in this case MMP13 affects cartilage directly.
Determining stages of diseases has important implication for therapeutic strategies, as potential targets may affect initiation, early and late stages of diseases differently. A recent review on standardizing OA definition highlights the need to stage disease, separating OA development, OA progression and early detection (Kraus et al. 2015). In addition, it was recently found different outcomes between short‐and long‐term inhibition of CCR2, suggesting this chemokine may play different roles during OA progression (Longobardi et al. 2016). For example, is the beneficial effect of a specific treatment due to a protection against initiation of AC lesions, or because of interference with a specific event involved in progression of joint degeneration? Furthermore, separating the different phases of OA development is likely to help in the search for markers of disease and allow for appropriate targeting of patients that are most likely to respond to specific treatment. What separates these stages and how to define them clinically is still largely unknown, although it is now well accepted that some disease stages show different cellular processes (Anderson et al. 2011; Jiang & Tuan 2015; Liu‐Bryan & Terkeltaub 2015). This review summarizes recent approaches to differentiate the stages of OA progression in both human and animal studies.
Defining the initiating event in human patients
A major issue with separating disease stages lies with the determination of the initiating factor. Although many risk factors for osteoarthritis development are well known, including ageing, genetics, obesity and mechanics, it is often impossible to determine the initiating event. Determining the initiating factors in many patients with osteoarthritis challenging as they present themselves at advanced stages of the disease; however, this might be possible in small subsets of patients. These small groups may then benefit from targeted therapies in the earlier stages of disease.
One such group is represented by those who have suffered from a severe joint injury. These are usually well monitored, with regular follow‐up, in particular at the earliest stages after the trauma. Specific examples include intra‐articular fractures, ligament and meniscal injuries. (For further detail of post‐traumatic OA and their animal models currently used, see recent reviews: Lohmander et al. (2007), Christiansen et al. (2015)).
Another possible group is linked to genetic predisposition to osteoarthritis. These specific genetic mutations that ‘guarantee’ OA development may include those that affect cartilage matrix genes, which are also associated with early‐onset osteoarthritis and joint chondrodysplasias (Hecht et al. 1995; Muragaki et al. 1996; Paassilta et al. 1999; Gleghorn et al. 2005; Rukavina et al. 2014; Hildebrand 2015). Genetic mutations that are associated with abnormalities in joint shape are also highly correlated with increased susceptibility to OA development (Baker‐Lepain & Lane 2010; Waarsing et al. 2011). Other genetic predisposition to OA development has been described (Tsezou 2014; Reynard 2017; Wang et al. 2016) but it remains largely unknown, however, whether these influence initiation and/or progression specifically.
Although trauma and injury may be the initiating factor leading to OA initiation in some groups, the development of OA is most likely due to the interaction between multiple risk factors. Further knowledge of how these interact to initiate disease may help differentiate between other patient subsets and may allow for more targeted therapy.
Separating initiation and progression phases using human tissues
Human samples are difficult to obtain, especially at different stages of disease. Samples can be obtained following total or partial joint replacement for severe/late OA, and controls normally from amputations from other non‐arthritic causes (such as trauma, necrosis from severe diabetes). But this has led to research concentrating on late disease because firstly it is difficult to define early OA clinically as there is currently a lack of effective early disease markers and secondly collecting cartilage from patients with early‐stage OA is highly invasive and may lead to an acceleration in disease progression. Some researchers have countered this issue using tissue from patients with osteoarthritis from different locations of the joint; indeed, these studies assumed that the tissue that is the closest to the lesion represents the most advanced OA, and thus, the further away from the lesion, the more ‘normal’ the tissue. Therefore, a gradation in disease severity in the same joint is used to determine the changes in cartilage and chondrocyte behaviour with disease advancement. This does not take into account that the joint environment in these OA joints may still be highly abnormal; thus, even ‘normal’‐looking tissue may still be compromised; for example, OA synovium has a very different composition, including high levels of degrading enzymes and cytokine levels (Struglics et al. 2015; Bigoni et al. 2016; Liu et al. 2016), which would affect the chondrocyte responses and expression profiles.
The use of such protocols has identified various pathways and molecular markers for early and late OA. Indeed, comparison between damaged and undamaged areas of the condyle, as described in (Snelling et al. 2014), shows expression patterns of genes involved in cell signalling, extracellular matrix remodelling and inflammatory responses; these include matrix metalloproteinases (MMPs), growth factor signalling proteins, collagens and SOX 9, amongst other common OA genes (Sato et al. 2006; Geyer et al. 2009). Fukui et al. (2008) have separated the severity of degradation by grouping the samples (from the same OA patients’ joint) into four distinct areas depending on the zones of the articular cartilage affected (preserved area, damaged with superficial, middle and deep zones present, damaged with middle and deep zones, damaged with deep zones only). They found that the profile of gene expression differed dependently on the zones and the extent of cartilage degeneration.
Other studies have analysed synovial fluid or cartilage tissue from normal compared with early and advanced OA. Heard et al. (2012) used criteria for the severity of disease based on arthroscopic examination and X‐ray analysis. Unfortunately, this study did not find any significant markers for early OA. Another study determined early OA as patients undergoing surgery for meniscal tears (during which surgery mild cartilage degeneration was observed), whereas late OA as patients undergoing total joint replacement (Ritter et al. 2013). This study found almost identical gene expression patterns between early OA and late OA compared with healthy controls. These and other similar studies (Gobezie et al. 2007) suggest that these criteria for early and late OA may represent the same phase of disease in progression, and are too late to identify initiation events.
In an attempt to identify markers of early and late diseases, one study used cartilage from sarcomas in lower extremities of patients and separated them into early‐, with signs of some fibrillation upon visual examination, and normal‐, as well as late‐stage OA following total knee replacement (Lorenzo et al. 2004). Proteomic analysis was performed on these tissues and showed that newly synthetized cartilage proteins were altered in a similar manner in early and late OA, including increased expression of cartilage oligomeric matrix protein (COMP), fibronectin and cartilage intermediate layer protein (CILP). This further suggests that early OA and late OA follow a common molecular pathway. These potential markers of disease were also confirmed in a mouse model with increased serum levels of COMP with onset of cartilage degeneration (Salminen et al. 2000).
Using time in well‐described animal models
Animal models are a great source of information to determine the sequence of events that take place during OA development in response to known initiating factors. Whilst model systems have been developed in a number of species, mice are especially widely used due to the abundant genetically modified strains available.
The Str/ort mouse shows progressive molecular changes with OA comparable to human disease
A well‐described model of spontaneous OA is the Str/ort mouse (Mason et al. 2001). This model shows similar patterns of disease as those described in human OA, including proteoglycan loss, AC degradation by A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS4‐5, major aggrecanases) and MMPs, and subchondral bone sclerosis. Disease development in this model has been well described, with 8‐week‐old joints showing no apparent disease, lesions appeared from 18 weeks of age and progressed further with age. This timing was used in a microarray study in which RNA gene expression in AC was assessed in 8 weeks (before apparent disease), 18 weeks (early signs of AC degradation) and 40 weeks of age (severe disease (Poulet et al. 2012)). Gene expression analysis showed 115 differentially regulated genes between 8 and 18 weeks, including genes involved in matrix synthesis and degradation, which could represent the switch between initiation and progression of AC degradation in OA. Interestingly, no changes were seen between early and late diseases, as seen in human studies mentioned above. Analysis of genes expression between Str/ort and CBA control mice at 8 weeks found that genes linked to the NFkB signalling pathway may be involved in initiation of disease in this model.
The surgical DMM model is the main post‐traumatic OA model in mouse studies
The most widely used model of OA in the mouse is the surgical model DMM (destabilization of the medial meniscus). In this case, the initiating factor is mechanical and starts with the destabilization of the joint. Changes as early as 6 hours following surgery, which included cartilage matrix proteases, were thought to be due to abnormal mechanical input induced in this model (Burleigh et al. 2012). Studies have used inhibitors to decrease disease severity showing potential for these targets as preventative and/or to slow progression; indeed, increasing the expression of PRG4 either before or after injury resulted with the same protection, suggesting the main effect is achieved during progression and not initiation (Ruan et al. 2013). Thus, this target could be used in later stages of OA. Similarly, Li et al. (2015) used resveratrol treatment beginning at 4 weeks after DMM surgery and still found a beneficial effect with a preservation of the AC and subchondral bone structural homeostasis, suggesting an effect on progression.
Reversible versus irreversible stages
Cartilage degradation reversibility as a hallmark of OA progression?
A main hallmark of OA is the active degradation of the extracellular matrix of AC, in particular of the two main components of this tissue, namely collagen type II and aggrecan. A molecular model of AC degradation reversibility has been put forward based on both in vitro and in vivo experiments. This model suggests that ADAMTS‐mediated aggrecan degradation was reversible, whereas MMP‐mediated aggrecan and collagen type II degradation was irreversible (van Meurs et al. 1999; Karsdal et al. 2008; Bay‐Jensen et al. 2010). This suggests that a switch in ADAMTS to MMP activity may be a marker of progression over initiation, by crossing a ‘point‐of‐no‐return’ discussed in a review by Bay‐Jensen et al. (2010). He suggests that this event of irreversibility may involve the change in phenotype of the chondrocyte from a ‘normal’ phenotype to a catabolic, hypertrophic and/or dedifferentiated phenotype. In this scenario, the chondrocyte is unable to maintain a healthy AC matrix. A marker of hypertrophic chondrocytes is the synthesis of MMP13, thus implying the possibility that chondrocyte phenotype change and MMP‐mediated degradation are linked.
Reversible changes in non‐cartilaginous tissue may be apparent from the transient nature of their changes
It is also important to remember that OA is a disease involving various tissues in the joint. Synovial fibrosis and hyperplasia have been shown to be transient in a murine model of non‐invasive trauma (Poulet et al. 2011) and in response to connective tissue growth factor treatment in vivo (Blaney Davidson et al. 2006), but fibrosis becomes permanent in response to TGFβ treatment. Thus, TGFβ activity in the synovium may represent a switch to irreversibility for OA‐related synovial changes. In addition, epiphyseal and subchondral bone changes can be reversed upon treatment in human and animal studies (Lv et al. 2014; Cui et al. 2015; Miller et al. 2015). Subchondral bone thickening with non‐repetitive regimes of mechanical stimulation in mice are transient changes, as opposed to bone changes induced by repetitive regimes (Poulet et al. 2015).
This suggests that the ‘point‐of‐no‐return’ that separates initiation and progression may be linked to chondrocyte hypertrophy, with increases in MMP13 expression and MMP‐mediated cartilage degradation, and with TGFβ activity.
Initiation does not necessarily mean progression
In the human population, a wide range of individuals show signs of cartilage defects with no other evidence of OA‐like pathology. Indeed, one small study reports that amongst 13 normal healthy individuals, nine had signs of focal cartilage abnormalities (Stahl et al. 2009). Another slightly bigger study (with 297 subjects, all with no significant current or past knee disease) found that 62% of the subjects had tibiofemoral cartilage defects (Racunica et al. 2007). Professional athletes are also prone to chondral defects, with 36% being affected in another study, whereas 14% of these same athletes were asymptomatic (Flanigan et al. 2010).
Animal models of trauma have also shown that articular cartilage lesions do not necessarily progress into osteoarthritis. Indeed, a single non‐invasive mechanical trauma protocol induced localized articular cartilage lesions, without ruptures or apparent tears in the ligaments or menisci, in the lateral femur of a CBA mouse knee joint (Poulet et al. 2011). Knees after this trauma regime were followed for 5 weeks, and up to 8 weeks, and did not show any worsening of the lesion severity. Thus, damage to the articulating surface alone, which cannot repair, is not sufficient to induce progressive OA degeneration.
Separating initiation and progression following joint trauma
A non‐invasive model of mouse knee trauma can distinguish between initiation and progression
Most animal models of OA used to date have allowed the definition of the main OA hallmarks and their mechanisms, and some were used to test possible candidates to slow disease progression. But most are not able to distinguish between initiation and progression. A recent model of mechanical trauma was able to induce reproducible AC lesions in the same mouse strain (CBA), which progress with time with a repetitive regime (Poulet et al. 2011). This same model was also used to induce AC lesions that do not worsen with time, using a single loading episode. Comparing these two regimes will allow us, for the first time, to define the mechanisms involved between initiation and progression. We have shown using this model that proteoglycan loss was one of these events that differentiate between progressive and non‐progressive lesions.
The conjunction of a spontaneous model (Str/ort) and non‐invasive trauma model of OA can further inform us on initiation and progression
This model was also used to determine the effect of mechanical loading on initiation and progression. The Str/ort mouse develops spontaneous OA lesions primarily in the medial tibia. Thus, mechanical loading of young Str/ort mice, in the early phases of OA development, resulted in an acceleration in progression of spontaneous OA lesions in the medial tibia (Poulet et al. 2013). This was marked by increased proteoglycan loss and collagen type II degradation by MMPs. The lateral femur, which is targeted in the trauma model, is the least affected by spontaneous OA in the Str/ort mouse. Initiation of mechanical lesions following trauma was reduced in this mouse, suggesting that mechanical loading can have different effects on different phases of disease and that the Str/ort mouse does not develop OA because of a major deficit in AC mechanical properties. The study of the specific cellular processes involved in the acceleration of OA lesions in the medial tibia will allow defining new targets for slowing the rate of progression.
Conclusions
Stratification of disease phases in osteoarthritis is necessary to define targets for therapy, as well as markers of disease severity. These will include the development of appropriate therapeutic strategies, such as prevention, blocking and slowing progression and repair, before the need for total joint replacement in end‐stage disease. The model of non‐invasive trauma described above (Poulet et al. 2011) allows for the distinction between initiation and progression to be made: comparing joints loaded with a single loading episode or repetitive regimes will permit to determine factors involved between initiation and progression phases. In other models, involving specific interventions to induce OA (such as surgery), the effects of specific targets or mechanisms on initiation and progression, respectively, can be achieved with different timings of treatment or gene deletions such as before and after surgery. This distinction becomes more challenging when using chronic models such as ageing‐associated Str/ort mouse: in this model, its well‐described timing of pathology can give clues as to which stage the disease might be at, and represents a good testing group for primary human OA. In the long term, this will further determine the effectiveness of the target in the each phase of OA for therapy as well as a marker of disease, leading to more patient‐specific time‐dependent treatments.
Conflict of interest
The author has no conflict of interest.
Funding source
Dr Poulet is funded by Arthritis Research UK (20859).
References
- Anderson D.D., Chubinskaya S., Guilak F. et al (2011) Post‐traumatic osteoarthritis: improved understanding and opportunities for early intervention. J. Orthop. Res. 29, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker‐Lepain J.C. & Lane N.E. (2010) Relationship between joint shape and the development of osteoarthritis. Curr. Opin. Rheumatol. 22, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay‐Jensen A.C., Hoegh‐Madsen S., Dam E. et al (2010) Which elements are involved in reversible and irreversible cartilage degradation in osteoarthritis? Rheumatol. Int. 30, 435–442. [DOI] [PubMed] [Google Scholar]
- Bigoni M., Turati M., Sacerdote P. et al (2016) Characterization of synovial fluid cytokine profiles in chronic meniscal tear of the knee. J. Orthop. Res. 35, 340–346. [DOI] [PubMed] [Google Scholar]
- Blaney Davidson E.N., Vitters E.L., Mooren F.M., Oliver N., Berg W.B., van der Kraan P.M. (2006) Connective tissue growth factor/CCN2 overexpression in mouse synovial lining results in transient fibrosis and cartilage damage. Arthritis Rheum., 54, 1653–1661. [DOI] [PubMed] [Google Scholar]
- Brandt K.D., Radin E.L., Dieppe P.A. & van de Putte L. (2006) Yet more evidence that osteoarthritis is not a cartilage disease. Ann. Rheum. Dis. 65, 1261–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh A., Chanalaris A., Gardiner M.D. et al (2012) Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheum. 64, 2278–2288. [DOI] [PubMed] [Google Scholar]
- Christiansen B.A., Guilak F., Lockwood K.A. et al (2015) Non‐invasive mouse models of post‐traumatic osteoarthritis. Osteoarthritis Cartilage 23, 1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z., Xu C., Li X., Song J. & Yu B. (2015) Treatment with recombinant lubricin attenuates osteoarthritis by positive feedback loop between articular cartilage and subchondral bone in ovariectomized rats. Bone 74, 37–47. [DOI] [PubMed] [Google Scholar]
- Flanigan D.C., Harris J.D., Trinh T.Q., Siston R.A. & Brophy R.H. (2010) Prevalence of chondral defects in athletes’ knees: a systematic review. Med. Sci. Sports Exerc. 42, 1795–1801. [DOI] [PubMed] [Google Scholar]
- Fukui N., Ikeda Y., Ohnuki T. et al (2008) Regional differences in chondrocyte metabolism in osteoarthritis: a detailed analysis by laser capture microdissection. Arthritis Rheum. 58, 154–163. [DOI] [PubMed] [Google Scholar]
- Geyer M., Grassel S., Straub R.H. et al (2009) Differential transcriptome analysis of intraarticular lesional vs intact cartilage reveals new candidate genes in osteoarthritis pathophysiology. Osteoarthritis Cartilage 17, 328–335. [DOI] [PubMed] [Google Scholar]
- Gleghorn L., Ramesar R., Beighton P. & Wallis G. (2005) A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobezie R., Kho A., Krastins B. et al (2007) High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis. Res. Ther. 9, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard B.J., Martin L., Rattner J.B., Frank C.B., Hart D.A. & Krawetz R. (2012) Matrix metalloproteinase protein expression profiles cannot distinguish between normal and early osteoarthritic synovial fluid. BMC Musculoskelet. Disord. 13, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht J.T., Nelson L.D., Crowder E. et al (1995) Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat. Genet. 10, 325–329. [DOI] [PubMed] [Google Scholar]
- Hildebrand B. (2015) Mutation in the type II collagen gene (COL2A1) as a cause of early osteoarthritis and juvenile idiopathic arthritis‐A pseudorheumatoid arthritis variant. Semin. Arthritis Rheum. 44, e22. [DOI] [PubMed] [Google Scholar]
- Jiang Y. & Tuan R.S. (2015) Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 11, 206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsdal M.A., Madsen S.H., Christiansen C., Henriksen K., Fosang A.J. & Sondergaard B.C. (2008) Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis. Res. Ther. 10, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus V.B., Blanco F.J., Englund M., Karsdal M.A. & Lohmander L.S. (2015) Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthritis Cartilage 23, 1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Cai L., Zhang Y., Cui L. & Shen G. (2015) Intra‐articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF‐2alpha. J. Orthop. Res. 33, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Little C.B. & Fosang A.J. (2010) Is cartilage matrix breakdown an appropriate therapeutic target in osteoarthritis–insights from studies of aggrecan and collagen proteolysis? Curr. Drug Targets 11, 561–575. [DOI] [PubMed] [Google Scholar]
- Little C.B. & Zaki S. (2012) What constitutes an “animal model of osteoarthritis”–the need for consensus? Osteoarthritis Cartilage 20, 261–267. [DOI] [PubMed] [Google Scholar]
- Little C.B., Barai A., Burkhardt D. et al (2009) Matrix metalloproteinase 13‐deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 60, 3723–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Goode A.P., Carter T.E. et al (2016) Matrix metalloproteinase activity and prostaglandin E2 are elevated in the synovial fluid of meniscus tear patients. Connect. Tissue Res. 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu‐Bryan R. & Terkeltaub R. (2015) Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 11, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmander L.S., Englund P.M., Dahl L.L. & Roos E.M. (2007) The long‐term consequence of anterior cruciate ligament and meniscus injuries: osteoarthritis. Am. J. Sports Med. 35, 1756–1769. [DOI] [PubMed] [Google Scholar]
- Longobardi L., Temple J.D., Tagliafierro L. et al (2016) Role of the C‐C chemokine receptor‐2 in a murine model of injury‐induced osteoarthritis. Osteoarthritis Cartilage pii: S1063‐4584(16)30399‐5. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo P., Bayliss M.T. & Heinegard D. (2004) Altered patterns and synthesis of extracellular matrix macromolecules in early osteoarthritis. Matrix Biol. 23, 381–391. [DOI] [PubMed] [Google Scholar]
- Lv Y., Xia J.Y., Chen J.Y. et al (2014) Effects of pamidronate disodium on the loss of osteoarthritic subchondral bone and the expression of cartilaginous and subchondral osteoprotegerin and RANKL in rabbits. BMC Musculoskelet. Disord. 15, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason R.M., Chambers M.G., Flannelly J., Gaffen J.D., Dudhia J. & Bayliss M.T. (2001) The STR/ort mouse and its use as a model of osteoarthritis. Osteoarthritis Cartilage 9, 85–91. [DOI] [PubMed] [Google Scholar]
- van Meurs J.B., van Lent P.L., Holthuysen A.E., Singer I.I., Bayne E.K. & van den Berg W.B. (1999) Kinetics of aggrecanase‐ and metalloproteinase‐induced neoepitopes in various stages of cartilage destruction in murine arthritis. Arthritis Rheum. 42, 1128–1139. [DOI] [PubMed] [Google Scholar]
- Miller L.E., Sode M., Fuerst T. & Block J.E. (2015) Joint unloading implant modifies subchondral bone trabecular structure in medial knee osteoarthritis: 2‐year outcomes of a pilot study using fractal signature analysis. Clin. Interv. Aging 10, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muragaki Y., Mariman E.C., van Beersum S.E. et al (1996) A mutation in the gene encoding the alpha 2 chain of the fibril‐associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nat. Genet. 12, 103–105. [DOI] [PubMed] [Google Scholar]
- Paassilta P., Lohiniva J., Annunen S. et al (1999) COL9A3: a third locus for multiple epiphyseal dysplasia. Am. J. Hum. Genet. 64, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet B., Hamilton R.W., Shefelbine S. & Pitsillides A.A. (2011) Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum. 63, 137–147. [DOI] [PubMed] [Google Scholar]
- Poulet B., Ulici V., Stone T.C. et al (2012) Time‐series transcriptional profiling yields new perspectives on susceptibility to osteoarthritis. Arthritis Rheum. 64, 3256–3266. [DOI] [PubMed] [Google Scholar]
- Poulet B., Westerhof T.A., Hamilton R.W., Shefelbine S.J. & Pitsillides A.A. (2013) Spontaneous osteoarthritis in Str/ort mice is unlikely due to greater vulnerability to mechanical trauma. Osteoarthritis Cartilage 21, 756–763. [DOI] [PubMed] [Google Scholar]
- Poulet B., de Souza R., Kent A.V. et al (2015) Intermittent applied mechanical loading induces subchondral bone thickening that may be intensified locally by contiguous articular cartilage lesions. Osteoarthritis Cartilage 23, 940–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racunica T.L., Teichtahl A.J., Wang Y. et al (2007) Effect of physical activity on articular knee joint structures in community‐based adults. Arthritis Rheum. 57, 1261–1268. [DOI] [PubMed] [Google Scholar]
- Reynard L.N. (2017) Analysis of genetics and DNA methylation in osteoarthritis: what have we learnt about the disease? Semin. Cell Dev. Biol. 62, 57–66. [DOI] [PubMed] [Google Scholar]
- Ritter S.Y., Subbaiah R., Bebek G. et al (2013) Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheum. 65, 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe M.A., Harper L.R., McNulty M.A. et al (2017) Deletion of macrophage migration inhibitory factor reduces severity of osteoarthritis in aged mice. Arthritis Rheumatol. 69, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan M.Z., Erez A., Guse K. et al (2013) Proteoglycan 4 expression protects against the development of osteoarthritis. Sci. Transl. Med. 5, 176ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukavina I., Mortier G., van Laer L., Frkovic M., Dapic T. & Jelusic M. (2014) Mutation in the type II collagen gene (COL2AI) as a cause of primary osteoarthritis associated with mild spondyloepiphyseal involvement. Semin. Arthritis Rheum. 44, 101–104. [DOI] [PubMed] [Google Scholar]
- Salminen H., Perala M., Lorenzo P. et al (2000) Up‐regulation of cartilage oligomeric matrix protein at the onset of articular cartilage degeneration in a transgenic mouse model of osteoarthritis. Arthritis Rheum. 43, 1742–1748. [DOI] [PubMed] [Google Scholar]
- Sato T., Konomi K., Yamasaki S. et al (2006) Comparative analysis of gene expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 54, 808–817. [DOI] [PubMed] [Google Scholar]
- Snelling S., Rout R., Davidson R. et al (2014) A gene expression study of normal and damaged cartilage in anteromedial gonarthrosis, a phenotype of osteoarthritis. Osteoarthritis Cartilage 22, 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl R., Luke A., Li X. et al (2009) T1rho, T2 and focal knee cartilage abnormalities in physically active and sedentary healthy subjects versus early OA patients–a 3.0‐Tesla MRI study. Eur. Radiol. 19, 132–143. [DOI] [PubMed] [Google Scholar]
- Struglics A., Larsson S., Kumahashi N., Frobell R. & Lohmander L.S. (2015) Changes in cytokines and aggrecan args neoepitope in synovial fluid and serum and in c‐terminal crosslinking telopeptide of type II collagen and n‐terminal crosslinking telopeptide of type I collagen in urine over five years after anterior cruciate ligament rupture: an exploratory analysis in the knee anterior cruciate ligament, nonsurgical versus surgical treatment trial. Arthritis Rheumatol 67, 1816–1825. [DOI] [PubMed] [Google Scholar]
- Tsezou A. (2014) Osteoarthritis year in review 2014: genetics and genomics. Osteoarthritis Cartilage 22, 2017–2024. [DOI] [PubMed] [Google Scholar]
- Usmani S.E., Ulici V., Pest M.A., Hill T.L., Welch I.D. & Beier F. (2016) Context‐specific protection of TGFα null mice from osteoarthritis. Sci. Rep. 6, 30434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde‐Franco G., Pelletier J.P., Fahmi H. et al (2012) In vivo bone‐specific EphB4 overexpression in mice protects both subchondral bone and cartilage during osteoarthritis. Arthritis Rheum. 64, 3614–3625. [DOI] [PubMed] [Google Scholar]
- Waarsing J.H., Kloppenburg M., Slagboom P.E. et al (2011) Osteoarthritis susceptibility genes influence the association between hip morphology and osteoarthritis. Arthritis Rheum. 63, 1349–1354. [DOI] [PubMed] [Google Scholar]
- Wang M., Sampson E.R., Jin H. et al (2013) MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis. Res. Ther. 15, R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Liang Y., Li H. et al. (2016) Single nucleotide polymorphisms and osteoarthritis: an Overview and a Meta‐Analysis. Medicine (Baltimore) 95, e2811. [DOI] [PMC free article] [PubMed] [Google Scholar]